
Pathophysiology of Prediabetes Hyperinsulinemia and Insulin Resistance in the Cardiovascular System



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
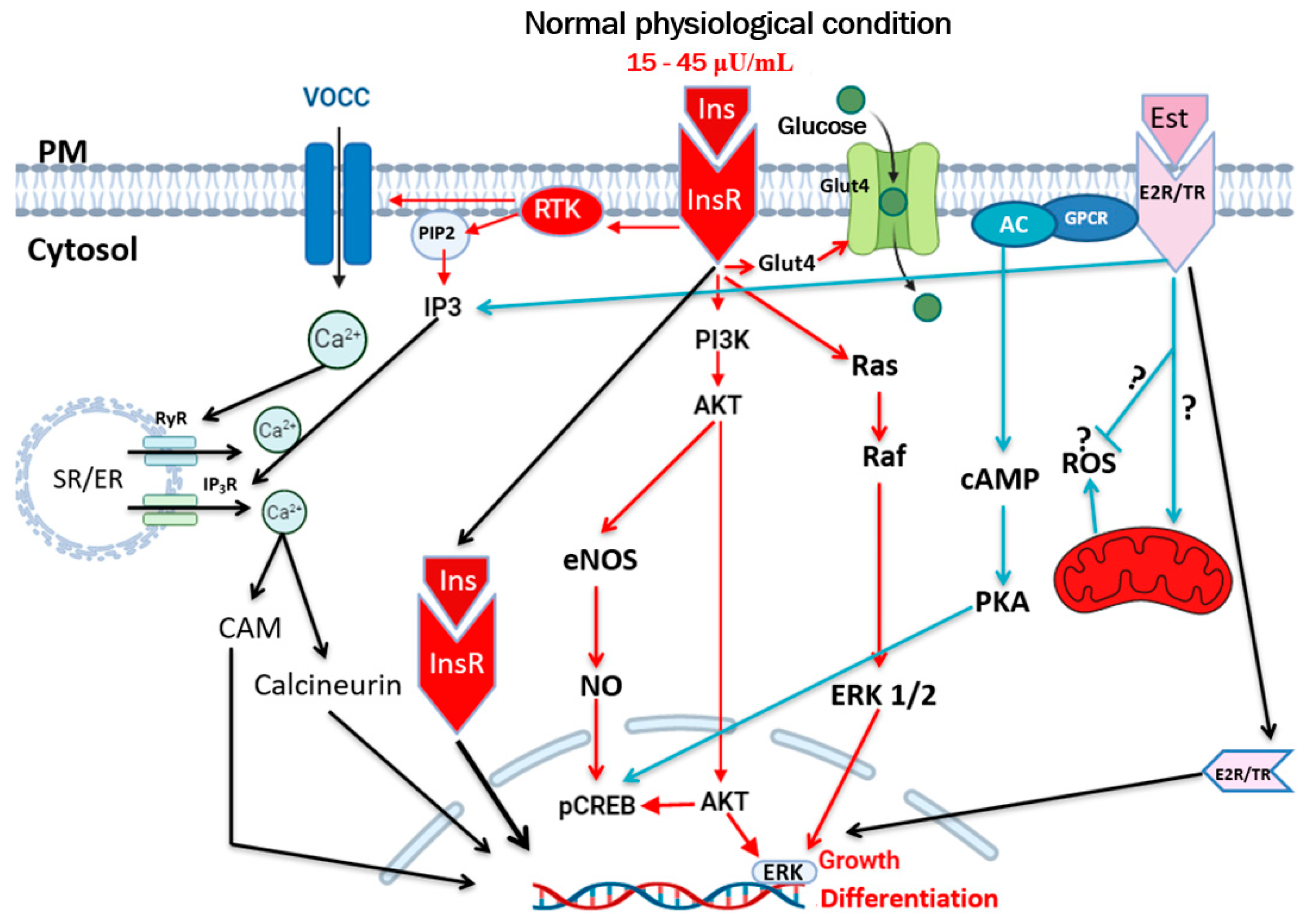
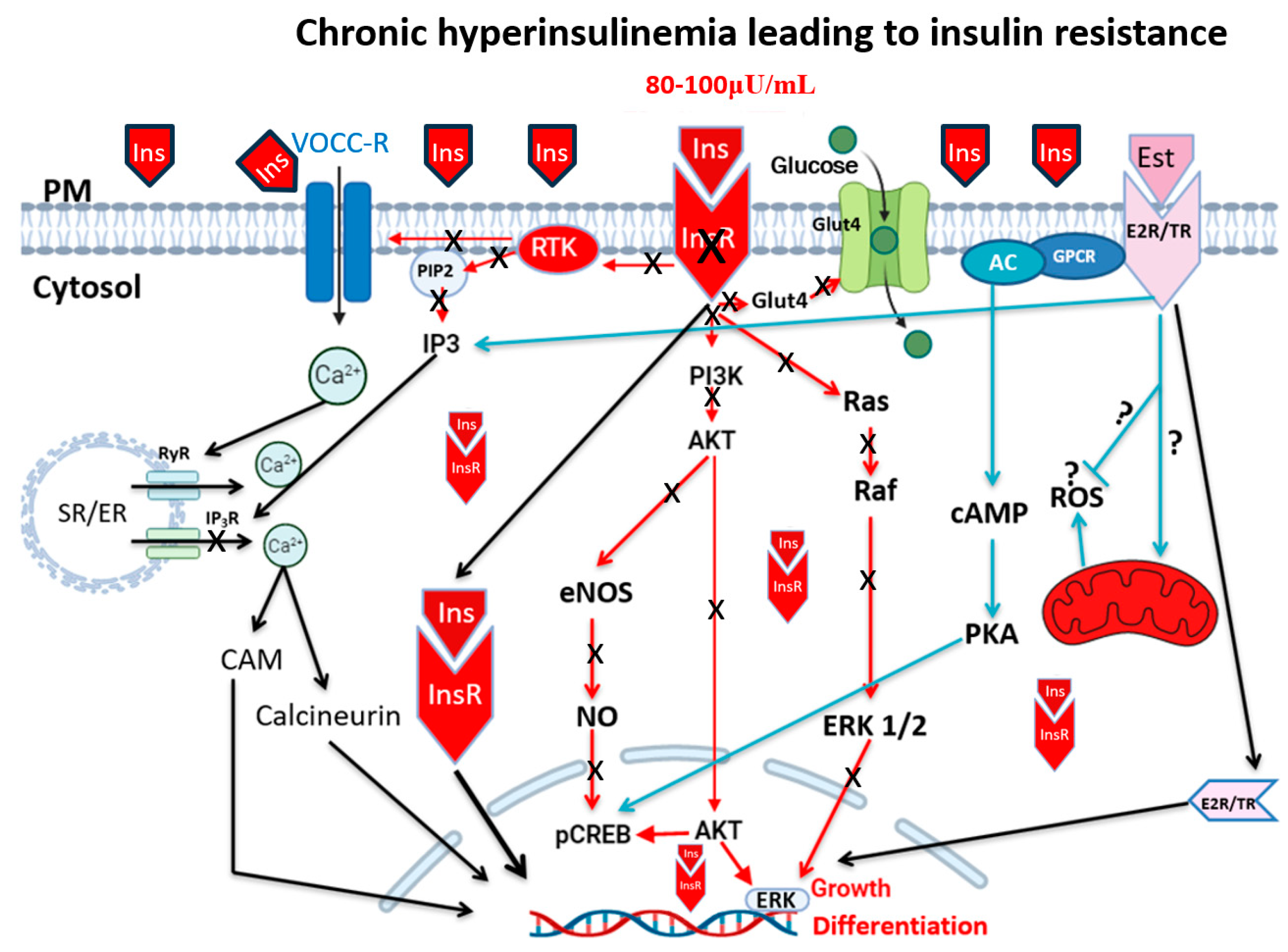
2. Hyperinsulinemia, Insulin Resistance, and Type 2 Diabetes
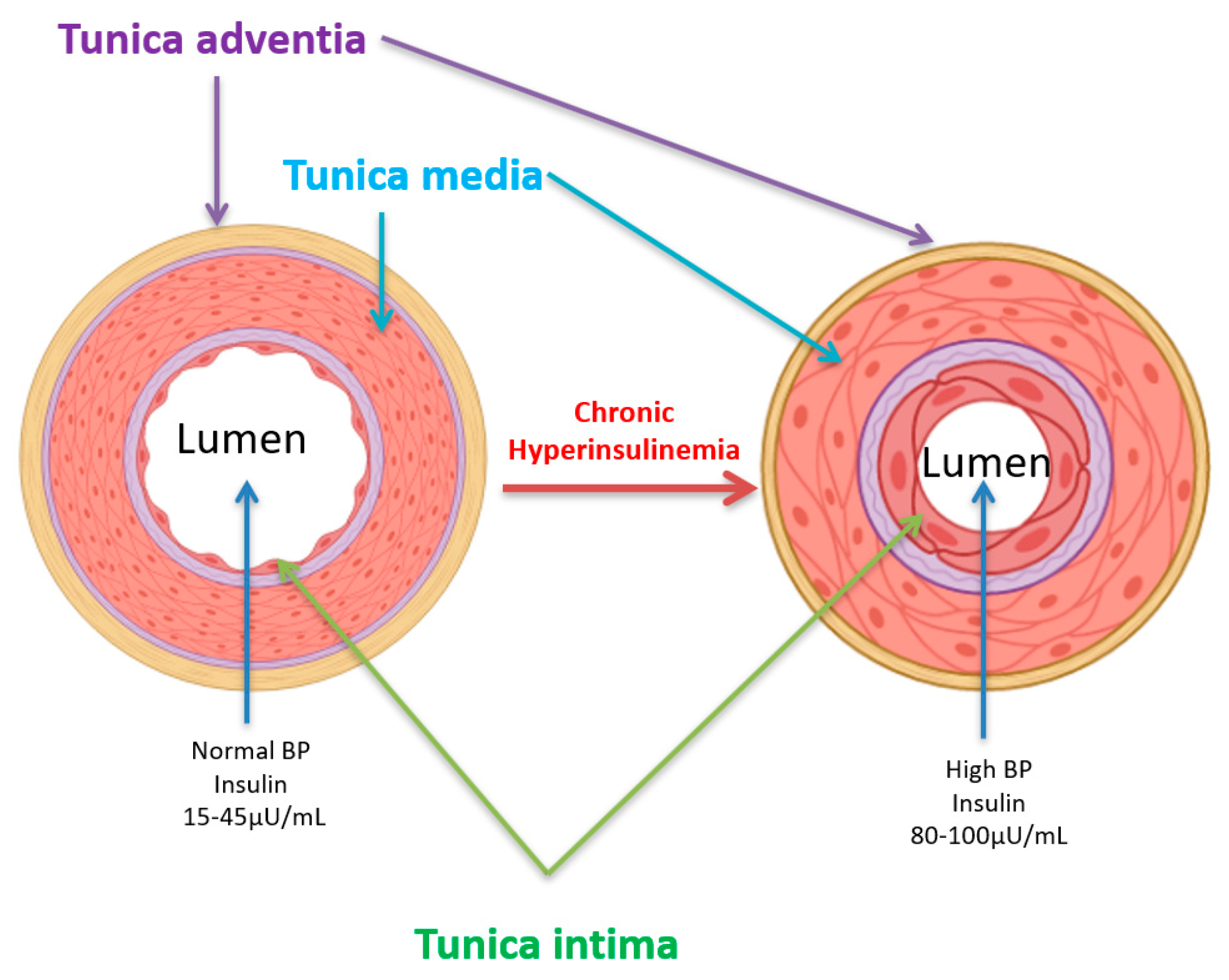
3. Hyperinsulinemia and Vascular Diseases
4. Hyperinsulinemia, Hypertension, and Atherosclerosis
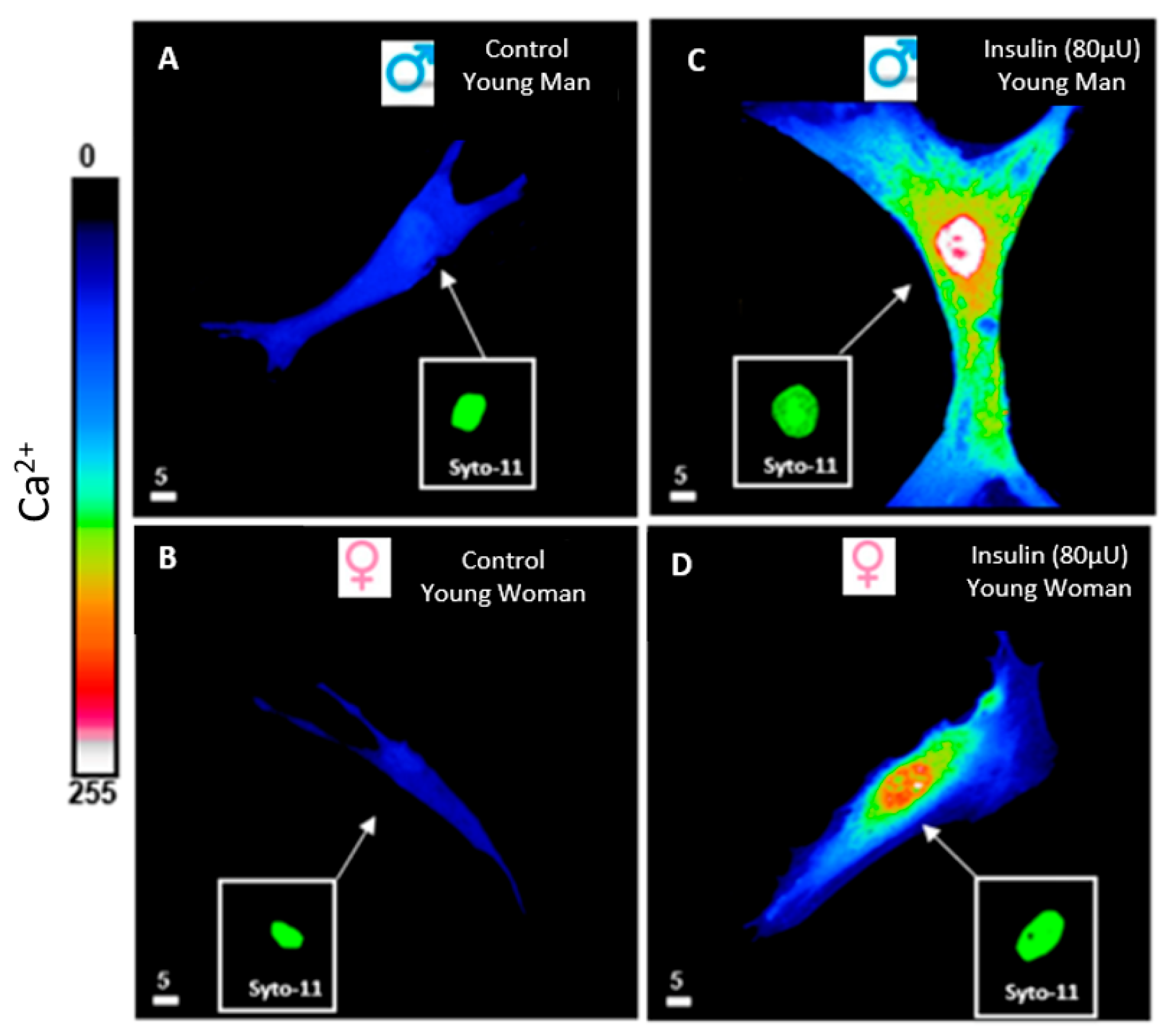
5. Hyperinsulinemia and Calcium Ionic Transporters
6. Hyperinsulinemia and Reactive Oxygen Species (ROS)
7. Discussion, Conclusions, and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Echouffo-Tcheugui, J.B.; Selvin, E. Prediabetes and What It Means: The Epidemiological Evidence. Annu. Rev. Public Health 2021, 42, 59–77. [Google Scholar] [CrossRef]
- Tabak, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimaki, M. Prediabetes: A high-risk state for diabetes development. Lancet 2012, 379, 2279–2290. [Google Scholar] [CrossRef]
- Papaetis, G.S.; Picolos, M.K.; Sacharidou, A. Pioglitazone with SGLT2 inhibitors or GLP-1 receptor agonists in patients with type 2 diabetes and non-alcoholic fatty liver disease: Could the combinations of an old friend with new players yield better outcomes? Arch. Med. Sci. Atheroscler. Dis. 2025, 10, e1–e15. [Google Scholar] [CrossRef] [PubMed]
- Despres, J.P. Body fat distribution and risk of cardiovascular disease: An update. Circulation 2012, 126, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Cecerska-Heryc, E.; Engwert, W.; Michalow, J.; Marciniak, J.; Birger, R.; Serwin, N.; Heryc, R.; Polikowska, A.; Goszka, M.; Wojciuk, B.; et al. Oxidative stress markers and inflammation in type 1 and 2 diabetes are affected by BMI, treatment type, and complications. Sci. Rep. 2025, 15, 23605. [Google Scholar] [CrossRef]
- Sodero, G.; Rigante, D.; Pane, L.C.; Sessa, L.; Quarta, L.; Candelli, M.; Cipolla, C. Cardiometabolic Risk Assessment in a Cohort of Children and Adolescents Diagnosed with Hyperinsulinemia. Diseases 2024, 12, 119. [Google Scholar] [CrossRef]
- Snider, K.E.; Becker, S.; Boyajian, L.; Shyng, S.L.; MacMullen, C.; Hughes, N.; Ganapathy, K.; Bhatti, T.; Stanley, C.A.; Ganguly, A. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J. Clin. Endocrinol. Metab. 2013, 98, E355–E363. [Google Scholar] [CrossRef]
- Janssen, J. Overnutrition, Hyperinsulinemia and Ectopic Fat: It Is Time for A Paradigm Shift in the Management of Type 2 Diabetes. Int. J. Mol. Sci. 2024, 25, 5488. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.; DeFronzo, R.A. Fasting Hyperinsulinemia and Obesity: Cause or Effect. J. Clin. Endocrinol. Metab. 2023, 108, e1151–e1152. [Google Scholar] [CrossRef]
- Arunagiri, A.; Haataja, L.; Pottekat, A.; Pamenan, F.; Kim, S.; Zeltser, L.M.; Paton, A.W.; Paton, J.C.; Tsai, B.; Itkin-Ansari, P.; et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. eLife 2019, 8, e44532. [Google Scholar] [CrossRef]
- Semple, R.K.; Savage, D.B.; Cochran, E.K.; Gorden, P.; O’Rahilly, S. Genetic syndromes of severe insulin resistance. Endocr. Rev. 2011, 32, 498–514. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Skrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. S2), S262–S268. [Google Scholar] [CrossRef]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, M.A.; Hannan, M.A.; Uddin, M.J.; Pang, M.G. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gay, M.A.; Ferraz-Amaro, I.; Castaneda, S.; Pinto Tasende, J.A.; Uriarte-Ecenarro, M.; Plaza, Z.; Sanchez-Alonso, F.; Garcia Gomez, C.; Gonzalez-Juanatey, C.; Llorca, J.; et al. The metabolic score for insulin resistance predicts the risk of cardiovascular disease in patients with psoriatic arthritis: Results from the 10-year prospective CARMA cohort. RMD Open 2025, 11, e005352. [Google Scholar] [CrossRef] [PubMed]
- Fazio, S.; Bellavite, P.; Affuso, F. Chronically Increased Levels of Circulating Insulin Secondary to Insulin Resistance: A Silent Killer. Biomedicines 2024, 12, 2416. [Google Scholar] [CrossRef]
- Accili, D.; Deng, Z.; Liu, Q. Insulin resistance in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2025, 21, 413–426. [Google Scholar] [CrossRef]
- Crofts, C.A.P.; Zinn, C.; Wheldon, M.; Schofield, G. Hyperinsulinemia: A unifying theory of chronic disease? Diabesity 2015, 1, 34–43. [Google Scholar] [CrossRef]
- Jazzar, A.; Jacques, D.; Abou-Aichi, A.; Bkaily, G. In Vitro Chronic Hyperinsulinemia Induces Remodelling of Vascular Smooth Muscle Cells from Young Men and Women in a Sex Hormone Independent Manner. Pathophysiology 2025, 32, 12. [Google Scholar] [CrossRef]
- Jia, G.; DeMarco, V.G.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2016, 12, 144–153. [Google Scholar] [CrossRef]
- Jha, D.; Prajapati, S.K.; Deb, P.K.; Jaiswal, M.; Mazumder, P.M. Madhuca longifolia-hydro-ethanolic-fraction reverses mitochondrial dysfunction and modulates selective GLUT expression in diabetic mice fed with high fat diet. Mol. Biol. Rep. 2024, 51, 209. [Google Scholar] [CrossRef]
- Bkaily, G.; Jacques, D. Calcium Homeostasis, Transporters, and Blockers in Health and Diseases of the Cardiovascular System. Int. J. Mol. Sci. 2023, 24, 8803. [Google Scholar] [CrossRef]
- Freychet, P.; Laudat, M.H.; Laudat, P.; Rosselin, G.; Kahn, C.R.; Gorden, P.; Roth, J. Impairment of insulin binding to the fat cell plasma membrane in the obese hyperglycemic mouse. FEBS Lett. 1972, 25, 339–342. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P. The Insulin Receptor and Its Signal Transduction Network. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Le, T.K.C.; Dao, X.D.; Nguyen, D.V.; Luu, D.H.; Bui, T.M.H.; Le, T.H.; Nguyen, H.T.; Le, T.N.; Hosaka, T.; Nguyen, T.T.T. Insulin signaling and its application. Front. Endocrinol. 2023, 14, 1226655. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of Hyperinsulinemia and Insulin Resistance in Hypertension: Metabolic Syndrome Revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef]
- Perng, W.; Kelsey, M.M.; Sauder, K.A.; Dabelea, D. How does exposure to overnutrition in utero lead to childhood adiposity? Testing the insulin hypersecretion hypothesis in the EPOCH cohort. Diabetologia 2021, 64, 2237–2246. [Google Scholar] [CrossRef]
- Marfella, R.; Amico, M.D.; Di Filippo, C.; Siniscalchi, M.; Sasso, F.C.; Ferraraccio, F.; Rossi, F.; Paolisso, G. The possible role of the ubiquitin proteasome system in the development of atherosclerosis in diabetes. Cardiovasc. Diabetol. 2007, 6, 35. [Google Scholar] [CrossRef]
- Jazzar, A.; Jacques, D.; Bkaily, G. Insulin-Induced Cardiomyocytes Hypertrophy That Is Prevented by Taurine via beta-alanine-Sensitive Na+-Taurine Symporter. Nutrients 2021, 13, 3686. [Google Scholar] [CrossRef]
- Lustig, R.H. Ultraprocessed Food: Addictive, Toxic, and Ready for Regulation. Nutrients 2020, 12, 3401. [Google Scholar] [CrossRef] [PubMed]
- Dabelea, D.; Crume, T. Maternal environment and the transgenerational cycle of obesity and diabetes. Diabetes 2011, 60, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Contractor, J.B.; Radha, V.; Shah, K.; Singh, P.; Tadepalli, S.; Nimbalkar, S.; Mohan, V.; Shah, P. Congenital Hyperinsulinism India Association: An Approach to Address the Challenges and Opportunities of a Rare Disease. Med. Sci. 2025, 13, 37. [Google Scholar] [CrossRef]
- Maly, J.; Urbanova, J.; Musil, V.; Broz, J.; Cerny, M.; Brunerova, L. Neonatal hypoglycaemia in the offsprings of parents with maturity-onset diabetes of the young (MODY). J. Pediatr. Endocrinol. Metab. 2025, 38, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N.; McConnachie, A.; Ford, I.; Gaw, A.; Cleland, S.J.; Forouhi, N.G.; McFarlane, P.; Shepherd, J.; Cobbe, S.; Packard, C. Serial metabolic measurements and conversion to type 2 diabetes in the west of Scotland coronary prevention study: Specific elevations in alanine aminotransferase and triglycerides suggest hepatic fat accumulation as a potential contributing factor. Diabetes 2007, 56, 984–991. [Google Scholar] [CrossRef]
- Jeon, Y.G.; Kim, Y.Y.; Lee, G.; Kim, J.B. Physiological and pathological roles of lipogenesis. Nat. Metab. 2023, 5, 735–759. [Google Scholar] [CrossRef]
- Kolb, H.; Stumvoll, M.; Kramer, W.; Kempf, K.; Martin, S. Insulin translates unfavourable lifestyle into obesity. BMC Med. 2018, 16, 232. [Google Scholar] [CrossRef]
- Heurtebize, M.A.; Faillie, J.L. Drug-induced hyperglycemia and diabetes. Therapie 2024, 79, 221–238. [Google Scholar] [CrossRef]
- Mendez-Barbero, N.; Gutierrez-Munoz, C.; Blanco-Colio, L.M. Cellular Crosstalk between Endothelial and Smooth Muscle Cells in Vascular Wall Remodeling. Int. J. Mol. Sci. 2021, 22, 7284. [Google Scholar] [CrossRef]
- Tan, C.M.J.; Lewandowski, A.J. The Transitional Heart: From Early Embryonic and Fetal Development to Neonatal Life. Fetal Diagn. Ther. 2020, 47, 373–386. [Google Scholar] [CrossRef]
- Gold, K.; Gaharwar, A.K.; Jain, A. Emerging trends in multiscale modeling of vascular pathophysiology: Organ-on-a-chip and 3D printing. Biomaterials 2019, 196, 2–17. [Google Scholar] [CrossRef]
- Camasao, D.B.; Mantovani, D. The mechanical characterization of blood vessels and their substitutes in the continuous quest for physiological-relevant performances. A critical review. Mater. Today Bio 2021, 10, 100106. [Google Scholar] [CrossRef] [PubMed]
- Bkaily, G.; Abou Abdallah, N.; Simon, Y.; Jazzar, A.; Jacques, D. Vascular smooth muscle remodeling in health and disease. Can. J. Physiol. Pharmacol. 2021, 99, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Reboussin, D.M.; Allen, N.B.; Griswold, M.E.; Guallar, E.; Hong, Y.; Lackland, D.T.; Miller, E.P.R., 3rd; Polonsky, T.; Thompson-Paul, A.M.; Vupputuri, S. Systematic Review for the 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, e116–e135. [Google Scholar] [PubMed]
- Chen, Z.; Li, Q.; Xu, T.; Zhou, X.; Shu, Y.; Guo, T.; Liang, F. An updated network meta-analysis of non-pharmacological interventions for primary hypertension in adults: Insights from recent studies. Syst. Rev. 2024, 13, 318. [Google Scholar] [CrossRef]
- Manosroi, W.; Williams, G.H. Genetics of Human Primary Hypertension: Focus on Hormonal Mechanisms. Endocr. Rev. 2019, 40, 825–856. [Google Scholar] [CrossRef]
- Azizi, Z.; Alipour, P.; Raparelli, V.; Norris, C.M.; Pilote, L. The role of sex and gender in hypertension. J. Hum. Hypertens. 2023, 37, 589–595. [Google Scholar] [CrossRef]
- Tousoulis, D.; Simopoulou, C.; Papageorgiou, N.; Oikonomou, E.; Hatzis, G.; Siasos, G.; Tsiamis, E.; Stefanadis, C. Endothelial dysfunction in conduit arteries and in microcirculation. Novel therapeutic approaches. Pharmacol. Ther. 2014, 144, 253–267. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Bousvarou, M.D.; Kostara, C.E.; Papakonstantinou, E.J.; Salamou, E.; Guzman, E. Insulin resistance and cardiovascular disease. J. Int. Med. Res. 2023, 51, 3000605231164548. [Google Scholar] [CrossRef]
- Tagi, V.M.; Mainieri, F.; Chiarelli, F. Hypertension in Patients with Insulin Resistance: Etiopathogenesis and Management in Children. Int. J. Mol. Sci. 2022, 23, 5814. [Google Scholar] [CrossRef]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights Into Pathogenesis and Treatment. Front. Pharmacol. 2021, 12, 787541. [Google Scholar] [CrossRef]
- Saxena, T.; Ali, A.O.; Saxena, M. Pathophysiology of essential hypertension: An update. Expert Rev. Cardiovasc. Ther. 2018, 16, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Role of insulin resistance in human disease (syndrome X): An expanded definition. Annu. Rev. Med. 1993, 44, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Mancusi, C.; Izzo, R.; di Gioia, G.; Losi, M.A.; Barbato, E.; Morisco, C. Insulin Resistance the Hinge Between Hypertension and Type 2 Diabetes. High Blood Press. Cardiovasc. Prev. 2020, 27, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Antkiewicz-Michaluk, L. Receptor and voltage-operated ion channels in the central nervous system. Pol. J. Pharmacol. 1995, 47, 253–264. [Google Scholar]
- Gok, C.; Main, A.; Gao, X.; Kerekes, Z.; Plain, F.; Kuo, C.W.; Robertson, A.D.; Fraser, N.J.; Fuller, W. Insights into the molecular basis of the palmitoylation and depalmitoylation of NCX1. Cell Calcium 2021, 97, 102408. [Google Scholar] [CrossRef]
- Orlowski, J.; Grinstein, S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflugers Arch. 2004, 447, 549–565. [Google Scholar] [CrossRef]
- Dibrov, P.; Fliegel, L. Comparative molecular analysis of Na+/H+ exchangers: A unified model for Na+/H+ antiport? FEBS Lett. 1998, 424, 1–5. [Google Scholar] [CrossRef]
- Nikolovska, K.; Seidler, U.E.; Stock, C. The Role of Plasma Membrane Sodium/Hydrogen Exchangers in Gastrointestinal Functions: Proliferation and Differentiation, Fluid/Electrolyte Transport and Barrier Integrity. Front. Physiol. 2022, 13, 899286. [Google Scholar] [CrossRef]
- Al Kury, L.T. Calcium Homeostasis in Ventricular Myocytes of Diabetic Cardiomyopathy. J. Diabetes Res. 2020, 2020, 1942086. [Google Scholar] [CrossRef]
- Jaque-Fernandez, F.; Beaulant, A.; Berthier, C.; Monteiro, L.; Allard, B.; Casas, M.; Rieusset, J.; Jacquemond, V. Preserved Ca2+ handling and excitation-contraction coupling in muscle fibres from diet-induced obese mice. Diabetologia 2020, 63, 2471–2481. [Google Scholar] [CrossRef] [PubMed]
- Bean, B.P. Two kinds of calcium channels in canine atrial cells. Differences in kinetics, selectivity, and pharmacology. J. Gen. Physiol. 1985, 86, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Hosey, M.M.; Lazdunski, M. Calcium channels: Molecular pharmacology, structure and regulation. J. Membr. Biol. 1988, 104, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Harding, E.K.; Boivin, B.; Salter, M.W. Intracellular Calcium Responses Encode Action Potential Firing in Spinal Cord Lamina I Neurons. J. Neurosci. 2020, 40, 4439–4456. [Google Scholar] [CrossRef]
- Bkaily, G. Regulation of Ca2+ channels in VSM by monocyte-released factors. In Ionic Channles in Vascular Smooth Muscle; Bkaily, G., Ed.; R.G. Landes Company: Austin, TX, USA, 1994; pp. 53–64. [Google Scholar]
- Kawano, S.; DeHaan, R.L. Low-threshold current is major calcium current in chick ventricle cells. Am. J. Physiol. 1989, 256 Pt 2, H1505–H1508. [Google Scholar] [CrossRef]
- Ghosh, D.; Syed, A.U.; Prada, M.P.; Nystoriak, M.A.; Santana, L.F.; Nieves-Cintron, M.; Navedo, M.F. Calcium Channels in Vascular Smooth Muscle. Adv. Pharmacol. 2017, 78, 49–87. [Google Scholar]
- Bkaily, G.; Economos, D.; Potvin, L.; Ardilouze, J.L.; Marriott, C.; Corcos, J.; Bonneau, D.; Fong, C.N. Blockade of insulin sensitive steady-state R-type Ca2+ channel by PN 200-110 in heart and vascular smooth muscle. Mol. Cell Biochem. 1992, 117, 93–106. [Google Scholar] [CrossRef]
- Bkaily, G.; Pothier, P.; D’Orléans-Juste, P.; Simaan, M.; Jacques, D.; Jaalouk, D.; Belzile, F.; Hassan, G.; Boutin, C.; Haddad, G.; et al. The use of confocal microscopy in the investigation of cell structure and function in the heart, vascular endothelium and smooth muscle cells. Mol. Cell Biochem. 1997, 172, 171–194. [Google Scholar] [CrossRef]
- Bkaily, G. Single heart cells as models for studying cardiac toxicology. In In Vitro Methods in Toxicology; Jolles, G., Cordier, A., Eds.; Academic Press: London, UK, 1992; pp. 289–334. [Google Scholar]
- Gok, C.; Plain, F.; Robertson, A.D.; Howie, J.; Baillie, G.S.; Fraser, N.J.; Fuller, W. Dynamic Palmitoylation of the Sodium-Calcium Exchanger Modulates Its Structure, Affinity for Lipid-Ordered Domains, and Inhibition by XIP. Cell Rep. 2020, 31, 107697. [Google Scholar] [CrossRef]
- Rout, P.; Jialal, I. Hyperphosphatemia. In StatPearls; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Sipido, K.R.; Volders, P.G.; Schoenmakers, M.; De Groot, S.H.; Verdonck, F.; Vos, M.A. Role of the Na/Ca exchanger in arrhythmias in compensated hypertrophy. Ann. N. Y. Acad. Sci. 2002, 976, 438–445. [Google Scholar] [CrossRef]
- Koster, S.; Pavkov-Keller, T.; Kuhlbrandt, W.; Yildiz, O. Structure of human Na+/H+ exchanger NHE1 regulatory region in complex with calmodulin and Ca2+. J. Biol. Chem. 2011, 286, 40954–40961. [Google Scholar] [CrossRef]
- Deisl, C.; Albano, G.; Fuster, D.G. Role of Na/H exchange in insulin secretion by islet cells. Curr. Opin. Nephrol. Hypertens. 2014, 23, 406–410. [Google Scholar] [CrossRef]
- Bkaily, G.; Avedanian, L.; Jacques, D. Nuclear membrane receptors and channels as targets for drug development in cardiovascular diseases. Can. J. Physiol. Pharmacol. 2009, 87, 108–119. [Google Scholar] [CrossRef]
- Bkaily, G.; Avedanian, L.; Al-Khoury, J.; Provost, C.; Nader, M.; D’Orléans-Juste, P.; Jacques, D. Nuclear membrane receptors for ET-1 in cardiovascular function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R251–R263. [Google Scholar] [CrossRef]
- Bkaily, G.; Avedanian, L.; Al-Khoury, J.; Ahmarani, L.; Perreault, C.; Jacques, D. Receptors and ionic transporters in nuclear membranes: New targets for therapeutical pharmacological interventions. Can. J. Physiol. Pharmacol. 2012, 90, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Sun, W. Insulin may promote SARS-CoV-2 cell entry and replication in diabetes patients. Med. Hypotheses 2023, 170, 110997. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Shen, Z.; Lu, Z. Increases in the expression of Na+/H+ exchanger 1 and 3 are associated with insulin signalling in the ruminal epithelium. J. Anim. Physiol. Anim. Nutr. 2018, 102, e569–e577. [Google Scholar] [CrossRef] [PubMed]
- Al-Shamasi, A.A.; Elkaffash, R.; Mohamed, M.; Rayan, M.; Al-Khater, D.; Gadeau, A.P.; Ahmed, R.; Hasan, A.; Eldassouki, H.; Yalcin, H.C.; et al. Crosstalk between Sodium-Glucose Cotransporter Inhibitors and Sodium-Hydrogen Exchanger 1 and 3 in Cardiometabolic Diseases. Int. J. Mol. Sci. 2021, 22, 12677. [Google Scholar] [CrossRef]
- Cojic, M.M.; Klisic, A.; Kocic, R.; Veljkovic, A.; Kocic, G. Data-Driven Cluster Analysis of Oxidative Stress Indexes in relation to Vitamin D Level, Age, and Metabolic Control in Patients with Type 2 Diabetes on Metformin Therapy. Oxid. Med. Cell Longev. 2021, 2021, 7942716. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Mechanistic Insight into Oxidative Stress-Triggered Signaling Pathways and Type 2 Diabetes. Molecules 2022, 27, 950. [Google Scholar] [CrossRef]
- Roy, B. Pathophysiological Mechanisms of Diabetes-Induced Macrovascular and Microvascular Complications: The Role of Oxidative Stress. Med. Sci. 2025, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Bkaily, G.; Jazzar, A.; Normand, A.; Simon, Y.; Al-Khoury, J.; Jacques, D. Taurine and cardiac disease: State of the art and perspectives. Can. J. Physiol. Pharmacol. 2020, 98, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, N.; Wolf, A.M.; Yokota, T.; Nito, C.; Takahashi, H.; Ohta, S. Transgenic type2 diabetes mouse models for in vivo redox measurement of hepatic mitochondrial oxidative stress. Biochim. Biophys. Acta Gen. Subj. 2023, 1867, 130302. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M. Exercise Amaliorates Metabolic Disturbances and Oxidative Stress in Diabetic Cardiomyopathy: Possible Underlying Mechanisms. Adv. Exp. Med. Biol. 2017, 999, 207–230. [Google Scholar]
- Gordeeva, A.V.; Zvyagilskaya, R.A.; Labas, Y.A. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry 2003, 68, 1077–1080. [Google Scholar] [CrossRef]
- Putney, J.W., Jr. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Wilkins, B.J.; Molkentin, J.D. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2004, 322, 1178–1191. [Google Scholar] [CrossRef]
- Choi, Y.S.; Lee, B.; Cho, H.Y.; Reyes, I.B.; Pu, X.A.; Saido, T.C.; Hoyt, K.R.; Obrietan, K. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington’s disease. Neurobiol. Dis. 2009, 36, 259–268. [Google Scholar] [CrossRef]
- Kautzky-Willer, A.; Leutner, M.; Harreiter, J. Sex differences in type 2 diabetes. Diabetologia 2023, 66, 986–1002. [Google Scholar] [CrossRef]
- Ayer, A.; Fazakerley, D.J.; James, D.E.; Stocker, R. The role of mitochondrial reactive oxygen species in insulin resistance. Free Radic. Biol. Med. 2022, 179, 339–362. [Google Scholar] [CrossRef]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef]
- Perszyk, R.E.; Zheng, Z.; Banke, T.G.; Zhang, J.; Xie, L.; McDaniel, M.J.; Katzman, B.M.; Pelly, S.C.; Yuan, H.; Liotta, D.C.; et al. The Negative Allosteric Modulator EU1794-4 Reduces Single-Channel Conductance and Ca2+ Permeability of GluN1/GluN2A N-Methyl-d-Aspartate Receptors. Mol. Pharmacol. 2021, 99, 399–411. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bkaily, G.; Jazzar, A.; Abou-Aichi, A.; Jacques, D. Pathophysiology of Prediabetes Hyperinsulinemia and Insulin Resistance in the Cardiovascular System. Biomedicines 2025, 13, 1842. https://doi.org/10.3390/biomedicines13081842
Bkaily G, Jazzar A, Abou-Aichi A, Jacques D. Pathophysiology of Prediabetes Hyperinsulinemia and Insulin Resistance in the Cardiovascular System. Biomedicines. 2025; 13(8):1842. https://doi.org/10.3390/biomedicines13081842
Chicago/Turabian StyleBkaily, Ghassan, Ashley Jazzar, Amira Abou-Aichi, and Danielle Jacques. 2025. "Pathophysiology of Prediabetes Hyperinsulinemia and Insulin Resistance in the Cardiovascular System" Biomedicines 13, no. 8: 1842. https://doi.org/10.3390/biomedicines13081842
APA StyleBkaily, G., Jazzar, A., Abou-Aichi, A., & Jacques, D. (2025). Pathophysiology of Prediabetes Hyperinsulinemia and Insulin Resistance in the Cardiovascular System. Biomedicines, 13(8), 1842. https://doi.org/10.3390/biomedicines13081842