
Role of Cellular Senescence in Parkinson’s Disease: Potential for Disease-Modification Through Senotherapy
Abstract
1. Introduction
2. Relationship Between PD and Aging
3. Vulnerability of SN DA Neurons to Cell Death
4. Pathogenesis of PD
4.1. Oxidative Stress
4.2. Diminished Anti-Oxidant Defenses
4.3. Mitochondrial Dysfunction
4.4. Protein Misfolding
4.5. Dysfunction of the ALP
4.6. Impaired UPS Function
4.7. Impaired Ca2+ Handling
4.8. Inflammation
4.9. Cellular Senescence
5. Mechanisms and Markers of Cellular Senescence
6. Role of Cellular Senescence in Aging
7. Effects of Senotherapeutics in PD Models
7.1. Effects of Curcumin in PD Models
7.2. Effects of Fisetin in PD Models
7.3. Effects of a Serum/Glucocorticoid-Related Kinase 1 Inhibitor in PD Models

{kind=link}
{kind=link}
{kind=link}
| Serotherapeutic | Main Finding(s) | Citation |
|---|---|---|
| Curcumin | Curcumin inhibited α-syn aggregation and increased α-syn solubility in SH-SY5Y cells. | [308] |
| Curcumin | Curcumin prevented LPS-induced astrocyte activation and upregulation of NFκB, pro-inflammatory cytokines, iNOS, and regulating molecules of the intrinsic apoptotic pathway in rats. Curcumin improved the glutathione system and prevented iron deposition and α-syn aggregation in DA neurons in rats. | [309] |
| Curcumin | Curcumin protected against 6-OHDA-induced death of SN DA neurons and decreased striatal DA content in rats. | [310] |
| Curcumin | Curcumin-loaded NPs restored TH to control levels, removed α-syn aggregates, reversed morphological alterations, and protected against cell death in MPP+-treated SH-SY5Y cells. Curcumin-loaded NPs reversed motor deficits and restored striatal DA levels and SN TH immunoreactivity in MPTP-treated mice. | [311] |
| Fisetin | Fisetin protected against 6-OHDA-induced elevation of oxidative stress-related genes, activation of caspase-3 and caspase-9, and cell death by activating PI3K-Akt signaling in SH-SY5Y cells. | [319] |
| Fisetin | Fisetin protected against MPTP-induced cell death, blocked MPTP-induced increases in NO and α-syn expression, attenuated MPTP-induced increased expression of pro-inflammatory cytokines, and suppressed pro-apoptotic signaling in rat PC12 cells. | [320] |
| Fisetin | Fisetin protected against MPTP-induced decreases in striatal DA and TH immunoreactivity in mice. | [321] |
| Fisetin | Fisetin attenuated rotenone-induced motor behavior deficits, decreases in striatal DA and TH immunoreactivity, deficits in midbrain mitochondrial function, and increases in oxidative stress markers in rats. | [322] |
| Fisetin | Fisetin prevented MPTP-induced motor deficits and protected SN DA neurons from apoptosis in mice. | [323] |
| GSK-650394 | Pharmacological inhibition of glial cell SGK1 in glia-mDA neuron co-cultures attenuated mDA neuron death and neurite degeneration. Glial cell SGK1 inhibition alleviated MPTP- and α-syn overexpression-induced behavioral deficits, SN mDA neuron loss, and striatal TH immunoreactivity in mice. | [330] |
| Astragaloside IV | Astragaloside IV prevented the induction of senescence in astrocytes in vitro. Astragaloside IV prevented MPTP-induced motor deficits, SNpc DA neuron loss, and accumulation of senescent astrocytes in the SNpc in mice. | [331] |
7.4. Effects of Astragaloside IV in PD Models
8. Challenges Associated with Senolytics and Senomorphics
9. Next Generation Senotherapeutics
9.1. Senolytic Peptides
9.2. Senoreverters
9.3. PROTACs
9.4. Pro-Drugs
9.5. Immunotherapy
9.6. NPs
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lebouvier, T.; Chaumette, T.; Paillusson, S.; Duyckaerts, C.; Bruley des Varannes, S.; Neunlist, M.; Derkinderen, P. The second brain and Parkinson’s disease. Eur. J. Neurosci. 2009, 30, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Park. Dis. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.; National Institute for Clinical Excellence. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Muleiro Alvarez, M.; Cano-Herrera, G.; Osorio Martinez, M.F.; Vega Gonzales-Portillo, J.; Monroy, G.R.; Murguiondo Perez, R.; Torres-Rios, J.A.; van Tienhoven, X.A.; Garibaldi Bernot, E.M.; Esparza Salazar, F.; et al. A Comprehensive Approach to Parkinson’s Disease: Addressing Its Molecular, Clinical, and Therapeutic Aspects. Int. J. Mol. Sci. 2024, 25, 7183. [Google Scholar] [CrossRef]
- Cilia, R.; Cereda, E.; Akpalu, A.; Sarfo, F.S.; Cham, M.; Laryea, R.; Obese, V.; Oppon, K.; Del Sorbo, F.; Bonvegna, S.; et al. Natural history of motor symptoms in Parkinson’s disease and the long-duration response to levodopa. Brain 2020, 143, 2490–2501. [Google Scholar] [CrossRef]
- Rascol, O.; Brooks, D.J.; Brunt, E.R.; Korczyn, A.D.; Poewe, W.H.; Stocchi, F. Ropinirole in the treatment of early Parkinson’s disease: A 6-month interim report of a 5-year levodopa-controlled study. 056 Study Group. Mov. Disord. 1998, 13, 39–45. [Google Scholar] [CrossRef]
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: A randomized controlled trial. Parkinson Study Group. JAMA 2000, 284, 1931–1938. [Google Scholar] [CrossRef]
- Rascol, O. L-dopa-induced peak-dose dyskinesias in patients with Parkinson’s disease: A clinical pharmacologic approach. Mov. Disord. 1999, 14 (Suppl. 1), 19–32. [Google Scholar]
- Bezard, E.; Brotchie, J.M.; Gross, C.E. Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies. Nat. Rev. Neurosci. 2001, 2, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. Second of two parts. N. Engl. J. Med. 1998, 339, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Manson, A.; Stirpe, P.; Schrag, A. Levodopa-induced-dyskinesias clinical features, incidence, risk factors, management and impact on quality of life. J. Park. Dis. 2012, 2, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Cenci, M.A.; Skovgard, K.; Odin, P. Non-dopaminergic approaches to the treatment of motor complications in Parkinson’s disease. Neuropharmacology 2022, 210, 109027. [Google Scholar] [CrossRef]
- Muller, T. Pharmacokinetic considerations for the use of levodopa in the treatment of Parkinson disease: Focus on levodopa/carbidopa/entacapone for treatment of levodopa-associated motor complications. Clin. Neuropharmacol. 2013, 36, 84–91. [Google Scholar] [CrossRef]
- Kataoka, H.; Sugie, K. Early-morning OFF in Parkinson’s disease: A systematic literature review and current therapeutics. Clin. Neurol. Neurosurg. 2024, 245, 108493. [Google Scholar] [CrossRef]
- Smith, Y.; Wichmann, T.; Factor, S.A.; DeLong, M.R. Parkinson’s disease therapeutics: New developments and challenges since the introduction of levodopa. Neuropsychopharmacology 2012, 37, 213–246. [Google Scholar] [CrossRef]
- Bodagh, I.Y.; Robertson, D.R. A risk-benefit assessment of drugs used in the management of Parkinson’s disease. Drug Saf. 1994, 11, 94–103. [Google Scholar] [CrossRef]
- Factor, S.A.; Molho, E.S.; Podskalny, G.D.; Brown, D. Parkinson’s disease: Drug-induced psychiatric states. Adv. Neurol. 1995, 65, 115–138. [Google Scholar]
- Senard, J.M.; Rai, S.; Lapeyre-Mestre, M.; Brefel, C.; Rascol, O.; Rascol, A.; Montastruc, J.L. Prevalence of orthostatic hypotension in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 63, 584–589. [Google Scholar] [CrossRef]
- Gentil, M.; Pollak, P.; Perret, J. [Parkinsonian dysarthria]. Rev. Neurol. 1995, 151, 105–112. [Google Scholar] [PubMed]
- Bloem, B.R.; van Vugt, J.P.; Beckley, D.J. Postural instability and falls in Parkinson’s disease. Adv. Neurol. 2001, 87, 209–223. [Google Scholar]
- Giladi, N.; Kao, R.; Fahn, S. Freezing phenomenon in patients with parkinsonian syndromes. Mov. Disord. 1997, 12, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Foltynie, T. The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat. Rev. Neurol. 2015, 11, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Goldman, S.M. Epidemiology of Parkinson’s disease. Neurol. Clin. 1996, 14, 317–335. [Google Scholar] [CrossRef]
- Bennett, D.A.; Beckett, L.A.; Murray, A.M.; Shannon, K.M.; Goetz, C.G.; Pilgrim, D.M.; Evans, D.A. Prevalence of parkinsonian signs and associated mortality in a community population of older people. N. Engl. J. Med. 1996, 334, 71–76. [Google Scholar] [CrossRef]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef]
- Buchman, A.S.; Shulman, J.M.; Nag, S.; Leurgans, S.E.; Arnold, S.E.; Morris, M.C.; Schneider, J.A.; Bennett, D.A. Nigral pathology and parkinsonian signs in elders without Parkinson disease. Ann. Neurol. 2012, 71, 258–266. [Google Scholar] [CrossRef]
- Ownby, R.L. Neuroinflammation and cognitive aging. Curr. Psychiatry Rep. 2010, 12, 39–45. [Google Scholar] [CrossRef]
- Zhu, B.; Park, J.M.; Coffey, S.R.; Russo, A.; Hsu, I.U.; Wang, J.; Su, C.; Chang, R.; Lam, T.T.; Gopal, P.P.; et al. Single-cell transcriptomic and proteomic analysis of Parkinson’s disease brains. Sci. Transl. Med. 2024, 16, eabo1997. [Google Scholar] [CrossRef] [PubMed]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Greenamyre, J.T. Environment, mitochondria, and Parkinson’s disease. Neuroscientist 2002, 8, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Zhang, Y.; Dawson, V.L.; Dawson, T.M. Oxidative stress and genetics in the pathogenesis of Parkinson’s disease. Neurobiol. Dis. 2000, 7, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Walther, D.M.; Kasturi, P.; Zheng, M.; Pinkert, S.; Vecchi, G.; Ciryam, P.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M.; Mann, M.; et al. Widespread Proteome Remodeling and Aggregation in Aging C. elegans. Cell 2015, 161, 919–932. [Google Scholar] [CrossRef]
- Nago, N.; Murata, S.; Tanaka, K.; Tanahashi, N. Changes in brain proteasome dynamics associated with aging. Genes. Cells 2024, 29, 438–445. [Google Scholar] [CrossRef]
- Zeng, B.Y.; Medhurst, A.D.; Jackson, M.; Rose, S.; Jenner, P. Proteasomal activity in brain differs between species and brain regions and changes with age. Mech. Ageing Dev. 2005, 126, 760–766. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; Dawson, V.L.; Dawson, T.M. The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci. 2001, 24, S7–S14. [Google Scholar] [CrossRef]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef]
- Raz, Y.; Guerrero-Ros, I.; Maier, A.; Slagboom, P.E.; Atzmon, G.; Barzilai, N.; Macian, F. Activation-Induced Autophagy Is Preserved in CD4+ T-Cells in Familial Longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Asghar, M.; Odeh, A.; Fattahi, A.J.; Henriksson, A.E.; Miglar, A.; Khosousi, S.; Svenningsson, P. Mitochondrial biogenesis, telomere length and cellular senescence in Parkinson’s disease and Lewy body dementia. Sci. Rep. 2022, 12, 17578. [Google Scholar] [CrossRef]
- Hornykiewicz, O. Dopamine miracle: From brain homogenate to dopamine replacement. Mov. Disord. 2002, 17, 501–508. [Google Scholar] [CrossRef]
- Marsden, C.D. Parkinson’s disease. Lancet 1990, 335, 948–952. [Google Scholar] [CrossRef]
- Brichta, L.; Greengard, P. Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: An update. Front. Neuroanat. 2014, 8, 152. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef]
- Moss, J.; Bolam, J.P. The Relationship between Dopaminergic Axons and Glutamatergic Synapses in the Striatum: Structural Considerations. In Dopamine Handbook; Oxford Academic: Oxford, UK, 2009. [Google Scholar]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Mercer, J.N.; Chan, C.S. Autonomous pacemakers in the basal ganglia: Who needs excitatory synapses anyway? Curr. Opin. Neurobiol. 2005, 15, 312–318. [Google Scholar] [CrossRef]
- Kampmann, M. Molecular and cellular mechanisms of selective vulnerability in neurodegenerative diseases. Nat. Rev. Neurosci. 2024, 25, 351–371. [Google Scholar] [CrossRef] [PubMed]
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of alpha-synuclein aggregation by exosomes. J. Biol. Chem. 2015, 290, 2969–2982. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M. Unraveling the role of defective genes in Parkinson’s disease. Park. Relat. Disord. 2007, 13 (Suppl. 3), S248–S249. [Google Scholar] [CrossRef]
- Olney, J.W.; Zorumski, C.F.; Stewart, G.R.; Price, M.T.; Wang, G.J.; Labruyere, J. Excitotoxicity of L-dopa and 6-OH-dopa: Implications for Parkinson’s and Huntington’s diseases. Exp. Neurol. 1990, 108, 269–272. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, T.; Barroso-Chinea, P.; De La Cruz Muros, I.; Del Mar Perez-Delgado, M.; Rodriguez, M. Expression of dopamine and vesicular monoamine transporters and differential vulnerability of mesostriatal dopaminergic neurons. J. Comp. Neurol. 2004, 479, 198–215. [Google Scholar] [CrossRef]
- Barroso-Chinea, P.; Cruz-Muros, I.; Aymerich, M.S.; Rodriguez-Diaz, M.; Afonso-Oramas, D.; Lanciego, J.L.; Gonzalez-Hernandez, T. Striatal expression of GDNF and differential vulnerability of midbrain dopaminergic cells. Eur. J. Neurosci. 2005, 21, 1815–1827. [Google Scholar] [CrossRef]
- Quartu, M.; Serra, M.P.; Boi, M.; Sestu, N.; Lai, M.L.; Del Fiacco, M. Tissue distribution of neurturin, persephin and artemin in the human brainstem at fetal, neonatal and adult age. Brain Res. 2007, 1143, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Nishio, T.; Furukawa, S.; Akiguchi, I.; Sunohara, N. Medial nigral dopamine neurons have rich neurotrophin support in humans. Neuroreport 1998, 9, 2847–2851. [Google Scholar] [CrossRef] [PubMed]
- Seroogy, K.B.; Lundgren, K.H.; Tran, T.M.; Guthrie, K.M.; Isackson, P.J.; Gall, C.M. Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J. Comp. Neurol. 1994, 342, 321–334. [Google Scholar] [CrossRef]
- Hurd, Y.L.; Pristupa, Z.B.; Herman, M.M.; Niznik, H.B.; Kleinman, J.E. The dopamine transporter and dopamine D2 receptor messenger RNAs are differentially expressed in limbic- and motor-related subpopulations of human mesencephalic neurons. Neuroscience 1994, 63, 357–362. [Google Scholar] [CrossRef]
- Mendez, I.; Sanchez-Pernaute, R.; Cooper, O.; Vinuela, A.; Ferrari, D.; Bjorklund, L.; Dagher, A.; Isacson, O. Cell type analysis of functional fetal dopamine cell suspension transplants in the striatum and substantia nigra of patients with Parkinson’s disease. Brain 2005, 128, 1498–1510. [Google Scholar] [CrossRef]
- Faucheux, B.A.; Nillesse, N.; Damier, P.; Spik, G.; Mouatt-Prigent, A.; Pierce, A.; Leveugle, B.; Kubis, N.; Hauw, J.J.; Agid, Y.; et al. Expression of lactoferrin receptors is increased in the mesencephalon of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1995, 92, 9603–9607. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Kordower, J.H.; Collier, T.J. Age-related changes in dopamine transporters and accumulation of 3-nitrotyrosine in rhesus monkey midbrain dopamine neurons: Relevance in selective neuronal vulnerability to degeneration. Eur. J. Neurosci. 2008, 27, 3205–3215. [Google Scholar] [CrossRef] [PubMed]
- Tran, J.; Anastacio, H.; Bardy, C. Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. NPJ Park. Dis. 2020, 6, 8. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative mechanisms in nigral cell death in Parkinson’s disease. Mov. Disord. 1998, 13 (Suppl. 1), 24–34. [Google Scholar]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Walker, A.J.; Berk, M.; Maes, M.; Puri, B.K. Cell Death Pathways: A Novel Therapeutic Approach for Neuroscientists. Mol. Neurobiol. 2018, 55, 5767–5786. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Bosco, D.A.; Fowler, D.M.; Zhang, Q.; Nieva, J.; Powers, E.T.; Wentworth, P., Jr.; Lerner, R.A.; Kelly, J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2006, 2, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef]
- Ferrer, I.; Martinez, A.; Blanco, R.; Dalfo, E.; Carmona, M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: Preclinical Parkinson disease. J. Neural Transm 2011, 118, 821–839. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef]
- Perier, C.; Bove, J.; Vila, M.; Przedborski, S. The rotenone model of Parkinson’s disease. Trends Neurosci. 2003, 26, 345–346. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Vergnes, L.; Franich, N.R.; Reue, K.; Chesselet, M.F. Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Westlund, K.N.; Denney, R.M.; Rose, R.M.; Abell, C.W. Localization of distinct monoamine oxidase A and monoamine oxidase B cell populations in human brainstem. Neuroscience 1988, 25, 439–456. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.J.; Double, K.L. Iron and dopamine: A toxic couple. Brain 2016, 139, 1026–1035. [Google Scholar] [CrossRef]
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef]
- Genoud, S.; Roberts, B.R.; Gunn, A.P.; Halliday, G.M.; Lewis, S.J.G.; Ball, H.J.; Hare, D.J.; Double, K.L. Subcellular compartmentalisation of copper, iron, manganese, and zinc in the Parkinson’s disease brain. Metallomics 2017, 9, 1447–1455. [Google Scholar] [CrossRef]
- Exner, N.; Lutz, A.K.; Haass, C.; Winklhofer, K.F. Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO J. 2012, 31, 3038–3062. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.A.; Augood, S.J.; Kingsbury, A.E.; Foster, O.J.; Emson, P.C. Dopamine transporter (Dat) and synaptic vesicle amine transporter (VMAT2) gene expression in the substantia nigra of control and Parkinson’s disease. Brain Res. Mol. Brain Res. 1996, 36, 157–162. [Google Scholar] [CrossRef]
- Nutt, J.G.; Carter, J.H.; Sexton, G.J. The dopamine transporter: Importance in Parkinson’s disease. Ann. Neurol. 2004, 55, 766–773. [Google Scholar] [CrossRef]
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef]
- Sidhu, A.; Wersinger, C.; Vernier, P. alpha-Synuclein regulation of the dopaminergic transporter: A possible role in the pathogenesis of Parkinson’s disease. FEBS Lett. 2004, 565, 1–5. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Fifita, J.A.; Freckleton, S.E.; Hare, D.J.; Lewis, S.J.G.; Halliday, G.M.; Blair, I.P.; Double, K.L. Accumulation of dysfunctional SOD1 protein in Parkinson’s disease is not associated with mutations in the SOD1 gene. Acta Neuropathol. 2018, 135, 155–156. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Razmpour, R.; Bernard, L.P. Glutathione and Parkinson’s disease: Is this the elephant in the room? Biomed. Pharmacother. 2008, 62, 236–249. [Google Scholar] [CrossRef]
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J.G.; et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain. Acta Neuropathol. 2017, 134, 113–127. [Google Scholar] [CrossRef]
- DelleDonne, A.; Klos, K.J.; Fujishiro, H.; Ahmed, Z.; Parisi, J.E.; Josephs, K.A.; Frigerio, R.; Burnett, M.; Wszolek, Z.K.; Uitti, R.J.; et al. Incidental Lewy body disease and preclinical Parkinson disease. Arch. Neurol. 2008, 65, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef]
- Le, W.; Wu, J.; Tang, Y. Protective Microglia and Their Regulation in Parkinson’s Disease. Front. Mol. Neurosci. 2016, 9, 89. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- Joe, E.H.; Choi, D.J.; An, J.; Eun, J.H.; Jou, I.; Park, S. Astrocytes, Microglia, and Parkinson’s Disease. Exp. Neurobiol. 2018, 27, 77–87. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinsons Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Mizuno, Y.; Ohta, S.; Tanaka, M.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Petit, P.X.; Zamzami, N.; Vayssiere, J.L.; Mignotte, B.; Kroemer, G.; Castedo, M. Implication of mitochondria in apoptosis. Mol. Cell Biochem. 1997, 174, 185–188. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Chartier-Harlin, M.C.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef]
- Ibanez, P.; Bonnet, A.M.; Debarges, B.; Lohmann, E.; Tison, F.; Pollak, P.; Agid, Y.; Durr, A.; Brice, A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004, 364, 1169–1171. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef]
- Li, L.; Nadanaciva, S.; Berger, Z.; Shen, W.; Paumier, K.; Schwartz, J.; Mou, K.; Loos, P.; Milici, A.J.; Dunlop, J.; et al. Human A53T alpha-synuclein causes reversible deficits in mitochondrial function and dynamics in primary mouse cortical neurons. PLoS ONE 2013, 8, e85815. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Casarejos, M.J.; Menendez, J.; Solano, R.M.; Rodriguez-Navarro, J.A.; Garcia de Yebenes, J.; Mena, M.A. Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J. Neurochem. 2006, 97, 934–946. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- Cleeter, M.W.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef]
- Larsen, S.B.; Hanss, Z.; Kruger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef]
- Lucchesi, M.; Biso, L.; Bonaso, M.; Longoni, B.; Buchignani, B.; Battini, R.; Santorelli, F.M.; Doccini, S.; Scarselli, M. Mitochondrial Dysfunction in Genetic and Non-Genetic Parkinson’s Disease. Int. J. Mol. Sci. 2025, 26, 4451. [Google Scholar] [CrossRef]
- Gautier, C.A.; Corti, O.; Brice, A. Mitochondrial dysfunctions in Parkinson’s disease. Rev. Neurol. 2014, 170, 339–343. [Google Scholar] [CrossRef]
- Rademacher, D.J. Potential for Therapeutic-Loaded Exosomes to Ameliorate the Pathogenic Effects of alpha-Synuclein in Parkinson’s Disease. Biomedicines 2023, 11, 1187. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell Biology and Pathophysiology of alpha-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of alpha-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef]
- Paciotti, S.; Bellomo, G.; Gatticchi, L.; Parnetti, L. Are We Ready for Detecting alpha-Synuclein Prone to Aggregation in Patients? The Case of “Protein-Misfolding Cyclic Amplification” and “Real-Time Quaking-Induced Conversion” as Diagnostic Tools. Front. Neurol. 2018, 9, 415. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef]
- Graham, S.F.; Rey, N.L.; Yilmaz, A.; Kumar, P.; Madaj, Z.; Maddens, M.; Bahado-Singh, R.O.; Becker, K.; Schulz, E.; Meyerdirk, L.K.; et al. Biochemical Profiling of the Brain and Blood Metabolome in a Mouse Model of Prodromal Parkinson’s Disease Reveals Distinct Metabolic Profiles. J. Proteome Res. 2018, 17, 2460–2469. [Google Scholar] [CrossRef]
- Frake, R.A.; Ricketts, T.; Menzies, F.M.; Rubinsztein, D.C. Autophagy and neurodegeneration. J. Clin. Investig. 2015, 125, 65–74. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Elazar, Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 2011, 80, 125–156. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Zalon, A.J.; Quiriconi, D.J.; Pitcairn, C.; Mazzulli, J.R. alpha-Synuclein: Multiple pathogenic roles in trafficking and proteostasis pathways in Parkinson’s disease. Neuroscientist 2024, 30, 612–635. [Google Scholar] [CrossRef]
- Ho, D.H.; Kim, H.; Nam, D.; Sim, H.; Kim, J.; Kim, H.G.; Son, I.; Seol, W. LRRK2 impairs autophagy by mediating phosphorylation of leucyl-tRNA synthetase. Cell Biochem. Funct. 2018, 36, 431–442. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef]
- Burbidge, K.; Rademacher, D.J.; Mattick, J.; Zack, S.; Grillini, A.; Bousset, L.; Kwon, O.; Kubicki, K.; Simon, A.; Melki, R.; et al. LGALS3 (galectin 3) mediates an unconventional secretion of SNCA/alpha-synuclein in response to lysosomal membrane damage by the autophagic-lysosomal pathway in human midbrain dopamine neurons. Autophagy 2022, 18, 1020–1048. [Google Scholar] [CrossRef]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26S proteasome: A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef]
- Sherman, M.Y.; Goldberg, A.L. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001, 29, 15–32. [Google Scholar] [CrossRef]
- Ii, K.; Ito, H.; Tanaka, K.; Hirano, A. Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J. Neuropathol. Exp. Neurol. 1997, 56, 125–131. [Google Scholar] [CrossRef]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef]
- Mizuno, Y.; Hattori, N.; Mori, H.; Suzuki, T.; Tanaka, K. Parkin and Parkinson’s disease. Curr. Opin. Neurol. 2001, 14, 477–482. [Google Scholar] [CrossRef]
- Mori, H.; Kondo, T.; Yokochi, M.; Matsumine, H.; Nakagawa-Hattori, Y.; Miyake, T.; Suda, K.; Mizuno, Y. Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 1998, 51, 890–892. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.; Yoshikawa, M.; Kitada, T.; Matsumine, H.; Asakawa, S.; Minoshima, S.; Yamamura, Y.; Shimizu, N.; et al. Immunohistochemical and subcellular localization of Parkin protein: Absence of protein in autosomal recessive juvenile parkinsonism patients. Ann. Neurol. 1999, 45, 668–672. [Google Scholar] [CrossRef]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Wilkinson, K.D.; Deshpande, S.; Larsen, C.N. Comparisons of neuronal (PGP 9.5) and non-neuronal ubiquitin C-terminal hydrolases. Biochem. Soc. Trans. 1992, 20, 631–637. [Google Scholar] [CrossRef]
- Lowe, J.; McDermott, H.; Landon, M.; Mayer, R.J.; Wilkinson, K.D. Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J. Pathol. 1990, 161, 153–160. [Google Scholar] [CrossRef]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Grace, A.A.; Bunney, B.S. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--1. Identification and characterization. Neuroscience 1983, 10, 301–315. [Google Scholar] [CrossRef]
- Foehring, R.C.; Zhang, X.F.; Lee, J.C.; Callaway, J.C. Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J. Neurophysiol. 2009, 102, 2326–2333. [Google Scholar] [CrossRef]
- Zampese, E.; Wokosin, D.L.; Gonzalez-Rodriguez, P.; Guzman, J.N.; Tkatch, T.; Kondapalli, J.; Surmeier, W.C.; D’Alessandro, K.B.; De Stefani, D.; Rizzuto, R.; et al. Ca2+ channels couple spiking to mitochondrial metabolism in substantia nigra dopaminergic neurons. Sci. Adv. 2022, 8, eabp8701. [Google Scholar] [CrossRef]
- Nath, S.; Goodwin, J.; Engelborghs, Y.; Pountney, D.L. Raised calcium promotes alpha-synuclein aggregate formation. Mol. Cell Neurosci. 2011, 46, 516–526. [Google Scholar] [CrossRef]
- Goodwin, J.; Nath, S.; Engelborghs, Y.; Pountney, D.L. Raised calcium and oxidative stress cooperatively promote alpha-synuclein aggregate formation. Neurochem. Int. 2013, 62, 703–711. [Google Scholar] [CrossRef]
- Jain, M.K.; Bhat, R. Modulation of human alpha-synuclein aggregation by a combined effect of calcium and dopamine. Neurobiol. Dis. 2014, 63, 115–128. [Google Scholar] [CrossRef]
- Caraveo, G.; Auluck, P.K.; Whitesell, L.; Chung, C.Y.; Baru, V.; Mosharov, E.V.; Yan, X.; Ben-Johny, M.; Soste, M.; Picotti, P.; et al. Calcineurin determines toxic versus beneficial responses to alpha-synuclein. Proc. Natl. Acad. Sci. USA 2014, 111, E3544–E3552. [Google Scholar] [CrossRef]
- Gomez-Sintes, R.; Ledesma, M.D.; Boya, P. Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 2016, 32, 150–168. [Google Scholar] [CrossRef]
- Yamada, T.; McGeer, P.L.; Baimbridge, K.G.; McGeer, E.G. Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 1990, 526, 303–307. [Google Scholar] [CrossRef]
- Epstein, B.J.; Vogel, K.; Palmer, B.F. Dihydropyridine calcium channel antagonists in the management of hypertension. Drugs 2007, 67, 1309–1327. [Google Scholar] [CrossRef]
- Becker, C.; Jick, S.S.; Meier, C.R. Use of antihypertensives and the risk of Parkinson disease. Neurology 2008, 70, 1438–1444. [Google Scholar] [CrossRef]
- Qu, Y.; Li, J.; Qin, Q.; Wang, D.; Zhao, J.; An, K.; Mao, Z.; Min, Z.; Xiong, Y.; Li, J.; et al. A systematic review and meta-analysis of inflammatory biomarkers in Parkinson’s disease. NPJ Park. Dis. 2023, 9, 18. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; St-Amour, I.; Saint-Pierre, M.; Lamontagne-Proulx, J.; Kriz, J.; Barker, R.A.; Cicchetti, F. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef]
- Conte, C.; Ingrassia, A.; Breve, J.; Bol, J.J.; Timmermans-Huisman, E.; van Dam, A.M.; Beccari, T.; van de Berg, W.D.J. Toll-like Receptor 4 Is Upregulated in Parkinson’s Disease Patients and Co-Localizes with pSer129alphaSyn: A Possible Link with the Pathology. Cells 2023, 12, 1368. [Google Scholar] [CrossRef]
- Grozdanov, V.; Bliederhaeuser, C.; Ruf, W.P.; Roth, V.; Fundel-Clemens, K.; Zondler, L.; Brenner, D.; Martin-Villalba, A.; Hengerer, B.; Kassubek, J.; et al. Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol. 2014, 128, 651–663. [Google Scholar] [CrossRef]
- Kosloski, L.M.; Kosmacek, E.A.; Olson, K.E.; Mosley, R.L.; Gendelman, H.E. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J. Neuroimmunol. 2013, 265, 1–10. [Google Scholar] [CrossRef]
- Saunders, J.A.; Estes, K.A.; Kosloski, L.M.; Allen, H.E.; Dempsey, K.M.; Torres-Russotto, D.R.; Meza, J.L.; Santamaria, P.M.; Bertoni, J.M.; Murman, D.L.; et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J. Neuroimmune Pharmacol. 2012, 7, 927–938. [Google Scholar] [CrossRef]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J. Neuroinflamm. 2018, 15, 205. [Google Scholar] [CrossRef]
- Holmans, P.; Moskvina, V.; Jones, L.; Sharma, M.; International Parkinson’s Disease Genomics Consortium (IPDGC); Vedernikov, A.; Buchel, F.; Saad, M.; Bras, J.M.; Bettella, F.; et al. A pathway-based analysis provides additional support for an immune-related genetic susceptibility to Parkinson’s disease. Hum. Mol. Genet. 2013, 22, 1039–1049. [Google Scholar] [CrossRef]
- Saiki, M.; Baker, A.; Williams-Gray, C.H.; Foltynie, T.; Goodman, R.S.; Taylor, C.J.; Compston, D.A.; Barker, R.A.; Sawcer, S.J.; Goris, A. Association of the human leucocyte antigen region with susceptibility to Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 890–891. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Benveniste, E.N. Inflammatory cytokines within the central nervous system: Sources, function, and mechanism of action. Am. J. Physiol. 1992, 263, C1–C16. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S.; Damier, P.; Faucheux, B. Glial cells and inflammation in Parkinson’s disease: A role in neurodegeneration? Ann. Neurol. 1998, 44, S115–S120. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Boka, G.; Anglade, P.; Wallach, D.; Javoy-Agid, F.; Agid, Y.; Hirsch, E.C. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci. Lett. 1994, 172, 151–154. [Google Scholar] [CrossRef]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef]
- Bogale, T.A.; Faustini, G.; Longhena, F.; Mitola, S.; Pizzi, M.; Bellucci, A. Alpha-Synuclein in the Regulation of Brain Endothelial and Perivascular Cells: Gaps and Future Perspectives. Front. Immunol. 2021, 12, 611761. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Brochard, V.; Combadiere, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Miklossy, J.; Doudet, D.D.; Schwab, C.; Yu, S.; McGeer, E.G.; McGeer, P.L. Role of ICAM-1 in persisting inflammation in Parkinson disease and MPTP monkeys. Exp. Neurol. 2006, 197, 275–283. [Google Scholar] [CrossRef]
- Liu, S.Y.; Qiao, H.W.; Song, T.B.; Liu, X.L.; Yao, Y.X.; Zhao, C.S.; Barret, O.; Xu, S.L.; Cai, Y.N.; Tamagnan, G.D.; et al. Brain microglia activation and peripheral adaptive immunity in Parkinson’s disease: A multimodal PET study. J. Neuroinflamm. 2022, 19, 209. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes. Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Kang, S.M.; Jung, H.S.; Kwon, M.J.; Lee, S.H.; Park, J.H. Testosterone Protects Pancreatic β-cells from Apoptosis and Stress-Induced Accelerated Senescence. World J. Mens. Health 2021, 39, 724–732. [Google Scholar] [CrossRef]
- Xue, L.; Wu, J.; Zheng, W.; Wang, P.; Li, J.; Zhang, Z.; Tong, T. Sp1 is involved in the transcriptional activation of p16INK4 by p21Waf1 in HeLa cells. FEBS Lett. 2004, 564, 199–204. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Deschenes-Simard, X.; Gaumont-Leclerc, M.F.; Bourdeau, V.; Lessard, F.; Moiseeva, O.; Forest, V.; Igelmann, S.; Mallette, F.A.; Saba-El-Leil, M.K.; Meloche, S.; et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes. Dev. 2013, 27, 900–915. [Google Scholar] [CrossRef]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef]
- Haugstetter, A.M.; Loddenkemper, C.; Lenze, D.; Grone, J.; Standfuss, C.; Petersen, I.; Dorken, B.; Schmitt, C.A. Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br. J. Cancer 2010, 103, 505–509. [Google Scholar] [CrossRef]
- Vernier, M.; Bourdeau, V.; Gaumont-Leclerc, M.F.; Moiseeva, O.; Begin, V.; Saad, F.; Mes-Masson, A.M.; Ferbeyre, G. Regulation of E2Fs and senescence by PML nuclear bodies. Genes. Dev. 2011, 25, 41–50. [Google Scholar] [CrossRef]
- Ancrile, B.; Lim, K.H.; Counter, C.M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes. Dev. 2007, 21, 1714–1719. [Google Scholar] [CrossRef]
- Ortiz-Montero, P.; Londono-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal 2017, 15, 17. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. Cellular senescence controls fibrosis in wound healing. Aging 2010, 2, 627–631. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; van de Berg, W.D. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 2013, 28, 747–754. [Google Scholar] [CrossRef]
- Blum-Degen, D.; Muller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef]
- Green, H.F.; Khosousi, S.; Svenningsson, P. Plasma IL-6 and IL-17A Correlate with Severity of Motor and Non-Motor Symptoms in Parkinson’s Disease. J. Park. Dis. 2019, 9, 705–709. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Choi, D.H.; Kim, Y.J.; Kim, Y.G.; Joh, T.H.; Beal, M.F.; Kim, Y.S. Role of matrix metalloproteinase 3-mediated alpha-synuclein cleavage in dopaminergic cell death. J. Biol. Chem. 2011, 286, 14168–14177. [Google Scholar] [CrossRef]
- Bae, E.J.; Choi, M.; Kim, J.T.; Kim, D.K.; Jung, M.K.; Kim, C.; Kim, T.K.; Lee, J.S.; Jung, B.C.; Shin, S.J.; et al. TNF-α promotes alpha-synuclein propagation through stimulation of senescence-associated lysosomal exocytosis. Exp. Mol. Med. 2022, 54, 788–800. [Google Scholar] [CrossRef]
- Wang, Y.; Kuca, K.; You, L.; Nepovimova, E.; Heger, Z.; Valko, M.; Adam, V.; Wu, Q.; Jomova, K. The role of cellular senescence in neurodegenerative diseases. Arch. Toxicol. 2024, 98, 2393–2408. [Google Scholar] [CrossRef]
- Gao, L.; Zheng, W.G.; Wu, X.K.; Du, G.H.; Qin, X.M. Baicalein Delays H2O2-Induced Astrocytic Senescence through Inhibition of Senescence-Associated Secretory Phenotype (SASP), Suppression of JAK2/STAT1/NF-κB Pathway, and Regulation of Leucine Metabolism. ACS Chem. Neurosci. 2021, 12, 2320–2335. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Liu, H.; Mi, Y.; Gou, X. Astrocyte Senescence and Alzheimer’s Disease: A Review. Front. Aging Neurosci. 2020, 12, 148. [Google Scholar] [CrossRef]
- Simmnacher, K.; Krach, F.; Schneider, Y.; Alecu, J.E.; Mautner, L.; Klein, P.; Roybon, L.; Prots, I.; Xiang, W.; Winner, B. Unique signatures of stress-induced senescent human astrocytes. Exp. Neurol. 2020, 334, 113466. [Google Scholar] [CrossRef]
- Kim, S.; Ock, J.; Kim, A.K.; Lee, H.W.; Cho, J.Y.; Kim, D.R.; Park, J.Y.; Suk, K. Neurotoxicity of microglial cathepsin D revealed by secretome analysis. J. Neurochem. 2007, 103, 2640–2650. [Google Scholar] [CrossRef]
- Chung, Y.C.; Kim, Y.S.; Bok, E.; Yune, T.Y.; Maeng, S.; Jin, B.K. MMP-3 contributes to nigrostriatal dopaminergic neuronal loss, BBB damage, and neuroinflammation in an MPTP mouse model of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 370526. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow. Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef]
- Cicchetti, F.; Brownell, A.L.; Williams, K.; Chen, Y.I.; Livni, E.; Isacson, O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur. J. Neurosci. 2002, 15, 991–998. [Google Scholar] [CrossRef]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef]
- Qian, L.; Flood, P.M. Microglial cells and Parkinson’s disease. Immunol. Res. 2008, 41, 155–164. [Google Scholar] [CrossRef]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Riessland, M.; Kolisnyk, B.; Kim, T.W.; Cheng, J.; Ni, J.; Pearson, J.A.; Park, E.J.; Dam, K.; Acehan, D.; Ramos-Espiritu, L.S.; et al. Loss of SATB1 Induces p21-Dependent Cellular Senescence in Post-mitotic Dopaminergic Neurons. Cell Stem Cell 2019, 25, 514–530.e8. [Google Scholar] [CrossRef]
- Shen, Q.Q.; Jv, X.H.; Ma, X.Z.; Li, C.; Liu, L.; Jia, W.T.; Qu, L.; Chen, L.L.; Xie, J.X. Cell senescence induced by toxic interaction between alpha-synuclein and iron precedes nigral dopaminergic neuron loss in a mouse model of Parkinson’s disease. Acta Pharmacol. Sin. 2024, 45, 268–281. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Zhang, C.; Sine, C.C.; van Deursen, E.J.; Jeganathan, K.B.; Hamada, N.; Grasic, J.; Friedman, D.; Stutchman, J.T.; Can, I.; et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 2021, 374, eabb3420. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Hampel, B.; Malisan, F.; Niederegger, H.; Testi, R.; Jansen-Durr, P. Differential regulation of apoptotic cell death in senescent human cells. Exp. Gerontol. 2004, 39, 1713–1721. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Sanokawa-Akakura, R.; Akakura, S.; Ostrakhovitch, E.A.; Tabibzadeh, S. Replicative senescence is distinguishable from DNA damage-induced senescence by increased methylation of promoter of rDNA and reduced expression of rRNA. Mech. Ageing Dev. 2019, 183, 111149. [Google Scholar] [CrossRef]
- Allsopp, R.C.; Chang, E.; Kashefi-Aazam, M.; Rogaev, E.I.; Piatyszek, M.A.; Shay, J.W.; Harley, C.B. Telomere shortening is associated with cell division in vitro and in vivo. Exp. Cell Res. 1995, 220, 194–200. [Google Scholar] [CrossRef]
- Gonzalez-Suarez, E.; Geserick, C.; Flores, J.M.; Blasco, M.A. Antagonistic effects of telomerase on cancer and aging in K5-mTert transgenic mice. Oncogene 2005, 24, 2256–2270. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Bhayadia, R.; Schmidt, B.M.; Melk, A.; Homme, M. Senescence-Induced Oxidative Stress Causes Endothelial Dysfunction. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 161–169. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Da Silva-Alvarez, S.; Picallos-Rabina, P.; Antelo-Iglesias, L.; Triana-Martinez, F.; Barreiro-Iglesias, A.; Sanchez, L.; Collado, M. The development of cell senescence. Exp. Gerontol. 2019, 128, 110742. [Google Scholar] [CrossRef]
- Song, T.; Gu, Y.; Hui, W.; Yang, X.; Liu, Y.; Chen, X. Oxygen-Glucose Deprivation Promoted Fibroblast Senescence and Collagen Expression via IL11. Int. J. Mol. Sci. 2022, 23, 12090. [Google Scholar] [CrossRef] [PubMed]
- Bielak-Zmijewska, A.; Mosieniak, G.; Sikora, E. Is DNA damage indispensable for stress-induced senescence? Mech. Ageing Dev. 2018, 170, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Tibbetts, R.S.; Brumbaugh, K.M.; Williams, J.M.; Sarkaria, J.N.; Cliby, W.A.; Shieh, S.Y.; Taya, Y.; Prives, C.; Abraham, R.T. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes. Dev. 1999, 13, 152–157. [Google Scholar] [CrossRef]
- Bieging-Rolett, K.T.; Johnson, T.M.; Brady, C.A.; Beaudry, V.G.; Pathak, N.; Han, S.; Attardi, L.D. p19(Arf) is required for the cellular response to chronic DNA damage. Oncogene 2016, 35, 4414–4421. [Google Scholar] [CrossRef]
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Cox, L.S. Multiple pathways control cell growth and transformation: Overlapping and independent activities of p53 and p21Cip1/WAF1/Sdi1. J. Pathol. 1997, 183, 134–140. [Google Scholar] [CrossRef]
- Macleod, K.F.; Sherry, N.; Hannon, G.; Beach, D.; Tokino, T.; Kinzler, K.; Vogelstein, B.; Jacks, T. p53-dependent and independent expression of p21 during cell growth, differentiation, and DNA damage. Genes. Dev. 1995, 9, 935–944. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Chetty, R. p16. J. Clin. Pathol. 2018, 71, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Grison, A.; Atanasoski, S. Cyclins, Cyclin-Dependent Kinases, and Cyclin-Dependent Kinase Inhibitors in the Mouse Nervous System. Mol. Neurobiol. 2020, 57, 3206–3218. [Google Scholar] [CrossRef]
- Yoon, M.H.; Kang, S.M.; Lee, S.J.; Woo, T.G.; Oh, A.Y.; Park, S.; Ha, N.C.; Park, B.J. p53 induces senescence through Lamin A/C stabilization-mediated nuclear deformation. Cell Death Dis. 2019, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalaf, H.H.; Aboussekhra, A. p16(INK4A) positively regulates p21WAF1 expression by suppressing AUF1-dependent mRNA decay. PLoS ONE 2013, 8, e70133. [Google Scholar] [CrossRef]
- Al-Khalaf, H.H.; Aboussekhra, A. p16 Controls p53 Protein Expression Through miR-dependent Destabilization of MDM2. Mol. Cancer Res. 2018, 16, 1299–1308. [Google Scholar] [CrossRef]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes. Dev. 2006, 20, 2913–2921. [Google Scholar] [CrossRef]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef]
- Yamamoto, H.; Schoonjans, K.; Auwerx, J. Sirtuin functions in health and disease. Mol. Endocrinol. 2007, 21, 1745–1755. [Google Scholar] [CrossRef]
- Chen, H.; Liu, X.; Zhu, W.; Chen, H.; Hu, X.; Jiang, Z.; Xu, Y.; Wang, L.; Zhou, Y.; Chen, P.; et al. SIRT1 ameliorates age-related senescence of mesenchymal stem cells via modulating telomere shelterin. Front. Aging Neurosci. 2014, 6, 103. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, J.H.; Lee, H.Y.; Min, K.J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34. [Google Scholar] [CrossRef]
- Sasca, D.; Hahnel, P.S.; Szybinski, J.; Khawaja, K.; Kriege, O.; Pante, S.V.; Bullinger, L.; Strand, S.; Strand, D.; Theobald, M.; et al. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood 2014, 124, 121–133. [Google Scholar] [CrossRef]
- Yuan, F.; Liu, L.; Lei, Y.; Tang, P. p53 inhibits the upregulation of sirtuin 1 expression induced by c-Myc. Oncol. Lett. 2017, 14, 4396–4402. [Google Scholar] [CrossRef]
- Lukasova, E.; Kovarik, A.; Kozubek, S. Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence. Cells 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Frediani, E.; Scavone, F.; Laurenzana, A.; Chillà, A.; Tortora, K.; Cimmino, I.; Leri, M.; Bucciantini, M.; Mangoni, M.; Fibbi, G.; et al. Olive phenols preserve lamin B1 expression reducing cGAS/STING/NFκB-mediated SASP in ionizing radiation-induced senescence. J. Cell Mol. Med. 2022, 26, 2337–2350. [Google Scholar] [CrossRef]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes. Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Ngai, Y.T.; Lau, D.; Mittal, P.; Hoffmann, P. Mini Review: Highlight of Recent Advances and Applications of MALDI Mass Spectrometry Imaging in 2024. Anal. Sci. Adv. 2025, 6, e70016. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, N.; Zhang, J.; Yang, B. Advances in Microfluidic Single-Cell RNA Sequencing and Spatial Transcriptomics. Micromachines 2025, 16, 426. [Google Scholar] [CrossRef] [PubMed]
- Dehkordi, S.K.; Walker, J.; Sah, E.; Bennett, E.; Atrian, F.; Frost, B.; Woost, B.; Bennett, R.E.; Orr, T.C.; Zhou, Y.; et al. Profiling senescent cells in human brains reveals neurons with CDKN2D/p19 and tau neuropathology. Nat. Aging 2021, 1, 1107–1116. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Lewén, A.; Matz, P.; Chan, P.H. Free radical pathways in CNS injury. J. Neurotrauma 2000, 17, 871–890. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Fan, Z.; Tong, Y.; Yang, Z.; Wang, S.; Huang, T.; Yang, D.; Ni, Q.; Zhang, M.; Li, D.; Yang, M.; et al. Inhibitor PF-04691502 works as a senolytic to regulate cellular senescence. Exp. Gerontol. 2024, 186, 112359. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Somogyvari, M.; Khatatneh, S.; Soti, C. Hsp90: From Cellular to Organismal Proteostasis. Cells 2022, 11, 2479. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. The Bcl-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Song, S.; Tchkonia, T.; Jiang, J.; Kirkland, J.L.; Sun, Y. Targeting Senescent Cells for a Healthier Aging: Challenges and Opportunities. Adv. Sci. 2020, 7, 2002611. [Google Scholar] [CrossRef]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Szaniawski, M.A.; Spivak, A.M. Senotherapeutics and HIV-1 Persistence. Curr. HIV/AIDS Rep. 2020, 17, 219–225. [Google Scholar] [CrossRef]
- Laberge, R.M.; Zhou, L.; Sarantos, M.R.; Rodier, F.; Freund, A.; de Keizer, P.L.; Liu, S.; Demaria, M.; Cong, Y.S.; Kapahi, P.; et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 2012, 11, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschenes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Park, H.; Kim, H.P. Effects of flavonoids on senescence-associated secretory phenotype formation from bleomycin-induced senescence in BJ fibroblasts. Biochem. Pharmacol. 2015, 96, 337–348. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef]
- Wan, M.; Gray-Gaillard, E.F.; Elisseeff, J.H. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 2021, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Alimbetov, D.; Davis, T.; Brook, A.J.; Cox, L.S.; Faragher, R.G.; Nurgozhin, T.; Zhumadilov, Z.; Kipling, D. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology 2016, 17, 305–315. [Google Scholar] [CrossRef]
- Schuliga, M.; Kanwal, A.; Read, J.; Blokland, K.E.C.; Burgess, J.K.; Prele, C.M.; Mutsaers, S.E.; Grainge, C.; Thomson, C.; James, A.; et al. A cGAS-dependent response links DNA damage and senescence in alveolar epithelial cells: A potential drug target in IPF. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L859–L871. [Google Scholar] [CrossRef]
- Shi, Y.; Zhao, L.; Wang, J.; Liu, S.; Zhang, Y.; Qin, Q. The selective NLRP3 inflammasome inhibitor MCC950 improves isoproterenol-induced cardiac dysfunction by inhibiting cardiomyocyte senescence. Eur. J. Pharmacol. 2022, 937, 175364. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Hughes, G.; Ledgerwood, E.C.; Gane, A.M.; Smith, R.A.; Murphy, M.P. Prevention of mitochondrial oxidative damage using targeted antioxidants. Ann. N. Y Acad. Sci. 2002, 959, 263–274. [Google Scholar] [CrossRef]
- Mantzorou, M.; Pavlidou, E.; Vasios, G.; Tsagalioti, E.; Giaginis, C. Effects of curcumin consumption on human chronic diseases: A narrative review of the most recent clinical data. Phytother. Res. 2018, 32, 957–975. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, J.; Prasad, S.; Aggarwal, B.B. Curcumin and cancer cells: How many ways can curry kill tumor cells selectively? AAPS J. 2009, 11, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Cherif, H.; Bisson, D.G.; Jarzem, P.; Weber, M.; Ouellet, J.A.; Haglund, L. Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro. J. Clin. Med. 2019, 8, 433. [Google Scholar] [CrossRef]
- Heger, M.; van Golen, R.F.; Broekgaarden, M.; Michel, M.C. The molecular basis for the pharmacokinetics and pharmacodynamics of curcumin and its metabolites in relation to cancer. Pharmacol. Rev. 2014, 66, 222–307. [Google Scholar] [CrossRef]
- Ragothaman, M.; Murgod, U.A.; Gururaj, G.; Kumaraswamy, S.D.; Muthane, U. Lower risk of Parkinson’s disease in an admixed population of European and Indian origins. Mov. Disord. 2003, 18, 912–914. [Google Scholar] [CrossRef] [PubMed]
- Commandeur, J.N.; Vermeulen, N.P. Cytotoxicity and cytoprotective activities of natural compounds. The case of curcumin. Xenobiotica 1996, 26, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Strider, J.; Nolan, W.C.; Yan, S.X.; Galvin, J.E. Curcumin inhibits aggregation of alpha-synuclein. Acta Neuropathol. 2008, 115, 479–489. [Google Scholar] [CrossRef]
- Sharma, N.; Nehru, B. Curcumin affords neuroprotection and inhibits alpha-synuclein aggregation in lipopolysaccharide-induced Parkinson’s disease model. Inflammopharmacology 2018, 26, 349–360. [Google Scholar] [CrossRef]
- Zbarsky, V.; Datla, K.P.; Parkar, S.; Rai, D.K.; Aruoma, O.I.; Dexter, D.T. Neuroprotective properties of the natural phenolic antioxidants curcumin and naringenin but not quercetin and fisetin in a 6-OHDA model of Parkinson’s disease. Free Radic. Res. 2005, 39, 1119–1125. [Google Scholar] [CrossRef]
- Zhang, N.; Yan, F.; Liang, X.; Wu, M.; Shen, Y.; Chen, M.; Xu, Y.; Zou, G.; Jiang, P.; Tang, C.; et al. Localized delivery of curcumin into brain with polysorbate 80-modified cerasomes by ultrasound-targeted microbubble destruction for improved Parkinson’s disease therapy. Theranostics 2018, 8, 2264–2277. [Google Scholar] [CrossRef]
- Arai, Y.; Watanabe, S.; Kimira, M.; Shimoi, K.; Mochizuki, R.; Kinae, N. Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL cholesterol concentration. J. Nutr. 2000, 130, 2243–2250. [Google Scholar] [CrossRef]
- Maher, P. How fisetin reduces the impact of age and disease on CNS function. Front. Biosci. (Sch. Ed.) 2015, 7, 58–82. [Google Scholar] [CrossRef]
- Khan, N.; Syed, D.N.; Ahmad, N.; Mukhtar, H. Fisetin: A dietary antioxidant for health promotion. Antioxid. Redox Signal 2013, 19, 151–162. [Google Scholar] [CrossRef]
- Pal, H.C.; Pearlman, R.L.; Afaq, F. Fisetin and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 2016, 928, 213–244. [Google Scholar] [CrossRef] [PubMed]
- Sundarraj, K.; Raghunath, A.; Perumal, E. A review on the chemotherapeutic potential of fisetin: In vitro evidences. Biomed. Pharmacother. 2018, 97, 928–940. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Watanabe, R.; Kurose, T.; Morishige, Y.; Fujimori, K. Protective Effects of Fisetin Against 6-OHDA-Induced Apoptosis by Activation of PI3K-Akt Signaling in Human Neuroblastoma SH-SY5Y Cells. Neurochem. Res. 2018, 43, 488–499. [Google Scholar] [CrossRef]
- Patel, M.Y.; Panchal, H.V.; Ghribi, O.; Benzeroual, K.E. The neuroprotective effect of fisetin in the MPTP model of Parkinson’s disease. J. Parkinsons Dis. 2012, 2, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. Protective effects of fisetin and other berry flavonoids in Parkinson’s disease. Food Funct. 2017, 8, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Alikatte, K.; Palle, S.; Rajendra Kumar, J.; Pathakala, N. Fisetin Improved Rotenone-Induced Behavioral Deficits, Oxidative Changes, and Mitochondrial Dysfunctions in Rat Model of Parkinson’s Disease. J. Diet. Suppl. 2021, 18, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.J.; Feng, Y.; Liu, T.; Wu, T.T.; Chen, Y.J.; Li, X.; Li, Q.; Wu, Y.C. Fisetin Regulates Gut Microbiota and Exerts Neuroprotective Effect on Mouse Model of Parkinson’s Disease. Front. Neurosci. 2020, 14, 549037. [Google Scholar] [CrossRef]
- Lang, F.; Cohen, P. Regulation and physiological roles of serum- and glucocorticoid-induced protein kinase isoforms. Sci. STKE 2001, 2001, re17. [Google Scholar] [CrossRef]
- Hartmann, A.; Veldhuis, J.D.; Deuschle, M.; Standhardt, H.; Heuser, I. Twenty-four hour cortisol release profiles in patients with Alzheimer’s and Parkinson’s disease compared to normal controls: Ultradian secretory pulsatility and diurnal variation. Neurobiol. Aging 1997, 18, 285–289. [Google Scholar] [CrossRef]
- Kaku, K.; Shikimi, T.; Kamisaki, Y.; Shinozuka, K.; Ishino, H.; Okunishi, H.; Takaori, S. Elevation of striatal interleukin-6 and serum corticosterone contents in MPTP-treated mice. Clin. Exp. Pharmacol. Physiol. 1999, 26, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Pratt, B.M.; McPherson, J.M. TGF-beta in the central nervous system: Potential roles in ischemic injury and neurodegenerative diseases. Cytokine Growth Factor. Rev. 1997, 8, 267–292. [Google Scholar] [CrossRef]
- Iwata, S.; Nomoto, M.; Morioka, H.; Miyata, A. Gene expression profiling in the midbrain of striatal 6-hydroxydopamine-injected mice. Synapse 2004, 51, 279–286. [Google Scholar] [CrossRef]
- Stichel, C.C.; Schoenebeck, B.; Foguet, M.; Siebertz, B.; Bader, V.; Zhu, X.R.; Lubbert, H. sgk1, a member of an RNA cluster associated with cell death in a model of Parkinson’s disease. Eur. J. Neurosci. 2005, 21, 301–316. [Google Scholar] [CrossRef]
- Kwon, O.C.; Song, J.J.; Yang, Y.; Kim, S.H.; Kim, J.Y.; Seok, M.J.; Hwang, I.; Yu, J.W.; Karmacharya, J.; Maeng, H.J.; et al. SGK1 inhibition in glia ameliorates pathologies and symptoms in Parkinson disease animal models. EMBO Mol. Med. 2021, 13, e13076. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.L.; Xie, X.H.; Ding, J.H.; Du, R.H.; Hu, G. Astragaloside IV inhibits astrocyte senescence: Implication in Parkinson’s disease. J. Neuroinflamm. 2020, 17, 105. [Google Scholar] [CrossRef]
- Cheng, X.; Gu, J.; Zhang, M.; Yuan, J.; Zhao, B.; Jiang, J.; Jia, X. Astragaloside IV inhibits migration and invasion in human lung cancer A549 cells via regulating PKC-alpha-ERK1/2-NF-κB pathway. Int. Immunopharmacol. 2014, 23, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Frei, B. Astragaloside IV inhibits NF-κB activation and inflammatory gene expression in LPS-treated mice. Mediators Inflamm. 2015, 2015, 274314. [Google Scholar] [CrossRef]
- Gui, D.; Huang, J.; Liu, W.; Guo, Y.; Xiao, W.; Wang, N. Astragaloside IV prevents acute kidney injury in two rodent models by inhibiting oxidative stress and apoptosis pathways. Apoptosis 2013, 18, 409–422. [Google Scholar] [CrossRef]
- Chan, W.S.; Durairajan, S.S.; Lu, J.H.; Wang, Y.; Xie, L.X.; Kum, W.F.; Koo, I.; Yung, K.K.; Li, M. Neuroprotective effects of Astragaloside IV in 6-hydroxydopamine-treated primary nigral cell culture. Neurochem. Int. 2009, 55, 414–422. [Google Scholar] [CrossRef]
- Novais, E.J.; Tran, V.A.; Johnston, S.N.; Darris, K.R.; Roupas, A.J.; Sessions, G.A.; Shapiro, I.M.; Diekman, B.O.; Risbud, M.V. Long-term treatment with senolytic drugs Dasatinib and Quercetin ameliorates age-dependent intervertebral disc degeneration in mice. Nat. Commun. 2021, 12, 5213. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Ait-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.M.; De Vos, J.; et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes. Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of senescent cells by beta-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Guiho, R.; Herranz, N.; Uren, A.; Withers, D.J.; Martinez-Barbera, J.P.; Tietze, L.F.; Gil, J. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 2020, 19, e13133. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gualda, E.; Paez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R., 3rd; Gonzalez-Lopez, C.; Ou, H.L.; Miron-Barroso, S.; Zhang, Z.; Lerida-Viso, A.; et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Noh, J.H.; Bodogai, M.; Martindale, J.L.; Yang, X.; Indig, F.E.; Basu, S.K.; Ohnuma, K.; Morimoto, C.; Johnson, P.F.; et al. Identification of senescent cell surface targetable protein DPP4. Genes. Dev. 2017, 31, 1529–1534. [Google Scholar] [CrossRef]
- Poblocka, M.; Bassey, A.L.; Smith, V.M.; Falcicchio, M.; Manso, A.S.; Althubiti, M.; Sheng, X.; Kyle, A.; Barber, R.; Frigerio, M.; et al. Targeted clearance of senescent cells using an antibody-drug conjugate against a specific membrane marker. Sci. Rep. 2021, 11, 20358. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Rovira, M.; Galiana, I.; Gimenez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Munoz-Martin, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10, e9355. [Google Scholar] [CrossRef]
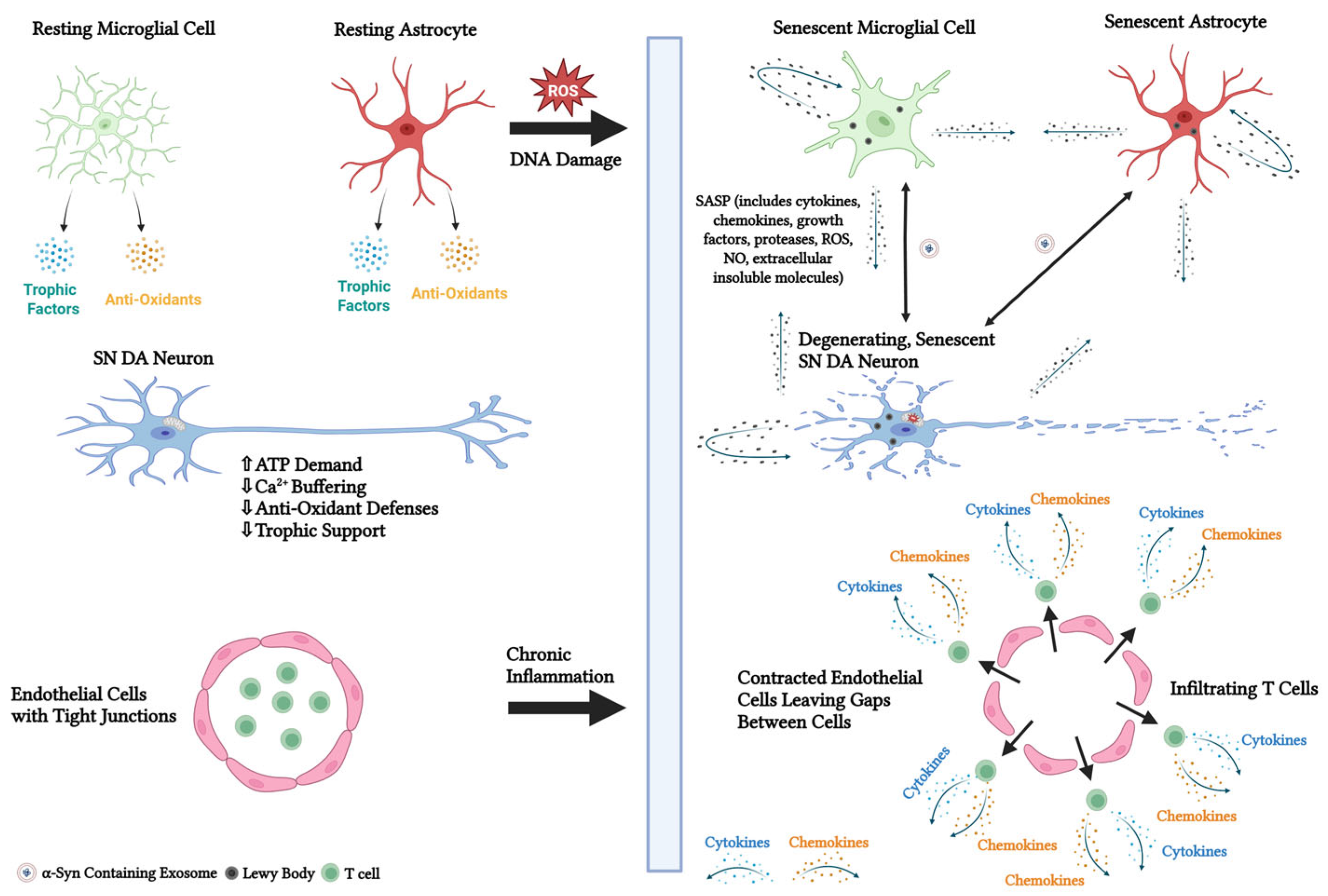
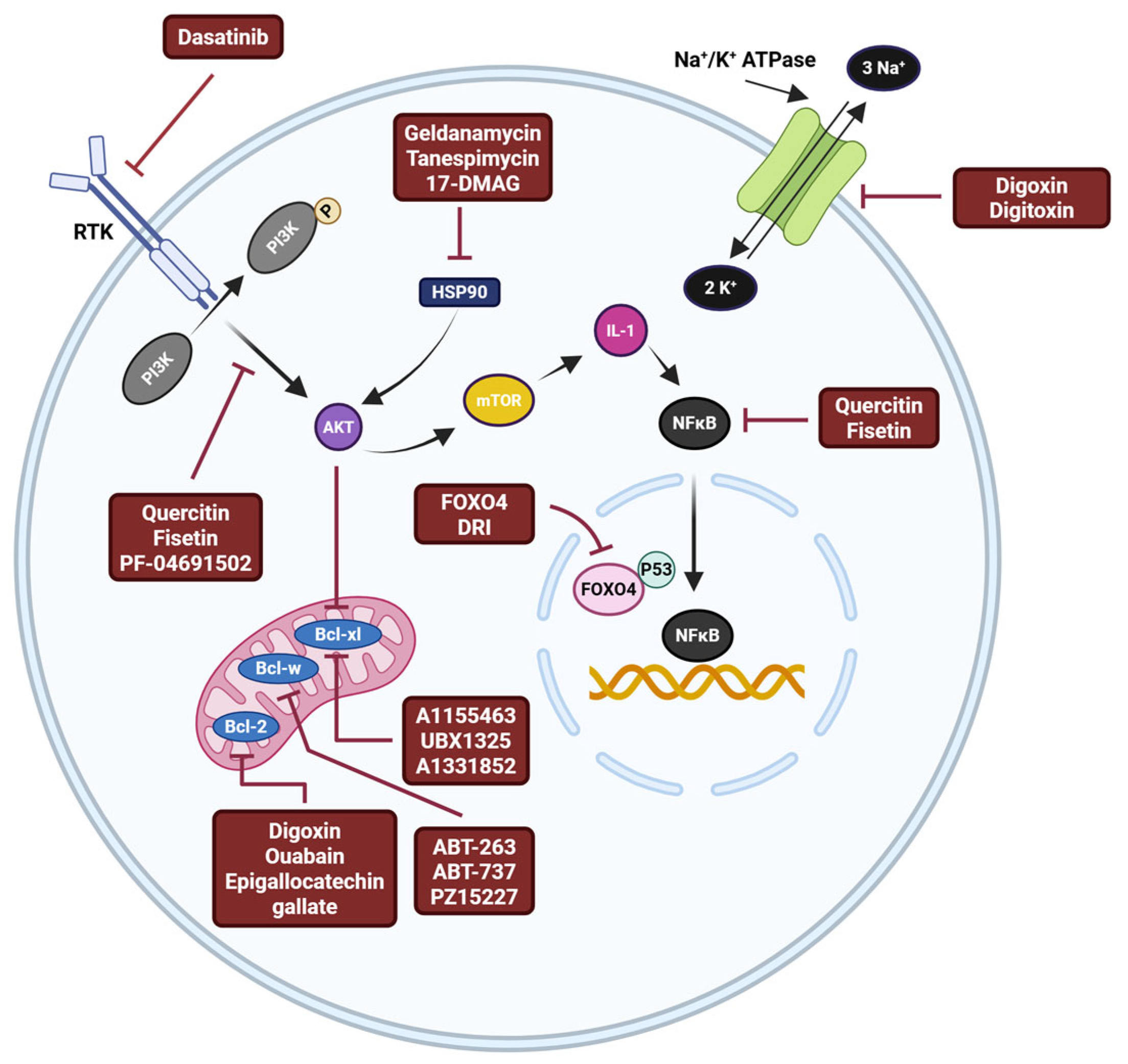
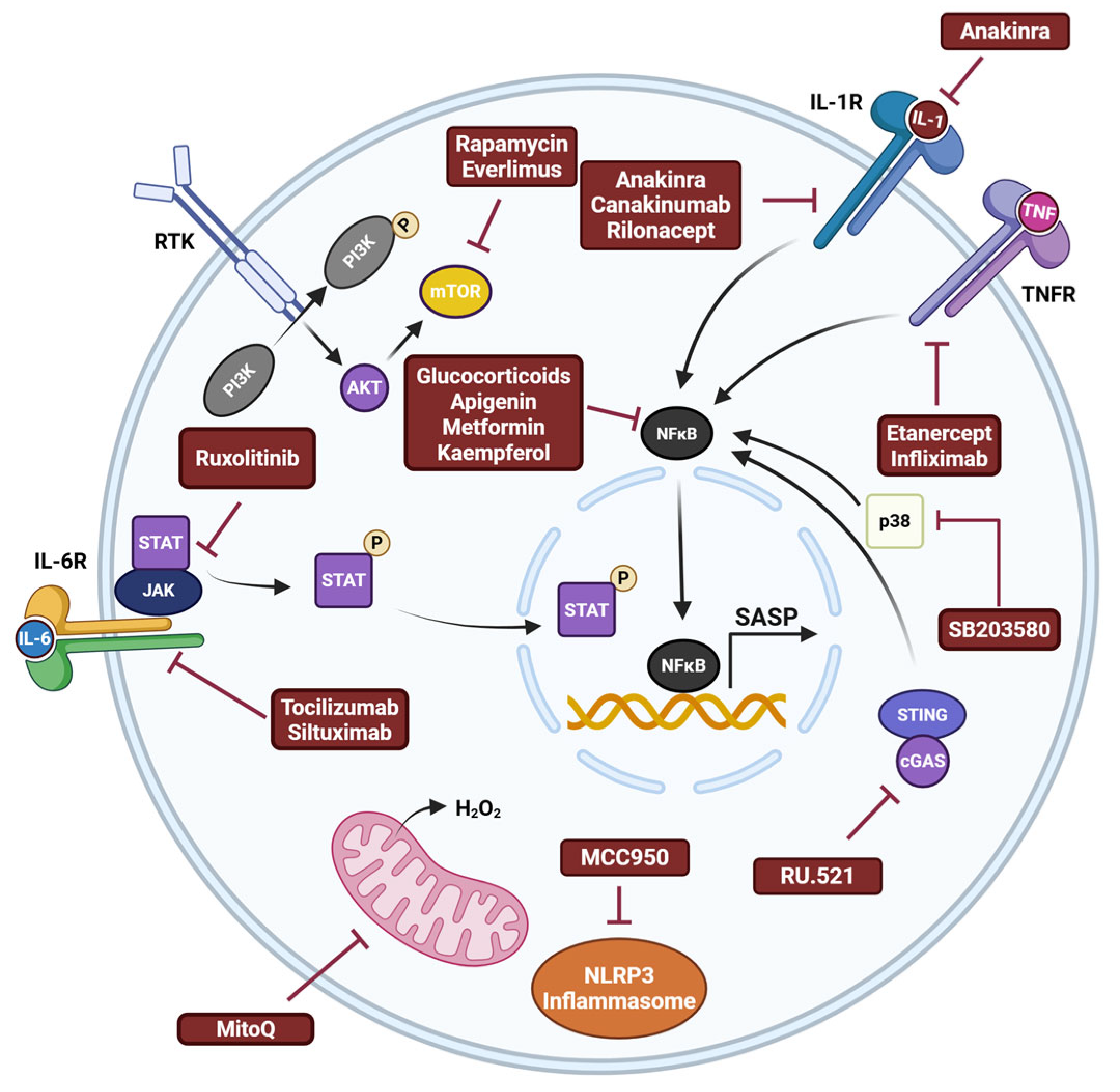
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rademacher, D.J.; Exline, J.E.; Foecking, E.M. Role of Cellular Senescence in Parkinson’s Disease: Potential for Disease-Modification Through Senotherapy. Biomedicines 2025, 13, 1400. https://doi.org/10.3390/biomedicines13061400
Rademacher DJ, Exline JE, Foecking EM. Role of Cellular Senescence in Parkinson’s Disease: Potential for Disease-Modification Through Senotherapy. Biomedicines. 2025; 13(6):1400. https://doi.org/10.3390/biomedicines13061400
Chicago/Turabian StyleRademacher, David J., Jacob E. Exline, and Eileen M. Foecking. 2025. "Role of Cellular Senescence in Parkinson’s Disease: Potential for Disease-Modification Through Senotherapy" Biomedicines 13, no. 6: 1400. https://doi.org/10.3390/biomedicines13061400
APA StyleRademacher, D. J., Exline, J. E., & Foecking, E. M. (2025). Role of Cellular Senescence in Parkinson’s Disease: Potential for Disease-Modification Through Senotherapy. Biomedicines, 13(6), 1400. https://doi.org/10.3390/biomedicines13061400