Abstract
Sepsis is a life-threatening syndrome caused by a dysregulated host response to infection. It follows a dynamic course in which early hyperinflammation coexists and overlaps with progressive immune suppression, a process best described as immunodynamic disruption. Key mechanisms include extensive lymphocyte death, expansion of regulatory T cells, impaired antigen presentation, and persistent activation of inhibitory checkpoints such as programmed cell death protein 1 (PD-1) and cytotoxic T lymphocyte–associated protein 4 (CTLA-4). These changes reduce immune competence and increase vulnerability to secondary infections. Clinically, reduced expression of Human Leukocyte Antigen–DR (HLA-DR) on monocytes and persistent lymphopenia have emerged as robust biomarkers for patient stratification and timing of immunomodulatory therapies. Beyond the acute phase, many survivors do not achieve full immune recovery but instead develop a Persistent Immune Remnant, defined as long-lasting immune, metabolic, and endothelial dysfunction despite apparent clinical resolution. Recognizing PIR emphasizes the need for long-term monitoring and biomarker-guided interventions to restore immune balance. To integrate these observations, we propose the SIMMP–Sepsis model (Sepsis-Associated Persistent Multiorgan Immunometabolic Syndrome), which links molecular dysfunction to clinical trajectories and provides a framework for developing precision immunotherapies. This perspective reframes sepsis not only as an acute crisis but also as a chronic immunometabolic syndrome, where survival marks the beginning of active immune restoration.
1. Introduction
Sepsis is a life-threatening clinical condition that triggers a complex, biphasic immune response. In the early phase, Pathogen-Associated Molecular Patterns (PAMPs) activate pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), initiating a cytokine storm (e.g., TNF-α, IL-1, IL-6). This hyperinflammatory surge drives endothelial dysfunction, vascular leakage, tissue hypoperfusion, and ultimately multiorgan injury [1,2,3,4].
This excessive activation is followed by immune exhaustion and suppression, marked by apoptosis of T cells and antigen-presenting cells, along with reduced expression of HLA-DR on monocytes, which impairs antigen presentation. Concurrently, inhibitory immune checkpoints such as programmed cell death protein 1 (PD-1) and cytotoxic T lymphocyte–associated protein 4 (CTLA-4) are upregulated, while anti-inflammatory cytokines, including IL-10 and TGF-β, dominate the microenvironment, establishing a state of immunoparalysis [5,6,7,8]. These changes increase susceptibility to secondary infections such as ventilator-associated pneumonia, invasive fungal disease, and viral reactivation [1,5,9].
In elderly patients, age-related immunosenescence exacerbates this suppression. Both innate (macrophage and neutrophil activity) and adaptive responses are diminished, prolonging the immunosuppressive phase and contributing to increased late mortality [5,9,10,11,12]. To stratify risk, biomarkers such as reduced monocyte HLA-DR expression and decreased TNF-α production after lipopolysaccharide (LPS) stimulation have been proposed. Immunorestorative interventions—including interferon-γ (IFN-γ), granulocyte-macrophage colony-stimulating factor (GM-CSF), and PD-1 blockade—have shown promise in selected cohorts [1,5,10,12].
A major clinical challenge lies in defining the optimal therapeutic window: early immune stimulation may exacerbate tissue injury, whereas delayed intervention may fail to restore competence. For this reason, sepsis is more accurately described as an evolving immunodynamic process, in which hyperinflammatory and immunosuppressive phases overlap rather than occur sequentially [5,10,12]. Recognizing this dynamic pattern is essential for designing personalized strategies that improve both survival and long-term outcomes.
2. Immunodynamic Disruption in Sepsis
Sepsis triggers a complex and dynamic immune response more accurately described as “immunodynamic disruption” rather than simply “immunoparalysis”. This concept captures the fluctuating interplay between uncontrolled inflammation and progressive suppression, avoiding an overly reductionist view of septic immunity.
In the early phase, pathogen recognition via toll-like receptors (TLRs) on innate immune cells induces a cytokine storm—including TNF-α, IL-1β, and IL-6—that drives vascular injury, coagulation activation, and multiorgan dysfunction [1,2,3,4]. Simultaneously, counter-regulatory mechanisms are initiated. As sepsis progresses, the immune system undergoes dynamic reprogramming rather than uniform suppression. This includes regulatory T cell (Treg) expansion, persistent expression of inhibitory checkpoints such as programmed cell death protein 1 (PD-1) and cytotoxic T lymphocyte–associated protein 4 (CTLA-4), and extensive apoptosis of T and B lymphocytes, ultimately reducing adaptive competence [5,10,11,12,13,14,15]. Antigen-presenting cell (APC) dysfunction—evidenced by reduced HLA-DR expression on monocytes and dendritic cell apoptosis—further impairs antigen presentation and increases susceptibility to secondary infections and viral reactivation [5,9,10,11,12]. In addition, low-density neutrophils (LDNs) exhibit impaired phagocytosis and high PD-L1 expression, contributing to tolerance rather than effective clearance [13,14,16,17].
The concept of immunodynamic disruption thus reflects the biphasic, heterogeneous, and evolving nature of immune responses in sepsis. It provides a more precise framework for understanding pathophysiology and guides the design of tailored therapies. Instead of generalized approaches that broadly stimulate or suppress immunity, recognizing this complexity enables personalized interventions according to each patient’s immunological phase and profile [18,19,20,21,22].
To support immunotherapeutic decisions, several biomarkers have been proposed, including surface markers, soluble mediators, and functional indicators of activation or suppression [18,19,20,21,22].
To support immunotherapeutic decisions, several biomarkers have been proposed to assess immune status in septic patients. These include surface markers, soluble mediators, and functional indicators of immune activation or suppression [18,19,20,21,22] (Table 1).

Table 1.
Immunological biomarkers in sepsis: clinical utility and limitations.
Each alteration has specific implications for monitoring and therapy. Lymphocyte apoptosis underscores the need for regular lymphocyte counts and supports trials with interleukin-7 (IL-7) aimed at restoring adaptive immunity [34,35]. Treg expansion and checkpoint overexpression (PD-1, CTLA-4) justify checkpoint expression as prognostic biomarkers and provide a rationale for selective inhibition [36]. Reduced monocyte HLA-DR expression, validated as a marker of immunoparalysis, guides stratification of patients who may benefit from interferon-γ (IFN-γ) or granulocyte-macrophage colony-stimulating factor (GM-CSF). Neutrophil dysfunction and LDN accumulation support monitoring of PD-L1 and chemotaxis to inform strategies targeting neutrophil survival and function [37]. Finally, evidence of mitochondrial dysfunction and epigenetic reprogramming highlights the value of metabolic biomarkers (e.g., circulating mitochondrial DNA [mtDNA], lactate levels) and supports the potential of metabolic modulators to restore homeostasis [37,38]. Linking mechanisms with measurable biomarkers strengthens translational value and facilitates precision immunotherapy [39].
Checkpoint Signaling and Treg Expansion
The immunodynamic collapse in sepsis involves profound remodeling of the adaptive system, driven by Treg expansion, checkpoint upregulation, and lymphocyte apoptosis. During the early-to-intermediate phase, Tregs expand disproportionately through persistent IL-2/STAT5 signaling and epigenetic reinforcement of FoxP3 expression [40,41,42]. This skewed repertoire suppresses effector responses and promotes tolerance, a phenomenon also observed in chronic viral infections and tumor microenvironments [43,44,45,46].
Concurrently, T cells upregulate checkpoints such as PD-1 and CTLA-4, which impair T cell receptor (TCR) signaling, reduce cytokine secretion, and hinder pathogen clearance. Experimental models show that sustained checkpoint engagement promotes exhaustion, reduces clonal diversity, and abrogates memory formation [6,47]. These alterations represent critical nodal points in sepsis-induced immunosuppression [23,32].
In parallel, widespread lymphocyte apoptosis—particularly of CD4+ and CD8+ subsets—depletes the adaptive reservoir, typically mediated via caspase-3 activation and mitochondrial cytochrome c release [26,48]. This triad—Treg-driven suppression, checkpoint-mediated exhaustion, and apoptotic attrition—produces a state of immune collapse marked by impaired antigen-specific responses, nosocomial infections, and viral reactivation. Clinically, this manifests as secondary pneumonia, cytomegalovirus (CMV) or herpes simplex virus (HSV) reactivation, and poor vaccine responsiveness [27,49].
These alterations are not transient. Immunophenotyping studies show persistent T cell anergy and epigenetic scarring weeks after sepsis [14,27,50]. Collectively, these mechanisms provide the rationale for targeted interventions such as IL-7, checkpoint blockade, and caspase inhibitors, which are under evaluation in early-phase trials. This constellation underscores the need for dynamic monitoring and personalized immunointerventions in sepsis survivors [27,50] (Table 1).
3. Mechanisms of Immune Dysfunction
The immunological dysfunction observed in sepsis involves both adaptive and innate compartments, affecting multiple cell types through distinct yet overlapping mechanisms. Table 2 summarizes the principal immune cells involved, the key dysregulated pathways, and the resulting clinical consequences.
Table 2.
Key immune cells and their principal dysfunctions in sepsis-induced immunosuppression, separated into adaptive and innate mechanisms and linked to their clinical relevance.
3.1. Adaptive Immune Exhaustion
The immunological dysfunction observed in sepsis involves both adaptive and innate compartments, affecting multiple cell types through distinct yet overlapping mechanisms [25,54]. These checkpoints impair T cell receptor (TCR) signaling, suppress cytokine production (e.g., IL-2, IFN-γ), and limit cytotoxic activity, resulting in a dysfunctional adaptive landscape. Elevated frequencies of PD-1+CD4+, PD-1+CD8+, and CTLA-4+CD8+ cells correlate with higher APACHE II and SOFA scores [25,54].
Mechanistically, PD-1 signaling promotes apoptosis and susceptibility to secondary infections [51,55,56], while Tim-3, through galectin-9 binding, induces tolerance in effector T cells. CTLA-4 dampens activation by competitively blocking CD28 interaction with CD80/CD86 on antigen-presenting cells [25,54]. These suppressive signals converge on mitochondrial apoptotic pathways, including cytochrome c release and caspase-9 activation, leading to profound depletion of the adaptive immune pool [25,54].
Loss of CD4+ T cells is consistently associated with poor outcomes, whereas their preservation predicts improved recovery. Notably, blockade of PD-1 or CTLA-4 has demonstrated efficacy in restoring T cell function and improving survival in preclinical sepsis models [25,54].
3.2. Innate Effector Failure
Monocytes and macrophages in sepsis exhibit marked downregulation of HLA-DR, impairing antigen presentation and sustaining immune anergy [35,45,46]. Dendritic cells undergo apoptosis and mitochondrial dysfunction [13,14,15,16], with reduced expression of co-stimulatory molecules (CD40, CD80, CD86), diminished IL-12p70 production, and increased IL-10 secretion, which promotes Treg expansion [15,57,58].
Neutrophils initially resist apoptosis and become hyperactivated, releasing proteolytic enzymes and Reactive Oxygen Species (ROS) that exacerbate tissue damage and multiorgan failure [27,59,60]. As sepsis progresses, they develop impaired chemotaxis and phagocytosis due to dysregulated integrin and adhesion molecule expression [31,52,61].
Low-density neutrophils (LDNs), characterized by a CD66b+CD63+CD11b+CD184+ phenotype, accumulate within the PBMC fraction. These cells express high PD-L1, resist apoptosis, exhibit poor antimicrobial activity, and suppress T cell proliferation, thereby contributing to increased secondary infections and mortality [15,57,58].
A central driver of innate dysfunction is aberrant activation of the cyclic GMP-AMP synthase (cGAS)–Stimulator of Interferon Genes (STING) pathway, triggered by cytoplasmic mitochondrial DNA (mtDNA) accumulation. This induces type I interferons and IL-10 through JAK–STAT3 signaling [15,57,58]. Experimental inhibition of this axis has been shown to restore dendritic cell function and promote effective T cell responses [30,62,63] (Table 3).
Table 3.
Innate and adaptive immune dysfunctions in sepsis.
Moreover, early inflammasome activation—marked by ASC-speck+ neutrophils and monocytes—is followed by a progressive decline in these cells during late-stage sepsis [30,62,63]. A sustained reduction below 1650 ASC-speck+ cells/mL by day six correlates with impaired phagocytosis, defective antigen presentation, and increased 90-day mortality [65,66]. This trajectory, characterized by persistent IL-18 elevation and declining IL-1β levels [65,67,68], reflects a reprogrammed inflammasome phenotype favoring immunosuppression, consolidating immunoparalysis as a hallmark of late-phase sepsis (Figure 1).
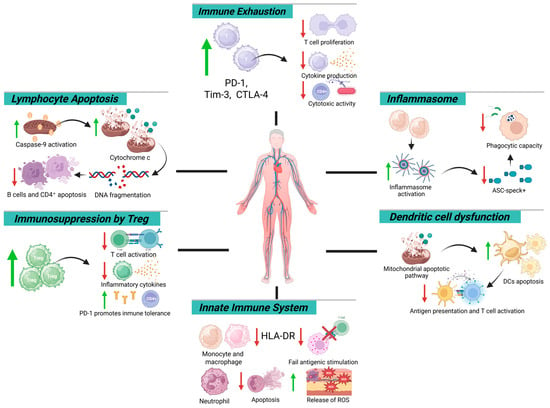
Figure 1.
Mechanistic map of immune dysfunction in sepsis: cellular and molecular targets. Green arrows indicate an increase, whereas red arrows indicate a decrease.
4. Mechanistic Ambiguities and Translational Targets
Although numerous immunological pathways involved in sepsis have been characterized in both experimental and clinical settings, several remain the subject of conflicting evidence regarding their translational relevance. For example, the PD-1/PD-L1 axis is a well-recognized marker of T cell exhaustion, and its blockade has demonstrated immunorestorative effects in murine models and ex vivo analyses. However, clinical trials in septic patients remain limited and show variable efficacy depending on disease stage and immune profile [33,60,69].
Likewise, NETosis exhibits a dual role: while essential for pathogen containment, excessive Neutrophil Extracellular Trap formation contributes to endothelial injury and thrombosis, complicating its therapeutic targeting [70]. The cGAS–STING pathway presents similar ambiguity—its activation promotes early pathogen sensing, yet persistent signaling, particularly in dendritic cells, fosters immunoparalysis. Thus, the timing and cellular context are critical determinants of its potential as a therapeutic target [29].
In contrast, pathways with more consistent evidence include IL-7–mediated lymphocyte restoration [33,60,69] and β1-adrenergic blockade for immunomodulatory cardiovascular regulation [71]. Both have shown promise in preclinical and early-phase studies and are currently under active investigation as viable therapeutic strategies [33,60,69].
5. Persistent Immune Remnant
In sepsis survivors, clinical recovery does not equate to full immunological restoration. Instead, a persistent state of cellular dysfunction emerges, referred to here as the Post-Sepsis Cellular Remnant (PSCR), a pathophysiological entity marked by epigenetic reprogramming, immunometabolic derangements, and maladaptive inflammatory memory. Multiple studies have documented sustained alterations in monocytes (↓HLA-DR), T lymphocytes (↑PD-1, B and T Lymphocyte Attenuator [BTLA]), and expansion of immunosuppressive populations such as myeloid-derived suppressor cells and regulatory T cells (Tregs) [72,73,74,75,76]. These cells demonstrate impaired cytokine production (IL-6, TNF-α, IFN-γ) in response to classical stimuli [18,77,78], alongside mitochondrial dysfunction and disrupted metabolic pathways [79].
5.1. Epigenetic Reprogramming
At the epigenetic level, persistent enrichment of histone marks, including H3K9ac, H3K27me3, and H3K4me3, has been observed at regulatory regions of genes such as IL-10, FPR1, HLA-DR, and NLRP3. These modifications persist for weeks after the septic episode and are more pronounced in non-survivors [80,81,82], suggesting a form of non-resolving immune memory directly linked to outcomes [18,77,78].
Recent evidence has also identified histone H3 lactylation at lysine 18 (H3K18la) as a durable epigenetic marker of trained immunity, induced by intracellular lactate accumulation during sepsis [83,84]. This modification promotes open chromatin architecture, enhances proinflammatory gene expression, and persists for up to 90 days post-activation [83,85].
5.2. Immunometabolic Alterations
The integration of metabolism and epigenetics is underscored by metabolites such as succinate, fumarate [64,86,87], and acetyl-CoA, which regulate the activity of histone- and DNA-modifying enzymes, establishing stable transcriptional programs in post-septic immune cells [88,89,90].
Accordingly, PSCR manifests as a state of metabolic dysfunction: monocytes exhibit persistent glycolytic activity [91,92], while lymphocytes display fragmented mitochondria and defective biosynthetic capacity [79]. These changes result in dysregulated cytokine release and impaired immune responsiveness [24,93,94].
Functional studies of monocyte-to-macrophage differentiation reveal sustained activation of proinflammatory enhancers enriched in H3K27ac and H3K4me3, marks associated with active transcription and trained immunity [95,96,97]. Unlike tolerance, this programming amplifies responses upon secondary stimulation, reinforcing maladaptive immune memory as a defining feature of PSCR [95,98].
Mitochondrial dysfunction represents a structural cornerstone of PSCR. Cells such as hepatocytes, lymphocytes, and monocytes previously exposed to sepsis exhibit reduced oxygen consumption, membrane depolarization, excessive ROS production, and depleted antioxidant reserves [28,38,99]. These abnormalities can be partially reversed by activating the AMPK–PGC-1α–autophagy axis, as shown in preclinical models with ginsenoside Rg3 [100,101,102,103]. Low expression of PGC-1α, a master regulator of mitochondrial biogenesis, perpetuates oxidative stress and sustains a proinflammatory basal state [100,101,103].
5.3. Endothelial Dysfunction and Immunothrombosis
At the endothelial level, sepsis leaves a pathological imprint characterized by persistent activation, with elevated angiopoietin-2, soluble vascular cell adhesion molecule-1 (sVCAM-1), endothelial microparticles, and impaired vascular integrity [104,105,106]. This dysfunction promotes a microenvironment conducive to immunothrombosis, involving neutrophil extracellular trap (NET) formation [105,107,108], tissue factor activation, and persistence of a procoagulant state even after clinical resolution [52,109].
Persistent endothelial activation is thus an integral component of PSCR, contributing to progressive vascular deterioration and chronic tissue hypoperfusion [105,107,108]. Collectively, these structural, functional, and metabolic alterations define PSCR as a clinically relevant post-inflammatory immunometabolic state [72,73,74,75,76]. This state is measurable through specific cellular and molecular biomarkers and carries significant implications for post-ICU monitoring, late infection risk, and long-term mortality [72,110]. Recognizing PSCR demands a paradigm shift in sepsis follow-up—from a survival-centric model to one focused on long-term immunometabolic recovery and reprogramming of innate immunity as therapeutic goals [111,112] (Table 4).
Table 4.
Immunocellular alterations in the Post-Sepsis Cellular Remnant (PSCR). This table outlines the principal immune and endothelial cell types affected in the PSCR, highlighting their functional impairments, clinical implications, and potential biomarkers for monitoring long-term immune dysfunction after sepsis.
5.4. Clinically Relevant Biomarkers of PSCR
Several biomarkers have emerged as predictors of immune dysfunction and adverse outcomes in sepsis survivors. Reduced HLA-DR expression on monocytes remains a key indicator of immunosuppression and correlates with increased risk of secondary infections and mortality [89,90,91].
Persistently elevated IL-6 during recovery is associated with ongoing lymphocyte dysfunction and poor long-term prognosis. Additional markers such as IL-10 and soluble TNF receptor 1 (sTNFR1) hold independent prognostic value, particularly in patients with protracted courses [92,93,94].
At the transcriptomic level, sustained upregulation of inhibitory receptors, including PD-1 and BTLA, on CD4+ and CD8+ T cells reflects an exhausted phenotype, predisposing to opportunistic infections and viral reactivations [95,96].
Collectively, these biomarkers provide mechanistic insights into persistent post-sepsis immunopathology and represent clinically actionable tools for patient stratification and the development of personalized immunomodulatory strategies during recovery [18,86,87].
6. Experimental Models of Immunodysfunction
6.1. Classical Animal and In Vitro Models
Experimental models have been central to advancing the understanding of immunoparalysis in sepsis. One of the most commonly used approaches is the one-hit model, which induces sepsis through a single insult—such as administration of lipopolysaccharide (LPS), injection of live bacteria, or surgical infection (most commonly via cecal ligation and puncture [CLP]) [65,66]. This model enables the study of early immune alterations, including lymphocyte apoptosis and the expansion of immunosuppressive populations like Tregs and MDSCs. However, it fails to replicate the temporal progression from hyperinflammation to sustained immunosuppression typically seen in human sepsis [65,66,113].
To address this limitation, the two-hit model was developed. Here, an initial septic event is followed by a secondary infection, commonly with pathogens such as Pseudomonas aeruginosa or Candida albicans [65]. This approach more accurately reflects the clinical course of sepsis in humans, reproducing features such as impaired resolution of inflammation, reduced lymphocyte proliferation, and elevated expression of inhibitory receptors. Despite its improved translational relevance, standardization remains challenging due to variability in the type and timing of the secondary insult [65,67,68].
Another widely studied method is the LPS tolerance model, which uses repeated sublethal doses of LPS to induce a hyporesponsive state in leukocytes [67,68]. This model simulates key features of immune reprogramming, including diminished cytokine production and functional suppression of innate responses. However, because it relies solely on endotoxin exposure and not on polymicrobial infection, it does not fully reflect the complexity of human sepsis [65,68].
To increase clinical relevance, newer models have incorporated aged animals, mice with comorbidities (e.g., obesity, cancer), or “dirty mice” exposed to natural microbiota. These modifications better capture the immunological heterogeneity observed in patients and demonstrate how baseline characteristics shape the trajectory of sepsis-induced immunosuppression. At the same time, they introduce additional variability that complicates reproducibility and cross-study comparison [65,114,115].
Despite these limitations, murine models remain essential tools in the study of immunoparalysis. They have facilitated the identification of key biomarkers and informed the development of targeted therapies. Nonetheless, the gap between preclinical models and human immune complexity underscores the urgent need for improved reproducibility, standardization, and translational alignment [113,114] (Table 5).
Table 5.
Summary of the main experimental models of immunoparalysis in sepsis. This table describes an overview of the principal models used to investigate immune collapse in sepsis. Each model replicates specific aspects of immunoparalysis, with implications for translational research and immunotherapeutic development.
6.2. Advanced and Translational Platforms
The elucidation of mechanisms underlying the Post-Sepsis Cellular Remnant (PSCR) has been supported by a diverse array of experimental models, each offering distinct methodological advantages and varying degrees of translational relevance.
The murine CLP model has been widely used to characterize sepsis-induced lymphocyte apoptosis and mitochondrial dysfunction. However, its translational value remains limited, as it does not replicate the complex epigenetic reprogramming observed in human sepsis survivors [116,117,118].
In contrast, ex vivo models using human monocytes have proven highly informative for uncovering persistent metabolic rewiring and stable epigenetic marks characteristic of PSCR. These models offer strong clinical relevance but are limited by the inability to support longitudinal, patient-specific follow-up.
Transcriptomic analyses—particularly RNA-seq and single-cell RNA-seq in critically ill patients—currently represent the translational gold standard. These technologies provide high-resolution characterization of immune phenotypes during and after sepsis [93,119,120]. Nevertheless, their application is restricted by high cost and the critical need for precisely timed sampling.
Meanwhile, 3D endothelial organoid models have shown value in studying chronic inflammation and vascular barrier dysfunction. However, their translational applicability remains moderate due to the absence of integrated immune cell interactions. Taken together, these platforms should be regarded as complementary rather than exclusive, each contributing to a more comprehensive understanding of PSCR pathophysiology [121,122,123].
6.3. Strategies to Enhance Translational Relevance of Experimental Models
Although the limitations of experimental models have been extensively discussed, bridging the gap with human sepsis requires clear strategies to enhance translational value. Current recommendations emphasize the adoption of standardized protocols, such as the MQTiPSS guidelines, to reduce variability across laboratories. In addition, the incorporation of clinically relevant variables—including age, sex, and comorbidities—into animal models is essential to reflect patient heterogeneity. The use of more complex systems, such as humanized or “dirty” mice exposed to natural microbiota, further improves biological relevance by better approximating human immune diversity [124]. Equally important is the alignment of experimental endpoints with clinically meaningful outcomes, including immune cell exhaustion, biomarker dynamics, and long-term mortality. Finally, harmonization of reporting standards and the establishment of shared data repositories are necessary to improve reproducibility and facilitate direct benchmarking across studies [125,126]. Together, these measures strengthen the translational relevance of preclinical research and accelerate the development of clinically applicable immunotherapies [124].
Bridging the gap between preclinical and clinical research requires aligning model-derived findings with patient data. For instance, lymphocyte apoptosis and HLA-DR downregulation in cecal ligation and puncture models mirror changes consistently seen in septic patients. Likewise, LPS tolerance models reproduce impaired TNF-α production and monocyte dysfunction validated in human cohorts. Such parallels reinforce the translational value of experimental models and support biomarker-based approaches that can be prospectively tested in clinical settings.
7. Adaptive Immune Collapse
7.1. Septic Lymphocytic Apoptosis
Lymphocyte apoptosis in sepsis represents one of the most profound and well-characterized disruptions of the adaptive immune system. Clinical and post-mortem studies consistently demonstrate a massive depletion of B lymphocytes and CD4+ T cells during the acute phase, primarily via mitochondrial apoptotic pathways involving caspase-9 [1,2]. Paradoxically, this occurs during active infection, when clonal expansion would normally be expected to reinforce host defense.
Human spleen analyses reveal that not all subsets are equally affected. CD8+ T cells and natural killer (NK) cells are relatively preserved, whereas B cells and CD4+ T cells—particularly follicular helper T cells (Tfh)—are severely reduced. Loss of Tfh cells compromises humoral immunity by disrupting germinal center formation, memory B cell development, and long-term antibody production [1,2].
Surviving lymphocytes often exhibit functional exhaustion, characterized by diminished cytokine secretion, increased expression of inhibitory receptors such as programmed PD-1, and impaired proliferative capacity. These alterations contribute to a state of adaptive immunosuppression that may persist for weeks after clinical resolution. From an immunodynamic perspective, apoptosis is not a passive consequence but an active driver of adaptive immune collapse. Early identification through immunophenotyping or tissue sampling may support risk stratification and guide targeted interventions such as IL-7 therapy, PD-1 blockade, or restorative strategies for B and CD4+ T cells [1,2].
7.2. Septic Treg Suppression
Regulatory Treg expansion in sepsis is marked by a highly suppressive phenotype defined by CD25^high and FoxP3+ expression, together with upregulation of inhibitory molecules including cytotoxic T lymphocyte–associated protein 4 (CTLA-4) and PD-1 [3,4,5]. These cells secrete Interleukin-10 (IL-10) and transforming growth factor beta (TGF-β), sustaining a profoundly immunosuppressive milieu.
In this context, dendritic cell maturation is impaired, CD4+ and CD8+ T cell proliferation is suppressed, and the phagocytic activity of macrophages and NK cells is diminished. Moreover, imbalance of the Th17/Treg axis—evidenced by reduced IL-17A and IL-22 production—further weakens mucosal and systemic defenses against bacterial and fungal infections [3,4]. Clinically, an elevated Treg/Th17 ratio correlates with higher Sequential Organ Failure Assessment (SOFA) scores, persistent infection beyond day 7, and increased 28-day mortality [3,4,5].
Tregs therefore act as both markers and drivers of immune dysregulation. Serial monitoring (CD4+CD25^highFoxP3+/CD127^low) and selective modulation via PD-1 inhibitors or IL-10 receptor blockade are emerging as promising strategies to recalibrate immune balance without triggering autoimmunity [3,4,5].
7.3. Adaptive Immune Resuscitation
Recent clinical trials with recombinant human IL-7 (CYT107) and the PD-1 inhibitor nivolumab have advanced personalized immunotherapy in sepsis. The IRIS-7 study demonstrated that IL-7 is safe and effective in patients with septic shock and severe lymphopenia, inducing a three- to four-fold increase in CD4+ and CD8+ T cell counts, enhancing proliferation, and restoring functional activity without precipitating cytokine storms or organ failure [88,89,90].
Similarly, nivolumab was well tolerated in patients with overt immunosuppression (lymphocyte count < 1.1 × 103/μL). Although the phase 1b trial did not assess efficacy, findings showed increased monocyte HLA-DR expression, sustained PD-1 receptor occupancy (>90% for at least 28 days), and no increase in pro-inflammatory cytokines. These results support checkpoint blockade as a viable strategy to restore T cell function and counteract immune paralysis [88,89,90].
Taken together, IL-7 and PD-1 blockade represent complementary approaches that may reverse immunodynamic disruption, reduce secondary infections, and prevent progression toward persistent immune dysfunction [88,89,90].
7.4. Sepsis Immune Phases
Sepsis is a dynamic condition in which pro-inflammatory and immunosuppressive pathways coexist, with their relative dominance determining clinical trajectory. In the early phase, hyperinflammation prevails, driven by massive cytokine release, endothelial activation, and multiorgan dysfunction [60]. Clinically, this phase manifests with fever, hemodynamic instability, and elevated acute-phase reactants.
In survivors of the initial insult, the immune response often shifts toward sustained immunosuppression. This phase is characterized by persistent lymphopenia, impaired TNF production after ex vivo stimulation, reduced monocyte HLA-DR expression, and increased susceptibility to secondary infections or viral reactivation [60,127]. Mechanistically, defects in both innate and adaptive immunity underlie this state, including apoptosis of effector cells, Treg expansion, T cell exhaustion, accumulation of myeloid-derived suppressor cells, and loss of co-stimulatory signaling [60,127].
Distinguishing between these phases requires integrating clinical features with laboratory parameters. Recognition of this temporal and mechanistic complexity is essential for tailoring interventions that avoid exacerbating inflammation in the early stage while restoring immune competence in the later phase [60,127].
8. SIMMP–Sepsis: Toward an Integrative Framework
Current understanding of sepsis increasingly recognizes that it extends beyond the classical definition of acute organ dysfunction triggered by infection [128,129]. Growing evidence shows that, in many patients, sepsis initiates a persistent immunometabolic syndrome that lasts well beyond apparent clinical recovery [96,98,130,131,132,133]. Nevertheless, most of the literature still fragments this process into acute and post-acute phases, lacking a unifying framework to explain their continuity.
To address this gap, we propose the SIMMP–Sepsis model (Sepsis-Associated Persistent Multiorgan Immunometabolic Syndrome), a conceptual framework that delineates five interconnected levels linking the initial immune response to the chronic dysfunction observed in survivors [96,98,131].
The first level represents the acute activation phase, initiated by recognition of PAMPs and DAMPs and characterized by hyperinflammation, NETosis, complement activation, mitochondrial injury, oxidative stress, and endothelial dysfunction. These processes converge to precipitate multiorgan failure that typically requires intensive support. However, resolution of this phase does not equate to true immunological recovery [132,134,135].
The second level corresponds to persistent cellular reprogramming during the post-critical period. Immunologically, it encompasses trained immunity and maladaptive tolerance, orchestrated by epigenetic modifications such as histone lactylation and H3K4me3 enrichment [123,135]. Metabolically, dysregulated pathways resembling the Warburg effect—marked by lactate and succinate accumulation and impaired tricarboxylic acid (TCA) cycle activity—sustain a pro-inflammatory environment and hinder the restoration of homeostasis [136,137,138].
The third level reflects sustained organ dysfunction. The endothelium remains activated, with increased expression of ICAM-1 and VCAM-1 and compromised vascular integrity. Simultaneously, mitochondrial fragmentation, incomplete mitophagy, and mitochondrial DNA (mtDNA) release exacerbate dysfunction [98,139,140,141]. In the central nervous system, persistent microglial activation maintains neuroinflammation [134]. At the systemic level, the gut–liver–brain axis is disrupted by dysbiosis, microbial translocation, and reduced short-chain fatty acid (SCFA) production, contributing to ongoing hepatic and systemic inflammation [98,139].
The fourth level represents the clinical manifestations of this biology, which include fatigue, myalgias, dysautonomia, cognitive decline, recurrent infections, and fibrotic remodeling across multiple organs [128,142]. These post-sepsis symptoms are often underdiagnosed and misinterpreted as isolated sequelae, rather than as evidence of persistent immunometabolic dysregulation. This highlights the need for long-term monitoring strategies capable of identifying and addressing sustained immune dysfunction [127,133,142].
Finally, the fifth level consolidates SIMMP–Sepsis as a distinct clinical entity, defined by the integration of immune, metabolic, vascular, neurological, and microbial alterations into a persistent syndrome. This framework provides a foundation for risk stratification, biomarker development, and the design of tailored immunometabolic interventions that extend beyond Intensive Care Unit (ICU) discharge [96,98,130,131,132,133].
Rather than replacing classical definitions of sepsis [143], SIMMP–Sepsis expands the conceptual framework by linking molecular mechanisms with clinical outcomes and therapeutic opportunities. This perspective promotes a shift toward precision immunology aimed not only at ensuring survival but also at restoring long-term immune and metabolic integrity [96,98,130,131,132,133] (Table 6).
Table 6.
Hierarchical cascade of the SIMMP–Sepsis model. This table shows progressive pathophysiological stages from acute immune activation to sustained immunometabolic dysfunction and clinical deterioration in sepsis survivors.
9. Clinical Implications of Sepsis-Induced Immunosuppression
9.1. Sepsis and Cancer Immunosurveillance
Sepsis induces a prolonged state of immunoparalysis that compromises both antimicrobial defense and tumor immunosurveillance. A key consequence is dysfunction of CD8+ T cells, which are central to identifying and eliminating malignant cells through cytokine secretion and cytotoxic activity [130,144]. Normally, these lymphocytes infiltrate tumor tissue, secrete IFN-γ, and engage in direct cytolytic killing. However, following sepsis, their number and functionality are markedly reduced. Expression of receptors such as PD-1 and LAG-3, necessary for effector function and retention within the tumor microenvironment, is also diminished [144,145,146].
In addition, sepsis impairs CD8+ T cell renewal and persistence in tissues. This coincides with reduced MHC-I expression on tumor cells, limiting antigen recognition and facilitating immune escape. Consequently, tumor progression accelerates in sepsis survivors, who face higher risks of de novo malignancies and worse trajectories of preexisting cancers [144,145,146]. Checkpoint inhibitor therapy may restore CD8+ T cell function in this context, but its efficacy depends on timely administration. Early intervention—before extensive lymphocyte depletion—can reestablish cytotoxic function and limit tumor growth. In contrast, delayed treatment is less effective due to irreversible immune depletion, underscoring the need for early immune profiling and targeted restoration strategies [50,144,146].
9.2. Pulmonary Immune Reprogramming
Long-term sepsis survivors frequently exhibit altered pulmonary immunity, which predisposes them to exaggerated inflammatory responses and increased susceptibility to lung injury. Upon subsequent exposure to LPS, these individuals display amplified cytokine release, enhanced neutrophilic infiltration, and aggravated alveolar damage mediated by overactive Ly6C^hi monocytes producing elevated TNF-α [147,148]. Notably, this primed state persists for weeks after clinical recovery and occurs even without active infection, reflecting a failure to restore pulmonary immune homeostasis.
This immune priming involves persistent epigenetic and metabolic reprogramming in myeloid cells. Murine models show that three weeks post-cecal ligation and puncture (CLP), lungs retain expanded Ly6C^hi monocyte populations and sustain inflammatory gene expression even in the absence of additional stimuli. Secondary LPS exposure triggers severe lung injury, including increased permeability, epithelial apoptosis, and excessive NLRP3 inflammasome activation with elevated TNF-α, IL-1β, and IL-6 [147,148,149].
At the molecular level, prolonged leukocyte expansion and persistently inflammatory transcriptomic profiles reinforce a “primed” immune state that coexists with systemic immunoparalysis, predisposing survivors to sterile inflammatory damage [147,150,151]. DAMPs such as S100A8/A9 (Calprotectin) remain elevated, promoting chronic inflammation and increasing vulnerability to secondary insults. In clinical studies, elevated plasma S100A8/A9 correlates with pulmonary complications, highlighting its translational relevance [147,148,152].
Circulating biomarkers such as S100A8/A9 therefore represent both early indicators and potential therapeutic targets for personalized interventions. Post-sepsis surveillance should incorporate dynamic immune profiling with emphasis on pulmonary-specific pathways. This challenges the conventional notion of uniform post-sepsis immunosuppression and supports a more nuanced, stratified, and proactive model of long-term care [150,151].
9.3. Calprotectin Endothelial Disruption
Calprotectin (S100A8/A9) is a heterodimeric protein complex released in large amounts by neutrophils and monocytes during intense inflammatory responses such as sepsis. Beyond serving as a biomarker, Calprotectin exerts direct pathogenic effects on the endothelium by inducing mitochondrial disruption. In endothelial cells, it interferes with components of mitochondrial respiratory chain complex I, reducing oxidative activity and the NAD+/NADH ratio. This suppresses the activity of Sirt1, a deacetylase that regulates mitochondrial biogenesis, fusion, fission, and degradation [153].
As a result, mitochondria undergo excessive fragmentation, mitophagy is blocked, and damaged organelles accumulate in the cytoplasm. This mitochondrial overload culminates in the release of mtDNA, which activates cytosolic receptors such as ZBP1, initiating PANoptosis—a form of programmed cell death integrating apoptosis, pyroptosis, and necroptosis. These processes destroy functional endothelium, exacerbate microvascular injury, and compromise tissue perfusion [153,154].
9.4. IL-36, Sepsis, and Lung Injury
Sepsis, a syndrome defined by a dysregulated host response to infection, disrupts immune homeostasis and contributes to multi-organ failure, notably in the lungs [155,156]. In this context, IL-36 cytokines—IL-36α, IL-36β, and IL-36γ—have been identified as key modulators of inflammation and epithelial integrity [155]. Their serum concentrations increase during the early septic phase, suggesting a role in the initial immune response; however, their precise contribution to the pathophysiology of sepsis and lung injury remains incompletely elucidated [74,75,76].
Clinically, elevated IL-36 levels correlate with sepsis severity and may serve as prognostic biomarkers. Lower IL-36 concentrations in non-survivors imply a protective immunomodulatory role, potentially through regulation of inflammatory cascades and preservation of pulmonary architecture [155,156].
The IL-36 receptor (IL-36R), expressed predominantly on epithelial cells and pulmonary fibroblasts, is essential for orchestrating local immune responses during systemic inflammation [155,156]. In CLP-induced sepsis models, IL-36R deficiency leads to impaired pathogen clearance, increased bacterial load, and aggravated organ damage. This is associated with diminished production of lipocalin-2 (LCN2), an antimicrobial protein that restricts bacterial proliferation in the lungs, thereby facilitating microbial dissemination [155,156,157].
Moreover, IL-36R deficiency exacerbates alveolar epithelial apoptosis, compromising barrier integrity and promoting neutrophil infiltration. This cascade culminates in sepsis-associated lung injury characterized by increased vascular permeability, alveolar edema, and impaired gas exchange [155,157].
9.5. Trained Immunity and IL-4
Trained immunity—the epigenetic and metabolic reprogramming of innate immune cells such as monocytes—offers a promising strategy to reverse sepsis-induced immunoparalysis [156,157]. IL-4 plays a dual immunomodulatory role [158,159]. Initially, it dampens inflammation by suppressing the production of proinflammatory cytokines such as TNF-α and IL-6 in activated monocytes [160,161], primarily via STAT6-dependent pathways that promote an anti-inflammatory macrophage phenotype [156,157].
Paradoxically, IL-4 also induces a long-lasting form of trained immunity through PI3K–mTOR signaling, enhancing monocyte responsiveness to secondary challenges. Upon restimulation, these reprogrammed cells produce elevated levels of IL-6 and TNF-α, thereby restoring innate immune competence and counteracting endotoxin-induced hyporesponsiveness [156,157].
To minimize systemic effects and improve delivery, an innovative nanotherapy has been developed [158,159]. This platform fuses IL-4 to apolipoprotein A1 (apoA1) and incorporates it into lipid nanoparticles, enabling targeted delivery to hematopoietic tissues such as the spleen and bone marrow. In preclinical models, this approach restored proinflammatory cytokine production and enhanced adaptive responses, effectively reversing immunoparalysis [156,157].
9.6. β1-Adrenergic Modulation
Sympathetic overactivation during sepsis promotes β1-adrenergic signaling, which amplifies Treg-mediated immunosuppression and attenuates CD4+ T cell proliferation. This immunologic shift is associated with reduced host capacity to control secondary infections and an increased risk of nosocomial complications [51,55]. β1-adrenergic stimulation exacerbates Treg-driven inhibition, suppressing proinflammatory responses and limiting effector T cell activation. Consequently, adrenergic signaling influences both cardiovascular dynamics and immune homeostasis in sepsis [51,55,56].
Pharmacological blockade of β1 receptors with esmolol [162], or genetic ablation in murine sepsis models, has been shown to reverse these immunosuppressive effects. Such interventions restore CD4+ T cell proliferation, reduce Treg frequency, and rebalance the immune response, as evidenced by decreased levels of both pro- and anti-inflammatory mediators [51,55,56]. Notably, β1 inhibition preserves cardiac efficiency despite heart rate reduction, thereby improving tissue perfusion without compromising hemodynamic stability [51,55,56].
9.7. Tachycardia and Immune Regulation
Tachycardia in sepsis reflects persistent sympathetic drive in response to systemic inflammation and hypoperfusion. While initially compensatory, sustained catecholaminergic stimulation impairs myocardial relaxation, reduces diastolic filling and coronary perfusion, and exacerbates septic cardiomyopathy through oxidative stress and contractile dysfunction [51,55,56].
This cardiovascular compromise further impairs immune responses by disrupting metabolic balance and reducing perfusion to immunologically active organs such as the spleen and bone marrow [51,163]. Simultaneously, adrenergic signaling promotes immune dysfunction by inducing inhibitory receptors—including PD-1, CTLA-4, and Tim-3—on T cells and monocytes [51,55,56]. This contributes to lymphocytopenia, reduced cytokine production, antigen-presenting cell dysfunction, and pathogen persistence—hallmarks of late-phase sepsis immunoparalysis [14,51,71,164,165].
Therapeutic modulation of heart rate offers dual benefits. β-blockers such as esmolol and landiolol stabilize hemodynamics and partially restore immune function by enhancing lymphocyte activity [162]. Ivabradine, a selective sinoatrial node inhibitor, lowers heart rate without affecting contractility, improving perfusion of immune-relevant tissues [14,51,71].
Together, these interventions emphasize the pathophysiological link between cardiovascular and immune dysfunction in sepsis. Heart rate modulation serves not only to optimize cardiac performance but also to promote immune recovery and reduce sepsis-related morbidity and mortality [14,51,71,164,165].
10. Ebola-Driven Immunoparalysis
The study of immune dysregulation in viral sepsis—particularly in infections caused by highly virulent pathogens such as the Ebola virus—reveals a dual profile of inflammatory hyperactivation combined with profound immunosuppression. Clinically, this presents as severe lymphocytopenia, loss of immune surveillance, progressive endothelial dysfunction, and multiorgan failure. At the cellular level, infected cells release large amounts of pyrogenic cytokines (IL-1β, TNF-α, IL-18) and DAMPs, which induce bystander cell death in uninfected immune cells through indirect inflammatory mechanisms. This process leads to apoptosis, functional exhaustion, or loss of viability in monocytes, T lymphocytes, and endothelial cells, independently of direct viral replication [166,167,168].
Within this inflammatory milieu, effector cells exhibit impaired cytotoxicity, reduced IFN-γ production, and diminished antigen-specific responses. In parallel, persistent metabolic and epigenetic alterations sustain an abnormal immune profile beyond the acute phase, aligning with the concept of a dysfunctional post-sepsis remnant characterized by chronic inflammation and progressive immune impairment [166,167,168]. Exaggerated inflammatory signaling also hampers lymphocyte regeneration and promotes the coexistence of both hyperinflammatory and immunosuppressive phenotypes, perpetuating immune paralysis. These mechanisms have been validated in vitro and are consistent with histopathological observations in patients with severe viral sepsis, including lymphoid tissue disruption, thymic atrophy, and widespread endothelial injury [166,167,168].
These insights provide a rationale for targeted immunotherapeutic approaches. Experimental inhibition of inflammasome activation, blockade of IL-1β, and modulation of non-apoptotic cell death pathways have shown beneficial effects, including cellular preservation, restoration of immune function, and reduction of systemic inflammation. Such strategies highlight the potential of phase-specific immunomodulation in viral sepsis [166,167,168].
The persistence of lymphocytopenia, residual inflammation, and endothelial dysfunction after resolution of acute viral sepsis underscores that sepsis is not a transient inflammatory event but a syndrome of sustained immunometabolic dysfunction. This post-inflammatory state increases susceptibility to reinfections and long-term complications, emphasizing the need for early recognition, longitudinal monitoring, and proactive immunologic management [166,167,168].
Overall, these findings strengthen the clinical and mechanistic framework of immunodynamic disruption in sepsis. They demonstrate that unresolved inflammation, secondary immune cell loss, and persistent epigenetic imprinting act as pathogenic cycles in Ebola-driven immunoparalysis, offering functional evidence to guide the design of personalized immunotherapeutic strategies aimed at restoring immune competence and preventing chronic inflammation induced by highly virulent viral pathogens [166,167,168].
11. Post-Inflammatory Oncologic Surveillance
A binational cohort study conducted in Finland and Sweden followed over 18,500 sepsis survivors for at least one year, revealing a significantly increased incidence of cancer compared to the general population. With a median follow-up of 3.5 years, standardized cancer incidence rates were markedly elevated in both men and women, particularly for gastrointestinal, urinary tract, and skin malignancies. These findings support the hypothesis that persistent immune dysfunction after sepsis contributes to oncogenesis through prolonged loss of immune surveillance and unresolved chronic inflammation [169,170].
Complementary evidence from a large epidemiological study based on U.S. healthcare records of older adults demonstrated a significantly elevated risk of developing at least fifteen distinct types of cancer following sepsis. This association persisted after adjusting for age, sex, comorbidities, and hospitalization frequency. The most frequently observed malignancies included lung, colorectal, bladder, and pancreatic cancers. Proposed mechanisms include sustained immunosuppression, epigenetic reprogramming, and microbiome dysbiosis, which may collectively facilitate malignant transformation [169,170].
These findings underscore a growing recognition of the link between sepsis and subsequent cancer development, reinforcing the need for proactive oncologic surveillance in post-sepsis care. Surviving sepsis may entail a significantly increased medium-term cancer risk, warranting implementation of early screening programs, risk stratification protocols, and preventive interventions as integral components of post-sepsis management [169,170].
12. Precision Immunotherapy After Sepsis
The use of immune checkpoint inhibitors in the post-sepsis setting has attracted growing interest; however, their efficacy depends strongly on timing and careful patient selection [171,172]. Preclinical studies in animal models demonstrate that delayed administration of anti–PD-1 antibodies during the immunosuppressive phase significantly improves survival, restores T cell function, and enhances pathogen clearance. These findings underscore that administration during the early hyperinflammatory phase may be ineffective or even harmful, whereas targeted use in the context of sustained immune dysfunction can yield meaningful clinical benefits [173,174].
Early-phase clinical trials with checkpoint inhibitors, such as nivolumab and anti–PD-L1 antibodies, have confirmed safety in patients with severe sepsis and established immunosuppression. Nonetheless, efficacy results remain inconsistent, highlighting the need for refined patient stratification. Rather than focusing exclusively on therapeutic blockade, these studies emphasize the importance of identifying reliable biomarkers—such as persistent lymphopenia, sustained PD-1 expression on T cells, and reduced HLA-DR levels on monocytes—to define the optimal therapeutic window. Incorporating these parameters into trial design is essential to move beyond safety assessment and determine whether precision immunotherapy can improve clinical outcomes [173,174].
Thus, post-sepsis immunotherapy should be conceptualized as a personalized strategy, guided by dynamic biomarkers and adapted to specific therapeutic windows [171,172]. Only a stratified approach can effectively restore immune competence while minimizing the risk of exacerbating inflammation or disrupting systemic homeostasis [173,174] (Table 7).
Table 7.
Summary of the biphasic immune response in sepsis. This table shows that the immune response in sepsis evolves from an initial hyperinflammatory surge to prolonged immunosuppressive dysfunction, shaping the timing and effectiveness of targeted immunotherapies.
13. Discussion
Sepsis is increasingly recognized not as a transient inflammatory event but as a persistent immunometabolic disorder with overlapping phases of hyperactivation and suppression [164]. While classical models described a linear progression from cytokine storm to immunoparalysis, our synthesis supports a more dynamic view in which immune activation, exhaustion, and tolerance coexist across anatomical and temporal compartments. This reconceptualization aligns with the concept of immunodynamic disruption, which provides a more integrative framework than the biphasic paradigm and accounts for long-term sequelae driven by epigenetic and metabolic reprogramming.
To bridge the gap between acute immune collapse and chronic dysfunction, we proposed the SIMMP–Sepsis framework. This model delineates five interconnected levels—ranging from acute activation to persistent clinical phenotypes—thereby integrating molecular events such as checkpoint overexpression (PD-1, CTLA-4), histone lactylation, and mitochondrial dysfunction with post-sepsis manifestations including recurrent infections, cognitive decline, and organ fibrosis [92,115,124]. As illustrated in Figure 2, SIMMP–Sepsis highlights the added value of dynamic immune profiling for precision interventions.
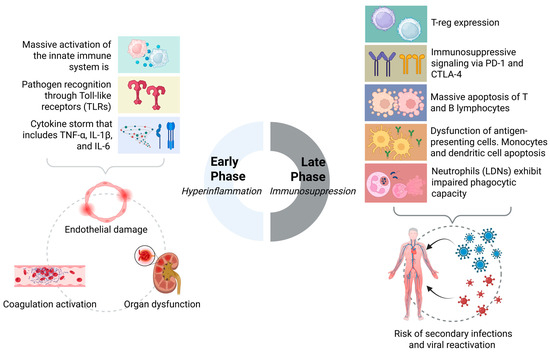
Figure 2.
Biphasic immunodynamic response in sepsis: from hyperinflammation to immunosuppression.
This conceptual evolution is summarized in Table 8, which contrasts classical and emerging models of septic immunopathology and highlights the added value of immunodynamic disruption in guiding biomarker-informed precision interventions.
Table 8.
Conceptual evolution of immunological models in sepsis and the clinical value of dynamic immunoprofiling.
Our analysis also contextualizes well-established mechanisms—including CD4+ and B cell apoptosis, Treg expansion, dendritic cell dysfunction, and neutrophil tolerance—within this longitudinal trajectory. Importantly, we identify the persistence of biomarkers such as S100A8/A9, IL-10, mtDNA, and histone lactylation as molecular signatures of a Post-Sepsis Cellular Remnant. Experimental evidence from two-hit and LPS tolerance models reinforces these findings, though translational gaps remain due to differences between murine and human immune complexity [65].
Therapeutic implications emerge from this framework. Promising interventions such as IL-7, PD-1 blockade, GM-CSF, β1-adrenergic inhibition, and metabolic modulators must be evaluated not as universal treatments but as phase-specific strategies guided by longitudinal biomarker monitoring (e.g., HLA-DR, PD-1, IL-6, mtDNA, S100A8/A9). In this regard, precision immunotherapy depends less on the availability of agents than on accurate patient stratification and timing [25,47,82,152]. The inconclusive evidence surrounding IL-36 exemplifies the need for cautious interpretation of exploratory mediators until validated in larger, controlled cohorts [155,175].
We believe the SIMMP–Sepsis model provides a testable and adaptable framework for advancing immunotherapeutic strategies into clinical practice—not as a replacement for current definitions but as a translational bridge linking molecular dysfunction to actionable interventions. Its validation requires prospective and longitudinal application in real-world cohorts, with integration of epigenetic, metabolic, and immunophenotypic markers to stratify patients according to their immune trajectory and therapeutic needs. In parallel, machine learning approaches hold promise for identifying sepsis endotypes with differential responses to treatment, thereby facilitating precision immunotherapy. Moreover, targeting metabolic pathways—such as enhancing mitochondrial biogenesis via PGC-1α activation or reducing lactate-driven immunosuppression—represents an innovative avenue to restore immune competence in sepsis survivors.
Ultimately, the central barrier is not the scarcity of mechanistic insights but the absence of conceptual integration. By framing sepsis as a persistent immunometabolic syndrome, the SIMMP–Sepsis model provides a multidimensional scaffold that links basic immunology with clinical practice. This perspective reframes post-ICU care, emphasizing that survival marks not the end of treatment but the beginning of long-term immune restoration.
Limitations
This work was conceived as a comprehensive review integrating mechanistic, experimental, and clinical perspectives on immunodynamic disruption in sepsis. Unlike systematic reviews, comprehensive reviews do not follow standardized frameworks such as PRISMA but instead aim for a broad synthesis of available evidence. While this allows the inclusion of diverse data—from preclinical models to emerging conceptual frameworks, it also entails inherent limitations.
First, the absence of a rigid methodology increases the risk of selection bias. Although targeted searches in PubMed, Scopus, and Web of Science were performed using specific keywords, the narrative approach cannot guarantee exhaustive coverage, and some relevant studies, particularly preprints or non-English publications, may have been missed.
Second, heterogeneity across studies limits comparability. Differences in study design, patient populations, timing of sampling, and laboratory techniques (e.g., HLA-DR measurement, transcriptomic protocols) hinder direct integration. Similarly, preclinical models such as cecal ligation and puncture or LPS tolerance capture only partial aspects of sepsis and do not fully reflect human immunology, restricting translational value.
Finally, this review prioritizes breadth over quantitative rigor. No formal risk-of-bias assessment or meta-analysis was conducted; instead, emphasis was placed on convergent findings, mechanistic plausibility, and translational implications.
Despite these limitations, the review provides an updated, integrative perspective linking molecular mechanisms, clinical implications, and therapeutic strategies, while proposing the SIMMP–Sepsis framework to advance the field.
14. Conclusions
Sepsis is a dynamic immunometabolic disorder that transcends the classical biphasic paradigm of hyperinflammation followed by immunosuppression. Instead, it involves overlapping and persistent immune dysfunctions—including lymphocyte apoptosis, Treg expansion, dendritic and neutrophil impairment, and checkpoint overexpression (e.g., PD-1, CTLA-4). This review supports the concept of immunodynamic disruption, which more accurately reflects the heterogeneous and evolving immune landscape of sepsis. Incorporating this perspective into clinical care requires dynamic immune profiling to guide personalized interventions.
Key biomarkers such as HLA-DR, PD-1/PD-L1, IL-6, IL-10, circulating mtDNA, and epigenetic signatures provide a foundation for stratifying patients according to immune phenotype and therapeutic need. We advocate for routine immune monitoring every 48–72 h in critical care, enabling timely and tailored immunomodulation. This approach may optimize immune balance, reduce harmful over- or under-stimulation, and improve survival and post-ICU outcomes. Importantly, immune-based therapies—such as IL-7, GM-CSF, or checkpoint inhibitors—should be deployed in a phase-specific manner rather than as one-size-fits-all strategies.
At the core of this model lies the Persistent Immune Remnant, a state defined by long-lasting epigenetic, mitochondrial, and metabolic dysfunctions that contribute to post-sepsis morbidity despite apparent clinical recovery. Recognizing sepsis as a persistent immunometabolic syndrome reframes post-acute care from passive observation to proactive immune restoration. The proposed SIMMP–Sepsis model offers a testable and translational scaffold that bridges molecular mechanisms with clinical decision-making, promoting the development of adaptive, biomarker-guided therapies. Ultimately, survival should be seen not as the end of treatment, but as the beginning of long-term immune recovery.
Author Contributions
Conceptualization: J.S.S.-T., M.V.P.-F. and J.S.I.-C.; Methodology: J.S.S.-T., M.V.P.-F., H.A.N.-C. and J.S.I.-C.; Resources: J.S.S.-T., M.V.P.-F., H.A.N.-C., V.C.C., L.C.L.M., J.E.G., D.T.-C., D.A.L.G., M.A.-I., A.T.-D.-l.-T. and A.G.-P.; Software: J.S.S.-T., M.A.-I., A.T.-D.-l.-T. and J.S.I.-C.; Validation: M.A.-I., A.G.-P. and J.S.I.-C.; Analysis: J.S.S.-T., M.V.P.-F. and J.S.I.-C.; Visualization: A.T.-D.-l.-T. and J.S.I.-C.; Writing—original draft: J.S.S.-T., M.V.P.-F., H.A.N.-C., V.C.C., L.C.L.M., J.E.G., D.T.-C., D.A.L.G., M.A.-I. and A.T.-D.-l.-T.; Writing—review and editing: A.G.-P. and J.S.I.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| APC | Antigen-presenting cell |
| ASC | Apoptosis-Associated Speck-Like Protein Containing a CARD |
| B cell | B lymphocyte |
| CD | Cluster of Differentiation |
| CLP | Cecal ligation and puncture |
| CTLA-4 | Cytotoxic T lymphocyte–associated protein 4 |
| DC | Dendritic cell |
| DAMP | Damage-Associated Molecular Pattern |
| HLA-DR | Human Leukocyte Antigen–DR isotype |
| IL | Interleukin |
| IL-7R | Interleukin-7 Receptor |
| LAG-3 | Lymphocyte Activation Gene-3 |
| LDN | Low-density neutrophil |
| LPS | Lipopolysaccharide |
| Ly6Chi | Lymphocyte Antigen 6 Complex, locus C high |
| MDSC | Myeloid-derived suppressor cell |
| mtDNA | Mitochondrial DNA |
| NETs | Neutrophil Extracellular Traps |
| NLRP3 | NOD-Like Receptor Family Pyrin Domain Containing 3 |
| PAMP | Pathogen-Associated Molecular Pattern |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| STING | Stimulator of Interferon Genes |
| SYRCLE | Systematic Review Centre for Laboratory Animal Experimentation |
| T cell | T lymphocyte |
| TCR | T cell receptor |
| Treg | Regulatory T cell |
| TNF | Tumor Necrosis Factor |
References
- Sundar, K.M.; Sires, M. Sepsis Induced Immunosuppression: Implications for Secondary Infections and Complications. Indian J. Crit. Care Med. 2013, 17, 162–169. [Google Scholar] [CrossRef]
- Baykara, N.; Akalın, H.; Arslantaş, M.K.; Hancı, V.; Çağlayan, Ç.; Kahveci, F.; Demirağ, K.; Baydemir, C.; Ünal, N.; Sepsis Study Group. Epidemiology of Sepsis in Intensive Care Units in Turkey: A Multicenter, Point-Prevalence Study. Crit. Care 2018, 22, 93. [Google Scholar] [CrossRef]
- Lundberg, O.H.M.; Lengquist, M.; Spångfors, M.; Annborn, M.; Bergmann, D.; Schulte, J.; Levin, H.; Melander, O.; Frigyesi, A.; Friberg, H. Circulating Bioactive Adrenomedullin as a Marker of Sepsis, Septic Shock and Critical Illness. Crit. Care 2020, 24, 636. [Google Scholar] [CrossRef]
- Ramakers, B.P.; Giamarellos-Bourboulis, E.J.; Tasioudis, C.; Coenen, M.J.H.; Kox, M.; Vermeulen, S.H.; Groothuismink, J.M.; van der Hoeven, J.G.; Routsi, C.; Savva, A.; et al. Effects of the 34C>T Variant of the AMPD1 Gene on Immune Function, Multi-Organ Dysfunction, and Mortality in Sepsis Patients. Shock 2015, 44, 542–547. [Google Scholar] [CrossRef]
- Labib, A. Sepsis Care Pathway 2019. Qatar Med. J. 2019, 2019, 4. [Google Scholar] [CrossRef]
- Jensen, I.J.; Sjaastad, F.V.; Griffith, T.S.; Badovinac, V.P. Sepsis-Induced T Cell Immunoparalysis: The Ins and Outs of Impaired T Cell Immunity. J. Immunol. 2018, 200, 1543–1553. [Google Scholar] [CrossRef]
- McLellan, A.D.; Terbeck, G.; Mengling, T.; Starling, G.C.; Kiener, P.A.; Gold, R.; Bröcker, E.-B.; Leverkus, M.; Kämpgen, E. Differential Susceptibility to CD95 (Apo-1/Fas) and MHC Class II-Induced Apoptosis during Murine Dendritic Cell Development. Cell Death Differ. 2000, 7, 933–938. [Google Scholar] [CrossRef]
- Wakamatsu, E.; Machiyama, H.; Toyota, H.; Takeuchi, A.; Hashimoto, R.; Kozono, H.; Yokosuka, T. Indirect Suppression of CD4 T Cell Activation through LAG-3-Mediated Trans-Endocytosis of MHC Class II. Cell Rep. 2024, 43, 114655. [Google Scholar] [CrossRef]
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T Cells in Health and Disease. Signal Transduct. Target. Ther. 2023, 8, 235. [Google Scholar] [CrossRef]
- He, W.; Xiao, K.; Fang, M.; Xie, L. Immune Cell Number, Phenotype, and Function in the Elderly with Sepsis. Aging Dis. 2021, 12, 277–296. [Google Scholar] [CrossRef]
- Nasa, P.; Juneja, D.; Singh, O. Severe Sepsis and Septic Shock in the Elderly: An Overview. World J. Crit. Care Med. 2012, 1, 23–30. [Google Scholar] [CrossRef]
- Martín, S.; Pérez, A.; Aldecoa, C. Sepsis and Immunosenescence in the Elderly Patient: A Review. Front. Med. 2017, 4, 20. [Google Scholar] [CrossRef]
- Cao, C.; Yu, M.; Chai, Y. Pathological Alteration and Therapeutic Implications of Sepsis-Induced Immune Cell Apoptosis. Cell Death Dis. 2019, 10, 782. [Google Scholar] [CrossRef]
- Delano, M.J.; Ward, P.A. The Immune System’s Role in Sepsis Progression, Resolution, and Long-term Outcome. Immunol. Rev. 2016, 274, 330–353. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Fleishman, J.S.; Liu, X.; Wang, H.; Huo, L. Targeting Novel Regulated Cell death: Ferroptosis, Pyroptosis, and Autophagy in Sepsis-Associated Encephalopathy. Biomed. Pharmacother. 2024, 174, 116453. [Google Scholar] [CrossRef]
- Wang, T.S.; Deng, J.C. Molecular and Cellular Aspects of Sepsis-Induced Immunosuppression. J. Mol. Med. 2008, 86, 495–506. [Google Scholar] [CrossRef]
- Patil, N.K.; Bohannon, J.K.; Sherwood, E.R. Immunotherapy: A Promising Approach to Reverse Sepsis-Induced Immunosuppression. Pharmacol. Res. 2016, 111, 688–702. [Google Scholar] [CrossRef]
- Cheng, S.-C.; Scicluna, B.P.; Arts, R.J.W.; Gresnigt, M.S.; Lachmandas, E.; Giamarellos-Bourboulis, E.J.; Kox, M.; Manjeri, G.R.; Wagenaars, J.A.L.; Cremer, O.L.; et al. Broad Defects in the Energy Metabolism of Leukocytes Underlie Immunoparalysis in Sepsis. Nat. Immunol. 2016, 17, 406–413. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory Caspases Are Innate Immune Receptors for Intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Fan, J. Neutrophil in Reverse Migration: Role in Sepsis. Front. Immunol. 2021, 12, 656039. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, J.; Berner, R.; Löser, T.; Schöll, E. Modeling Tumor Disease and Sepsis by Networks of Adaptively Coupled Phase Oscillators. Front. Netw. Physiol. 2021, 1, 730385. [Google Scholar] [CrossRef]
- Engelmann, B.; Massberg, S. Thrombosis as an Intravascular Effector of Innate Immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Tinsley, K.W.; Swanson, P.E.; Chang, K.C.; Cobb, J.P.; Buchman, T.G.; Korsmeyer, S.J.; Karl, I.E. Prevention of Lymphocyte Cell Death in Sepsis Improves Survival in Mice. Proc. Natl. Acad. Sci. USA 1999, 96, 14541–14546. [Google Scholar] [CrossRef]
- Joshi, I.; Carney, W.P.; Rock, E.P. Utility of Monocyte HLA-DR and Rationale for Therapeutic GM-CSF in Sepsis Immunoparalysis. Front. Immunol. 2023, 14, 1130214. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Z.; Chen, X.; Yu, F.; Huang, H.; Shen, X.; Tan, Y.; Wu, Q. Relationship Between the Expression of PD-1 and CTLA-4 on T Lymphocytes and the Severity and Prognosis of Sepsis. Int. J. Gen. Med. 2023, 16, 1513–1525. [Google Scholar] [CrossRef]
- Padovani, C.M.; Yin, K. Immunosuppression in Sepsis: Biomarkers and Specialized Pro-Resolving Mediators. Biomedicines 2024, 12, 175. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Huang, S.-Y.; Sun, J.-H.; Zhang, H.-C.; Cai, Q.-L.; Gao, C.; Li, L.; Cao, J.; Xu, F.; Zhou, Y.; et al. Sepsis-Induced Immunosuppression: Mechanisms, Diagnosis and Current Treatment Options. Mil. Med. Res. 2022, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Sheeja Prabhakaran, H.; Zhang, Y.-Y.; Luo, G.; He, W.; Liou, Y.-C. Mitochondrial Dysfunction in Sepsis: Mechanisms and Therapeutic Perspectives. Crit. Care 2024, 28, 292. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, H.; Li, G.; Wang, D.; Zhou, Q.; Wu, J.; Zheng, J.; Huang, J.; Slade, D.A.; Wu, X.; et al. STING-Mediated Intestinal Barrier Dysfunction Contributes to Lethal Sepsis. EBioMedicine 2019, 41, 497–508. [Google Scholar] [CrossRef]
- Cui, J.; Oehrl, S.; Ahmad, F.; Brenner, T.; Uhle, F.; Nusshag, C.; Rupp, C.; Funck, F.; Meisel, S.; Weigand, M.A.; et al. Detection of In Vivo Inflammasome Activation for Predicting Sepsis Mortality. Front. Immunol. 2020, 11, 613745. [Google Scholar] [CrossRef]
- Gierlikowska, B.; Stachura, A.; Gierlikowski, W.; Demkow, U. The Impact of Cytokines on Neutrophils’ Phagocytosis and NET Formation during Sepsis-A Review. Int. J. Mol. Sci. 2022, 23, 5076. [Google Scholar] [CrossRef]
- Girardot, T.; Rimmelé, T.; Venet, F.; Monneret, G. Apoptosis-Induced Lymphopenia in Sepsis and Other Severe Injuries. Apoptosis 2017, 22, 295–305. [Google Scholar] [CrossRef]
- Francois, B.; Jeannet, R.; Daix, T.; Walton, A.H.; Shotwell, M.S.; Unsinger, J.; Monneret, G.; Rimmelé, T.; Blood, T.; Morre, M.; et al. Interleukin-7 Restores Lymphocytes in Septic Shock: The IRIS-7 Randomized Clinical Trial. JCI Insight 2018, 3, e98960. [Google Scholar] [CrossRef]
- Lélu, K.; Dubois, C.; Evlachev, A.; Crausaz, M.; Baldazza, M.; Kehrer, N.; Brandely, R.; Schlesinger, Y.; Silvestre, N.; Marchand, J.-B.; et al. Viral Delivery of IL-7 Is a Potent Immunotherapy Stimulating Innate and Adaptive Immunity and Confers Survival in Sepsis Models. J. Immunol. 2022, 209, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.; Horie, S.; Laffey, J.G. Role of the Adaptive Immune Response in Sepsis. Intensive Care Med. Exp. 2020, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, E.; D’Angerio, M.; Ciobanu, A.L.; Masini, L.; Lo Tartaro, D.; Coloretti, I.; Busani, S.; Rubio, I.; Meschiari, M.; Franceschini, E.; et al. Advances and Challenges in Sepsis Management: Modern Tools and Future Directions. Cells 2024, 13, 439. [Google Scholar] [CrossRef]
- Cui, J.; Cai, W.; Zhang, L.; Wu, Y.; Huang, Y.; Zhao, W. Decreased Monocytic HLA-DR in Patients with Sepsis: Prediction of Diagnosis, Severity and Prognosis. Clin. Biochem. 2025, 135, 110851. [Google Scholar] [CrossRef]
- Nedel, W.; Deutschendorf, C.; Portela, L.V.C. Sepsis-Induced Mitochondrial Dysfunction: A Narrative Review. World J. Crit. Care Med. 2023, 12, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cai, S.; Li, X.; Wu, G. Sepsis-Induced Immunosuppression: Mechanisms, Biomarkers and Immunotherapy. Front. Immunol. 2025, 16, 1577105. [Google Scholar] [CrossRef]
- Sun, H.; Lee, H.-S.; Kim, S.H.-J.; Fernandes de Lima, M.; Gingras, A.R.; Du, Q.; McLaughlin, W.; Ablack, J.; Lopez-Ramirez, M.A.; Lagarrigue, F.; et al. IL-2 Can Signal via Chemokine Receptors to Promote Regulatory T Cells’ Suppressive Function. Cell Rep. 2023, 42, 112996. [Google Scholar] [CrossRef]
- Jones, D.M.; Read, K.A.; Oestreich, K.J. Dynamic Roles for IL-2-STAT5 Signaling in Effector and Regulatory CD4+ T Cell Populations. J. Immunol. 2020, 205, 1721–1730. [Google Scholar] [CrossRef]
- Mahmud, S.A.; Manlove, L.S.; Farrar, M.A. Interleukin-2 and STAT5 in Regulatory T Cell Development and Function. JAK-STAT 2013, 2, e23154. [Google Scholar] [CrossRef] [PubMed]
- Garduno, A.; Martín-Loeches, I. Targeting Sepsis: Disease Tolerance, Immune Resilience, and Compartmentalized Immunity. Biomedicines 2024, 12, 2420. [Google Scholar] [CrossRef]
- Shankar-Hari, M.; Calandra, T.; Soares, M.P.; Bauer, M.; Wiersinga, W.J.; Prescott, H.C.; Knight, J.C.; Baillie, K.J.; Bos, L.D.J.; Derde, L.P.G.; et al. Reframing Sepsis Immunobiology for Translation: Towards Informative Subtyping and Targeted Immunomodulatory Therapies. Lancet Respir. Med. 2024, 12, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Rudick, C.P.; Cornell, D.L.; Agrawal, D.K. Single versus Combined Immunoregulatory Approach Using PD-1 and CTLA-4 Modulators in Controlling Sepsis. Expert. Rev. Clin. Immunol. 2017, 13, 907–919. [Google Scholar] [CrossRef]
- Izquierdo-Condoy, J.S.; Vásconez-Gonzáles, J.; Morales-Lapo, E.; Tello-De-la-Torre, A.; Naranjo-Lara, P.; Fernández, R.; Hidalgo, M.R.; Escobar, A.; Yépez, V.H.; Díaz, A.M.; et al. Beyond the Acute Phase: A Comprehensive Literature Review of Long-Term Sequelae Resulting from Infectious Diseases. Front. Cell. Infect. Microbiol. 2024, 14, 1293782. [Google Scholar] [CrossRef]
- Silva, E.E.; Skon-Hegg, C.; Badovinac, V.P.; Griffith, T.S. The Calm after the Storm: Implications of Sepsis Immunoparalysis on Host Immunity. J. Immunol. 2023, 211, 711–719. [Google Scholar] [CrossRef]
- Lee, S.I.; Kim, N.Y.; Chung, C.; Park, D.; Kang, D.H.; Kim, D.K.; Yeo, M.-K.; Sun, P.; Lee, J.E. IL-6 and PD-1 Antibody Blockade Combination Therapy Regulate Inflammation and T Lymphocyte Apoptosis in Murine Model of Sepsis. BMC Immunol. 2025, 26, 3. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-L.; Yao, Y.; Zhang, X.; Chen, F.; Meng, X.-L.; Chen, X.-S.; Wang, C.-L.; Liu, Y.-C.; Tian, X.; Shou, S.-T.; et al. Regulatory T Cells: Angels or Demons in the Pathophysiology of Sepsis? Front. Immunol. 2022, 13, 829210. [Google Scholar] [CrossRef]
- Jensen, I.J.; Li, X.; McGonagill, P.W.; Shan, Q.; Fosdick, M.G.; Tremblay, M.M.; Houtman, J.C.; Xue, H.-H.; Griffith, T.S.; Peng, W.; et al. Sepsis Leads to Lasting Changes in Phenotype and Function of Memory CD8 T Cells. eLife 2021, 10, e70989. [Google Scholar] [CrossRef]
- Durand, M.; Hagimont, E.; Louis, H.; Asfar, P.; Frippiat, J.-P.; Singer, M.; Gauchotte, G.; Labat, C.; Lacolley, P.; Levy, B.; et al. The β 1-Adrenergic Receptor Contributes to Sepsis-Induced Immunosuppression Through Modulation of Regulatory T-Cell Inhibitory Function. Crit. Care Med. 2022, 50, e707–e718. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Qu, M.; Li, W.; Wu, D.; Cata, J.P.; Miao, C. Neutrophil, Neutrophil Extracellular Traps and Endothelial Cell Dysfunction in Sepsis. Clin. Transl. Med. 2023, 13, e1170. [Google Scholar] [CrossRef]
- Tu, Q.; Li, Y.; Zhu, J.; Guo, L.; Liu, C.; Liu, L.; Yuan, Y.; Zou, Y.; Chen, F.; Yao, L.; et al. Mitochondrial DNA Mediates Immunoparalysis of Dendritic Cells in Sepsis via STING Signalling. Cell Prolif. 2022, 55, e13328. [Google Scholar] [CrossRef]
- Gao, D.-N.; Yang, Z.-X.; Qi, Q.-H. Roles of PD-1, Tim-3 and CTLA-4 in Immunoregulation in Regulatory T Cells among Patients with Sepsis. Int. J. Clin. Exp. Med. 2015, 8, 18998–19005. [Google Scholar] [PubMed]
- Nascimento, D.C.; Alves-Filho, J.C.; Sônego, F.; Fukada, S.Y.; Pereira, M.S.; Benjamim, C.; Zamboni, D.S.; Silva, J.S.; Cunha, F.Q. Role of Regulatory T Cells in Long-Term Immune Dysfunction Associated with Severe Sepsis. Crit. Care Med. 2010, 38, 1718–1725. [Google Scholar] [CrossRef]
- Kimmoun, A.; Louis, H.; Al Kattani, N.; Delemazure, J.; Dessales, N.; Wei, C.; Marie, P.Y.; Issa, K.; Levy, B. β1-Adrenergic Inhibition Improves Cardiac and Vascular Function in Experimental Septic Shock. Crit. Care Med. 2015, 43, e332–e340. [Google Scholar] [CrossRef]
- Zheng, L.; Duan, Y.; He, P.; Wu, M.; Wei, S.; Du, X.; Yao, R.; Yao, Y. Dysregulated Dendritic Cells in Sepsis: Functional Impairment and Regulated Cell Death. Cell Mol. Biol. Lett. 2024, 29, 81. [Google Scholar] [CrossRef]
- Lu, J.; Sun, K.; Yang, H.; Fan, D.; Huang, H.; Hong, Y.; Wu, S.; Zhou, H.; Fang, F.; Li, Y.; et al. Sepsis Inflammation Impairs the Generation of Functional Dendritic Cells by Targeting Their Progenitors. Front. Immunol. 2021, 12, 732612. [Google Scholar] [CrossRef]
- Venet, F.; Chung, C.-S.; Kherouf, H.; Geeraert, A.; Malcus, C.; Poitevin, F.; Bohé, J.; Lepape, A.; Ayala, A.; Monneret, G. Increased Circulating Regulatory T Cells (CD4+CD25+CD127−) Contribute to Lymphocyte Anergy in Septic Shock Patients. Intensive Care Med. 2009, 35, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-Induced Immunosuppression: From Cellular Dysfunctions to Immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Koike, Y.; Shimura, T.; Okigami, M.; Ide, S.; Toiyama, Y.; Okugawa, Y.; Inoue, Y.; Araki, T.; Uchida, K.; et al. In Vivo Characterization of Neutrophil Extracellular Traps in Various Organs of a Murine Sepsis Model. PLoS ONE 2014, 9, e111888. [Google Scholar] [CrossRef] [PubMed]
- Chandrabhatla, B.; A V, A.; Puvvula, L.S.; Gopal, P.B. Decoding Inflammation: Predicting Sepsis in the ICU. Cureus 2024, 16, e75256. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, J.; Sun, Y.; Ding, X.; Zhang, X.; Liu, S.; Han, B.; Wang, H.; Duan, X.; Sun, T. A Machine Learning Model for Accurate Prediction of Sepsis in ICU Patients. Front. Public Health 2021, 9, 754348. [Google Scholar] [CrossRef]
- Cavaillon, J.-M.; Adib-Conquy, M. Bench-to-Bedside Review: Endotoxin Tolerance as a Model of Leukocyte Reprogramming in Sepsis. Crit. Care 2006, 10, 233. [Google Scholar] [CrossRef][Green Version]
- Wang, N.; Lu, Y.; Zheng, J.; Liu, X. Of Mice and Men: Laboratory Murine Models for Recapitulating the Immunosuppression of Human Sepsis. Front. Immunol. 2022, 13, 956448. [Google Scholar] [CrossRef] [PubMed]
- Stortz, J.A.; Raymond, S.L.; Mira, J.C.; Moldawer, L.L.; Mohr, A.M.; Efron, P.A. Murine Models of Sepsis and Trauma: Can We Bridge the Gap? ILAR J. 2017, 58, 90–105. [Google Scholar] [CrossRef]
- Muenzer, J.T.; Davis, C.G.; Dunne, B.S.; Unsinger, J.; Dunne, W.M.; Hotchkiss, R.S. Pneumonia after Cecal Ligation and Puncture: A Clinically Relevant “Two-Hit” Model of Sepsis. Shock 2006, 26, 565–570. [Google Scholar] [CrossRef]
- Dejager, L.; Pinheiro, I.; Dejonckheere, E.; Libert, C. Cecal Ligation and Puncture: The Gold Standard Model for Polymicrobial Sepsis? Trends Microbiol. 2011, 19, 198–208. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, D.-Z.; Zhu, C.-L.; Ren, S.-C.; Sun, C.-Y.; Wang, Y.; Wang, J.-F. The Implication of Targeting PD-1:PD-L1 Pathway in Treating Sepsis through Immunostimulatory and Anti-Inflammatory Pathways. Front. Immunol. 2023, 14, 1323797. [Google Scholar] [CrossRef]
- Kumar, S.; Payal, N.; Srivastava, V.K.; Kaushik, S.; Saxena, J.; Jyoti, A. Neutrophil Extracellular Traps and Organ Dysfunction in Sepsis. Clin. Chim. Acta 2021, 523, 152–162. [Google Scholar] [CrossRef]
- Morelli, A.; Ertmer, C.; Westphal, M.; Rehberg, S.; Kampmeier, T.; Ligges, S.; Orecchioni, A.; D’Egidio, A.; D’Ippoliti, F.; Raffone, C.; et al. Effect of Heart Rate Control with Esmolol on Hemodynamic and Clinical Outcomes in Patients with Septic Shock: A Randomized Clinical Trial. JAMA 2013, 310, 1683–1691. [Google Scholar] [CrossRef]
- Darden, D.B.; Kelly, L.S.; Fenner, B.P.; Moldawer, L.L.; Mohr, A.M.; Efron, P.A. Dysregulated Immunity and Immunotherapy after Sepsis. J. Clin. Med. 2021, 10, 1742. [Google Scholar] [CrossRef] [PubMed]
- Arens, C.; Bajwa, S.A.; Koch, C.; Siegler, B.H.; Schneck, E.; Hecker, A.; Weiterer, S.; Lichtenstern, C.; Weigand, M.A.; Uhle, F. Sepsis-Induced Long-Term Immune Paralysis--Results of a Descriptive, Explorative Study. Crit. Care 2016, 20, 93. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.K.; Ghita, G.L.; Wang, Z.; Ozrazgat-Baslanti, T.; Raymond, S.L.; Mankowski, R.T.; Brumback, B.A.; Efron, P.A.; Bihorac, A.; Moore, F.A.; et al. The Development of Chronic Critical Illness Determines Physical Function, Quality of Life, and Long-Term Survival Among Early Survivors of Sepsis in Surgical ICUs. Crit. Care Med. 2019, 47, 566–573. [Google Scholar] [CrossRef]
- Huang, Z.; Fu, Z.; Huang, W.; Huang, K. Prognostic Value of Neutrophil-to-Lymphocyte Ratio in Sepsis: A Meta-Analysis. Am. J. Emerg. Med. 2020, 38, 641–647. [Google Scholar] [CrossRef]
- Coudereau, R.; Haem Rahimi, M.; Lukaszewicz, A.-C.; Cour, M.; Bidar, F.; Argaud, L.; Venet, F.; Monneret, G. Immature Neutrophils and Myeloid-Derived Suppressor Cells in Sepsis: Differences in Occurrence Kinetics. Crit. Care 2024, 28, 7. [Google Scholar] [CrossRef] [PubMed]
- Mira, J.C.; Brakenridge, S.C.; Moldawer, L.L.; Moore, F.A. Persistent Inflammation, Immunosuppression and Catabolism Syndrome. Crit. Care Clin. 2017, 33, 245–258. [Google Scholar] [CrossRef]
- Malavika, M.; Sanju, S.; Poorna, M.R.; Vishnu Priya, V.; Sidharthan, N.; Varma, P.; Mony, U. Role of Myeloid Derived Suppressor Cells in Sepsis. Int. Immunopharmacol. 2022, 104, 108452. [Google Scholar] [CrossRef]
- Maestraggi, Q.; Lebas, B.; Clere-Jehl, R.; Ludes, P.-O.; Chamaraux-Tran, T.-N.; Schneider, F.; Diemunsch, P.; Geny, B.; Pottecher, J. Skeletal Muscle and Lymphocyte Mitochondrial Dysfunctions in Septic Shock Trigger ICU-Acquired Weakness and Sepsis-Induced Immunoparalysis. Biomed. Res. Int. 2017, 2017, 7897325. [Google Scholar] [CrossRef]
- Falcão-Holanda, R.B.; Leite, G.G.F.; Brunialti, M.K.C.; Jasiulionis, M.G.; Salomão, R. ALTERED LEVELS OF H3K9AC, H3K4ME3, AND H3K27ME3 IN PROMOTERS OF DIFFERENTIALLY EXPRESSED GENES RELATED TO INNATE IMMUNE RESPONSE IN SEPTIC PATIENTS WITH DIFFERENT CLINICAL OUTCOMES. Shock 2023, 59, 882–891. [Google Scholar] [CrossRef]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 Acetylation Co-Occur at Many Gene Regulatory Elements, While H3K14ac Marks a Subset of Inactive Inducible Promoters in Mouse Embryonic Stem Cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef]
- Bermick, J.R.; Lambrecht, N.J.; denDekker, A.D.; Kunkel, S.L.; Lukacs, N.W.; Hogaboam, C.M.; Schaller, M.A. Neonatal Monocytes Exhibit a Unique Histone Modification Landscape. Clin. Epigenetics 2016, 8, 99. [Google Scholar] [CrossRef]
- Ziogas, A.; Novakovic, B.; Ventriglia, L.; Galang, N.; Tran, K.A.; Li, W.; Matzaraki, V.; van Unen, N.; Schlüter, T.; Ferreira, A.V.; et al. Long-Term Histone Lactylation Connects Metabolic and Epigenetic Rewiring in Innate Immune Memory. Cell 2025, 188, 2992–3012.e16. [Google Scholar] [CrossRef]
- Cai, H.; Chen, X.; Liu, Y.; Chen, Y.; Zhong, G.; Chen, X.; Rong, S.; Zeng, H.; Zhang, L.; Li, Z.; et al. Lactate Activates Trained Immunity by Fueling the Tricarboxylic Acid Cycle and Regulating Histone Lactylation. Nat. Commun. 2025, 16, 3230. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Shi, Y.; Zhang, H.; Miao, C. Epigenetic Mechanisms of Immune Remodeling in Sepsis: Targeting Histone Modification. Cell Death Dis. 2023, 14, 112. [Google Scholar] [CrossRef]
- Manzoli, T.F.; Troster, E.J.; Ferranti, J.F.; Sales, M.M. Prolonged Suppression of Monocytic Human Leukocyte Antigen-DR Expression Correlates with Mortality in Pediatric Septic Patients in a Pediatric Tertiary Intensive Care Unit. J. Crit. Care 2016, 33, 84–89. [Google Scholar] [CrossRef]
- Lee, M.J.; Bae, J.; Lee, J.H.; Park, Y.J.; Lee, H.A.R.; Mun, S.; Kim, Y.-S.; Yune, C.J.; Chung, T.N.; Kim, K. Serial Change of Endotoxin Tolerance in a Polymicrobial Sepsis Model. Int. J. Mol. Sci. 2022, 23, 6581. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, G.; Wang, X.; Liu, D. Metabolic Reprogramming Consequences of Sepsis: Adaptations and Contradictions. Cell Mol. Life Sci. 2022, 79, 456. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Xuan, Y.; Zhang, X.; Liu, Y.; Yang, S.; Yang, K. Immune Cell Metabolism and Metabolic Reprogramming. Mol. Biol. Rep. 2022, 49, 9783–9795. [Google Scholar] [CrossRef]
- Willmann, K.; Moita, L.F. Physiologic Disruption and Metabolic Reprogramming in Infection and Sepsis. Cell Metab. 2024, 36, 927–946. [Google Scholar] [CrossRef]
- Gritte, R.B.; Souza-Siqueira, T.; da Silva, E.B.; Oliveira, L.C.d.S.d.; Borges, R.C.; Alves, H.H.d.O.; Masi, L.N.; Murata, G.M.; Gorjão, R.; Levada-Pires, A.C.; et al. Evidence for Monocyte Reprogramming in a Long-Term Postsepsis Study. Crit. Care Explor. 2022, 4, e0734. [Google Scholar] [CrossRef]
- de Azambuja Rodrigues, P.M.; Valente, R.H.; Brunoro, G.V.F.; Nakaya, H.T.I.; Araújo-Pereira, M.; Bozza, P.T.; Bozza, F.A.; Trugilho, M.R. de O. Proteomics Reveals Disturbances in the Immune Response and Energy Metabolism of Monocytes from Patients with Septic Shock. Sci. Rep. 2021, 11, 15149. [Google Scholar] [CrossRef]
- Caldwell, B.A.; Wu, Y.; Wang, J.; Li, L. Altered DNA Methylation Underlies Monocyte Dysregulation and Immune Exhaustion Memory in Sepsis. Cell Rep. 2024, 43, 113894. [Google Scholar] [CrossRef]
- Ferreira, B.L.; Sousa, M.B.; Leite, G.G.F.; Brunialti, M.K.C.; Nishiduka, E.S.; Tashima, A.K.; van der Poll, T.; Salomão, R. Glucose Metabolism Is Upregulated in the Mononuclear Cell Proteome during Sepsis and Supports Endotoxin-Tolerant Cell Function. Front. Immunol. 2022, 13, 1051514. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368.E14. [Google Scholar] [CrossRef] [PubMed]
- Bordon, Y. Macrophages: Innate Memory Training. Nat. Rev. Immunol. 2014, 14, 713. [Google Scholar] [CrossRef]
- Rusek, P.; Wala, M.; Druszczyńska, M.; Fol, M. Infectious Agents as Stimuli of Trained Innate Immunity. Int. J. Mol. Sci. 2018, 19, 456. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.-C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic Programming of Monocyte-to-Macrophage Differentiation and Trained Innate Immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef]
- Rahmel, T.; Marko, B.; Nowak, H.; Bergmann, L.; Thon, P.; Rump, K.; Kreimendahl, S.; Rassow, J.; Peters, J.; Singer, M.; et al. Mitochondrial Dysfunction in Sepsis Is Associated with Diminished Intramitochondrial TFAM despite Its Increased Cellular Expression. Sci. Rep. 2020, 10, 21029. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Yang, L.; Peng, Y.; Wang, Q.; Gao, M.; Yang, M.; Xiao, X. Ginsenoside Rg3 Attenuates Sepsis-Induced Injury and Mitochondrial Dysfunction in Liver via AMPK-Mediated Autophagy Flux. Biosci. Rep. 2017, 37, BSR20170934. [Google Scholar] [CrossRef]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. [Google Scholar] [CrossRef]
- Qian, L.; Zhu, Y.; Deng, C.; Liang, Z.; Chen, J.; Chen, Y.; Wang, X.; Liu, Y.; Tian, Y.; Yang, Y. Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 (PGC-1) Family in Physiological and Pathophysiological Process and Diseases. Signal Transduct. Target. Ther. 2024, 9, 50. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Maneta, E.; Aivalioti, E.; Tual-Chalot, S.; Emini Veseli, B.; Gatsiou, A.; Stamatelopoulos, K.; Stellos, K. Endothelial Dysfunction and Immunothrombosis in Sepsis. Front. Immunol. 2023, 14, 1144229. [Google Scholar] [CrossRef] [PubMed]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Dolmatova, E.V.; Wang, K.; Mandavilli, R.; Griendling, K.K. The Effects of Sepsis on Endothelium and Clinical Implications. Cardiovasc. Res. 2021, 117, 60–73. [Google Scholar] [CrossRef]
- Tuculeanu, G.; Barbu, E.C.; Lazar, M.; Chitu-Tisu, C.E.; Moisa, E.; Negoita, S.I.; Ion, D.A. Coagulation Disorders in Sepsis and COVID-19-Two Sides of the Same Coin? A Review of Inflammation-Coagulation Crosstalk in Bacterial Sepsis and COVID-19. J. Clin. Med. 2023, 12, 601. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Inflammation and Thrombosis: Roles of Neutrophils, Platelets and Endothelial Cells and Their Interactions in Thrombus Formation during Sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef]
- Zou, S.; Jie, H.; Han, X.; Wang, J. The Role of Neutrophil Extracellular Traps in Sepsis and Sepsis-Related Acute Lung Injury. Int. Immunopharmacol. 2023, 124, 110436. [Google Scholar] [CrossRef]
- Cajander, S.; Kox, M.; Scicluna, B.P.; Weigand, M.A.; Mora, R.A.; Flohé, S.B.; Martin-Loeches, I.; Lachmann, G.; Girardis, M.; Garcia-Salido, A.; et al. Profiling the Dysregulated Immune Response in Sepsis: Overcoming Challenges to Achieve the Goal of Precision Medicine. Lancet Respir. Med. 2024, 12, 305–322. [Google Scholar] [CrossRef]
- Watkins, R.R.; Bonomo, R.A.; Rello, J. Managing Sepsis in the Era of Precision Medicine: Challenges and Opportunities. Expert. Rev. Anti Infect. Ther. 2022, 20, 871–880. [Google Scholar] [CrossRef]
- Mankowski, R.T.; Anton, S.D.; Ghita, G.L.; Brumback, B.; Darden, D.B.; Bihorac, A.; Leeuwenburgh, C.; Moldawer, L.L.; Efron, P.A.; Brakenridge, S.C.; et al. Older Adults Demonstrate Biomarker Evidence of the Persistent Inflammation, Immunosuppression, and Catabolism Syndrome (PICS) After Sepsis. J. Gerontol. Ser. A 2022, 77, 188–196. [Google Scholar] [CrossRef]
- Chen, P.; Stanojcic, M.; Jeschke, M.G. Differences between Murine and Human Sepsis. Surg. Clin. N. Am. 2014, 94, 1135–1149. [Google Scholar] [CrossRef]
- Masopust, D.; Sivula, C.P.; Jameson, S.C. Of Mice, Dirty Mice, and Men: Using Mice To Understand Human Immunology. J. Immunol. 2017, 199, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Sans-Fons, M.G.; Yeramian, A.; Pereira-Lopes, S.; Santamaría-Babi, L.F.; Modolell, M.; Lloberas, J.; Celada, A. Arginine Transport Is Impaired in C57Bl/6 Mouse Macrophages as a Result of a Deletion in the Promoter of Slc7a2 (CAT2), and Susceptibility to Leishmania Infection Is Reduced. J. Infect. Dis. 2013, 207, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human Monocytes Undergo Functional Re-Programming during Sepsis Mediated by Hypoxia-Inducible Factor-1α. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Vardon-Bounes, F.; Merlet-Dupuy, V.; Conil, J.-M.; Buléon, M.; Fourcade, O.; Tack, I.; Minville, V. Sepsis Modeling in Mice: Ligation Length Is a Major Severity Factor in Cecal Ligation and Puncture. Intensive Care Med. Exp. 2016, 4, 22. [Google Scholar] [CrossRef]
- Wen, H. Sepsis Induced by Cecal Ligation and Puncture. Methods Mol. Biol. 2013, 1031, 117–124. [Google Scholar] [CrossRef]
- Chen, I.-C.; Chen, H.-H.; Jiang, Y.-H.; Hsiao, T.-H.; Ko, T.-M.; Chao, W.-C. Whole Transcriptome Analysis to Explore the Impaired Immunological Features in Critically Ill Elderly Patients with Sepsis. J. Transl. Med. 2023, 21, 141. [Google Scholar] [CrossRef]
- Caldwell, B.A.; Li, L. Epigenetic Regulation of Innate Immune Dynamics during Inflammation. J. Leukoc. Biol. 2024, 115, 589–606. [Google Scholar] [CrossRef]
- Meijer, E.M.; Koch, S.E.; van Dijk, C.G.M.; Maas, R.G.C.; Chrifi, I.; Szymczyk, W.; Besseling, P.J.; Pomp, L.; Koomen, V.J.C.H.; Buikema, J.W.; et al. 3D Human iPSC Blood Vessel Organoids as a Source of Flow-Adaptive Vascular Cells for Creating a Human-Relevant 3D-Scaffold Based Macrovessel Model. Adv. Biol. 2023, 7, e2200137. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, L.; Tong, X.; He, H.; Zheng, Y.; Xia, Z. Sepsis-Induced Endothelial Dysfunction: Permeability and Regulated Cell Death. J. Inflamm. Res. 2024, 17, 9953–9973. [Google Scholar] [CrossRef]
- Liu, D.; Langston, J.C.; Prabhakarpandian, B.; Kiani, M.F.; Kilpatrick, L.E. The Critical Role of Neutrophil-Endothelial Cell Interactions in Sepsis: New Synergistic Approaches Employing Organ-on-Chip, Omics, Immune Cell Phenotyping and in Silico Modeling to Identify New Therapeutics. Front. Cell Infect. Microbiol. 2023, 13, 1274842. [Google Scholar] [CrossRef] [PubMed]
- Osuchowski, M.F.; Ayala, A.; Bahrami, S.; Bauer, M.; Boros, M.; Cavaillon, J.-M.; Chaudry, I.H.; Coopersmith, C.M.; Deutschman, C.; Drechsler, S.; et al. Minimum Quality Threshold in Pre-Clinical Sepsis Studies (MQTiPSS): An International Expert Consensus Initiative for Improvement of Animal Modeling in Sepsis. Infection 2018, 46, 687–691. [Google Scholar] [CrossRef]
- Martin-Loeches, I.; Singer, M.; Leone, M. Sepsis: Key Insights, Future Directions, and Immediate Goals. A Review and Expert Opinion. Intensive Care Med. 2024, 50, 2043–2049. [Google Scholar] [CrossRef]
- Xiong, W.; Zhan, Y.; Xiao, R.; Liu, F. Advancing Sepsis Diagnosis and Immunotherapy Machine Learning-Driven Identification of Stable Molecular Biomarkers and Therapeutic Targets. Sci. Rep. 2025, 15, 8333. [Google Scholar] [CrossRef]
- van der Slikke, E.C.; An, A.Y.; Hancock, R.E.W.; Bouma, H.R. Exploring the Pathophysiology of Post-Sepsis Syndrome to Identify Therapeutic Opportunities. EBioMedicine 2020, 61, 103044. [Google Scholar] [CrossRef]
- Huang, C.Y.; Daniels, R.; Lembo, A.; Hartog, C.; O’Brien, J.; Heymann, T.; Reinhart, K.; Nguyen, H.B.; Sepsis Survivors Engagement Project (SSEP). Life after Sepsis: An International Survey of Survivors to Understand the Post-Sepsis Syndrome. Int. J. Qual. Health Care 2019, 31, 191–198. [Google Scholar] [CrossRef]
- Torres, J.S.S.; Tamayo-Giraldo, F.J.; Bejarano-Zuleta, A.; Nati-Castillo, H.A.; Quintero, D.A.; Ospina-Mejía, M.J.; Salazar-Santoliva, C.; Suárez-Sangucho, I.; Ortiz-Prado, E.; Izquierdo-Condoy, J.S. Sepsis and Post-Sepsis Syndrome: A Multisystem Challenge Requiring Comprehensive Care and Management—A Review. Front. Med. 2025, 12, 1560737. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.-L. Sepsis and Septic Shock. Nat. Rev. Dis. Prim. 2016, 2, 16045. [Google Scholar] [CrossRef] [PubMed]
- Divangahi, M.; Aaby, P.; Khader, S.A.; Barreiro, L.B.; Bekkering, S.; Chavakis, T.; van Crevel, R.; Curtis, N.; DiNardo, A.R.; Dominguez-Andres, J.; et al. Trained Immunity, Tolerance, Priming and Differentiation: Distinct Immunological Processes. Nat. Immunol. 2021, 22, 2–6. [Google Scholar] [CrossRef]
- Im, G.-B.; Melero-Martin, J.M. Mitochondrial Transfer in Endothelial Cells and Vascular Health. Trends Cell Biol. 2025, 1–11. [Google Scholar] [CrossRef] [PubMed]
- van der Slikke, E.C.; Beumeler, L.F.E.; Holmqvist, M.; Linder, A.; Mankowski, R.T.; Bouma, H.R. Understanding Post-Sepsis Syndrome: How Can Clinicians Help? Infect. Drug Resist. 2023, 16, 6493–6511. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; Hashimoto, K.; Yuan, S.; Zhang, J. The Gut-Liver Axis in Sepsis: Interaction Mechanisms and Therapeutic Potential. Crit. Care 2022, 26, 213. [Google Scholar] [CrossRef]
- Saavedra-Torres, J.S.; Pinzón-Fernández, M.V.; Ocampo-Posada, M.; Nati-Castillo, H.A.; Jiménez Hincapie, L.A.; Cadrazo-Gil, E.J.; Arias-Intriago, M.; Rojas-Cadena, M.; Tello-De-la-Torre, A.; Osejos, W.; et al. Inflammasomes and Signaling Pathways: Key Mechanisms in the Pathophysiology of Sepsis. Cells 2025, 14, 930. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhang, K.; Wang, Y.; Gu, Y. Comprehensive Review of Histone Lactylation: Structure, Function, and Therapeutic Targets. Biochem. Pharmacol. 2024, 225, 116331. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Du, J. Histone and Non-Histone Lactylation: Molecular Mechanisms, Biological Functions, Diseases, and Therapeutic Targets. Mol. Biomed. 2025, 6, 38. [Google Scholar] [CrossRef]
- Li, Y.; Wan, D.; Luo, X.; Song, T.; Wang, Y.; Yu, Q.; Jiang, L.; Liao, R.; Zhao, W.; Su, B. Circulating Histones in Sepsis: Potential Outcome Predictors and Therapeutic Targets. Front. Immunol. 2021, 12, 650184. [Google Scholar] [CrossRef] [PubMed]
- Srdić, T.; Đurašević, S.; Lakić, I.; Ružičić, A.; Vujović, P.; Jevđović, T.; Dakić, T.; Đorđević, J.; Tosti, T.; Glumac, S.; et al. From Molecular Mechanisms to Clinical Therapy: Understanding Sepsis-Induced Multiple Organ Dysfunction. Int. J. Mol. Sci. 2024, 25, 7770. [Google Scholar] [CrossRef]
- Caraballo, C.; Jaimes, F. Organ Dysfunction in Sepsis: An Ominous Trajectory From Infection To Death. Yale J. Biol. Med. 2019, 92, 629–640. [Google Scholar]
- Fry, D.E. Sepsis, Systemic Inflammatory Response, and Multiple Organ Dysfunction: The Mystery Continues. Am. Surg. 2012, 78, 1–8. [Google Scholar] [CrossRef]
- Qazi Arisar, F.A.; Abid, S.; Shaikh, P.A.; Awan, S. Impact of Sepsis and Non-Communicable Diseases on Prognostic Models to Predict the Outcome of Hospitalized Chronic Liver Disease Patients. World J. Hepatol. 2018, 10, 944–955. [Google Scholar] [CrossRef] [PubMed]
- DeMerle, K.M.; Angus, D.C.; Baillie, J.K.; Brant, E.; Calfee, C.S.; Carcillo, J.; Chang, C.-C.H.; Dickson, R.; Evans, I.; Gordon, A.C.; et al. Sepsis Subclasses: A Framework for Development and Interpretation. Crit. Care Med. 2021, 49, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Danahy, D.B.; Kurup, S.P.; Winborn, C.S.; Jensen, I.J.; Harty, J.T.; Griffith, T.S.; Badovinac, V.P. Sepsis-Induced State of Immunoparalysis Is Defined by Diminished CD8 T Cell-Mediated Antitumor Immunity. J. Immunol. 2019, 203, 725–735. [Google Scholar] [CrossRef]
- Danahy, D.B.; Jensen, I.J.; Griffith, T.S.; Badovinac, V.P. Cutting Edge: Polymicrobial Sepsis Has the Capacity to Reinvigorate Tumor-Infiltrating CD8 T Cells and Prolong Host Survival. J. Immunol. 2019, 202, 2843–2848. [Google Scholar] [CrossRef] [PubMed]
- Condotta, S.A.; Khan, S.H.; Rai, D.; Griffith, T.S.; Badovinac, V.P. Polymicrobial Sepsis Increases Susceptibility to Chronic Viral Infection and Exacerbates CD8+ T Cell Exhaustion. J. Immunol. 2015, 195, 116–125. [Google Scholar] [CrossRef]
- Denstaedt, S.J.; Bustamante, A.C.; Newstead, M.W.; Moore, B.B.; Standiford, T.J.; Zemans, R.L.; Singer, B.H. Long-Term Survivors of Murine Sepsis Are Predisposed to Enhanced LPS-Induced Lung Injury and Proinflammatory Immune Reprogramming. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L451–L465. [Google Scholar] [CrossRef]
- Janicova, A.; Becker, N.; Xu, B.; Wutzler, S.; Vollrath, J.T.; Hildebrand, F.; Ehnert, S.; Marzi, I.; Störmann, P.; Relja, B. Endogenous Uteroglobin as Intrinsic Anti-Inflammatory Signal Modulates Monocyte and Macrophage Subsets Distribution Upon Sepsis Induced Lung Injury. Front. Immunol. 2019, 10, 2276. [Google Scholar] [CrossRef]
- Pinzón-Fernández, M.V.; Saavedra T, J.S.; López Garzón, N.A.; Pachón Bueno, J.S.; Tamayo-Giraldo, F.J.; Rojas Gomez, M.C.; Arias-Intriago, M.; Gaibor-Pazmiño, A.; López-Cortés, A.; Izquierdo-Condoy, J.S. NLRP3 and Beyond: Inflammasomes as Central Cellular Hub and Emerging Therapeutic Target in Inflammation and Disease. Front. Immunol. 2025, 16, 1624770. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, B.; Pi, Q.; Zhou, G.; Cheng, Z.; Qu, C.; Wang, X.; Kong, L.; Luo, S.; Du, D.; et al. Deficiency of S100A8/A9 Attenuates Pulmonary Microvascular Leakage in Septic Mice. Respir. Res. 2023, 24, 288. [Google Scholar] [CrossRef]
- Xu, J.; Gao, Y.; Huang, X.; Li, J.; Sun, T.; Wang, X.; Zhao, Y.; Wang, T. S100A9 in Sepsis: A Biomarker for Inflammation and a Mediator of Organ Damage. Biochem. Biophys. Res. Commun. 2025, 752, 151484. [Google Scholar] [CrossRef] [PubMed]
- Denstaedt, S.J.; Spencer-Segal, J.L.; Newstead, M.; Laborc, K.; Zeng, X.; Standiford, T.J.; Singer, B.H. Persistent Neuroinflammation and Brain-Specific Immune Priming in a Novel Survival Model of Murine Pneumosepsis. Shock 2020, 54, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Y.; Shao, Y.; Lu, X.; Zhang, H.; Miao, C. S100A8/A9hi Neutrophils Induce Mitochondrial Dysfunction and PANoptosis in Endothelial Cells via Mitochondrial Complex I Deficiency during Sepsis. Cell Death Dis. 2024, 15, 462. [Google Scholar] [CrossRef]
- Cai, X.-Y.; Huang, G.-Q.; Zhou, Y.-M.; Li, D.-J. Targeting Calprotectin S100A8/A9 to Overcome AML Progression in DNMT3A-Mutant Cells. Curr. Med. Sci. 2025, 45, 458–468. [Google Scholar] [CrossRef]
- Wang, H.; Wang, M.; Chen, J.; Hou, H.; Guo, Z.; Yang, H.; Tang, H.; Chen, B. Interleukin-36 Is Overexpressed in Human Sepsis and IL-36 Receptor Deletion Aggravates Lung Injury and Mortality through Epithelial Cells and Fibroblasts in Experimental Murine Sepsis. Crit. Care 2023, 27, 490. [Google Scholar] [CrossRef]
- Scheibe, K.; Backert, I.; Wirtz, S.; Hueber, A.; Schett, G.; Vieth, M.; Probst, H.C.; Bopp, T.; Neurath, M.F.; Neufert, C. IL-36R Signalling Activates Intestinal Epithelial Cells and Fibroblasts and Promotes Mucosal Healing in Vivo. Gut 2017, 66, 823–838. [Google Scholar] [CrossRef]
- Wu, C.; Ma, J.; Yang, H.; Zhang, J.; Sun, C.; Lei, Y.; Liu, M.; Cao, J. Interleukin-37 as a Biomarker of Mortality Risk in Patients with Sepsis. J. Infect. 2021, 82, 346–354. [Google Scholar] [CrossRef]
- Wang, A.; Zhang, S.; Peng, G.; Tang, Y.; Yang, Y. ICU and Sepsis: Role of Myeloid and Lymphocyte Immune Cells. J. Oncol. 2022, 2022, 7340266. [Google Scholar] [CrossRef] [PubMed]
- Schrijver, D.P.; Röring, R.J.; Deckers, J.; de Dreu, A.; Toner, Y.C.; Prevot, G.; Priem, B.; Munitz, J.; Nugraha, E.G.; van Elsas, Y.; et al. Resolving Sepsis-Induced Immunoparalysis via Trained Immunity by Targeting Interleukin-4 to Myeloid Cells. Nat. Biomed. Eng. 2023, 7, 1097–1112. [Google Scholar] [CrossRef]
- Dong, C. Cytokine Regulation and Function in T Cells. Annu. Rev. Immunol. 2021, 39, 51–76. [Google Scholar] [CrossRef]
- Reddy, H.; Javvaji, C.K.; Malali, S.; Kumar, S.; Acharya, S.; Toshniwal, S. Navigating the Cytokine Storm: A Comprehensive Review of Chemokines and Cytokines in Sepsis. Cureus 2024, 16, e54275. [Google Scholar] [CrossRef]
- Jing, D.; Xiong, L.; Zhang, R.; Fang, H.; Chen, L. Esmolol Improves Sepsis Outcomes through Cardiovascular and Immune Modulation. Front. Pharmacol. 2025, 16, 1498227. [Google Scholar] [CrossRef]
- Sanfilippo, F.; Santonocito, C.; Morelli, A.; Foex, P. Beta-Blocker Use in Severe Sepsis and Septic Shock: A Systematic Review. Curr. Med. Res. Opin. 2015, 31, 1817–1825. [Google Scholar] [CrossRef]
- He, J.; Yang, J.; Liu, J. Early Heart Rate Fluctuation and Outcomes in Critically Ill Patients with Sepsis: A Retrospective Cohort Study of the MIMIC-IV Database. Heliyon 2023, 9, e20898. [Google Scholar] [CrossRef]
- Ning, Y.-L.; Li, W.-J.; Lu, X.; Zhang, Y.; Zhang, J.-W.; Zhou, J.-H. Association between Heart Rate and Mortality in Patients with Septic Shock: An Analysis Revealed by Time Series Data. BMC Infect. Dis. 2024, 24, 1088. [Google Scholar] [CrossRef] [PubMed]
- Iampietro, M.; Amurri, L.; Reynard, O.; Bukreyev, A. Interplay of Ebola Virus With Immune Cells Leading to Their Death by Diverse Mechanisms. J. Infect. Dis. 2023, 228, S582–S586. [Google Scholar] [CrossRef] [PubMed]
- Danahy, D.B.; Anthony, S.M.; Jensen, I.J.; Hartwig, S.M.; Shan, Q.; Xue, H.-H.; Harty, J.T.; Griffith, T.S.; Badovinac, V.P. Polymicrobial Sepsis Impairs Bystander Recruitment of Effector Cells to Infected Skin despite Optimal Sensing and Alarming Function of Skin Resident Memory CD8 T Cells. PLoS Pathog. 2017, 13, e1006569. [Google Scholar] [CrossRef]
- Ficenec, S.; Bond, N.; Zifodya, J.; Schieffelin, J. A Systematic Review of the Immuno-Inflammatory Dysfunction Secondary to Viral Hemorrhagic Fevers; Ebola and Lassa Fever. PLoS Negl. Trop. Dis. 2025, 19, e0013230. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Mahale, P.; Engels, E.A. Sepsis and Risk of Cancer Among Elderly Adults in the United States. Clin. Infect. Dis. 2019, 68, 717–724. [Google Scholar] [CrossRef]
- Hästbacka, J.; But, A.; Strandberg, G.; Lipcsey, M. Risk of Malignant Disease in 1-Year Sepsis Survivors, a Registry-Based Nationwide Follow-up Study. Crit. Care 2023, 27, 376. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Colston, E.; Yende, S.; Angus, D.C.; Moldawer, L.L.; Crouser, E.D.; Martin, G.S.; Coopersmith, C.M.; Brakenridge, S.; Mayr, F.B.; et al. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized, Placebo-Controlled, Single Ascending Dose Study of Antiprogrammed Cell Death-Ligand 1 Antibody (BMS-936559). Crit. Care Med. 2019, 47, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Heipertz, E.L.; Walker, W.E. Creation of BLT Humanized Mice for Sepsis Studies. Methods Mol. Biol. 2021, 2321, 137–154. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Colston, E.; Yende, S.; Crouser, E.D.; Martin, G.S.; Albertson, T.; Bartz, R.R.; Brakenridge, S.C.; Delano, M.J.; Park, P.K.; et al. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Nivolumab. Intensive Care Med. 2019, 45, 1360–1371. [Google Scholar] [CrossRef]
- Laudanski, K. Humanized Mice as a Tool to Study Sepsis-More Than Meets the Eye. Int. J. Mol. Sci. 2021, 22, 2403. [Google Scholar] [CrossRef] [PubMed]
- Murrieta-Coxca, J.M.; Rodríguez-Martínez, S.; Cancino-Diaz, M.E.; Markert, U.R.; Favaro, R.R.; Morales-Prieto, D.M. IL-36 Cytokines: Regulators of Inflammatory Responses and Their Emerging Role in Immunology of Reproduction. Int. J. Mol. Sci. 2019, 20, 1649. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).