
A Consolidated Review of Contemporary Targeted and Immunotherapeutic Options for Melanoma
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. BRAF-MEK Inhibitor Combination Therapy
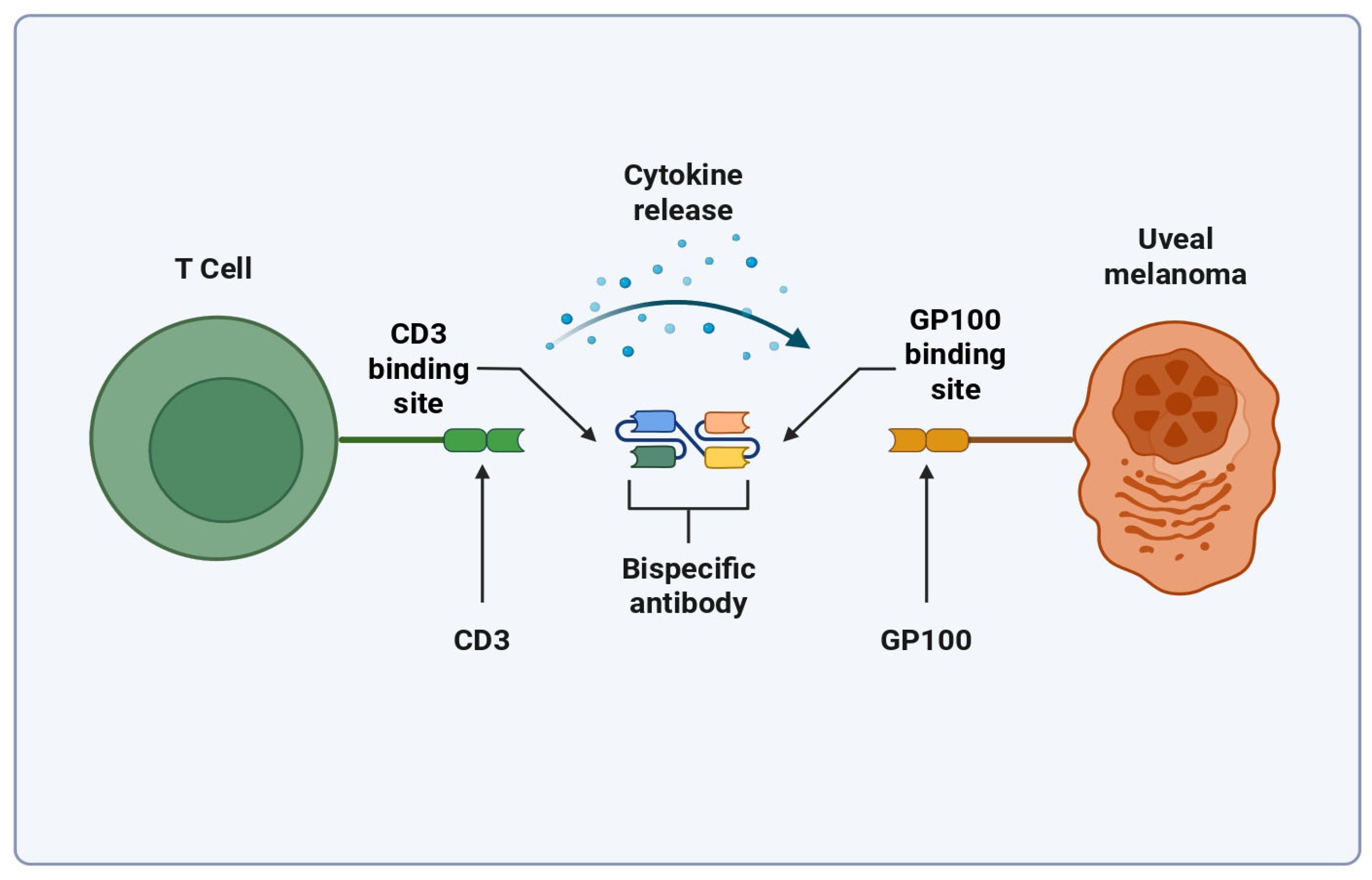
3. Bispecific T Cell Engagers
4. Personalized Neoantigen Vaccines
5. Tumor-Infiltrating Lymphocytes
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Androgen receptor |
| BRAFi | BRAF inhibitor |
| CRS | Cytokine release syndrome |
| ctDNA | Circulating tumor DNA |
| CTLs | Cytotoxic T lymphocytes |
| FUT4 | Fucosyltransferase 4 |
| GP100 | Glycoprotein-100 |
| HLA | Human leukocyte antigen |
| ICI | Immune checkpoint inhibitor |
| IFN | Interferon |
| L1CAM | L1 cell adhesion molecule |
| mAb | Monoclonal antibody |
| MAPK | Mitogen-activated protein kinase |
| MDA | Melanocyte differentiation antigens |
| MEKi | MEK inhibitor |
| MHC | Major histocompatibility complex |
| OS | Overall survival |
| PFS | Progression free survival |
| PNV | Personalized neoantigen vaccine |
| RTK | Receptor tyrosine kinase |
| TAA | Tumor-associated antigen |
| TAM | Tumor-associated macrophages |
| TCE | Bispecific T cell engager |
| TIL | Tumor-infiltrating lymphocyte |
| TME | Tumor microenvironment |
| TSA | Tumor-specific antigen |
References
- Strashilov, S.; Yordanov, A. Aetiology and Pathogenesis of Cutaneous Melanoma: Current Concepts and Advances. Int. J. Mol. Sci. 2021, 22, 6395. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, B.; Hill, C.E.; Pollack, B.P. Vemurafenib enhances MHC induction in BRAF(V600E) homozygous melanoma cells. Oncoimmunology 2013, 2, e22890. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.; Rodrigues, C.M.P.; Gaspar, M.M.; Reis, C.P. Melanoma Management: From Epidemiology to Treatment and Latest Advances. Cancers 2022, 14, 4652. [Google Scholar] [CrossRef]
- National Cancer Institute. SEER Cancer Stat Facts: Melanoma of the Skin. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 21 April 2025).
- Tang, T.; Eldabaje, R.; Yang, L. Current Status of Biological Therapies for the Treatment of Metastatic Melanoma. Anticancer Res. 2016, 36, 3229–3241. [Google Scholar]
- Li, Z.; Fang, Y.; Chen, H.; Zhang, T.; Yin, X.; Man, J.; Yang, X.; Lu, M. Spatiotemporal trends of the global burden of melanoma in 204 countries and territories from 1990 to 2019: Results from the 2019 global burden of disease study. Neoplasia 2022, 24, 12–21. [Google Scholar] [CrossRef]
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495–503. [Google Scholar] [CrossRef]
- Robert, C.; Grob Jean, J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Rauschenberg, R.; Garzarolli, M.; Dietrich, U.; Beissert, S.; Meier, F. Systemic therapy of metastatic melanoma. JDDG J. Der Dtsch. Dermatol. Ges. 2015, 13, 1223–1237. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao Christopher, D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Terheyden, P.; Krackhardt, A.; Eigentler, T. The Systemic Treatment of Melanoma. Dtsch. Arztebl. Int. 2019, 116, 497–504. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Ivashko, I.N.; Kolesar, J.M. Pembrolizumab and nivolumab: PD-1 inhibitors for advanced melanoma. Am. J. Health Syst. Pharm. 2016, 73, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients With Previously Untreated BRAF Wild-Type Advanced Melanoma Treated With Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Carlino, M.S.; Long, G.V.; Schadendorf, D.; Robert, C.; Ribas, A.; Richtig, E.; Nyakas, M.; Caglevic, C.; Tarhini, A.; Blank, C.; et al. Outcomes by line of therapy and programmed death ligand 1 expression in patients with advanced melanoma treated with pembrolizumab or ipilimumab in KEYNOTE-006: A randomised clinical trial. Eur. J. Cancer 2018, 101, 236–243. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef]
- Inamdar, G.S.; Madhunapantula, S.V.; Robertson, G.P. Targeting the MAPK pathway in melanoma: Why some approaches succeed and other fail. Biochem. Pharmacol. 2010, 80, 624–637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Ullah, R.; Yin, Q.; Snell, A.H.; Wan, L. RAF-MEK-ERK pathway in cancer evolution and treatment. Semin. Cancer Biol. 2022, 85, 123–154. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Queirolo, P.; Spagnolo, F. BRAF plus MEK-targeted drugs: A new standard of treatment for BRAF-mutant advanced melanoma. Cancer Metastasis Rev. 2017, 36, 35–42. [Google Scholar] [CrossRef]
- Cohen, J.V.; Sullivan, R.J. Developments in the Space of New MAPK Pathway Inhibitors for BRAF-Mutant Melanoma. Clin. Cancer Res. 2019, 25, 5735–5742. [Google Scholar] [CrossRef] [PubMed]
- Long Georgina, V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob Jean, J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Castellani, G.; Buccarelli, M.; Arasi, M.B.; Rossi, S.; Pisanu, M.E.; Bellenghi, M.; Lintas, C.; Tabolacci, C. BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers. Cancers 2023, 15, 4026. [Google Scholar] [CrossRef] [PubMed]
- Lelliott, E.J.; McArthur, G.A.; Oliaro, J.; Sheppard, K.E. Immunomodulatory Effects of BRAF, MEK, and CDK4/6 Inhibitors: Implications for Combining Targeted Therapy and Immune Checkpoint Blockade for the Treatment of Melanoma. Front. Immunol. 2021, 12, 661737. [Google Scholar] [CrossRef]
- Durrant, D.E.; Morrison, D.K. Targeting the Raf kinases in human cancer: The Raf dimer dilemma. Br. J. Cancer 2018, 118, 3–8. [Google Scholar] [CrossRef]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente, M.I.; Rodon Ahnert, J.; Yaeger, R.; Tsai, F.Y.-C.; Janku, F.; Butowski, N.A.; Allen, C.E.; Ammakkanavar, N.R.; Taylor, J.W.; Michelson, G.; et al. Safety and efficacy of the novel BRAF inhibitor FORE8394 in patients with advanced solid and CNS tumors: Results from a phase 1/2a study. J. Clin. Oncol. 2023, 41, 3006. [Google Scholar] [CrossRef]
- Yaeger, R.; McKean, M.A.; Haq, R.; Beck, J.T.; Taylor, M.H.; Cohen, J.E.; Bowles, D.W.; Gadgeel, S.M.; Mihalcioiu, C.; Papadopoulos, K.P.; et al. A Next-Generation BRAF Inhibitor Overcomes Resistance to BRAF Inhibition in Patients with BRAF-Mutant Cancers Using Pharmacokinetics-Informed Dose Escalation. Cancer Discov. 2024, 14, 1599–1611. [Google Scholar] [CrossRef]
- Cotto-Rios, X.M.; Agianian, B.; Gitego, N.; Zacharioudakis, E.; Giricz, O.; Wu, Y.; Zou, Y.; Verma, A.; Poulikakos, P.I.; Gavathiotis, E. Inhibitors of BRAF dimers using an allosteric site. Nat. Commun. 2020, 11, 4370. [Google Scholar] [CrossRef]
- Beck, J.T.; Shepard, D.R.; Dumas, O.; Pezo, R.C.; Rose, A.A.N.; Ong, M.; Saleh, R.R.; Iwamoto, F.; Gray, J.; Wollenberg, L.; et al. A phase 1, open-label, dose escalation and dose expansion study to evaluate the safety, tolerability, pharmacokinetics, and antitumor activity of PF-07799544 (ARRY-134) as a single agent and in combination with PF-07799933 BRAF dimer inhibitor, in participants 16 years and older with advanced solid tumors. J. Clin. Oncol. 2024, 42, TPS3180. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [PubMed]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.-N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E Inhibition Enhances T-Cell Recognition of Melanoma without Affecting Lymphocyte Function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef]
- Sanlorenzo, M.; Vujic, I.; Floris, A.; Novelli, M.; Gammaitoni, L.; Giraudo, L.; Macagno, M.; Leuci, V.; Rotolo, R.; Donini, C.; et al. BRAF and MEK Inhibitors Increase PD-1-Positive Melanoma Cells Leading to a Potential Lymphocyte-Independent Synergism with Anti–PD-1 Antibody. Clin. Cancer Res. 2018, 24, 3377–3385. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. FDA Approves Atezolizumab for BRAF V600 Unresectable or Metastatic Melanoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-braf-v600-unresectable-or-metastatic-melanoma (accessed on 27 April 2025).
- Ascierto, P.A.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Overall survival with first-line atezolizumab in combination with vemurafenib and cobimetinib in BRAFV600 mutation-positive advanced melanoma (IMspire150): Second interim analysis of a multicentre, randomised, phase 3 study. Lancet Oncol. 2023, 24, 33–44. [Google Scholar] [CrossRef]
- Dummer, R.; Long, G.V.; Robert, C.; Tawbi, H.A.; Flaherty, K.T.; Ascierto, P.A.; Nathan, P.D.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; et al. Randomized Phase III Trial Evaluating Spartalizumab Plus Dabrafenib and Trametinib for BRAF V600-Mutant Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2022, 40, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Novartis Pharmaceuticals. A Randomized, Double-Blind, Placebo-Controlled, Phase III Study Comparing the Combination of PDR001, Dabrafenib and Trametinib Versus Placebo, Dabrafenib and Trametinib in Previously Untreated Patients With Unresectable or Metastatic BRAF V600 Mutant Melanoma. ClinicalTrials.gov Identifier: NCT02967692. Available online: https://clinicaltrials.gov/study/NCT02967692 (accessed on 21 April 2025).
- Joosse, A.; de Vries, E.; Eckel, R.; Nijsten, T.; Eggermont, A.M.M.; Hölzel, D.; Coebergh, J.W.W.; Engel, J. Gender Differences in Melanoma Survival: Female Patients Have a Decreased Risk of Metastasis. J. Investig. Dermatol. 2011, 131, 719–726. [Google Scholar] [CrossRef]
- Di Donato, M.; Cristiani, C.M.; Capone, M.; Garofalo, C.; Madonna, G.; Passacatini, L.C.; Ottaviano, M.; Ascierto, P.A.; Auricchio, F.; Carbone, E.; et al. Role of the androgen receptor in melanoma aggressiveness. Cell Death Dis. 2025, 16, 34. [Google Scholar] [CrossRef]
- Wang, Y.; Ou, Z.; Sun, Y.; Yeh, S.; Wang, X.; Long, J.; Chang, C. Androgen receptor promotes melanoma metastasis via altering the miRNA-539-3p/USP13/MITF/AXL signals. Oncogene 2017, 36, 1644–1654. [Google Scholar] [CrossRef]
- Liu, Q.; Adhikari, E.; Lester, D.K.; Fang, B.; Johnson, J.O.; Tian, Y.; Mockabee-Macias, A.T.; Izumi, V.; Guzman, K.M.; White, M.G.; et al. Androgen drives melanoma invasiveness and metastatic spread by inducing tumorigenic fucosylation. Nat. Commun. 2024, 15, 1148. [Google Scholar] [CrossRef] [PubMed]
- GlaxoSmithKline. Neoadjuvant and Adjuvant Dabrafenib and Trametinib in Patients With Clinical Stage III Melanoma (Combi-Neo). ClinicalTrials.gov Identifier: NCT02231775. Available online: https://clinicaltrials.gov/study/NCT02231775 (accessed on 21 April 2025).
- Vellano, C.P.; White, M.G.; Andrews, M.C.; Chelvanambi, M.; Witt, R.G.; Daniele, J.R.; Titus, M.; McQuade, J.L.; Conforti, F.; Burton, E.M.; et al. Androgen receptor blockade promotes response to BRAF/MEK-targeted therapy. Nature 2022, 606, 797–803. [Google Scholar] [CrossRef]
- Ma, M.; Ghosh, S.; Tavernari, D.; Katarkar, A.; Clocchiatti, A.; Mazzeo, L.; Samarkina, A.; Epiney, J.; Yu, Y.R.; Ho, P.C.; et al. Sustained androgen receptor signaling is a determinant of melanoma cell growth potential and tumorigenesis. J. Exp. Med. 2021, 218, e20201137. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef]
- Mann, J.E.; Hasson, N.; Su, D.G.; Adeniran, A.J.; Smalley, K.S.M.; Djureinovic, D.; Jilaveanu, L.B.; Schoenfeld, D.A.; Kluger, H.M. GP100 expression is variable in intensity in melanoma. Cancer Immunol. Immunother. 2024, 73, 191. [Google Scholar] [CrossRef]
- Sacco, J.J.; Kirk, P.; Leach, E.; Shoushtari, A.N.; Carvajal, R.D.; Britton-Rivet, C.; Khakoo, S.; Collins, L.; de la Cruz-Merino, L.; Eroglu, Z.; et al. Evolution of the tumor immune landscape during treatment with tebentafusp, a T cell receptor-CD3 bispecific. Cell Rep. Med. 2025, 6, 102076. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tebentafusp-tebn-unresectable-or-metastatic-uveal-melanoma (accessed on 24 April 2025).
- Carvajal, R.D.; Butler, M.O.; Shoushtari, A.N.; Hassel, J.C.; Ikeguchi, A.; Hernandez-Aya, L.; Nathan, P.; Hamid, O.; Piulats, J.M.; Rioth, M.; et al. Clinical and molecular response to tebentafusp in previously treated patients with metastatic uveal melanoma: A phase 2 trial. Nat. Med. 2022, 28, 2364–2373. [Google Scholar] [CrossRef] [PubMed]
- Lazaro Sanchez, A.D.; Benitez Fuentes, J.D.; Gil, G.L.; Garcia, M.T.G.; Moreno, E.F.; Zamora, P.C.; Yago, J.B.; Mohamed, K.M.; Arroyo Rodriguez, A.B. Clinical and Proteomic Insights into a Cytokine Release Syndrome Triggered by Tebentafusp in a Metastatic Uveal Melanoma Patient: Case Report. J. Clin. Med. 2025, 14, 1333. [Google Scholar] [CrossRef]
- Hassel, J.C.; Stanhope, S.; Greenshields-Watson, A.; Machiraju, D.; Enk, A.; Holland, C.; Abdullah, S.E.; Benlahrech, A.; Orloff, M.; Nathan, P.; et al. Tebentafusp Induces a T-Cell-Driven Rash in Melanocyte-Bearing Skin as an Adverse Event Consistent with the Mechanism of Action. J. Investig. Dermatol. 2025, 145, 559–572 e559. [Google Scholar] [CrossRef]
- Rodriguez, I.; Norman, T.; Guenther, J.; Smart, K.; Kwong, A.; Berry, J.; In, G.K.; Worswick, S. Cutaneous adverse effects induced by tebentafusp in patients with metastatic uveal melanoma: A case series and treatment insights. Clin. Exp. Dermatol. 2024, 49, 392–394. [Google Scholar] [CrossRef]
- Pan, M.; Wei, X.; Xiang, X.; Liu, Y.; Zhou, Q.; Yang, W. Targeting CXCL9/10/11-CXCR3 axis: An important component of tumor-promoting and antitumor immunity. Clin. Transl. Oncol. 2023, 25, 2306–2320. [Google Scholar] [CrossRef] [PubMed]
- Micevic, G.; Daniels, A.; Flem-Karlsen, K.; Park, K.; Talty, R.; McGeary, M.; Mirza, H.; Blackburn, H.N.; Sefik, E.; Cheung, J.F.; et al. IL-7R licenses a population of epigenetically poised memory CD8+ T cells with superior antitumor efficacy that are critical for melanoma memory. Proc. Natl. Acad. Sci. USA 2023, 120, e2304319120. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-gamma in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef]
- Anbari, S.; Wang, H.; Arulraj, T.; Nickaeen, M.; Pilvankar, M.; Wang, J.; Hansel, S.; Popel, A.S. Identifying biomarkers for treatment of uveal melanoma by T cell engager using a QSP model. NPJ Syst. Biol. Appl. 2024, 10, 108. [Google Scholar] [CrossRef]
- Zhou, Q.; Fang, T.; Wei, S.; Chai, S.; Yang, H.; Tao, M.; Cao, Y. Macrophages in melanoma: A double-edged sword and targeted therapy strategies (Review). Exp. Ther. Med. 2022, 24, 640. [Google Scholar] [CrossRef]
- Guc, E.; Treveil, A.; Leach, E.; Broomfield, A.; Camera, A.; Clubley, J.; Nieto Garcia, P.; Kazachenka, A.; Khanolkar, R.; Del Carpio, L.; et al. Tebentafusp, a T cell engager, promotes macrophage reprogramming and in combination with IL-2 overcomes macrophage immunosuppression in cancer. Nat. Commun. 2025, 16, 2374. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.; Malakoutikhah, Z.; Rahimmanesh, I.; Ferns, G.A.; Nedaeinia, R.; Ishaghi, S.M.M.; Dana, N.; Haghjooy Javanmard, S. The nexus of natural killer cells and melanoma tumor microenvironment: Crosstalk, chemotherapeutic potential, and innovative NK cell-based therapeutic strategies. Cancer Cell Int. 2023, 23, 312. [Google Scholar] [CrossRef]
- Tarazona, R.; Duran, E.; Solana, R. Natural Killer Cell Recognition of Melanoma: New Clues for a More Effective Immunotherapy. Front. Immunol. 2015, 6, 649. [Google Scholar] [CrossRef]
- Mehta, A.; Motavaf, M.; Nebo, I.; Luyten, S.; Osei-Opare, K.D.; Gru, A.A. Advancements in Melanoma Treatment: A Review of PD-1 Inhibitors, T-VEC, mRNA Vaccines, and Tumor-Infiltrating Lymphocyte Therapy in an Evolving Landscape of Immunotherapy. J. Clin. Med. 2025, 14, 1200. [Google Scholar] [CrossRef]
- Peng, W.; Liu, C.; Xu, C.; Lou, Y.; Chen, J.; Yang, Y.; Yagita, H.; Overwijk, W.W.; Lizee, G.; Radvanyi, L.; et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. 2012, 72, 5209–5218. [Google Scholar] [CrossRef]
- Hamid, O.; Hassel, J.C.; Shoushtari, A.N.; Meier, F.; Bauer, T.M.; Salama, A.K.S.; Kirkwood, J.M.; Ascierto, P.A.; Lorigan, P.C.; Mauch, C.; et al. Tebentafusp in combination with durvalumab and/or tremelimumab in patients with metastatic cutaneous melanoma: A phase 1 study. J. Immunother. Cancer 2023, 11, e006747. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, L.; Vaubel, J.; Mohr, P.; Hauschild, A.; Utikal, J.; Simon, J.; Garbe, C.; Herbst, R.; Enk, A.; Kampgen, E.; et al. Phase II DeCOG-study of ipilimumab in pretreated and treatment-naive patients with metastatic uveal melanoma. PLoS ONE 2015, 10, e0118564. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, S.; Shan, B.; Li, B.; Li, F. A PD-L1xCD3 bispecific nanobody as a novel T-cell engager in treating PD-L1 overexpression melanoma. Mol. Immunol. 2023, 163, 20–27. [Google Scholar] [CrossRef]
- Cho, J.; Tae, N.; Song, Y.; Kim, C.W.; Lee, S.J.; Ahn, J.H.; Lee, K.H.; Lee, B.H.; Kim, B.S.; Chang, S.Y.; et al. The expression of PD-L1 on tumor-derived exosomes enhances infiltration and anti-tumor activity of alphaCD3 x alphaPD-L1 bispecific antibody-armed T cells. Cancer Immunol. Immunother. 2024, 73, 196. [Google Scholar] [CrossRef] [PubMed]
- Gary Schwartz, O.S. Methods of treating ocular cancer using anti-met antibodies and bispecific antigen binding molecules that bind met. U.S. Patent Application 16/796,380, 10 February 2022. [Google Scholar]
- Apavaloaei, A.; Hardy, M.P.; Thibault, P.; Perreault, C. The Origin and Immune Recognition of Tumor-Specific Antigens. Cancers 2020, 12, 2607. [Google Scholar] [CrossRef]
- Bezu, L.; Kepp, O.; Cerrato, G.; Pol, J.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Peptide-based vaccines in anticancer therapy. Oncoimmunology 2018, 7, e1511506. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, J.; Dang, Q.; Liu, L.; Weng, S.; Wang, L.; Zhou, Z.; Kong, Y.; Li, H.; Han, Y.; et al. Engineering neoantigen vaccines to improve cancer personalized immunotherapy. Int. J. Biol. Sci. 2022, 18, 5607–5623. [Google Scholar] [CrossRef]
- Khaddour, K.; Buchbinder, E.I. Individualized Neoantigen-Directed Melanoma Therapy. Am. J. Clin. Dermatol. 2025, 26, 225–235. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Danelli, L. Personalized neoantigen therapy for melanoma immunotherapy. Nat. Cancer 2024, 5, 1783. [Google Scholar] [CrossRef] [PubMed]
- Mørk, S.K.; Kadivar, M.; Bol, K.F.; Draghi, A.; Westergaard, M.C.W.; Skadborg, S.K.; Overgaard, N.; Sørensen, A.B.; Rasmussen, I.S.; Andreasen, L.V.; et al. Personalized therapy with peptide-based neoantigen vaccine (EVX-01) including a novel adjuvant, CAF®09b, in patients with metastatic melanoma. Oncoimmunology 2022, 11, 2023255. [Google Scholar] [CrossRef] [PubMed]
- Tretiakova, D.S.; Vodovozova, E.L. Liposomes as Adjuvants and Vaccine Delivery Systems. Biochem. (Mosc.) Suppl. Ser. A Membr. Cell Biol. 2022, 16, 1–20. [Google Scholar] [CrossRef] [PubMed]
- D’Alise, A.M.; Leoni, G.; Cotugno, G.; Siani, L.; Vitale, R.; Ruzza, V.; Garzia, I.; Antonucci, L.; Micarelli, E.; Venafra, V.; et al. Phase I Trial of Viral Vector-Based Personalized Vaccination Elicits Robust Neoantigen-Specific Antitumor T-Cell Responses. Clin. Cancer Res. 2024, 30, 2412–2423. [Google Scholar] [CrossRef]
- Vaitiekus, D.; Juozaityte, E.; Puzauskienė, L.; Tulyte, S.; Gatijatullin, L.; Platten, M.; Poschke, I.; Hulsmeyer, I.; Kuhn, A.; Aranguren, A.; et al. 160P Oral DNA vaccination targeting personalised neoantigens in immune checkpoint-inhibitor treated solid tumor patients: Interim results. Immuno-Oncol. Technol. 2024, 24, 100789. [Google Scholar] [CrossRef]
- Latifyan, S.; Haanen, J.B. Melanoma neoantigen vaccines: Are we getting more personal now? Med 2024, 5, 288–290. [Google Scholar] [CrossRef]
- Galuppini, F.; Dal Pozzo, C.; Deckert, J.; Loupakis, F.; Fassan, M.; Baffa, R. Tumor mutation burden: From comprehensive mutational screening to the clinic. Cancer Cell Int. 2019, 19, 209. [Google Scholar] [CrossRef]
- Davis, L.; Tarduno, A.; Lu, Y.-C. Neoantigen-Reactive T Cells: The Driving Force behind Successful Melanoma Immunotherapy. Cancers 2021, 13, 6061. [Google Scholar] [CrossRef]
- Zhao, Y.; Deng, J.; Rao, S.; Guo, S.; Shen, J.; Du, F.; Wu, X.; Chen, Y.; Li, M.; Chen, M.; et al. Tumor Infiltrating Lymphocyte (TIL) Therapy for Solid Tumor Treatment: Progressions and Challenges. Cancers 2022, 14, 4160. [Google Scholar] [CrossRef]
- Maibach, F.; Sadozai, H.; Seyed Jafari, S.M.; Hunger, R.; Schenk, M. Tumor-Infiltrating Lymphocytes and Their Prognostic Value in Cutaneous Melanoma. Front. Immunol. 2020, 11, 2105. [Google Scholar] [CrossRef]
- Oble, D.A.; Loewe, R.; Yu, P.; Mihm Jr, M.C. Focus on TILs: Prognostic significance of tumor infiltrating lymphocytes in human melanoma. Cancer Immun. 2009, 9, 3. [Google Scholar] [PubMed]
- Martens, A.; Wistuba-Hamprecht, K.; Yuan, J.; Postow, M.; Wong, P.; Capone, M.; Madonna, G.; Khammari, A.; Schilling, B.; Sucker, A.; et al. Increases in Absolute Lymphocytes and Circulating CD4+ and CD8+ T Cells Are Associated with Positive Clinical Outcome of Melanoma Patients Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 4848–4858. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Margolin, K. Tumor-Infiltrating Lymphocytes in Melanoma. Curr. Oncol. Rep. 2012, 14, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.; Borch, T.; van den Berg, J.; Met, Ö.; Kessels, R.; Geukes Foppen, M.; Stoltenborg Granhøj, J.; Nuijen, B.; Nijenhuis, C.; Jedema, I.; et al. Tumor-Infiltrating Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. N. Engl. J. Med. (NEJM) 2022, 387, 2113–2125. [Google Scholar] [CrossRef]
- Geukes Foppen, M.H.; Donia, M.; Svane, I.M.; Haanen, J.B.A.G. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol. Oncol. 2015, 9, 1918–1935. [Google Scholar] [CrossRef]
- Cheng, L.E.; Ohlén, C.; Nelson, B.H.; Greenberg, P.D. Enhanced signaling through the IL-2 receptor in CD8+ T cells regulated by antigen recognition results in preferential proliferation and expansion of responding CD8+ T cells rather than promotion of cell death. Proc. Natl. Acad. Sci. USA 2002, 99, 3001–3006. [Google Scholar] [CrossRef]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef]
- Dudley, M.E.; Gross, C.A.; Langhan, M.M.; Garcia, M.R.; Sherry, R.M.; Yang, J.C.; Phan, G.Q.; Kammula, U.S.; Hughes, M.S.; Citrin, D.E. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin. Cancer Res. 2010, 16, 6122–6131. [Google Scholar] [CrossRef]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.l.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.-C.; Klebanoff, C.A.; Overwijk, W.W. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Pilon-Thomas, S.; Kuhn, L.; Ellwanger, S.; Janssen, W.; Royster, E.; Marzban, S.; Kudchadkar, R.; Zager, J.; Gibney, G.; Sondak, V.K.; et al. Efficacy of Adoptive Cell Transfer of Tumor-infiltrating Lymphocytes After Lymphopenia Induction for Metastatic Melanoma. J. Immunother. 2012, 35, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Saibil, S.; Sotov, V.; Le, M.; Khoja, L.; Ghazarian, D.; Bonilla, L.; Majeed, H.; Hogg, D.; Joshua, A.; et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol. Immunother. 2019, 68, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Dalle, S.; Sanmamed, M.F.; Wilson, M.; Hassel, J.C.; Kluger, H.; Orloff, M.; Weber, J.S.; Graf Finckenstein, F.; Hari, P.; et al. 844P Efficacy and safety of lifileucel, an investigational autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma previously treated with anti-LAG3 antibody. Ann. Oncol. 2022, 33, S935–S936. [Google Scholar] [CrossRef]
- Seitter, S.; Sherry, R.; Yang, J.; Robbins, P.; Shindorf, M.; Copeland, A.; McGowan, C.; Epstein, M.; Shelton, T.; Langhan, M.; et al. Impact of Prior Treatment on the Efficacy of Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Patients with Metastatic Melanoma. Clin. Cancer Res. 2021, 27, 5289–5298. [Google Scholar] [CrossRef]
- Amaria, R.; McQuade, J.; Davies, M.; Glitza, I.; Jose, S.; Cressman, E.; Clausell, A.; Bassett, R.; Patel, S.; Diab, A.; et al. OBX-115, an interleukin 2 (IL2)-sparing engineered tumor-infiltrating lymphocyte (TIL) cell therapy, in patients (pts) with immune checkpoint inhibitor (ICI)-resistant unresectable or metastatic melanoma. J. Clin. Oncol. (JCO) 2024, 42, 9515. [Google Scholar] [CrossRef]
- Thomas, S.; Hong, Y.; Samhouri, Y.; Larkin, J.; Olson, D.; In, G.; Atkinson, V.; Lammers, P.; Furness, A.; Martin-Liberal, J.; et al. Abstract CT286: TILVANCE-301, a phase 3 study of lifileucel tumor-infiltrating lymphocyte (TIL) cell therapy in combination with pembrolizumab vs pembrolizumab alone in treatment-naive unresectable or metastatic melanoma. Cancer Res. 2024, 84, CT286. [Google Scholar] [CrossRef]
- Thomas, S.; Gogas, H.; Hong, Y.; In, G.; Doger de Spéville Uribe, B.; Furness, A.S.; García Castaño, A.; Haefliger, S.; He, K.; Medina, T.; et al. Efficacy and safety of lifileucel, an autologous tumor-infiltrating lymphocyte cell therapy, and pembrolizumab in patients with immune checkpoint inhibitor-naive unresectable or metastatic melanoma: Updated results from IOV-COM-202 cohort 1A. J. Clin. Oncol. (JCO) 2024, 42, 9505. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Champion, P.J.; Bluestein, J.R.; Quinn, A.E.; Bell, S.D.; Kiley, J.H.; Wakefield, M.R.; Fang, Y. A Consolidated Review of Contemporary Targeted and Immunotherapeutic Options for Melanoma. Biomedicines 2025, 13, 1388. https://doi.org/10.3390/biomedicines13061388
Champion PJ, Bluestein JR, Quinn AE, Bell SD, Kiley JH, Wakefield MR, Fang Y. A Consolidated Review of Contemporary Targeted and Immunotherapeutic Options for Melanoma. Biomedicines. 2025; 13(6):1388. https://doi.org/10.3390/biomedicines13061388
Chicago/Turabian StyleChampion, Parker J., Jacob R. Bluestein, Anthony E. Quinn, Scott D. Bell, Josiah H. Kiley, Mark R. Wakefield, and Yujiang Fang. 2025. "A Consolidated Review of Contemporary Targeted and Immunotherapeutic Options for Melanoma" Biomedicines 13, no. 6: 1388. https://doi.org/10.3390/biomedicines13061388
APA StyleChampion, P. J., Bluestein, J. R., Quinn, A. E., Bell, S. D., Kiley, J. H., Wakefield, M. R., & Fang, Y. (2025). A Consolidated Review of Contemporary Targeted and Immunotherapeutic Options for Melanoma. Biomedicines, 13(6), 1388. https://doi.org/10.3390/biomedicines13061388