
Aquaporins in Acute Brain Injury: Insights from Clinical and Experimental Studies
 , , , ,
, , , ,  ,
,  ,
,  , and
, and
Abstract
1. Introduction
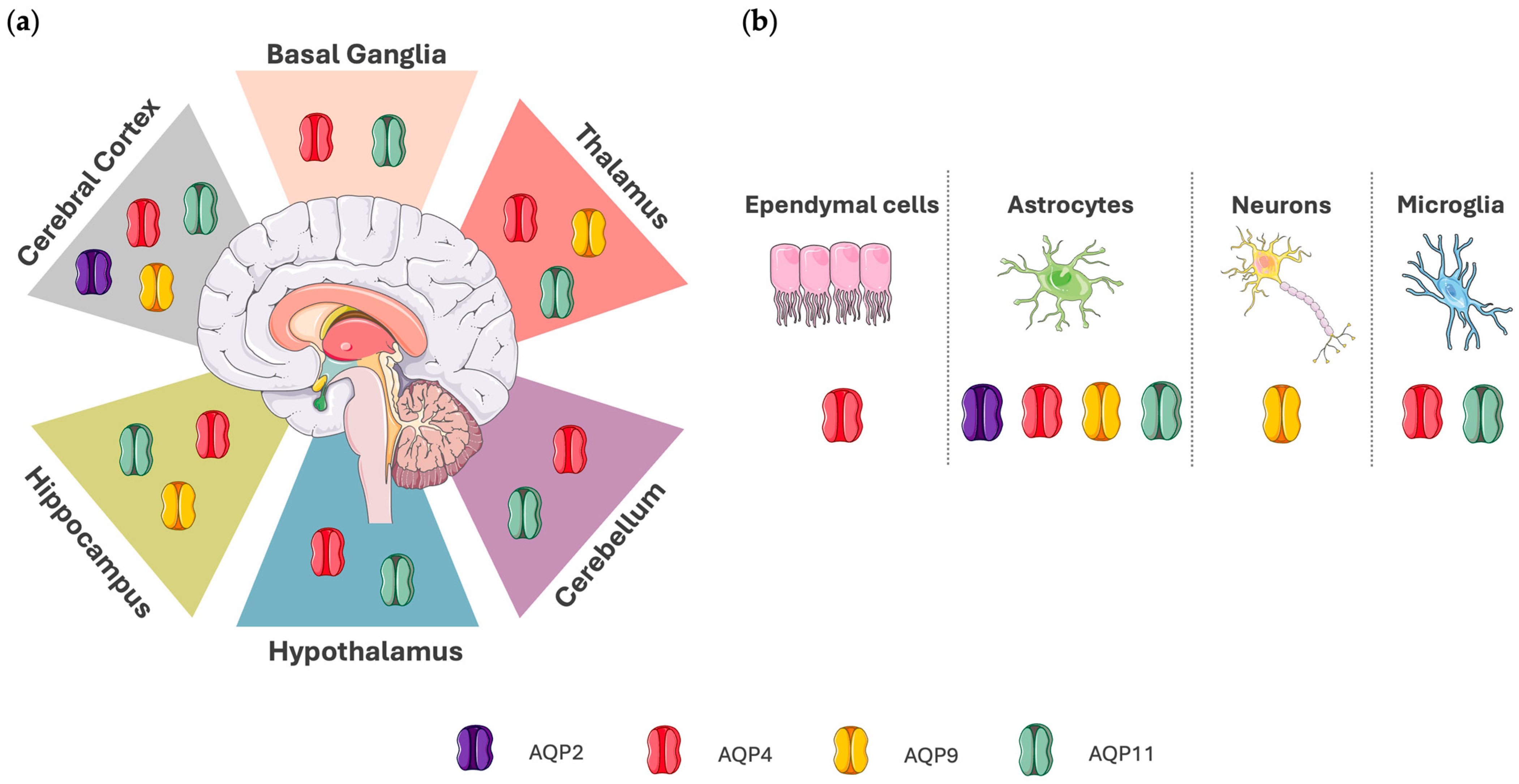
2. Expression and Function of Aquaporins in the Brain
3. Clinical Studies of Aquaporins in Acute Brain Injury
3.1. Aquaporins in Traumatic Brain Injury
3.2. Aquaporins in Acute Ischemic Stroke
3.3. Aquaporins in Aneurysmal Subarachnoid Hemorrhage and Intracerebral Hemorrhage
4. Experimental Studies of Aquaporins in Acute Brain Injury
4.1. AQP4
4.1.1. AQP4 in Traumatic Brain Injury
Methodological Differences of Experimental Studies on the Role of AQP4 in TBI
AQP4 as a Therapeutic Target in Traumatic Brain Injury
4.1.2. AQP4 in Ischemic Stroke
Methodological Differences in Experimental Studies Regarding the Role of AQP4 in Ischemic Stroke
AQP4 as a Therapeutic Target in Ischemic Stroke
Critical Assessment of Studies Concerning AQP4 in TBI and Ischemic Stroke
- Shared Mechanisms of AQP4 in TBI and Ischemic Stroke in Experimental Studies
- Divergent Mechanisms of AQP4 in TBI and Ischemic Stroke in Experimental Studies
4.1.3. AQP4 in Experimental Subarachnoid Hemorrhage
Methodological Differences Among Experimental Studies Regarding the Role of AQP4 in Subarachnoid Hemorrhage
AQP4 as a Therapeutic Target in Subarachnoid Hemorrhage
4.1.4. AQP4 in Experimental Intracerebral Hemorrhage
Methodological Differences Among Experimental Studies Regarding the Role of AQP4 in Intracerebral Hemorrhage
AQP4 as a Therapeutic Target in Intracerebral Hemorrhage
4.2. AQP2
4.2.1. AQP2 in Traumatic Brain Injury
4.2.2. AQP2 in Intracerebral Hemorrhage
4.2.3. AQP2 in Inflammation
4.3. AQP9
4.3.1. AQP9 in Traumatic Brain Injury
4.3.2. AQP9 in Ischemic Stroke
4.3.3. AQP9 in Intracerebral Hemorrhage
4.3.4. AQP9 in Hyperosmotic Stress
Methodological Differences Among Experimental Studies Regarding the Role of AQP9 in Acute Brain Injuries
4.4. AQP11
4.4.1. AQP11 in Ischemic Stroke
4.4.2. AQP11 in Intracerebral Hemorrhage
- While AQP11 is expressed in the brain and may influence water transport mechanisms, its specific role in brain injury has not been revealed. Further research is needed to elucidate its potential involvement in neuropathological conditions and possible therapeutic implications.
5. Aquaporins as Targets in Brain Injuries—Translational Perspectives
5.1. Roles of AQPs in Clinical vs. Experimental Acute Brain Injuries
5.1.1. Traumatic Brain Injury
5.1.2. Ischemic Stroke
5.1.3. Subarachnoid Hemorrhage
5.1.4. Intracerebral Hemorrhage
5.2. Mechanisms of AQP Regulation in Acute Brain Injuries
5.3. Targeting Aquaporins in Acute Brain Injuries: Perspectives and Limitations
5.3.1. Therapeutic Strategies
5.3.2. Therapeutic Limitations
5.3.3. Future Directions
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galgano, M.; Toshkezi, G.; Qiu, X.; Russell, T.; Chin, L.; Zhao, L.R. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transplant. 2017, 26, 1118–1130. [Google Scholar] [CrossRef]
- Maas, A.I.R.; Menon, D.K.; Manley, G.T.; Abrams, M.; Åkerlund, C.; Andelic, N.; Aries, M.; Bashford, T.; Bell, M.J.; Bodien, Y.G.; et al. Traumatic brain injury: Progress and challenges in prevention, clinical care, and research. Lancet Neurol. 2022, 21, 1004–1060. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Osgood, M.L. Aneurysmal Subarachnoid Hemorrhage: Review of the Pathophysiology and Management Strategies. Curr. Neurol. Neurosci. Rep. 2021, 21, 50. [Google Scholar] [CrossRef]
- Thilak, S.; Brown, P.; Whitehouse, T.; Gautam, N.; Lawrence, E.; Ahmed, Z.; Veenith, T. Diagnosis and management of subarachnoid haemorrhage. Nat. Commun. 2024, 15, 1850. [Google Scholar] [CrossRef] [PubMed]
- Seiffge, D.J.; Fandler-Höfler, S.; Du, Y.; Goeldlin, M.B.; Jolink, W.M.T.; Klijn, C.J.M.; Werring, D.J. Intracerebral haemorrhage-mechanisms, diagnosis and prospects for treatment and prevention. Nat. Rev. Neurol. 2024, 20, 708–723. [Google Scholar] [CrossRef]
- Denker, B.M.; Smith, B.L.; Kuhajda, F.P.; Agre, P. Identification, purification, and partial characterization of a novel Mr 28,000 integral membrane protein from erythrocytes and renal tubules. J. Biol. Chem. 1988, 263, 15634–15642. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Agre, P. Isolation of the cDNA for erythrocyte integral membrane protein of 28 kilodaltons: Member of an ancient channel family. Proc. Natl. Acad. Sci. USA 1991, 88, 11110–11114. [Google Scholar] [CrossRef]
- Preston, G.M.; Carroll, T.P.; Guggino, W.B.; Agre, P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science 1992, 256, 385–387. [Google Scholar] [CrossRef]
- Vrettou, C.S.; Issaris, V.; Kokkoris, S.; Poupouzas, G.; Keskinidou, C.; Lotsios, N.S.; Kotanidou, A.; Orfanos, S.E.; Dimopoulou, I.; Vassiliou, A.G. Exploring Aquaporins in Human Studies: Mechanisms and Therapeutic Potential in Critical Illness. Life 2024, 14, 1688. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin-4 and brain edema. Pediatr. Nephrol. 2007, 22, 778–784. [Google Scholar] [CrossRef]
- Szu, J.I.; Binder, D.K. The Role of Astrocytic Aquaporin-4 in Synaptic Plasticity and Learning and Memory. Front. Integr. Neurosci. 2016, 10, 8. [Google Scholar] [CrossRef]
- Verkman, A.S. More than just water channels: Unexpected cellular roles of aquaporins. J. Cell Sci. 2005, 118, 3225–3232. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Mammalian aquaporins: Diverse physiological roles and potential clinical significance. Expert. Rev. Mol. Med. 2008, 10, e13. [Google Scholar] [CrossRef]
- Bihlmaier, R.; Deffner, F.; Mattheus, U.; Neckel, P.H.; Hirt, B.; Mack, A.F. Aquaporin-1 and Aquaporin-4 Expression in Ependyma, Choroid Plexus and Surrounding Transition Zones in the Human Brain. Biomolecules 2023, 13, 212. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Gunnarson, E. Potassium dependent regulation of astrocyte water permeability is mediated by cAMP signaling. PLoS ONE 2012, 7, e34936. [Google Scholar] [CrossRef]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Kurimoto, T.; Miki, A.; Maeda, H.; Kusuhara, S.; Nakamura, M. Aqp9 Gene Deletion Enhances Retinal Ganglion Cell (RGC) Death and Dysfunction Induced by Optic Nerve Crush: Evidence that Aquaporin 9 Acts as an Astrocyte-to-Neuron Lactate Shuttle in Concert with Monocarboxylate Transporters To Support RGC Function and Survival. Mol. Neurobiol. 2020, 57, 4530–4548. [Google Scholar] [CrossRef]
- Amiry-Moghaddam, M.; Lindland, H.; Zelenin, S.; Roberg, B.A.; Gundersen, B.B.; Petersen, P.; Rinvik, E.; Torgner, I.A.; Ottersen, O.P. Brain mitochondria contain aquaporin water channels: Evidence for the expression of a short AQP9 isoform in the inner mitochondrial membrane. FASEB J. 2005, 19, 1459–1467. [Google Scholar] [CrossRef]
- Trillo-Contreras, J.L.; Ramírez-Lorca, R.; Villadiego, J.; Echevarría, M. Cellular Distribution of Brain Aquaporins and Their Contribution to Cerebrospinal Fluid Homeostasis and Hydrocephalus. Biomolecules 2022, 12, 530. [Google Scholar] [CrossRef]
- Deng, S.; Chen, X.; Lei, Q.; Lu, W. AQP2 Promotes Astrocyte Activation by Modulating the TLR4/NFkappaB-p65 Pathway Following Intracerebral Hemorrhage. Front. Immunol. 2022, 13, 847360. [Google Scholar] [CrossRef]
- Czyżewski, W.; Korulczyk, J.; Szymoniuk, M.; Sakwa, L.; Litak, J.; Ziemianek, D.; Czyżewska, E.; Mazurek, M.; Kowalczyk, M.; Turek, G.; et al. Aquaporin 2 in Cerebral Edema: Potential Prognostic Marker in Craniocerebral Injuries. Int. J. Mol. Sci. 2024, 25, 6617. [Google Scholar] [CrossRef]
- Noda, Y.; Sasaki, S. Updates and Perspectives on Aquaporin-2 and Water Balance Disorders. Int. J. Mol. Sci. 2021, 22, 12950. [Google Scholar] [CrossRef]
- Bestetti, S.; Galli, M.; Sorrentino, I.; Pinton, P.; Rimessi, A.; Sitia, R.; Medraño-Fernandez, I. Human aquaporin-11 guarantees efficient transport of H(2)O(2) across the endoplasmic reticulum membrane. Redox Biol. 2020, 28, 101326. [Google Scholar] [CrossRef]
- Zador, Z.; Stiver, S.; Wang, V.; Manley, G.T. Role of aquaporin-4 in cerebral edema and stroke. In Aquaporins; Handbook of Experimental Pharmacology ((HEP, Volume 190)); Springer: Berlin/Heidelberg, Germany, 2009; pp. 159–170. [Google Scholar] [CrossRef]
- Huang, Y.; Li, S.N.; Zhou, X.Y.; Zhang, L.X.; Chen, G.X.; Wang, T.H.; Xia, Q.J.; Liang, N.; Zhang, X. The Dual Role of AQP4 in Cytotoxic and Vasogenic Edema Following Spinal Cord Contusion and Its Possible Association with Energy Metabolism via COX5A. Front. Neurosci. 2019, 13, 584. [Google Scholar] [CrossRef]
- Badaut, J. Aquaglyceroporin 9 in brain pathologies. Neuroscience 2010, 168, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.M.; Badaut, J. Aquaporin 4: A player in cerebral edema and neuroinflammation. J. Neuroinflamm. 2012, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Zahl, S.; Skauli, N.; Stahl, K.; Prydz, A.; Frey, M.M.; Dissen, E.; Ottersen, O.P.; Amiry-Moghaddam, M. Aquaporin-9 in the Brain Inflammatory Response: Evidence from Mice Injected with the Parkinsonogenic Toxin MPP(). Biomolecules 2023, 13, 588. [Google Scholar] [CrossRef]
- Frühbeck, G.; Balaguer, I.; Méndez-Giménez, L.; Valentí, V.; Becerril, S.; Catalán, V.; Gómez-Ambrosi, J.; Silva, C.; Salvador, J.; Calamita, G.; et al. Aquaporin-11 Contributes to TGF-β1-Induced Endoplasmic Reticulum Stress in Human Visceral Adipocytes: Role in Obesity-Associated Inflammation. Cells 2020, 9, 1403. [Google Scholar] [CrossRef]
- Ishibashi, K.; Tanaka, Y.; Morishita, Y. The role of mammalian superaquaporins inside the cell. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 1507–1512. [Google Scholar] [CrossRef]
- Ishibashi, K.; Tanaka, Y.; Morishita, Y. The role of mammalian superaquaporins inside the cell: An update. Biochim. Biophys. Acta (BBA) Biomembr. 2021, 1863, 183617. [Google Scholar] [CrossRef] [PubMed]
- Katada, R.; Akdemir, G.; Asavapanumas, N.; Ratelade, J.; Zhang, H.; Verkman, A.S. Greatly improved survival and neuroprotection in aquaporin-4-knockout mice following global cerebral ischemia. FASEB J. 2014, 28, 705–714. [Google Scholar] [CrossRef]
- Badaut, J.; Brunet, J.F.; Petit, J.M.; Guérin, C.F.; Magistretti, P.J.; Regli, L. Induction of brain aquaporin 9 (AQP9) in catecholaminergic neurons in diabetic rats. Brain Res. 2008, 1188, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Amro, Z.; Collins-Praino, L.E.; Yool, A.J. Protective roles of peroxiporins AQP0 and AQP11 in human astrocyte and neuronal cell lines in response to oxidative and inflammatory stressors. Biosci. Rep. 2024, 44, BSR20231725. [Google Scholar] [CrossRef]
- Tang, G.; Yang, G.Y. Aquaporin-4: A Potential Therapeutic Target for Cerebral Edema. Int. J. Mol. Sci. 2016, 17, 1413. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, A.; Halici, H.; Yayla, M. Aquaporins: Potential Targets in Inflammatory Diseases. Eurasian J. Med. 2023, 55, 106–113. [Google Scholar] [CrossRef]
- da Silva, I.V.; Garra, S.; Calamita, G.; Soveral, G. The Multifaceted Role of Aquaporin-9 in Health and Its Potential as a Clinical Biomarker. Biomolecules 2022, 12, 897. [Google Scholar] [CrossRef]
- Radin, M.J.; Yu, M.J.; Stoedkilde, L.; Miller, R.L.; Hoffert, J.D.; Frokiaer, J.; Pisitkun, T.; Knepper, M.A. Aquaporin-2 regulation in health and disease. Vet. Clin. Pathol. 2012, 41, 455–470. [Google Scholar] [CrossRef]
- Lo Pizzo, M.; Schiera, G.; Di Liegro, I.; Di Liegro, C.M.; Pál, J.; Czeiter, E.; Sulyok, E.; Dóczi, T. Aquaporin-4 distribution in control and stressed astrocytes in culture and in the cerebrospinal fluid of patients with traumatic brain injuries. Neurol. Sci. 2013, 34, 1309–1314. [Google Scholar] [CrossRef]
- Aoki, K.; Uchihara, T.; Tsuchiya, K.; Nakamura, A.; Ikeda, K.; Wakayama, Y. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol. 2003, 106, 121–124. [Google Scholar] [CrossRef]
- Badaut, J.; Brunet, J.F.; Grollimund, L.; Hamou, M.F.; Magistretti, P.J.; Villemure, J.G.; Regli, L. Aquaporin 1 and aquaporin 4 expression in human brain after subarachnoid hemorrhage and in peritumoral tissue. Acta Neurochir. Suppl. 2003, 86, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Hirt, L.; Price, M.; Mastour, N.; Brunet, J.F.; Barrière, G.; Friscourt, F.; Badaut, J. Increase of aquaporin 9 expression in astrocytes participates in astrogliosis. J. Neurosci. Res. 2018, 96, 194–206. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Huang, X.; Zhang, H.; Guan, Y.; Zhang, X. Role of aquaporins in brain water transport and edema. Front. Neurosci. 2025, 19, 1518967. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Elahi, F.M.; Mustapic, M.; Kapogiannis, D.; Pryhoda, M.; Gilmore, A.; Gorgens, K.A.; Davidson, B.; Granholm, A.C.; Ledreux, A. Altered levels of plasma neuron-derived exosomes and their cargo proteins characterize acute and chronic mild traumatic brain injury. FASEB J. 2019, 33, 5082–5088. [Google Scholar] [CrossRef]
- Neri, M.; Frati, A.; Turillazzi, E.; Cantatore, S.; Cipolloni, L.; Di Paolo, M.; Frati, P.; La Russa, R.; Maiese, A.; Scopetti, M.; et al. Immunohistochemical Evaluation of Aquaporin-4 and its Correlation with CD68, IBA-1, HIF-1α, GFAP, and CD15 Expressions in Fatal Traumatic Brain Injury. Int. J. Mol. Sci. 2018, 19, 3544. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Okuda, M.; Asai, J.; Nagashima, G.; Itokawa, H.; Matsunaga, A.; Fujimoto, T.; Suzuki, T. Astrocytes co-express aquaporin-1, -4, and vascular endothelial growth factor in brain edema tissue associated with brain contusion. Acta Neurochir. Suppl. 2006, 96, 398–401. [Google Scholar] [CrossRef]
- Yang, Z.H.; Yin, X.J.; Fu, G.Y. The correlation between CT findings of diffuse axonal injury and the expression of neuronal aquaporin in patients with craniocerebral injury. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 6871–6878. [Google Scholar] [CrossRef] [PubMed]
- Laird, M.D.; Shields, J.S.; Sukumari-Ramesh, S.; Kimbler, D.E.; Fessler, R.D.; Shakir, B.; Youssef, P.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 2014, 62, 26–38. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Peltz, C.B.; Mustapic, M.; Kapogiannis, D.; Yaffe, K. Neuron-Derived Plasma Exosome Proteins after Remote Traumatic Brain Injury. J. Neurotrauma 2020, 37, 382–388. [Google Scholar] [CrossRef]
- Braun, M.; Sevao, M.; Keil, S.A.; Gino, E.; Wang, M.X.; Lee, J.; Haveliwala, M.A.; Klein, E.; Agarwal, S.; Pedersen, T.; et al. Macroscopic changes in aquaporin-4 underlie blast traumatic brain injury-related impairment in glymphatic function. Brain 2024, 147, 2214–2229. [Google Scholar] [CrossRef]
- Wang, M.; Yu, X.; Li, B.; Gao, C.; Chen, Y.; Zhang, X.; Li, W.; Yang, L.; Fan, Z. miR-211-5p targeting MMP9 regulates the expressions of AQP4 in traumatic brain injury. Acta Neurol. Belg. 2023, 123, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Paterakis, K.; Tsivgoulis, G.; Tsintou, M.; Hadjigeorgiou, G.F.; Dardioti, M.; Grigoriadis, S.; Simeonidou, C.; Komnos, A.; Kapsalaki, E.; et al. AQP4 tag single nucleotide polymorphisms in patients with traumatic brain injury. J. Neurotrauma 2014, 31, 1920–1926. [Google Scholar] [CrossRef]
- Romeiro, R.R.; Romano-Silva, M.A.; De Marco, L.; Teixeira, A.L., Jr.; Correa, H. Can variation in aquaporin 4 gene be associated with different outcomes in traumatic brain edema? Neurosci. Lett. 2007, 426, 133–134. [Google Scholar] [CrossRef] [PubMed]
- Nekludov, M.; Bellander, B.M.; Gryth, D.; Wallen, H.; Mobarrez, F. Brain-Derived Microparticles in Patients with Severe Isolated TBI. Brain Inj. 2017, 31, 1856–1862. [Google Scholar] [CrossRef]
- Zhao, Z.A.; Li, P.; Ye, S.Y.; Ning, Y.L.; Wang, H.; Peng, Y.; Yang, N.; Zhao, Y.; Zhang, Z.H.; Chen, J.F.; et al. Perivascular AQP4 dysregulation in the hippocampal CA1 area after traumatic brain injury is alleviated by adenosine A(2A) receptor inactivation. Sci. Rep. 2017, 7, 2254. [Google Scholar] [CrossRef]
- Nazzi, C.; Avenanti, A.; Battaglia, S. The Involvement of Antioxidants in Cognitive Decline and Neurodegeneration: Mens Sana in Corpore Sano. Antioxidants 2024, 13, 701. [Google Scholar] [CrossRef]
- Battaglia, S.; Fazio, C.D.; Borgomaneri, S.; Avenanti, A. Cortisol Imbalance and Fear Learning in PTSD: Therapeutic Approaches to Control Abnormal Fear Responses. Curr. Neuropharmacol. 2025, 23, 835–846. [Google Scholar] [CrossRef]
- Stokum, J.A.; Mehta, R.I.; Ivanova, S.; Yu, E.; Gerzanich, V.; Simard, J.M. Heterogeneity of aquaporin-4 localization and expression after focal cerebral ischemia underlies differences in white versus grey matter swelling. Acta Neuropathol. Commun. 2015, 3, 61. [Google Scholar] [CrossRef] [PubMed]
- Ramiro, L.; Simats, A.; Penalba, A.; Garcia-Tornel, A.; Rovira, A.; Mancha, F.; Bustamante, A.; Montaner, J. Circulating Aquaporin-4 as A biomarker of early neurological improvement in stroke patients: A pilot study. Neurosci. Lett. 2020, 714, 134580. [Google Scholar] [CrossRef]
- He, Y.; Yang, Q.; Liu, H.; Jiang, L.; Liu, Q.; Lian, W.; Huang, J. Effect of blood pressure on early neurological deterioration of acute ischemic stroke patients with intravenous rt-PA thrombolysis may be mediated through oxidative stress induced blood-brain barrier disruption and AQP4 upregulation. J. Stroke Cerebrovasc. Dis. 2020, 29, 104997. [Google Scholar] [CrossRef]
- Kleffner, I.; Bungeroth, M.; Schiffbauer, H.; Schäbitz, W.R.; Ringelstein, E.B.; Kuhlenbäumer, G. The role of aquaporin-4 polymorphisms in the development of brain edema after middle cerebral artery occlusion. Stroke 2008, 39, 1333–1335. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Yan, L.; Wang, C.; Han, A.; Qin, Y.; Cui, L.; Xiang, Q. Associations between Aquaglyceroporin Gene Polymorphisms and Risk of Stroke among Patients with Hypertension. BioMed Res. Int. 2020, 2020, 9358290. [Google Scholar] [CrossRef]
- Takagi, J.; Otake, K.; Nakao, N.; Takashashi, M.; Hirooka, Y. Urinary excretion of aquaporin-2 and inappropriate secretion of vasopressin in hyponatremic patients after cerebral infarction. Horm. Metab. Res. 2003, 35, 62–66. [Google Scholar] [CrossRef]
- Yao, X.; Derugin, N.; Manley, G.T.; Verkman, A.S. Reduced brain edema and infarct volume in aquaporin-4 deficient mice after transient focal cerebral ischemia. Neurosci. Lett. 2015, 584, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Hirt, L.; Fukuda, A.M.; Ambadipudi, K.; Rashid, F.; Binder, D.; Verkman, A.; Ashwal, S.; Obenaus, A.; Badaut, J. Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. J. Cereb. Blood Flow Metab. 2017, 37, 277–290. [Google Scholar] [CrossRef]
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Castellanos Rivera, R.M.; Simon, M.J.; et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. eLife 2018, 7, e40070. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Yao, H.T.; Zhang, W.P.; Zhang, L.; Ding, W.; Zhang, S.H.; Chen, Z.; Wei, E.Q. Increased expression of aquaporin-4 in human traumatic brain injury and brain tumors. J. Zhejiang Univ. Sci. B 2005, 6, 33–37. [Google Scholar] [CrossRef]
- Wu, H.; Zhao, R.; Qi, J.; Cong, Y.; Wang, D.; Liu, T.; Gu, Y.; Ban, X.; Huang, Q. The expression and the role of protease nexin-1 on brain edema after intracerebral hemorrhage. J. Neurol. Sci. 2008, 270, 172–183. [Google Scholar] [CrossRef]
- Appelboom, G.; Bruce, S.; Duren, A.; Piazza, M.; Monahan, A.; Christophe, B.; Zoller, S.; LoPresti, M.; Connolly, E.S. Aquaporin-4 gene variant independently associated with oedema after intracerebral haemorrhage. Neurol. Res. 2015, 37, 657–661. [Google Scholar] [CrossRef]
- Dardiotis, E.; Siokas, V.; Marogianni, C.; Aloizou, A.M.; Sokratous, M.; Paterakis, K.; Dardioti, M.; Grigoriadis, S.; Brotis, A.; Kapsalaki, E.; et al. AQP4 tag SNPs in patients with intracerebral hemorrhage in Greek and Polish population. Neurosci. Lett. 2019, 696, 156–161. [Google Scholar] [CrossRef]
- Czyżewski, W.; Litak, J.; Sobstyl, J.; Mandat, T.; Torres, K.; Staśkiewicz, G. Aquaporins: Gatekeepers of Fluid Dynamics in Traumatic Brain Injury. Int. J. Mol. Sci. 2024, 25, 6553. [Google Scholar] [CrossRef] [PubMed]
- Kiening, K.L.; van Landeghem, F.K.; Schreiber, S.; Thomale, U.W.; von Deimling, A.; Unterberg, A.W.; Stover, J.F. Decreased hemispheric Aquaporin-4 is linked to evolving brain edema following controlled cortical impact injury in rats. Neurosci. Lett. 2002, 324, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Cartagena, C.M.; Phillips, K.L.; Tortella, F.C.; Dave, J.R.; Schmid, K.E. Temporal alterations in aquaporin and transcription factor HIF1α expression following penetrating ballistic-like brain injury (PBBI). Mol. Cell. Neurosci. 2014, 60, 81–87. [Google Scholar] [CrossRef]
- Xiong, A.; Xiong, R.; Yu, J.; Liu, Y.; Liu, K.; Jin, G.; Xu, J.; Yan, J. Aquaporin-4 is a potential drug target for traumatic brain injury via aggravating the severity of brain edema. Burns Trauma 2021, 9, tkaa050. [Google Scholar] [CrossRef]
- Kapoor, S.; Kim, S.M.; Farook, J.M.; Mir, S.; Saha, R.; Sen, N. Foxo3a transcriptionally upregulates AQP4 and induces cerebral edema following traumatic brain injury. J. Neurosci. 2013, 33, 17398–17403. [Google Scholar] [CrossRef]
- Ren, Z.; Iliff, J.J.; Yang, L.; Yang, J.; Chen, X.; Chen, M.J.; Giese, R.N.; Wang, B.; Shi, X.; Nedergaard, M. ‘Hit & Run’ model of closed-skull traumatic brain injury (TBI) reveals complex patterns of post-traumatic AQP4 dysregulation. J. Cereb. Blood Flow Metab. 2013, 33, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qiu, G.; Zhuo, F.; Yu, W.; Sun, S.; Li, F.; Yang, M. Lost Polarization of Aquaporin4 and Dystroglycan in the Core Lesion after Traumatic Brain Injury Suggests Functional Divergence in Evolution. BioMed Res. Int. 2015, 2015, 471631. [Google Scholar] [CrossRef]
- Yao, X.; Uchida, K.; Papadopoulos, M.C.; Zador, Z.; Manley, G.T.; Verkman, A.S. Mildly Reduced Brain Swelling and Improved Neurological Outcome in Aquaporin-4 Knockout Mice following Controlled Cortical Impact Brain Injury. J. Neurotrauma 2015, 32, 1458–1464. [Google Scholar] [CrossRef]
- Liu, X.; Xie, Y.; Wan, X.; Wu, J.; Fan, Z.; Yang, L. Protective Effects of Aquaporin-4 Deficiency on Longer-term Neurological Outcomes in a Mouse Model. Neurochem. Res. 2021, 46, 1380–1389. [Google Scholar] [CrossRef]
- Chen, J.Q.; Zhang, C.C.; Jiang, S.N.; Lu, H.; Wang, W. Effects of Aquaporin 4 Knockdown on Brain Edema of the Uninjured Side After Traumatic Brain Injury in Rats. Med. Sci. Monit. 2016, 22, 4809–4819. [Google Scholar] [CrossRef]
- Taya, K.; Marmarou, C.R.; Okuno, K.; Prieto, R.; Marmarou, A. Effect of secondary insults upon aquaporin-4 water channels following experimental cortical contusion in rats. J. Neurotrauma 2010, 27, 229–239. [Google Scholar] [CrossRef]
- Rao, K.V.; Reddy, P.V.; Curtis, K.M.; Norenberg, M.D. Aquaporin-4 expression in cultured astrocytes after fluid percussion injury. J. Neurotrauma 2011, 28, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Finnie, J.W.; Blumbergs, P.C.; Manavis, J. Aquaporin-4 expression after experimental contusional injury in an ovine impact-acceleration head injury model. J. Clin. Neurosci. 2011, 18, 947–950. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, J.; Lu, H. Expression of aquaporin-4 and pathological characteristics of brain injury in a rat model of traumatic brain injury. Mol. Med. Rep. 2015, 12, 7351–7357. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Zhang, J.; Xu, J.; Sheng, G.; Huang, G. Propofol administration modulates AQP-4 expression and brain edema after traumatic brain injury. Cell Biochem. Biophys. 2013, 67, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhang, H.; Chen, H.; Lv, Q.; Jin, X. Hypertonic Saline Alleviates Brain Edema After Traumatic Brain Injury via Downregulation of Aquaporin 4 in Rats. Med. Sci. Monit. 2018, 24, 1863–1870. [Google Scholar] [CrossRef]
- Zhang, M.; Cui, Z.; Cui, H.; Cao, Y.; Zhong, C.; Wang, Y. Astaxanthin alleviates cerebral edema by modulating NKCC1 and AQP4 expression after traumatic brain injury in mice. BMC Neurosci. 2016, 17, 60. [Google Scholar] [CrossRef]
- Jin, H.; Li, W.; Dong, C.; Ma, L.; Wu, J.; Zhao, W. Effects of Different Doses of Levetiracetam on Aquaporin 4 Expression in Rats with Brain Edema Following Fluid Percussion Injury. Med. Sci. Monit. 2016, 22, 678–686. [Google Scholar] [CrossRef]
- Fukuda, A.M.; Adami, A.; Pop, V.; Bellone, J.A.; Coats, J.S.; Hartman, R.E.; Ashwal, S.; Obenaus, A.; Badaut, J. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J. Cereb. Blood Flow Metab. 2013, 33, 1621–1632. [Google Scholar] [CrossRef]
- Guan, Y.; Li, L.; Chen, J.; Lu, H. Effect of AQP4-RNAi in treating traumatic brain edema: Multi-modal MRI and histopathological changes of early stage edema in a rat model. Exp. Ther. Med. 2020, 19, 2029–2036. [Google Scholar] [CrossRef]
- Lu, H.; Zhan, Y.; Ai, L.; Chen, H.; Chen, J. AQP4-siRNA alleviates traumatic brain edema by altering post-traumatic AQP4 polarity reversal in TBI rats. J. Clin. Neurosci. 2020, 81, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Glober, N.K.; Sprague, S.; Ahmad, S.; Mayfield, K.G.; Fletcher, L.M.; Digicaylioglu, M.H.; Sayre, N.L. Acetazolamide Treatment Prevents Redistribution of Astrocyte Aquaporin 4 after Murine Traumatic Brain Injury. Neurosci. J. 2019, 2019, 2831501. [Google Scholar] [CrossRef]
- Xing, R.; Cheng, J.; Yu, J.; Li, S.; Ma, H.; Zhao, Y. Trifluoperazine reduces apoptosis and inflammatory responses in traumatic brain injury by preventing the accumulation of Aquaporin4 on the surface of brain cells. Int. J. Med. Sci. 2023, 20, 797–809. [Google Scholar] [CrossRef]
- Shi, Z.F.; Zhao, W.J.; Xu, L.X.; Dong, L.P.; Yang, S.H.; Yuan, F. Downregulation of Aquaporin 4 Expression through Extracellular Signal-regulated Kinases1/2 Activation in Cultured Astrocytes Following Scratch-injury. Biomed. Environ. Sci. 2015, 28, 199–205. [Google Scholar] [CrossRef]
- Tomura, S.; Nawashiro, H.; Otani, N.; Uozumi, Y.; Toyooka, T.; Ohsumi, A.; Shima, K. Effect of decompressive craniectomy on aquaporin-4 expression after lateral fluid percussion injury in rats. J. Neurotrauma 2011, 28, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Szczygielski, J.; Glameanu, C.; Müller, A.; Klotz, M.; Sippl, C.; Hubertus, V.; Schäfer, K.H.; Mautes, A.E.; Schwerdtfeger, K.; Oertel, J. Changes in Posttraumatic Brain Edema in Craniectomy-Selective Brain Hypothermia Model Are Associated With Modulation of Aquaporin-4 Level. Front. Neurol. 2018, 9, 799. [Google Scholar] [CrossRef] [PubMed]
- Ghabriel, M.N.; Thomas, A.; Vink, R. Magnesium restores altered aquaporin-4 immunoreactivity following traumatic brain injury to a pre-injury state. Acta Neurochir. Suppl. 2006, 96, 402–406. [Google Scholar] [CrossRef]
- Guo, Q.; Sayeed, I.; Baronne, L.M.; Hoffman, S.W.; Guennoun, R.; Stein, D.G. Progesterone administration modulates AQP4 expression and edema after traumatic brain injury in male rats. Exp. Neurol. 2006, 198, 469–478. [Google Scholar] [CrossRef]
- Karasu, A.; Aras, Y.; Sabancı, P.A.; Sağlam, G.; Izgi, N.; Biltekin, B.; Barak, T.; Hepgül, K.T.; Kaya, M.; Bilir, A. The effects of protein kinase C activator phorbol dibutyrate on traumatic brain edema and aquaporin-4 expression. Turk. J. Trauma Emerg. Surg./Ulus. Travma Acil Cerrahi Derg. 2010, 16, 390–394. [Google Scholar]
- Lv, Q.; Fan, X.; Xu, G.; Liu, Q.; Tian, L.; Cai, X.; Sun, W.; Wang, X.; Cai, Q.; Bao, Y.; et al. Intranasal delivery of nerve growth factor attenuates aquaporins-4-induced edema following traumatic brain injury in rats. Brain Res. 2013, 1493, 80–89. [Google Scholar] [CrossRef]
- Ribeiro Mde, C.; Hirt, L.; Bogousslavsky, J.; Regli, L.; Badaut, J. Time course of aquaporin expression after transient focal cerebral ischemia in mice. J. Neurosci. Res. 2006, 83, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Ashwal, S.; Tone, B.; Regli, L.; Tian, H.R.; Obenaus, A. Temporal and regional evolution of aquaporin-4 expression and magnetic resonance imaging in a rat pup model of neonatal stroke. Pediatr. Res. 2007, 62, 248–254. [Google Scholar] [CrossRef]
- Hirt, L.; Ternon, B.; Price, M.; Mastour, N.; Brunet, J.F.; Badaut, J. Protective role of early aquaporin 4 induction against postischemic edema formation. J. Cereb. Blood Flow Metab. 2009, 29, 423–433. [Google Scholar] [CrossRef]
- Friedman, B.; Schachtrup, C.; Tsai, P.S.; Shih, A.Y.; Akassoglou, K.; Kleinfeld, D.; Lyden, P.D. Acute vascular disruption and aquaporin 4 loss after stroke. Stroke 2009, 40, 2182–2190. [Google Scholar] [CrossRef]
- Frydenlund, D.S.; Bhardwaj, A.; Otsuka, T.; Mylonakou, M.N.; Yasumura, T.; Davidson, K.G.; Zeynalov, E.; Skare, O.; Laake, P.; Haug, F.M.; et al. Temporary loss of perivascular aquaporin-4 in neocortex after transient middle cerebral artery occlusion in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 13532–13536. [Google Scholar] [CrossRef] [PubMed]
- Zeynalov, E.; Chen, C.H.; Froehner, S.C.; Adams, M.E.; Ottersen, O.P.; Amiry-Moghaddam, M.; Bhardwaj, A. The perivascular pool of aquaporin-4 mediates the effect of osmotherapy in postischemic cerebral edema. Crit. Care Med. 2008, 36, 2634–2640. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, K.; Liu, Y.; Erokwu, B.O.; Zhao, P.; Flask, C.A.; Ramos-Estebanez, C.; Farr, G.W.; LaManna, J.C.; Boron, W.F.; et al. Increased cerebral vascularization and decreased water exchange across the blood-brain barrier in aquaporin-4 knockout mice. PLoS ONE 2019, 14, e0218415. [Google Scholar] [CrossRef]
- Murata, Y.; Sugimoto, K.; Yang, C.; Harada, K.; Gono, R.; Harada, T.; Miyashita, Y.; Higashisaka, K.; Katada, R.; Tanaka, J.; et al. Activated microglia-derived macrophage-like cells exacerbate brain edema after ischemic stroke correlate with astrocytic expression of aquaporin-4 and interleukin-1 alpha release. Neurochem. Int. 2020, 140, 104848. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.Z.; Ma, Y.M.; Zhang, X.P.; Dong, L.D.; Jing, L.; Zhang, J.Z. Region-specific changes in aquaporin 4 induced by hyperglycemia underlie the differences in cell swelling in the cortex and striatum after cerebral ischemia-reperfusion. Neurosci. Lett. 2021, 754, 135885. [Google Scholar] [CrossRef]
- Yang, M.; Gao, F.; Liu, H.; Yu, W.H.; Sun, S.Q. Temporal changes in expression of aquaporin-3, -4, -5 and -8 in rat brains after permanent focal cerebral ischemia. Brain Res. 2009, 1290, 121–132. [Google Scholar] [CrossRef]
- Banitalebi, S.; Skauli, N.; Geiseler, S.; Ottersen, O.P.; Amiry-Moghaddam, M. Disassembly and Mislocalization of AQP4 in Incipient Scar Formation after Experimental Stroke. Int. J. Mol. Sci. 2022, 23, 1117. [Google Scholar] [CrossRef] [PubMed]
- Mages, B.; Aleithe, S.; Blietz, A.; Krueger, M.; Härtig, W.; Michalski, D. Simultaneous alterations of oligodendrocyte-specific CNP, astrocyte-specific AQP4 and neuronal NF-L demarcate ischemic tissue after experimental stroke in mice. Neurosci. Lett. 2019, 711, 134405. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.C.; Chen, A.Y.; Hung, V.K.; Yaw, L.P.; Fung, M.K.; Ho, M.C.; Tsang, M.C.; Chung, S.S.; Chung, S.K. Endothelin-1 overexpression leads to further water accumulation and brain edema after middle cerebral artery occlusion via aquaporin 4 expression in astrocytic end-feet. J. Cereb. Blood Flow Metab. 2005, 25, 998–1011. [Google Scholar] [CrossRef]
- Cheng, Z.J.; Dai, T.M.; Shen, Y.Y.; He, J.L.; Li, J.; Tu, J.L. Atorvastatin Pretreatment Attenuates Ischemic Brain Edema by Suppressing Aquaporin 4. J. Stroke Cerebrovasc. Dis. 2018, 27, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Lin, L.; Yin, L.; Hao, X.; Tian, J.; Zhang, X.; Ren, Y.; Li, C.; Yang, Y. Acutely Inhibiting AQP4 with TGN-020 Improves Functional Outcome by Attenuating Edema and Peri-Infarct Astrogliosis After Cerebral Ischemia. Front. Immunol. 2022, 13, 870029. [Google Scholar] [CrossRef]
- Liu, M.; Li, H.; Fan, L.; Yan, W.; Yan, Y.F. Treatment Effects of Acetazolamide on Ischemic Stroke: A Meta-Analysis and Systematic Review. World Neurosurg. 2024, 185, e750–e757. [Google Scholar] [CrossRef]
- Kikuchi, K.; Tancharoen, S.; Matsuda, F.; Biswas, K.K.; Ito, T.; Morimoto, Y.; Oyama, Y.; Takenouchi, K.; Miura, N.; Arimura, N.; et al. Edaravone attenuates cerebral ischemic injury by suppressing aquaporin-4. Biochem. Biophys. Res. Commun. 2009, 390, 1121–1125. [Google Scholar] [CrossRef]
- Li, S.; Hu, X.; Zhang, M.; Zhou, F.; Lin, N.; Xia, Q.; Zhou, Y.; Qi, W.; Zong, Y.; Yang, H.; et al. Remote ischemic post-conditioning improves neurological function by AQP4 down-regulation in astrocytes. Behav. Brain Res. 2015, 289, 1–8. [Google Scholar] [CrossRef]
- Ji, F.T.; Liang, J.J.; Miao, L.P.; Wu, Q.; Cao, M.H. Propofol post-conditioning protects the blood brain barrier by decreasing matrix metalloproteinase-9 and aquaporin-4 expression and improves the neurobehavioral outcome in a rat model of focal cerebral ischemia-reperfusion injury. Mol. Med. Rep. 2015, 12, 2049–2055. [Google Scholar] [CrossRef]
- Yang, D.; Ma, L.; Wang, P.; Yang, D.; Zhang, Y.; Zhao, X.; Lv, J.; Zhang, J.; Zhang, Z.; Gao, F. Normobaric oxygen inhibits AQP4 and NHE1 expression in experimental focal ischemic stroke. Int. J. Mol. Med. 2019, 43, 1193–1202. [Google Scholar] [CrossRef]
- Jin, J.; Wang, H.; Zheng, X.; Xie, S.; Zheng, L.; Zhan, R. Inhibition of LncRNA MALAT1 Attenuates Cerebral Ischemic Reperfusion Injury via Regulating AQP4 Expression. Eur. Neurol. 2020, 83, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zheng, X.; Jin, J.; Zheng, L.; Guan, T.; Huo, Y.; Xie, S.; Wu, Y.; Chen, W. LncRNA MALAT1 silencing protects against cerebral ischemia-reperfusion injury through miR-145 to regulate AQP4. J. Biomed. Sci. 2020, 27, 40. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.; Ma, Y.; Tang, G.; Liu, Y.; Chen, X.; Zhang, Z.; Zeng, L.; Wang, Y.; Ouyang, Y.B.; et al. MicroRNA-29b is a therapeutic target in cerebral ischemia associated with aquaporin 4. J. Cereb. Blood Flow Metab. 2015, 35, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.X.; Lu, C.; Xia, C.M.; Qiao, Z.W.; Zhu, D.N. Simvastatin pretreatment protects cerebrum from neuronal injury by decreasing the expressions of phosphor-CaMK II and AQP4 in ischemic stroke rats. J. Mol. Neurosci. 2014, 54, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.Q.; He, X.Y.; Yang, X.; Xiao, Y.C.; Duan, S.Q.; Wang, H.; Bai, H.; Zhang, Y.; Shi, J.Y.; Zhu, X.L.; et al. Acetazolamide Alleviate Cerebral Edema Induced by Ischemic Stroke Through Inhibiting the Expression of AQP4 mRNA. Neurocrit. Care 2022, 36, 97–105. [Google Scholar] [CrossRef]
- He, Z.; Wang, X.; Wu, Y.; Jia, J.; Hu, Y.; Yang, X.; Li, J.; Fan, M.; Zhang, L.; Guo, J.; et al. Treadmill pre-training ameliorates brain edema in ischemic stroke via down-regulation of aquaporin-4: An MRI study in rats. PLoS ONE 2014, 9, e84602. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Lan, Y.P.; Tang, H.F.; Zhu, S.M. Propofol pretreatment attenuates aquaporin-4 over-expression and alleviates cerebral edema after transient focal brain ischemia reperfusion in rats. Anesth. Analg. 2008, 107, 2009–2016. [Google Scholar] [CrossRef]
- Pirici, I.; Balsanu, T.A.; Bogdan, C.; Margaritescu, C.; Divan, T.; Vitalie, V.; Mogoanta, L.; Pirici, D.; Carare, R.O.; Muresanu, D.F. Inhibition of Aquaporin-4 Improves the Outcome of Ischaemic Stroke and Modulates Brain Paravascular Drainage Pathways. Int. J. Mol. Sci. 2017, 19, 46. [Google Scholar] [CrossRef]
- Shi, Z.F.; Fang, Q.; Chen, Y.; Xu, L.X.; Wu, M.; Jia, M.; Lu, Y.; Wang, X.X.; Wang, Y.J.; Yan, X.; et al. Methylene blue ameliorates brain edema in rats with experimental ischemic stroke via inhibiting aquaporin 4 expression. Acta Pharmacol. Sin. 2021, 42, 382–392. [Google Scholar] [CrossRef]
- Rutkowsky, J.M.; Wallace, B.K.; Wise, P.M.; O’Donnell, M.E. Effects of estradiol on ischemic factor-induced astrocyte swelling and AQP4 protein abundance. Am. J. Physiol. Cell Physiol. 2011, 301, C204–C212. [Google Scholar] [CrossRef]
- Shin, J.A.; Choi, J.H.; Choi, Y.H.; Park, E.M. Conserved aquaporin 4 levels associated with reduction of brain edema are mediated by estrogen in the ischemic brain after experimental stroke. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, T.; Chang, X.; Xing, J. Long Noncoding RNA SNHG14 Promotes Ischemic Brain Injury via Regulating miR-199b/AQP4 Axis. Neurochem. Res. 2021, 46, 1280–1290. [Google Scholar] [CrossRef]
- Zheng, L.; Cheng, W.; Wang, X.; Yang, Z.; Zhou, X.; Pan, C. Overexpression of MicroRNA-145 Ameliorates Astrocyte Injury by Targeting Aquaporin 4 in Cerebral Ischemic Stroke. BioMed Res. Int. 2017, 2017, 9530951. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, C.; Chen, M.; Pei, A.; Xie, L.; Zhu, S. miR-29a ameliorates ischemic injury of astrocytes in vitro by targeting the water channel protein aquaporin 4. Oncol. Rep. 2019, 41, 1707–1717. [Google Scholar] [CrossRef]
- Farr, G.W.; Hall, C.H.; Farr, S.M.; Wade, R.; Detzel, J.M.; Adams, A.G.; Buch, J.M.; Beahm, D.L.; Flask, C.A.; Xu, K.; et al. Functionalized Phenylbenzamides Inhibit Aquaporin-4 Reducing Cerebral Edema and Improving Outcome in Two Models of CNS Injury. Neuroscience 2019, 404, 484–498. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.L.; Qi, S.H. ALDH2 Protects Against Ischemic Stroke in Rats by Facilitating 4-HNE Clearance and AQP4 Down-Regulation. Neurochem. Res. 2018, 43, 1339–1347. [Google Scholar] [CrossRef]
- Liu, X.; Nakayama, S.; Amiry-Moghaddam, M.; Ottersen, O.P.; Bhardwaj, A. Arginine-vasopressin V1 but not V2 receptor antagonism modulates infarct volume, brain water content, and aquaporin-4 expression following experimental stroke. Neurocrit. Care 2010, 12, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Migliati, E.; Meurice, N.; DuBois, P.; Fang, J.S.; Somasekharan, S.; Beckett, E.; Flynn, G.; Yool, A.J. Inhibition of aquaporin-1 and aquaporin-4 water permeability by a derivative of the loop diuretic bumetanide acting at an internal pore-occluding binding site. Mol. Pharmacol. 2009, 76, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Nishigami, C.; Irie, K.; Shigemori, Y.; Sano, K.; Yamashita, Y.; Myose, T.; Tominaga, K.; Matsuo, K.; Nakamura, Y.; et al. Goreisan Prevents Brain Edema after Cerebral Ischemic Stroke by Inhibiting Aquaporin 4 Upregulation in Mice. J. Stroke Cerebrovasc. Dis. 2018, 27, 758–763. [Google Scholar] [CrossRef]
- Tang, G.; Liu, Y.; Zhang, Z.; Lu, Y.; Wang, Y.; Huang, J.; Li, Y.; Chen, X.; Gu, X.; Wang, Y.; et al. Mesenchymal stem cells maintain blood-brain barrier integrity by inhibiting aquaporin-4 upregulation after cerebral ischemia. Stem Cells 2014, 32, 3150–3162. [Google Scholar] [CrossRef]
- Xiong, X.X.; Gu, L.J.; Shen, J.; Kang, X.H.; Zheng, Y.Y.; Yue, S.B.; Zhu, S.M. Probenecid protects against transient focal cerebral ischemic injury by inhibiting HMGB1 release and attenuating AQP4 expression in mice. Neurochem. Res. 2014, 39, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.H.; Khatibi, N.H.; Han, H.B.; Hu, Q.; Chen, C.H.; Li, L.; Yang, X.M.; Zhou, C.M. p53-induced uncoupling expression of aquaporin-4 and inwardly rectifying K + 4.1 channels in cytotoxic edema after subarachnoid hemorrhage. CNS Neurosci. Ther. 2012, 18, 334–342. [Google Scholar] [CrossRef]
- Long, C.Y.; Huang, G.Q.; Du, Q.; Zhou, L.Q.; Zhou, J.H. The dynamic expression of aquaporins 1 and 4 in rats with hydrocephalus induced by subarachnoid haemorrhage. Folia Neuropathol. 2019, 57, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Shi, H.; Ren, R.; Huang, L.; Okada, T.; Lenahan, C.; Gamdzyk, M.; Travis, Z.D.; Lu, Q.; Tang, L.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide Attenuates Brain Edema by Protecting Blood-Brain Barrier and Glymphatic System After Subarachnoid Hemorrhage in Rats. Neurotherapeutics 2020, 17, 1954–1972. [Google Scholar] [CrossRef]
- Liu, E.; Peng, X.; Ma, H.; Zhang, Y.; Yang, X.; Zhang, Y.; Sun, L.; Yan, J. The Involvement of Aquaporin-4 in the Interstitial Fluid Drainage Impairment Following Subarachnoid Hemorrhage. Front. Aging Neurosci. 2020, 12, 611494. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Li, J.; Wang, B.; Liu, Q.; Zhao, Y.; Zhang, H.; Wang, W.; Ren, W.; Cui, X.; Yang, X. Dynamic Evolution of the Glymphatic System at the Early Stages of Subarachnoid Hemorrhage. Front. Neurol. 2022, 13, 924080. [Google Scholar] [CrossRef]
- Anzabi, M.; Ardalan, M.; Iversen, N.K.; Rafati, A.H.; Hansen, B.; Østergaard, L. Hippocampal Atrophy Following Subarachnoid Hemorrhage Correlates with Disruption of Astrocyte Morphology and Capillary Coverage by AQP4. Front. Cell. Neurosci. 2018, 12, 19. [Google Scholar] [CrossRef]
- Tait, M.J.; Saadoun, S.; Bell, B.A.; Verkman, A.S.; Papadopoulos, M.C. Increased brain edema in aqp4-null mice in an experimental model of subarachnoid hemorrhage. Neuroscience 2010, 167, 60–67. [Google Scholar] [CrossRef]
- Luo, C.; Yao, X.; Li, J.; He, B.; Liu, Q.; Ren, H.; Liang, F.; Li, M.; Lin, H.; Peng, J.; et al. Paravascular pathways contribute to vasculitis and neuroinflammation after subarachnoid hemorrhage independently of glymphatic control. Cell Death Dis. 2016, 7, e2160. [Google Scholar] [CrossRef]
- Pu, T.; Zou, W.; Feng, W.; Zhang, Y.; Wang, L.; Wang, H.; Xiao, M. Persistent Malfunction of Glymphatic and Meningeal Lymphatic Drainage in a Mouse Model of Subarachnoid Hemorrhage. Exp. Neurobiol. 2019, 28, 104–118. [Google Scholar] [CrossRef]
- Liu, E.; Sun, L.; Zhang, Y.; Wang, A.; Yan, J. Aquaporin4 Knockout Aggravates Early Brain Injury Following Subarachnoid Hemorrhage Through Impairment of the Glymphatic System in Rat Brain. Acta Neurochir. Suppl. 2020, 127, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Meng, C.J.; Shen, X.M.; Shu, Z.; Ma, C.; Zhu, G.Q.; Liu, H.X.; He, W.C.; Sun, X.B.; Huo, L.; et al. Potential contribution of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 to blood-brain barrier disruption and brain edema after experimental subarachnoid hemorrhage. J. Mol. Neurosci. 2012, 48, 273–280. [Google Scholar] [CrossRef]
- Cao, S.; Zhu, P.; Yu, X.; Chen, J.; Li, J.; Yan, F.; Wang, L.; Yu, J.; Chen, G. Hydrogen sulfide attenuates brain edema in early brain injury after subarachnoid hemorrhage in rats: Possible involvement of MMP-9 induced blood-brain barrier disruption and AQP4 expression. Neurosci. Lett. 2016, 621, 88–97. [Google Scholar] [CrossRef]
- Shi, S.S.; Zhang, H.B.; Wang, C.H.; Yang, W.Z.; Liang, R.S.; Chen, Y.; Tu, X.K. Propofol Attenuates Early Brain Injury After Subarachnoid Hemorrhage in Rats. J. Mol. Neurosci. 2015, 57, 538–545. [Google Scholar] [CrossRef]
- Chen, J.H.; Yang, L.K.; Chen, L.; Wang, Y.H.; Wu, Y.; Jiang, B.J.; Zhu, J.; Li, P.P. Atorvastatin ameliorates early brain injury after subarachnoid hemorrhage via inhibition of AQP4 expression in rabbits. Int. J. Mol. Med. 2016, 37, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Cao, D.; Li, Y.; Peng, A.; Wang, Y.; Gao, K.; Tao, C.; Wu, Y. Atorvastatin ameliorates early brain injury through inhibition of apoptosis and ER stress in a rat model of subarachnoid hemorrhage. Biosci. Rep. 2018, 38, BSR20171035. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, Y.; Lu, J.; Zhang, W.; Yan, J.; Yang, L.; Zhou, C.; Liu, R.; Chen, C. Salvinorin A ameliorates cerebral vasospasm through activation of endothelial nitric oxide synthase in a rat model of subarachnoid hemorrhage. Microcirculation 2018, 25, e12442. [Google Scholar] [CrossRef]
- Zhang, H.B.; Tu, X.K.; Song, S.W.; Liang, R.S.; Shi, S.S. Baicalin Reduces Early Brain Injury after Subarachnoid Hemorrhage in Rats. Chin. J. Integr. Med. 2020, 26, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.Y.; Chen, Y.R.; Lee, J.E.; Chen, K.W.; Luh, H.T.; Chen, Y.T.; Wang, K.C.; Hsieh, S.T. Dental Pulp Stem Cell-Derived Conditioned Medium Alleviates Subarachnoid Hemorrhage-Induced Microcirculation Impairment by Promoting M2 Microglia Polarization and Reducing Astrocyte Swelling. Transl. Stroke Res. 2023, 14, 688–703. [Google Scholar] [CrossRef]
- Zhang, C.; Jiang, M.; Wang, W.Q.; Zhao, S.J.; Yin, Y.X.; Mi, Q.J.; Yang, M.F.; Song, Y.Q.; Sun, B.L.; Zhang, Z.Y. Selective mGluR1 Negative Allosteric Modulator Reduces Blood-Brain Barrier Permeability and Cerebral Edema After Experimental Subarachnoid Hemorrhage. Transl. Stroke Res. 2020, 11, 799–811. [Google Scholar] [CrossRef]
- Tan, X.; Li, X.; Li, R.; Meng, W.; Xie, Z.; Li, J.; Pang, Y.; Huang, G.; Li, L.; Li, H. β-hydroxybutyrate alleviates neurological deficits by restoring glymphatic and inflammation after subarachnoid hemorrhage in mice. Exp. Neurol. 2024, 378, 114819. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Sun, S.Q. [Expression of aquaporin-4 protein in brain from rats with hemorrhagic edema]. Zhongguo Wei Zhong Bing. Ji Jiu Yi Xue 2003, 15, 538–541. [Google Scholar]
- Wu, H.; Zhang, Z.; Li, Y.; Zhao, R.; Li, H.; Song, Y.; Qi, J.; Wang, J. Time course of upregulation of inflammatory mediators in the hemorrhagic brain in rats: Correlation with brain edema. Neurochem. Int. 2010, 57, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Qiu, G.P.; Huang, J.; Zhang, B.; Sun, S.Q.; Gan, S.W.; Lu, W.T.; Wang, K.J.; Huang, S.Q.; Zhu, S.J. Internalization of aquaporin-4 after collagenase-induced intracerebral hemorrhage. Anat. Rec. 2015, 298, 554–561. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, P.; Su, J.; Xiang, J.; Cai, D.; Dong, Q. Effects of Aquaporin-4 on edema formation following intracerebral hemorrhage. Exp. Neurol. 2010, 223, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.D.; Chen, C.C.; Shen, C.C.; Chin, L.T.; Ma, H.I.; Chuang, H.Y.; Cho, D.Y.; Chu, C.H.; Chang, C. Hyperglycemia exacerbates intracerebral hemorrhage via the downregulation of aquaporin-4: Temporal assessment with magnetic resonance imaging. Stroke 2013, 44, 1682–1689. [Google Scholar] [CrossRef]
- Chu, H.; Xiang, J.; Wu, P.; Su, J.; Ding, H.; Tang, Y.; Dong, Q. The role of aquaporin 4 in apoptosis after intracerebral hemorrhage. J. Neuroinflammation 2014, 11, 184. [Google Scholar] [CrossRef]
- Jeon, H.; Kim, M.; Park, W.; Lim, J.S.; Lee, E.; Cha, H.; Ahn, J.S.; Kim, J.H.; Hong, S.H.; Park, J.E.; et al. Upregulation of AQP4 Improves Blood-Brain Barrier Integrity and Perihematomal Edema Following Intracerebral Hemorrhage. Neurotherapeutics 2021, 18, 2692–2706. [Google Scholar] [CrossRef]
- Liu, X.; Wu, G.; Tang, N.; Li, L.; Liu, C.; Wang, F.; Ke, S. Glymphatic Drainage Blocking Aggravates Brain Edema, Neuroinflammation via Modulating TNF-α, IL-10, and AQP4 After Intracerebral Hemorrhage in Rats. Front. Cell. Neurosci. 2021, 15, 784154. [Google Scholar] [CrossRef]
- Xue, F.; Chen, W.C.; Lian, X.; He, G.H.; Tian, J.Y.; Liu, Y.H.; Wang, G.Q. The Regulatory Role and Mechanism of Circadian Rhythm in Hemoglobin Co-cultured Neurovascular Unit. Biomed. Environ. Sci. 2024, 37, 726–738. [Google Scholar] [CrossRef]
- Jia, P.; He, J.; Li, Z.; Wang, J.; Jia, L.; Hao, R.; Lai, J.; Zang, W.; Chen, X.; Wang, J. Profiling of Blood-Brain Barrier Disruption in Mouse Intracerebral Hemorrhage Models: Collagenase Injection vs. Autologous Arterial Whole Blood Infusion. Front. Cell. Neurosci. 2021, 15, 699736. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liang, C.; Peng, S.; Bao, S.; Xue, F.; Lian, X.; Liu, Y.; Wang, G. Aquaporin-4 activation facilitates glymphatic system function and hematoma clearance post-intracerebral hemorrhage. Glia 2025, 73, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.P.; Xu, J.; Zhuo, F.; Sun, S.Q.; Liu, H.; Yang, M.; Huang, J.; Lu, W.T.; Huang, S.Q. Loss of AQP4 polarized localization with loss of β-dystroglycan immunoreactivity may induce brain edema following intracerebral hemorrhage. Neurosci. Lett. 2015, 588, 42–48. [Google Scholar] [CrossRef]
- Tang, Y.P.; Cai, D.F.; Liu, J. [Research on acting mechanism of rhubarb on aquaporin-4 in rats with blood-brain barrier injury after acute cerebral hemorrhage]. Zhongguo Zhong Xi Yi Jie He Za Zhi 2006, 26, 152–156. [Google Scholar]
- Gu, Y.T.; Zhang, H.; Xue, Y.X. Dexamethasone treatment modulates aquaporin-4 expression after intracerebral hemorrhage in rats. Neurosci. Lett. 2007, 413, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhao, Z.; Zhao, S.; Sheng, Y.; Zhao, Z.; Gao, C.; Li, J.; Liu, X. Recombinant hirudin treatment modulates aquaporin-4 and aquaporin-9 expression after intracerebral hemorrhage in vivo. Mol. Biol. Rep. 2009, 36, 1119–1127. [Google Scholar] [CrossRef]
- Qing, W.G.; Dong, Y.Q.; Ping, T.Q.; Lai, L.G.; Fang, L.D.; Min, H.W.; Xia, L.; Heng, P.Y. Brain edema after intracerebral hemorrhage in rats: The role of iron overload and aquaporin 4. J. Neurosurg. 2009, 110, 462–468. [Google Scholar] [CrossRef]
- Manaenko, A.; Fathali, N.; Khatibi, N.H.; Lekic, T.; Hasegawa, Y.; Martin, R.; Tang, J.; Zhang, J.H. Arginine-vasopressin V1a receptor inhibition improves neurologic outcomes following an intracerebral hemorrhagic brain injury. Neurochem. Int. 2011, 58, 542–548. [Google Scholar] [CrossRef]
- Gao, D.; Ding, F.; Lei, G.; Luan, G.; Zhang, S.; Li, K.; Wang, D.; Zhang, L.; Dai, D. Effects of focal mild hypothermia on thrombin-induced brain edema formation and the expression of protease activated receptor-1, matrix metalloproteinase-9 and aquaporin 4 in rats. Mol. Med. Rep. 2015, 11, 3009–3014. [Google Scholar] [CrossRef]
- Fang, J.; Li, H.; Li, G.; Wang, L. Effect of hyperbaric oxygen preconditioning on peri-hemorrhagic focal edema and aquaporin-4 expression. Exp. Ther. Med. 2015, 10, 699–704. [Google Scholar] [CrossRef]
- Huang, P.; He, X.Y.; Xu, M. Protease-activated receptor 1 inhibitor improves brain edema in rats with intracerebral hemorrhage. Bratisl. Lek. Listy 2020, 121, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Jiang, F.; Mao, Y.; Wei, W.; Song, J.; Jia, F.; Du, X.; Zhong, D.; Li, G. Disulfiram attenuates cell and tissue damage and blood–brain barrier dysfunction after intracranial haemorrhage by inhibiting the classical pyroptosis pathway. Sci. Rep. 2024, 14, 21860. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.N.; Li, Y.B.; Li, Y. [Effects of herbs capable of activating blood circulation or inducing diuresis on the expressions of tumor necrosis factor-alpha, nuclear factor-kappaB, and aquaporin-4 in rats with intracerebral hemorrhage]. Zhongguo Zhong Xi Yi Jie He Za Zhi 2012, 32, 203–208. [Google Scholar] [PubMed]
- Wang, B.F.; Cui, Z.W.; Zhong, Z.H.; Sun, Y.H.; Sun, Q.F.; Yang, G.Y.; Bian, L.G. Curcumin attenuates brain edema in mice with intracerebral hemorrhage through inhibition of AQP4 and AQP9 expression. Acta Pharmacol. Sin. 2015, 36, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Yang, X.; Huang, Y.; Qiu, H.; Huang, G.; Xiao, H.; Kuai, J. The neuroprotective effect of apelin-13 in a mouse model of intracerebral hemorrhage. Neurosci. Lett. 2016, 628, 219–224. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Wang, Z.; Wang, S.; Gao, M.; Xu, R.; Liang, C.; Zhang, H. Attenuation of Acute Phase Injury in Rat Intracranial Hemorrhage by Cerebrolysin that Inhibits Brain Edema and Inflammatory Response. Neurochem. Res. 2016, 41, 748–757. [Google Scholar] [CrossRef]
- Zhong, Z.; Wang, B.; Dai, M.; Sun, Y.; Sun, Q.; Yang, G.; Bian, L. Carvacrol alleviates cerebral edema by modulating AQP4 expression after intracerebral hemorrhage in mice. Neurosci. Lett. 2013, 555, 24–29. [Google Scholar] [CrossRef]
- Guo, F.; Xu, D.; Lin, Y.; Wang, G.; Wang, F.; Gao, Q.; Wei, Q.; Lei, S. Chemokine CCL2 contributes to BBB disruption via the p38 MAPK signaling pathway following acute intracerebral hemorrhage. FASEB J. 2020, 34, 1872–1884. [Google Scholar] [CrossRef]
- Xi, Z.; Chen, X.; Xu, C.; Wang, B.; Zhong, Z.; Sun, Q.; Sun, Y.; Bian, L. Protocatechuic acid attenuates brain edema and blood-brain barrier disruption after intracerebral hemorrhage in mice by promoting Nrf2/HO-1 pathway. Neuroreport 2020, 31, 1274–1282. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Lian, L.; Zhang, C.; He, Z. Glycine-Histidine-Lysine (GHK) Alleviates Astrocytes Injury of Intracerebral Hemorrhage via the Akt/miR-146a-3p/AQP4 Pathway. Front. Neurosci. 2020, 14, 576389. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, W.; Xu, N. Effects of butyphthalide on microglia polarization after intracerebral hemorrhage and the underlying mechanisms. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2022, 47, 717–729. [Google Scholar] [CrossRef]
- Michel-Monigadon, D.; Bonny, C.; Hirt, L. c-Jun N-terminal kinase pathway inhibition in intracerebral hemorrhage. Cerebrovasc. Dis. 2010, 29, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Tang, Y.; Dong, Q. Protection of Vascular Endothelial Growth Factor to Brain Edema Following Intracerebral Hemorrhage and Its Involved Mechanisms: Effect of Aquaporin-4. PLoS ONE 2013, 8, e66051. [Google Scholar] [CrossRef]
- Chu, H.; Tang, Y.; Dong, Q. Protection of granulocyte-colony stimulating factor to hemorrhagic brain injuries and its involved mechanisms: Effects of vascular endothelial growth factor and aquaporin-4. Neuroscience 2014, 260, 59–72. [Google Scholar] [CrossRef]
- Chu, H.; Ding, H.; Tang, Y.; Dong, Q. Erythropoietin protects against hemorrhagic blood-brain barrier disruption through the effects of aquaporin-4. Lab. Invest. 2014, 94, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Su, C.; Zhang, Y.; Guo, Y.; Liu, Y.; Liu, Y.; Yong, V.W.; Xue, M. Adjudin protects blood-brain barrier integrity and attenuates neuroinflammation following intracerebral hemorrhage in mice. Int. Immunopharmacol. 2024, 132, 111962. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yue, J.; Tao, Y.; Zhao, G.; Yi, X.; Zhang, M.; Huang, N.; Cheng, Y. Neutrophil Extracellular Traps Induce Brain Edema Around Intracerebral Hematoma via ERK-Mediated Regulation of MMP9 and AQP4. Transl. Stroke Res. 2024. [Google Scholar] [CrossRef]
- Suda, S.; Yang, B.; Schaar, K.; Xi, X.; Pido, J.; Parsha, K.; Aronowski, J.; Savitz, S.I. Autologous Bone Marrow Mononuclear Cells Exert Broad Effects on Short- and Long-Term Biological and Functional Outcomes in Rodents with Intracerebral Hemorrhage. Stem Cells Dev. 2015, 24, 2756–2766. [Google Scholar] [CrossRef]
- Huang, T.; Chen, B.; Zeng, Z.M. [VEGF-transfected hBMSCs Aggravate Early Brain Edema in Cerebral Hemorrhage Rats]. Sichuan Da Xue Xue Bao Yi Xue Ban 2020, 51, 622–629. [Google Scholar] [CrossRef]
- Zhang, Y.; Deng, H.; Hu, Y.; Pan, C.; Wu, G.; Li, Q.; Tang, Z. Adipose-derived mesenchymal stem cells stereotactic transplantation alleviate brain edema from intracerebral hemorrhage. J. Cell. Biochem. 2019, 120, 14372–14382. [Google Scholar] [CrossRef]
- Chen, K.H.; Chai, H.T.; Lin, K.C.; Chiang, J.Y.; Sung, P.H.; Chen, C.H.; Yip, H.K. Dose-dependent benefits of iron-magnetic nanoparticle-coated human umbilical-derived mesenchymal stem cell treatment in rat intracranial hemorrhage model. Stem Cell Res. Ther. 2022, 13, 265. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Wang, Z.; Liao, Y.; Ye, Q.; Tang, S.; Wei, T.; Xiao, P.; Huang, J.; Lu, W. Edaravone Maintains AQP4 Polarity Via OS/MMP9/β-DG Pathway in an Experimental Intracerebral Hemorrhage Mouse Model. Mol. Neurobiol. 2024, 61, 7639–7658. [Google Scholar] [CrossRef]
- Geng, X.; Ren, C.; Wang, T.; Fu, P.; Luo, Y.; Liu, X.; Yan, F.; Ling, F.; Jia, J.; Du, H.; et al. Effect of remote ischemic postconditioning on an intracerebral hemorrhage stroke model in rats. Neurol. Res. 2012, 34, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Liu, R.; Zhang, F.; Yao, Z.; Zhou, M.L.; Jiang, X.; Ling, S. Roles of mechanosensitive ion channel PIEZO1 in the pathogenesis of brain injury after experimental intracerebral hemorrhage. Neuropharmacology 2024, 251, 109896. [Google Scholar] [CrossRef]
- Kwon, T.H.; Frøkiær, J.; Nielsen, S. Regulation of aquaporin-2 in the kidney: A molecular mechanism of body-water homeostasis. Kidney Res. Clin. Pract. 2013, 32, 96–102. [Google Scholar] [CrossRef]
- Ma, T.H.; Gao, H.W.; Fang, X.D.; Yang, H. Expression and function of aquaporins in peripheral nervous system. Acta Pharmacol. Sin. 2011, 32, 711–715. [Google Scholar] [CrossRef]
- Mobasheri, A.; Wray, S.; Marples, D. Distribution of AQP2 and AQP3 water channels in human tissue microarrays. J. Mol. Histol. 2005, 36, 1–14. [Google Scholar] [CrossRef]
- Penkowa, M.; Cáceres, M.; Borup, R.; Nielsen, F.C.; Poulsen, C.B.; Quintana, A.; Molinero, A.; Carrasco, J.; Florit, S.; Giralt, M.; et al. Novel roles for metallothionein-I + II (MT-I + II) in defense responses, neurogenesis, and tissue restoration after traumatic brain injury: Insights from global gene expression profiling in wild-type and MT-I + II knockout mice. J. Neurosci. Res. 2006, 84, 1452–1474. [Google Scholar] [CrossRef] [PubMed]
- Borsani, E.; Bernardi, S.; Albertini, R.; Rezzani, R.; Rodella, L.F. Alterations of AQP2 expression in trigeminal ganglia in a murine inflammation model. Neurosci. Lett. 2009, 449, 183–188. [Google Scholar] [CrossRef]
- Ding, J.Y.; Kreipke, C.W.; Speirs, S.L.; Schafer, P.; Schafer, S.; Rafols, J.A. Hypoxia-inducible factor-1α signaling in aquaporin upregulation after traumatic brain injury. Neurosci. Lett. 2009, 453, 68–72. [Google Scholar] [CrossRef]
- Shenaq, M.; Kassem, H.; Peng, C.; Schafer, S.; Ding, J.Y.; Fredrickson, V.; Guthikonda, M.; Kreipke, C.W.; Rafols, J.A.; Ding, Y. Neuronal damage and functional deficits are ameliorated by inhibition of aquaporin and HIF1alpha after traumatic brain injury (TBI). J. Neurol. Sci. 2012, 323, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.A., Jr.; Kang, Y.; Truettner, J.S.; Sanchez-Molano, J.; Furones, C.; Yool, A.J.; Atkins, C.M. Fluid-percussion brain injury induces changes in aquaporin channel expression. Neuroscience 2011, 180, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, M.; Qiu, G.P.; Zhuo, F.; Yu, W.H.; Sun, S.Q.; Xiu, Y. Aquaporin 9 in rat brain after severe traumatic brain injury. Arq. Neuropsiquiatr. 2012, 70, 214–220. [Google Scholar] [CrossRef]
- Ali, A.; Konakondla, S.; Zwagerman, N.T.; Peng, C.; Schafer, S.; Ding, J.Y.; Dornbos, D.; Sikharam, C.; Geng, X.; Guthikonda, M.; et al. Glycerol accumulation in edema formation following diffuse traumatic brain injury. Neurol. Res. 2012, 34, 462–468. [Google Scholar] [CrossRef]
- Wang, T.; Chou, D.Y.; Ding, J.Y.; Fredrickson, V.; Peng, C.; Schafer, S.; Guthikonda, M.; Kreipke, C.; Rafols, J.A.; Ding, Y. Reduction of brain edema and expression of aquaporins with acute ethanol treatment after traumatic brain injury. J. Neurosurg. 2013, 118, 390–396. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, Y.W.; Kim, J.H.; Lee, W.T.; Park, K.A.; Lee, J.E. Agmatine Attenuates Brain Edema and Apoptotic Cell Death after Traumatic Brain Injury. J. Korean Med. Sci. 2015, 30, 943–952. [Google Scholar] [CrossRef]
- Badaut, J.; Hirt, L.; Granziera, C.; Bogousslavsky, J.; Magistretti, P.J.; Regli, L. Astrocyte-specific expression of aquaporin-9 in mouse brain is increased after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2001, 21, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Higashida, T.; Peng, C.; Li, J.; Dornbos, D.; Teng, K.; Li, X.; Kinni, H.; Guthikonda, M.; Ding, Y. Hypoxia-inducible factor-1alpha contributes to brain edema after stroke by regulating aquaporins and glycerol distribution in brain. Curr. Neurovasc. Res. 2011, 8, 44–51. [Google Scholar] [CrossRef]
- Zeng, X.; Asmaro, K.; Ren, C.; Gao, M.; Peng, C.; Ding, J.Y.; Fredrickson, V.; Ji, X.; Ding, Y. Acute ethanol treatment reduces blood-brain barrier dysfunction following ischemia/reperfusion injury. Brain Res. 2012, 1437, 127–133. [Google Scholar] [CrossRef]
- Wei, X.; Ren, X.; Jiang, R.; Li, H.; Gao, F.; Chen, Y.; Hou, J.; Liu, X.; Sun, S.; Yang, M. Phosphorylation of p38 MAPK mediates aquaporin 9 expression in rat brains during permanent focal cerebral ischaemia. J. Mol. Histol. 2015, 46, 273–281. [Google Scholar] [CrossRef]
- Wu, H.; Tang, C.; Tai, L.W.; Yao, W.; Guo, P.; Hong, J.; Yang, X.; Li, X.; Jin, Z.; Ke, J.; et al. Flurbiprofen axetil attenuates cerebral ischemia/reperfusion injury by reducing inflammation in a rat model of transient global cerebral ischemia/reperfusion. Biosci. Rep. 2018, 38, BSR20171562. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Wang, J.; Xu, J.; Zhao, X.; Xu, X.; Lu, X. Lack of Aquaporin 9 Reduces Brain Angiogenesis and Exaggerates Neuronal Loss in the Hippocampus Following Intracranial Hemorrhage in Mice. J. Mol. Neurosci. 2017, 61, 351–358. [Google Scholar] [CrossRef]
- Fan, Z.; Yuan, Y.; Wang, F.; Qi, Y.; Han, H.; Wu, J.; Zhang, G.; Yang, L. Diabetes mitigates the recovery following intracranial hemorrhage in rats. Behav. Brain Res. 2017, 320, 412–419. [Google Scholar] [CrossRef]
- Arima, H.; Yamamoto, N.; Sobue, K.; Umenishi, F.; Tada, T.; Katsuya, H.; Asai, K. Hyperosmolar mannitol simulates expression of aquaporins 4 and 9 through a p38 mitogen-activated protein kinase-dependent pathway in rat astrocytes. J. Biol. Chem. 2003, 278, 44525–44534. [Google Scholar] [CrossRef]
- Xi, T.; Jin, F.; Zhu, Y.; Wang, J.; Tang, L.; Wang, Y.; Liebeskind, D.S.; Scalzo, F.; He, Z. miR-27a-3p protects against blood-brain barrier disruption and brain injury after intracerebral hemorrhage by targeting endothelial aquaporin-11. J. Biol. Chem. 2018, 293, 20041–20050. [Google Scholar] [CrossRef] [PubMed]
- Amro, Z.; Ryan, M.; Collins-Praino, L.E.; Yool, A.J. Unexpected Classes of Aquaporin Channels Detected by Transcriptomic Analysis in Human Brain Are Associated with Both Patient Age and Alzheimer’s Disease Status. Biomedicines 2023, 11, 770. [Google Scholar] [CrossRef]
- Milton, N.G. Role of hydrogen peroxide in the aetiology of Alzheimer’s disease: Implications for treatment. Drugs Aging 2004, 21, 81–100. [Google Scholar] [CrossRef]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflammation 2019, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Ferdinand, P.; Roffe, C. Hypoxia after stroke: A review of experimental and clinical evidence. Exp. Transl. Stroke Med. 2016, 8, 9. [Google Scholar] [CrossRef]
- Bao, M.; Liang, M.; Sun, X.; Mohyuddin, S.G.; Chen, S.; Wen, J.; Yong, Y.; Ma, X.; Yu, Z.; Ju, X.; et al. Baicalin Alleviates LPS-Induced Oxidative Stress via NF-κB and Nrf2-HO1 Signaling Pathways in IPEC-J2 Cells. Front. Vet. Sci. 2021, 8, 808233. [Google Scholar] [CrossRef]
- Coimbra-Costa, D.; Alva, N.; Duran, M.; Carbonell, T.; Rama, R. Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox Biol. 2017, 12, 216–225. [Google Scholar] [CrossRef]
- Gu, Y.; Zhou, C.; Piao, Z.; Yuan, H.; Jiang, H.; Wei, H.; Zhou, Y.; Nan, G.; Ji, X. Cerebral edema after ischemic stroke: Pathophysiology and underlying mechanisms. Front. Neurosci. 2022, 16, 988283. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, K.; Opiełka, M.; Gozdalski, J.; Radziwon, J.; Dańczyszyn, A.; Aitken, A.V.; Biancardi, V.C.; Winklewski, P.J. The Role of Arginine-Vasopressin in Stroke and the Potential Use of Arginine-Vasopressin Type 1 Receptor Antagonists in Stroke Therapy: A Narrative Review. Int. J. Mol. Sci. 2023, 24, 2119. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Regli, L. Distribution and possible roles of aquaporin 9 in the brain. Neuroscience 2004, 129, 971–981. [Google Scholar] [CrossRef]
- Ren, H.; Han, R.; Chen, X.; Liu, X.; Wan, J.; Wang, L.; Yang, X.; Wang, J. Potential therapeutic targets for intracerebral hemorrhage-associated inflammation: An update. J. Cereb. Blood Flow Metab. 2020, 40, 1752–1768. [Google Scholar] [CrossRef]
- Tamma, G.; Valenti, G.; Grossini, E.; Donnini, S.; Marino, A.; Marinelli, R.A.; Calamita, G. Aquaporin Membrane Channels in Oxidative Stress, Cell Signaling, and Aging: Recent Advances and Research Trends. Oxid. Med. Cell. Longev. 2018, 2018, 1501847. [Google Scholar] [CrossRef]
- da Silva, I.V.; Mlinarić, M.; Lourenço, A.R.; Pérez-Garcia, O.; Čipak Gašparović, A.; Soveral, G. Peroxiporins and Oxidative Stress: Promising Targets to Tackle Inflammation and Cancer. Int. J. Mol. Sci. 2024, 25, 8381. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef]
- Rizwan Siddiqui, M.; Attar, F.; Mohanty, V.; Kim, K.S.; Shekhar Mayanil, C.; Tomita, T. Erythropoietin-mediated activation of aquaporin-4 channel for the treatment of experimental hydrocephalus. Child’s Nerv. Syst. 2018, 34, 2195–2202. [Google Scholar] [CrossRef]

{kind=link}
| Characteristic | AQP2 | AQP4 | AQP9 | AQP11 |
|---|---|---|---|---|
| Location | Kidney collecting ducts, minimal expression in the brain astrocytes [22] | Astrocyte end-feet, ependymal cells [11,12,13,14] | Astrocytes, neurons [18] | Endoplasmic reticulum (ER) of astrocytes and glial cells [24] |
| Primary Function | Water reabsorption in response to ADH [21] | Water transport, brain water homeostasis [11,12,13,14,15] | Glycerol, lactate, and water transport [17] | Intracellular water regulation, ER stress response [24] |
| Role in brain injury | Indirect role via systemic water balance, affected by SIADH or DI [23] | Regulates brain edema formation and resolution [25] | Supports metabolic adaptation by shuttling lactate and glycerol for energy [19,20] | Protects against ER stress and oxidative damage [24] |
| Involvement in edema | ADH-induced upregulation may exacerbate fluid retention and increase ICP [10,22] | Major contributor to cytotoxic and vasogenic edema [26] | Indirect role, may contribute to cell swelling [27] | Minimal role in extracellular edema formation |
| Inflammatory response | Limited direct involvement | Can exacerbate inflammation during edema [11,28] | May amplify inflammation through metabolic byproducts [29] | May reduce inflammation by alleviating ER stress [30,31,32] |
| Neuroprotection | Not directly involved in neuroprotection | Can be detrimental if overexpressed during injury [33] | Provides metabolic support for neurons [34] | May protect cells by maintaining ER function [35] |
| Therapeutic potential | Managed through ADH modulation in systemic disorders [23] | Target for edema management using AQP4 inhibitors [36] | Potential target for reducing inflammation and metabolic imbalance [37,38] | Investigational target for reducing ER stress-related damage [32,35] |
| Clinical relevance | Important in brain injury with secondary endocrine issues (e.g., SIADH, DI) [39] | Widely studied in stroke, TBI, and hemorrhages [40,41,42] | Relevant in ischemia and stroke [43] | Limited research, emerging interest in neuroprotection [44] |
| Disease | Aquaporin | Main Findings | Ref. |
|---|---|---|---|
| TBI | AQP4 | AQP4 increases from 15 h to 8-days post-injury; strongly expressed in astrocytomas and peritumoral tissue. | [68] |
| AQP4 | AQP4 and VEGF co-expressed in astrocytes in edematous tissue; may regulate water flow. | [47] | |
| AQP4 | No exon 4 mutation found in AQP4 gene among TBI patients; further genetic studies needed. | [54] | |
| AQP4 | CSF AQP4 elevated post-TBI; levels reflect water metabolism and correlate with ICP control. | [40] | |
| AQP4 | HMGB1-TLR4 signaling in microglia promotes IL-6 release, increasing astrocytic AQP4 expression and edema. | [49] | |
| AQP4 | SNPs in the AQP4 gene (rs3763043) associated with 6-month outcome; influence recovery post-TBI. | [53] | |
| AQP4 | Elevated AQP4 microparticles in TBI patient blood; potential non-invasive biomarker. | [55] | |
| AQP4 | AQP4 upregulation peaks at 7–30-days post-TBI; associated with hypoxia and neuroinflammation. | [46] | |
| AQP4 | NDEs show 8.9-fold AQP4 increase in acute mTBI; 3.6-fold in chronic mTBI; phase-specific biomarker. | [45] | |
| AQP4 | Chronic TBI with CI shows elevated IL-6 and tau biomarkers; AQP4 not elevated in CI cases. | [50] | |
| AQP4 | Increased AQP4 expression correlates with DAI severity and brain edema in CT and pathology. | [48] | |
| AQP4 | miR-211-5p suppresses MMP9 and AQP4; reduced levels linked to increased AQP4 in TBI. | [52] | |
| AQP2, AQP4, AQP9 | AQP2 levels correlated with chronic SDH volume and midline shift; no correlation found in acute cases. | [22] | |
| AQP4 | Blast-related mTBI alters AQP4 expression and glymphatic function; associated with neurodegeneration. | [51] | |
| AIS | AQP2 | Elevated urinary AQP2 in hyponatremia post-stroke; not solely AVP-dependent. | [64] |
| AQP4 | AQP4 upregulation near ischemic foci linked to edema development via astrocytic transport routes. | [41] | |
| AQP4 | The AQP4 SNP rs9951307 is associated with reduced risk of severe brain edema. | [62] | |
| AQP4 | White matter shows increased AQP4 expression and edema compared to cortex post-stroke. | [59] | |
| AQP4 | Higher baseline serum AQP4 predicts reduced infarct growth and better recovery. | [60] | |
| AQP4 | High SBP post-thrombolysis linked to AQP4 upregulation and early neurological deterioration. | [61] | |
| AQP9 | AQP9 SNPs affect stroke risk in hypertensive patients. | [63] | |
| aSAH | AQP4 | Upregulation of AQP4 in astrocytic processes; loss of polarization; no neuronal expression; implicated in edema dynamics. | [42] |
| ICH | AQP2 | Lower serum AQP2 levels associated with worse 90-day outcomes; AQP2 overexpression promotes astrocyte activation and pro-inflammatory signaling. | [21] |
| AQP4 | AQP4 and thrombin contribute to cerebral edema; findings differ between humans and rats; need for human-based data. | [69] | |
| AQP4 | The AQP4 SNP rs1058427 is associated with increased hematoma and perihematomal edema volume. | [70] | |
| AQP4 | AQP4 SNPs may influence ICH susceptibility and age of onset, though findings did not remain significant after correction. | [71] |
| Experimental Model | Main Findings | Ref. |
|---|---|---|
| Rat model of penetrating ballistic-like brain injury via rapid balloon inflation/deflation | Global AQP4 mRNA decreased at 24 h; significant reductions in AQP4 M1 and isoform 3 at 3–7 days. | [74] |
| Rat CCI | Brain edema peaked at 24 h; global AQP4 protein expression was reduced by 48 h, despite only transient reductions in cortical perfusion. | [73] |
| Murine CCI | TBI triggered nuclear translocation of Foxo3a in astrocytes, which increased AQP4 expression, leading to cytotoxic edema and memory deficits; depletion of Foxo3a prevented AQP4 upregulation and rescued edema. | [76] |
| Adult male Wistar rat TBI model assessing hippocampal proteins | Hippocampal AQP4 increased starting at 1 h, peaking at 12 and 72 h, closely correlating with brain water content and edema severity. | [75] |
| Rat CCI-induced TBI with intraventricular siRNA infusion | In both mild and severe TBI, AQP4 expression increased in contralateral brain tissue over different time courses; AQP4 knockdown reduced brain water content. | [81] |
| Murine CCI comparing AQP4 knockout and wild-type mice | AQP4 deficiency reduced brain edema, intracranial pressure, and neuroinflammation; it improved BBB integrity, enhanced amyloid β clearance, and led to better cognitive outcomes. | [80] |
| In vitro FPI in cultured astrocytes | FPI induced a significant upregulation of AQP4 in the astrocyte plasma membrane via new protein synthesis; AQP4 knockdown markedly reduced trauma induced astrocyte swelling. | [83] |
| Murine CCI comparing AQP4+/+ and AQP4–/– mice | AQP4–/– mice showed reduced injury volume, intracranial pressure, and brain water accumulation, as well as ultrastructural changes that contributed to improved neurological outcomes. | [79] |
| Rat TBI model of contusional injury | Early after TBI, AQP4 and DG maintained perivascular polarization; later, loss of polarization (with upregulation of AQP4 isoforms M1 and M23) correlated with severe cytotoxic edema. | [78] |
| Murine closed skull “Hit and Run” TBI model | Global AQP4 increased post-TBI, but a prominent loss of polarized localization at astrocyte end-feet peaked at 7-days, suggesting a compensatory mechanism for edema resolution. | [77] |
| Murine TBI model focusing on the hippocampal CA1 region with adenosine A2A receptor inactivation | TBI impaired perivascular AQP4 polarization in the hippocampal CA1 area; A2AR knockout alleviated these abnormalities, suggesting A2AR involvement in AQP4 dysregulation. | [56] |
| Ovine impact acceleration head injury model of closed head contusional injury | Within contusions, AQP4 expression was heterogeneous: some astrocytes in the core were non-viable (AQP4 negative), whereas pericontusional astrocytes showed robust AQP4 expression, suggesting regional differences in edema regulation. | [84] |
| Rat CCI-induced TBI with analysis of both injured and contralateral hemispheres | In the injured hemisphere, vasogenic edema occurred first followed by cellular edema (with AQP4 downregulated during vasogenic and upregulated during cellular edema); the contralateral side showed a delayed pathological progression. | [85] |
| Rat cortical contusion injury model with secondary insults (hypoxia and hypotension) | Secondary insults at 5 h post-injury significantly worsened BBB function and blunted the normal upregulation of AQP4, thereby exacerbating brain edema and ionic imbalance. | [82] |
| Molecule/Intervention | Experimental Model | Main Findings | Ref. |
|---|---|---|---|
| Propofol | Rat CCI | Reduced brain edema, reduced AQP4 expression; blocked IL-1β/TNF-α-induced AQP4 via NF-κB/p38. | [86] |
| AQP4-siRNA | CCI in post-natal day-17 rats | Reduced edema, increased motor/cognitive recovery, reduced neuronal death, 30% reduction in AQP4 expression. | [90] |
| AQP4-siRNA | Rat TBI (unspecified method) + multimodal MRI | Reduced AQP4 expression and edema at 6–12 h post-TBI; validated MRI for edema tracking. | [91] |
| AQP4-siRNA | Rat TBI (unspecified method) | Prevented AQP4 polarity reversal (astrocytic vs. perivascular); reduced edema. | [92] |
| Magnesium sulfate | Rat diffuse TBI (impact-acceleration) | Restored AQP4 polarity (perivascular localization), reduced edema. | [98] |
| Acetazolamide | Murine/human astrocyte TBI models (unspecified) | Prevented AQP4 redistribution post-TBI; reduced cytotoxic edema. | [93] |
| Progesterone | Rat bilateral medial frontal cortex contusion | Reduced brain water content; region-specific AQP4 reduction (peri-contusion) and increase (third ventricle). | [99] |
| Levetiracetam | Rat FPI | Dose-dependent decrease in AQP4 mRNA/protein and edema; high dose most effective. | [89] |
| Phorbol dibutyrate | Rat diffuse TBI (unspecified) | Reduced brain water content and AQP4 upregulation post-TBI. | [100] |
| Intranasal delivery of nerve growth factor (NGF) | Rat TBI (weight-drop model) | Reduced brain edema, reduced expression of AQP4, IL-1β/TNF-α, and reduced apoptosis. | [101] |
| U0126 (ERK1/2 inhibitor) | Rat astrocyte scratch-injury model | ERK1/2 activation reduced AQP4; U0126 restored AQP4 levels. | [95] |
| Decompressive craniectomy (DC) + hypothermia | Murine TBI (unspecified) + MRI | DC + hypothermia reduced AQP4 expression and edema volume; AQP4 correlated with edema. | [97] |
| Decompressive craniectomy | Rat FPI | DC reduced cortical AQP4 expression and water content at 48 h post-TBI. | [96] |
| Trifluoperazine | Rat TBI (unspecified method) | Reduced AQP4 accumulation on astrocyte end-feet, reduced apoptosis/inflammation, increased recovery. | [94] |
| 3% Hypertonic saline | Rat CCI | Reduced edema, AQP4, TNF-α, IL-1β, and caspase-3-mediated apoptosis. | [87] |
| Astaxanthin + Bumetanide | Murine CCI | Reduced edema, BBB disruption, and AQP4/NKCC1 expression; bumetanide reduced AQP4. | [88] |
| Experimental Model | Main Findings | Ref. |
|---|---|---|
| Murine model of transient focal cerebral ischemia (occlusion followed by reperfusion) | Two peaks of maximal hemispheric swelling were observed at 1 h and 48 h after ischemia. At 1 h, AQP4 expression was significantly increased on astrocyte end-feet in both the core and border of the lesion; at 48 h, AQP4 was elevated throughout astrocytes in the border. | [102] |
| Rat model of neonatal stroke (using high-field 11.7 T MRI and immunohistochemistry) | At 24 h, MRI findings indicated edema, coinciding with significant increases in AQP4 expression on astrocytic end feet in the lesion border. At 72 h, imaging findings persisted with a slow normalization of AQP4 in the border, and by 28-days, AQP4 expression normalized. | [103] |
| Mice with thrombin preconditioning subjected to ischemia (early reperfusion phase) | Early induction of AQP4 coincides with initial tissue swelling and may facilitate water clearance—limiting edema formation, although it did not prevent BBB disruption. | [104] |
| Rats subjected to permanent MCAO (analyzed up to 24 h) | AQP4 expression continuously increased in both the ischemic core and border regions up to 24 h, correlating with brain swelling. | [111] |
| Adult male rats subjected to transient MCAO (1–8 h) with 30 min reperfusion | Focal loss of AQP4 immunoreactivity in regions with high vascular permeability (indicated by fluorescein-dextran uptake) despite unchanged overall AQP4 mRNA/protein levels; minimal astrocyte death observed. | [105] |
| Mice subjected to 90 min MCAO followed by reperfusion (assessed at 24- and 72 h) | Significant loss of perivascular AQP4 in the striatal core at 24 h (with no recovery) and partial recovery in neocortex by 72 h; cortical border zones showed a slight increase in AQP4. | [106] |
| Rodent model of discrete cortical ischemia (examined one-week post-insult) | Loss of AQP4 from astrocytic end-feet, disassembly of supramolecular AQP4 complexes, and downregulation of the AQP4 ex isoform, suggesting a role in facilitating astrocyte mobility during incipient scar formation. | [112] |
| Mice subjected to permanent MCAO for 4 and 24 h | AQP4 immunoreactivity decreased in the striatum and varied in the cortex, delineating ischemic tissue. | [113] |
| Rat focal cerebral ischemia model | Cortical (grey matter) regions exhibited reduced perivascular AQP4 and ~9% swelling, while white matter showed increased AQP4 (2.2–6.2× higher) with ~40% swelling, indicating regional heterogeneity in edema formation. | [59] |
| AQP4 knockout vs. wild-type mice subjected to 1 h transient MCAO with 23 h reperfusion | AQP4 deficiency resulted in a 39% reduction in infarct volume, a 23% reduction in cerebral edema, and a 31% decrease in BBB leakage, with diffusion MRI showing lesser ADC reduction around the occlusion site. | [65] |
| AQP4 knockout mice vs. wild-type controls subjected to transient MCAO (3–14 days follow-up) | AQP4 deletion resulted in reduced lesion volume, decreased neuronal cell death and neuroinflammation, improved motor recovery, and lower mortality. | [66] |
| AQP4 knockout mice assessed via oxygen-17 MRI and immunohistochemistry | AQP4 deletion led to significantly reduced water exchange across the BBB and a 22% increase in cortical capillary density, suggesting an adaptive vascular response to chronic AQP4 loss. | [108] |
| Wild-type and α-syntrophin knockout mice (lacking perivascular AQP4) subjected to 90-min MCAO, treated with hypertonic saline for 48 h | Hypertonic saline reduced brain water content and mitigated BBB disruption in wild-type mice but had no effect in α-syntrophin knockout mice, indicating that the perivascular AQP4 pool is essential for the anti-edema effect of osmotherapy. | [107] |
| Rodent model of cerebral ischemia-reperfusion under hyperglycemic conditions | Hyperglycemia disrupted the continuity of perivascular AQP4 in the cortical penumbra and reduced fluorescence intensity and polarity of AQP4 in the striatal penumbra, leading to increased cellular swelling in the striatum. | [110] |
| Transgenic mice overexpressing ET-1 in astrocytes, subjected to transient MCAO | Overexpression of ET-1 exacerbated neurological deficits, increased infarct size, worsened BBB disruption, and elevated brain water content with enhanced AQP4 expression in astrocytic end feet. | [114] |
| Rat transient MCAO model and primary astrocyte cultures (evaluated at 3–7 days post-reperfusion) | Elevated AQP4 expression in peri-infarct and core regions was closely correlated with increased inflammatory markers (e.g., IL-1α); IL-1α from microglia-derived cells was shown to upregulate astrocytic AQP4, thereby exacerbating edema. | [109] |
| Molecule/Intervention | Experimental Model | Main Findings | Ref. |
|---|---|---|---|
| Atorvastatin | Rats; permanent MCAO via intraluminal suture (with sham, MCAO, and pretreatment groups) | Reduced infarct volume and brain water content, improved neurological deficits, and downregulated AQP4. | [115] |
| Simvastatin | Rats; MCAO model | Decreased neuronal degeneration and brain edema; lowered phospho-CaMK II and AQP4 expression. | [125] |
| Acetazolamide | Rats; ischemic stroke induced by bilateral carotid artery ligation | Reduced brain water content and AQP4 mRNA/protein expression, thereby alleviating cerebral edema and dysfunction. | [126] |
| Acetazolamide (meta-analysis) | Various animal models of ischemic stroke (systematic review/meta-analysis) | Inhibited AQP4 expression and reduced brain edema in the early stages post-stroke, though neurological benefits remain uncertain. | [117] |
| Treadmill pre-training | Rats; transient MCAO following 2-weeks of treadmill exercise | Downregulated AQP4, reduced brain edema, and improved BBB integrity and neurological scores. | [127] |
| Remote ischemic post-conditioning | Rats; transient MCAO with intermittent hindlimb occlusion | Improved neurological function, reduced infarct volume and edema, and decreased AQP4 expression in astrocytes. | [119] |
| Propofol (post-conditioning) | Rats; transient MCAO-induced ischemia/reperfusion injury | Reduced brain edema and BBB damage by decreasing MMP9 and AQP4 expression, leading to improved neurobehavioral outcomes. | [120] |
| Propofol (pre-treatment) | Rats; transient MCAO with 90-min occlusion | Attenuated cerebral edema and reduced AQP4 overexpression in the ischemic border zone; no significant change in infarct volume. | [128] |
| TGN-020 (AQP4 inhibitor) | Rats; non-reperfusion ischemia induced by MCAO | Reduced edema, gliosis, albumin extravasation, and apoptosis; improved overall outcome. | [129] |
| TGN-020 (AQP4 inhibitor) | Rats; MCAO model with MRI follow-up | Reduced brain swelling, lesion volume, and peri-infarct astrogliosis; improved functional recovery. | [116] |
| Methylene blue | Rats; transient MCAO with MRI evaluation | Ameliorated cytotoxic and vasogenic edema by blocking AQP4 upregulation and ERK1/2 activation. | [130] |
| Estradiol | In vitro cultured astrocytes exposed to ischemic factors (hypoxia, AVP, OGD) | Abolished astrocyte swelling induced by AVP/hypoxia and reduced AQP4 abundance with prolonged exposure. | [131] |
| Estradiol | Mice; MCAO model comparing males, females, and ovariectomized females | Preserved AQP4 levels and reduced brain edema in females; effect reversed by estrogen receptor antagonism. | [132] |
| LncRNA MALAT silencing (via miR-145) | In vitro OGD/reoxygenation and mouse MCAO model | Reduced AQP4 expression via miR-145 modulation, leading to decreased infarct area and neuronal injury. | [123] |
| LncRNA SNHG14 knockdown | Mice (MCAO) and BV2 cells under OGD | Reduced inflammation and oxidative stress, decreased AQP4 via miR-199b modulation, attenuating ischemic injury. | [133] |
| LncRNA MALAT1 silencing (via siRNA) | Rats (MCAO) and in vitro OGD/reoxygenation model | Decreased AQP4 expression, enhanced cell viability, and reduced apoptosis. | [122] |
| MicroRNA-145 overexpression | Primary cultured astrocytes under OGD | Promoted astrocyte health, reduced apoptosis, and decreased AQP4 expression by directly targeting AQP4. | [134] |
| MicroRNA-29b overexpression | Patients and mice with ischemic stroke; in vivo experiments | Reduced infarct volume, edema, and BBB disruption; downregulated AQP4 expression. | [124] |
| MicroRNA-29a overexpression | Primary cultured astrocytes subjected to OGD | Reduced LDH release, apoptosis, and AQP4 expression, protecting astrocytes against ischemic injury. | [135] |
| AER-270/AER-271 (functionalized phenylbenzamides) | Models of CNS injury: water intoxication and MCAO-induced ischemic stroke | Inhibited AQP4-mediated water permeability, reduced cerebral edema, and improved neurological outcomes. | [136] |
| Edaravone | Rats; transient focal ischemia (MCAO model) | Reduced infarct area and neurological deficits while markedly lowering AQP4 immunoreactivity. | [118] |
| ALDH2 activation (via Alda-1)/inhibition (via cyanamide) | Rats; MCAO-induced ischemic stroke | Alda-1 improved neurological deficits and reduced infarct size, edema, and AQP4 expression; cyanamide worsened outcomes. | [137] |
| AVP V1 receptor antagonist | Mice; 60-min MCAO model | Attenuated infarct volume and brain edema; modulated AQP4 expression; V2 receptor antagonist showed no benefit. | [138] |
| Bumetanide | Mice (WT and α-Syn−/−) subjected to 90-min MCAO with 24–48 h reperfusion | Reduced infarct volume and brain edema in WT mice, associated with decreased AQP4 expression; effect absent in α-Syn−/− mice | [139] |
| Goreisan | Mice; 4 h MCAO | Decreased brain water content and AQP4 upregulation, with improved motor coordination post-stroke. | [140] |
| Mesenchymal stem cells (MSCs) | Mice; 90-min transient MCAO with intracranial MSC transplantation | Improved neurological scores, reduced brain edema and BBB leakage, and inhibited AQP4 upregulation | [141] |
| Probenecid | Mice; transient MCAO (1 h) | Reduced infarct size and brain edema, inhibited HMGB1 release, and attenuated AQP4 expression. | [142] |
| Normobaric Oxygen (NBO) | Rats; transient MCAO (120-min) with 48 h reperfusion | Improved neurological scores and reduced edema by decreasing AQP4 expression; 100% NBO more effective than 60%. | [121] |
| Experimental Model | Main Findings | Ref. |
|---|---|---|
| Murine model of SAH induced by blood injection into the basal cisterns. Wild-type and AQP−/− mice were used. | AQP4-null mice exhibited greater brain edema than wild-type mice, followed by higher intracranial pressure and worse neurological deficits. | [149] |
| Rat SAH induced by a suture in the middle and anterior cerebral artery. Sham rats used; p53 inhibitor used. | p53 mediates cytotoxic edema following SAH via upregulation of AQP4. This expression was regulated by p38 MAPK. | [143] |
| SAH induced via injection of autologous blood into the cisterna magna. Sham rats used. Hydrocephalus diagnosed by histological identification. | Higher AQP4 levels in SAH-induced hydrocephalus and correlation with its severity. | [144] |
| Rat SAH induced via endovascular perforation of the circle of Willis. Sham rats used. | Loss of capillary coverage by AQP4-positive astrocytes’ end-feet at 4 days after SAH and astrocyte cell swelling. | [148] |
| Murine SAH was induced by injection of fresh unheparinized arterial blood into the cisterna magna; AQP4−/− and wild-type mice. | After SAH, AQP4-null mice had a decreased blood diffusion from the perivascular space to the brain parenchyma. No neurological deficits compared with the sham group. | [150] |
| Perforation of the bifurcation of the anterior and middle cerebral arteries in mice inducing SAH. Sham mice used. | High AQP4 levels 6 to 72 h after SAH. Reduction of perivascular localization of AQP4. Stromal AQP4 expression was higher. PACAP treatment promoted perivascular AQP4 polarization 24 h after SAH by SUR-1 downregulation. | [145] |
| Murine SAH model induced by autologous blood injection into the cisterna magna. | SAH increased hippocampal AQP4 and decreased the polarization of astrocyte AQP4. | [151] |
| Rat SAH model induced via endovascular perforation. AQP4 knockout rats were compared to wild-type. | AQP4 knockout aggravated the function of the glymphatic system. The water content in the whole brain increased and the neurological deficits were more intense. | [146] |
| Murine SAH model induced by transfusing blood into the cisterna magna. | Increased levels of AQP4 24 h after SAH. AQP4 expression varied at different cortical sites. Depolarization was observed at all time points and correlation between AQP4 and the amount of DAC partial protein expression. AQP4 levels in the anterior cortex were significantly higher. | [147] |
| Molecule/Intervention | Experimental Model | Main Findings | Ref. |
|---|---|---|---|
| Hypoxia-inducible factor 1α (HIF-1α) | Prechiasmatic cistern perforation SAH model in rats | Reduced brain edema via inhibition of AQP4. | [153] |
| Hydrogen sulfide (H2S) | Endovascular perforation SAH model in rats | Downregulation of AQP4; reduced edema. | [154] |
| Atorvastatin | Artery blood was injected into the cisterna magna in a rabbit SAH model; sham group used | Downregulation of AQP4 at 72 h; reduced edema. | [156] |
| Atorvastatin | Rat SAH. Endovascular perforation | Downregulation of AQP4 in a dose-dependent manner; reduced edema. | [157] |
| Salvinorin A | Rat SAH. Cerebral artery perforation; sham group used | Downregulation of AQP4 in the basilar artery and hippocampus. | [158] |
| Glutamate | Rat SAH. Cerebral artery perforation; sham group used | Glutamate elevated further AQP4 expression and edema following SAH. | [161] |
| Baicalin | Rat SAH. Cerebral artery perforation; sham group used | Baicalin alleviated SAH-induced early brain injury via activation of the Nrf2/HO-1 pathway and suppression of MMP9 and AQP4. | [159] |
| Dental pulp stem cell conditioned medium (DPSC-CM) | Rat SAH by autologous blood injection into the cisterna magna; sham group used | AQP4 downregulation; effect reversed after exposure to IGF-1. | [160] |
| β-hydroxybutyrate (BHB) | Murine SAH. Autologous blood injection into the cisterna magna; sham group used | Following SAH, SNTA1 levels decreased, leading to AQP4 depolarization. This action was reversed with BHB treatment. | [162] |
| Experimental Model | Main Findings | Ref. |
|---|---|---|
| Rat model of ICH by injecting quantitative collagenase into the left caudate nuclei | High perihematomal AQP4 levels at 6 h up to 1 week after ICH and correlation between AQP4 and brain water. | [163] |
| Mixed model; rat model of ICH by autologous blood injection and post-mortem human brains with ICH | The expression of AQP4 differs between human and rat post-ICH. | [69] |
| Murine ICH model induced by autologous whole blood into the striatum of AQP4+/+ and AQP4−/− mice | AQP4 overexpression in AQP4+/+ mice. Increased edema formation, BBB disruption, and elevated neuronal death in AQP-null mice. | [166] |
| Rat model; ICH induced by infusing autologous blood into the striatum | AQP4 protein expression peaked at 5 days after ICH while mRNA peaked at 12 h; weak correlation between brain edema and AQP4 levels. | [164] |
| Rat model; ICH induced by infusing collagenase/heparin into the striatum | Hyperglycemia induced brain edema aggravation and significant downregulation of AQP4. | [167] |
| Rat model; collagenase-induced ICH; sham group used | Perihematomal AQP4 upregulation was time-dependent following ICH; AQP4 internalized to endosomes undergoing degradation into lysosomes. | [165] |
| Murine ICH model induced by autologous whole blood into the striatum of AQP4+/+ and AQP4−/− mice | Increased apoptosis in AQP4-null mice post-ICH; higher levels of apoptosis-related proteins and worse neurologic deficits and brain edema in AQP4 deletion. | [168] |
| Rat model; ICH induced by autologous whole blood into the right caudate nucleus | Downregulation of β-DG leads to depolarization of astrocyte AQP4 and worse brain edema. | [174] |
| Murine ICH model comparing autologous whole blood injection vs. collagen-induced ICH | BBB leakage and brain edema due to AQP4 mRNA and MMP9 upregulation; tight junctions proteins decreased; above effects noticed in c-ICH on day 3 and on day 5 in b-ICH. | [172] |
| Murine ICH model induced by autologous whole blood in AQP4+/+ and AQP4−/− mice | ROS from ICH downregulated AQP4 resulting in increased BBB permeability; AQP4-null mice had worse edema. | [169] |
| Rat model; collagenase-induced ICH | Glymphatic system blockage resulted in downregulation of AQP4, cell apoptosis, and greater brain edema. | [170] |
| Murine collagenase-induced ICH model in AQP4+/+ and AQP4−/− mice | Improved glymphatic system function by AQP4 activation and hematoma reduction. Opposite effects in AQP4-null mice. | [173] |
| In vitro neurovascular unit model by co-culturing hemoglobin; circadian rhythm stimulation by short-wavelength blue-light exposure | Circadian rhythm stimulation mitigated the reduction in AQP4 expression; plausible effect in brain edema after ICH. | [171] |
| Molecule/Intervention | Experimental Model | Main Findings | Ref. |
|---|---|---|---|
| Rhubarb | Rat ICH model induced by stereospecific injection of auto-blood into caudate nucleus | Alleviated cerebral edema by reducing BBB tight junction damage and astrocyte end-feet process swelling; inhibition of transcription and translation of the AQP4 gene. | [175] |
| Dexamethasone | Rat autologous blood brain injection; sham group used | AQP4 mRNA reduced levels in perihematomal area and increased levels in brain area surrounding the third ventricle on day 3 post-ICH; brain edema reduction. | [176] |
| Recombinant herudin | Rat whole blood injection in caudate nucleus | Inhibition of AQP4; thrombin regulation of AQP4; decreased brain edema. | [177] |
| Deferoxamine | Rat ICH autologous blood injection in right caudate nucelous; healthy rats as controls | Reduced brain edema; downregulation of AQP4 by reduced iron overload. | [178] |
| XG-102 | Murine ICH; intrastriatal collagenase injection | Increased AQP4 levels; reduced edema. | [193] |
| AVP V1a receptor inhibitor | Collagenase-induced ICH murine model; sham group used | Reduced AQP4 levels; reduced brain edema. | [179] |
| Remote ischemic post-conditioning | Collagenase-induced ICH rat model | No difference in AQP4 expression or edema. | [204] |
| Chinese herbs | Rat whole blood injection in caudate nucleus | Reduced brain water content and AQP4 levels. | [184] |
| VEGF | Murine autologous blood brain injection model; AQP4+/+ and AQP4−/− were used | AQP4 upregulation resulted in decreased brain edema; more severe brain edema in AQP4−/− mice. | [194] |
| G-CSF | Murine autologous blood brain injection model; AQP4+/+ and AQP4−/− were used | Upregulation of perihematomal AQP4 in a VEGF-independent manner; worse edema in AQP4 null mice; G-CSF reduce edema AQP4-dependant. | [195] |
| Carvacol | Murine collagenase-induced ICH; AQP4+/+ and AQP4−/− were used | Downregulation of AQP4 mRNA at 24 h and perihematomal protein levels in a dose-dependent manner; reduced edema. | [188] |
| Erythropoietin (EPO) | Murine autologous blood brain injection model; sham group used | Upregulation of perihematomal AQP4; brain edema reduction; tight junction and BBB prevention; EPO effects associated with AQP4. | [196] |
| Focal mild hypothermia | Rat model; thrombin-induced ICH | Downregulation of AQP4 and brain edema reduction. | [180] |
| Curcumin | Murine autologous blood brain injection model; sham group used | AQP4 downregulation in a dose-dependent manner; edema reduction. | [185] |
| Cerebrolysin | Collagenase-induced ICH rat model; sham used | Reduced edema, proinflammatory factors and AQP4 expression; upregulation of tight junction proteins. | [187] |
| Hyperbaric oxygen preconditioning | Autologous blood injection-induced ICH in rats; sham group used | Reduced edema and AQP4 expression in perihematomal site. | [181] |
| Autologous bone marrow-derived mononuclear cells (MNCs) | Rat model; ICH induced by autologous whole blood injection in left striatum | Reduced edema and AQP4 expression in perihematomal site. | [199] |
| Apelin-13 | Collagenase-induced ICH in mice; sham used | AQP4, brain edema-associated and apoptosis-related proteins downregulation; brain edema reduction. | [186] |
| Adipose-derived mesenchymal stromal cells (ADSCs) | Collagenase-induced ICH in mice; sham used | Reduced edema and AQP4 expression in perihematomal site. | [201] |
| Propagermanium | Collagenase-induced ICH in rats; sham used | Brain edema and neurological deficits reduction; BBB integrity prevention; AQP4 downregulation. | [189] |
| PAR-1 inhibitor | Autologous blood injection-induced ICH in rats; sham group used | Reduced edema and AQP4 m RNA levels in perihematomal site. | [182] |
| Human bone marrow mesenchymal stem cells (HBMSCs) | Rat ICH; type I collagenase and heparin brain injection | AQP4, MMP9, VEGF protein reduction; reduced edema; opposite actions and edema aggravation in the hBMSC/VEGF transfection group. | [200] |
| Protocatechuic acid | Collagenase-induced ICH in mice; sham used | Brain edema and BBB disruption alleviation; downregulation of AQP4 protein levels. | [190] |
| GHK | Collagenase-induced ICH in rats; sham used | Upregulation of miR-146a-3p and downregulation of AQP4; edema reduction. | [191] |
| Iron-magnetic nanoparticle-coated human umbilical-derived mesenchymal stem cells (hUC-MSCs) | Collagenase-induced ICH in rats; sham used | Dose-dependent edema reduction and AQP4 downregulation. | [202] |
| Butyphthalide | Rat collagenase-induced ICH model | AQP4 downregulation; BBB integrity prevention; neurological defects improvement. | [192] |
| Edaravone; MMP9-IN-1 | Autologous blood injection-induced ICH in mice | AQP4 polarization maintenance; brain edema alleviation and BBB integrity maintenance. | [203] |
| GsMTx4 (Piezo1 blocker) | Murine ICH injected with autologous arterial blood into the basal ganglia | Reduced the upregulated levels of AQP4 after ICH; positive correlation of AQP4 and Piezo1. | [205] |
| Adjudin | Collagenase-induced ICH in mice; sham group used | Increased AQP4, tight junction and adherens junction protein levels; BBB permeability and brain cell apoptosis decreased. | [197] |
| Disulfiram | Collagenase-induced ICH in mice; sham group used | AQP4, MMP9, and apoptosis proteins downregulation; BBB structural proteins upregulation; edema reduction. | [183] |
| NETs | Rat ICH model | Aggravation of BBB integrity; tight junction proteins decreased; brain edema; increased neuronal apoptosis; perihematomal AQP4 downregulation; inhibition had opposite effects. | [198] |
| Aquaporin | Experimental Model | Main Findings | Ref. |
|---|---|---|---|
| AQP2 | Cryolesion-induced TBI in mice | Increased AQP2 post-TBI; decreased AQP2 expression in Mt1+2 knockout mice post-TBI. | [209] |
| AQP2 | Collagenase-induced rat ICH model | ICH induced AQP2 upregulation in rat astrocytes and microglia both in vitro and in vivo. AQP2 promoted astrocyte activation and indirectly enhanced microglia transition to the M1 phenotype. | [21] |
| AQP2 | Murine model of perioral acute inflammatory pain induced by subcutaneous injection of formalin | Increased AQP2 protein levels and altered distribution in the trigeminal ganglia. | [210] |
| AQP9 | Closed head trauma model in rats | Elevated AQP9 mRNA and protein levels as early as 1 h post-TBI. HIF-1α inhibitor treatment reversed AQP9 upregulation. | [211] |
| AQP9 | Modified Marmarou rat acceleration impact model | AQP9 inhibition ameliorated brain edema, neuronal damage, and improved neurobehavioural outcomes post-TBI. HIF-1α inhibition reduced both mRNA and protein levels of AQP9. | [212] |
| AQP9 | Moderate parasagittal fluid-percussion brain injury (FPI) in rats | AQP9 mRNA and protein expression was elevated following FPI, with sustained elevation observed in both the ipsilateral parietal cortex and hippocampus. | [213] |
| AQP9 | Severe TBI in rats | AQP9 protein and mRNA expression levels increased, reaching a maximum at 6 h post-TBI, followed by a minor reduction at 12 h. | [214] |
| AQP9 | Modified impact/head acceleration model of diffuse TBI in rats | Increased AQP9 and HIF-1α protein levels post-TBI. Inhibition of HIF-1α reduced the TBI-induced AQP9 upregulation. | [215] |
| AQP9 | Modified Marmarou TBI rat model (closed head trauma model) | Ethanol doses significantly decreased the elevated AQP9 mRNA and protein levels induced by TBI. | [216] |
| AQP9 | TBI model of cold injury to the primary motor cortex in rats | AQP9 expression was reduced in agmatine-treated rats 7 days post-TBI. | [217] |
| AQP9 | Penetrating ballistic-like brain injury (PBBI) in rats | Decreased AQP9 mRNA levels within the first 24 h post-PBBI. AQP9 protein levels decreased at 3 days post-injury. | [74] |
| AQP9 | Focal cerebral ischemia was induced in male B6CF1 mice by MCAO | Increased AQP9 protein expression in the infract zone, including the cortex, the ventral pallidum, and the nuclei of the amygdala on reactive astrocytes. | [218] |
| AQP9 | Transient focal ischemia was induced in male ICR-CD1 mice by MCAO | AQP9 protein expression increased with time post-ischemia, independently from swelling in mice. | [102] |
| AQP9 | MCAO followed by reperfusion in male Sprague Dawley rats | Inhibition of either HIF-1α or AQP9 halted the progression of edema, while increasing intracellular glycerol in rats. | [219] |
| AQP9 | Ischemic stroke model generated by MCAO in male Sprague Dawley rats | Ethanol administration post-stroke reduced the expression of AQP9, MMP2, and MMP9, while simultaneously ameliorating brain edema and BBB leakage in rats. | [220] |
| AQP9 | Rats with permanent MCAO | Inhibition of p38 with SB203580 prior to injury resulted in decreased levels of both AQP9 and phosphorylated p38 post-MCAO in rats. | [221] |
| AQP9 | Global cerebral ischemia was achieved in rats by occlusion of bilateral common carotid arteries combined with hypotension for 20-min followed by reperfusion for 72 h | Pre-treatment of rats with flurbiprofen reduced AQP9 mRNA expression in comparison to the I/R group, while demonstrating a dose-dependent effect up to 72 h post-injury. | [222] |
| AQP9 | ICH was induced in rats by whole blood injection in the caudate nucleus | Downregulation of AQP9 expression after ICH, indicating that thrombin might play a key role in AQP9 regulation. | [177] |
| AQP9 | ICH was induced in mice by autologous blood infusion | Curcumin suppressed elevated brain AQP9 mRNA levels and protein levels in astrocytes of ICH mice. | [185] |
| AQP9 | Collagenase-induced ICH in mice | AQP9-null ICH mice demonstrate decreased neovascularization and brain cell proliferation, and greater behavioral dysfunction in comparison to wild-type ICH mice. | [223] |
| AQP9 | Collagenase-induced ICH in male Sprague Dawley rats | Increased hippocampal AQP9 protein levels. AQP9 negatively correlated with brain angiogenesis, neuronal survival, and BBB function. | [224] |
| AQP9 | Mannitol-induced hyperosmotic stress in rats | Increased both mRNA and protein expression levels of AQP9. | [225] |
| AQP11 | The established cell lines for astroglia (1321N1) and neurons (SHSY5Y) were studied in response to inflammation (LPS, 10–100 ng/mL, 24 h) and hypoxia (5 min N2, followed by 0 to 24 h normoxia) | AQP11 transcripts were upregulated in astroglia and neurons. Increased AQP11 expression reduced subsequent H2O2-induced MDA responses compared to controls. | [35] |
| AQP11 | Collagenase-induced ICH rat model | The miR-27a-3p mimic effectively suppressed AQP11 and mitigated post-ICH complications. | [226] |
| AQP | Expression in the Brain | Injury Context | Therapeutic Potential | Limitations |
|---|---|---|---|---|
| AQP4 | Astrocyte end-feet (BBB, glia limitans) | TBI, stroke, vasogenic/cytotoxic edema | Edema control (phase-specific); glymphatic clearance; neuroprotection | Dual role in edema; timing critical; limited drug options |
| AQP2 | Low/induced in hypothalamus (mostly renal) | Hyponatremia/DI post-injury (SIADH, trauma) | Indirect control of systemic water balance via vasopressin axis | Not a CNS target; systemic effects only |
| AQP9 | Astrocytes, some neurons | Stroke, epilepsy, hypoxia | Support energy metabolism (glycerol/lactate transport); cell survival | Less studied; few tools to modulate expression/function |
| AQP11 | Low expression; intracellular (ER of glia/neuron) | Ischemia, TBI, oxidative stress | Reduce ER stress and astrocyte inflammation; experimental neuroprotection | Poorly characterized; intracellular; no specific modulators |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokkoris, S.; Vrettou, C.S.; Lotsios, N.S.; Issaris, V.; Keskinidou, C.; Papavassiliou, K.A.; Papavassiliou, A.G.; Kotanidou, A.; Dimopoulou, I.; Vassiliou, A.G. Aquaporins in Acute Brain Injury: Insights from Clinical and Experimental Studies. Biomedicines 2025, 13, 1406. https://doi.org/10.3390/biomedicines13061406
Kokkoris S, Vrettou CS, Lotsios NS, Issaris V, Keskinidou C, Papavassiliou KA, Papavassiliou AG, Kotanidou A, Dimopoulou I, Vassiliou AG. Aquaporins in Acute Brain Injury: Insights from Clinical and Experimental Studies. Biomedicines. 2025; 13(6):1406. https://doi.org/10.3390/biomedicines13061406
Chicago/Turabian StyleKokkoris, Stelios, Charikleia S. Vrettou, Nikolaos S. Lotsios, Vasileios Issaris, Chrysi Keskinidou, Kostas A. Papavassiliou, Athanasios G. Papavassiliou, Anastasia Kotanidou, Ioanna Dimopoulou, and Alice G. Vassiliou. 2025. "Aquaporins in Acute Brain Injury: Insights from Clinical and Experimental Studies" Biomedicines 13, no. 6: 1406. https://doi.org/10.3390/biomedicines13061406
APA StyleKokkoris, S., Vrettou, C. S., Lotsios, N. S., Issaris, V., Keskinidou, C., Papavassiliou, K. A., Papavassiliou, A. G., Kotanidou, A., Dimopoulou, I., & Vassiliou, A. G. (2025). Aquaporins in Acute Brain Injury: Insights from Clinical and Experimental Studies. Biomedicines, 13(6), 1406. https://doi.org/10.3390/biomedicines13061406