
Integrative Analysis of Neutrophil-Associated Genes Reveals Prognostic Significance and Immune Microenvironment Modulation in Cervical Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
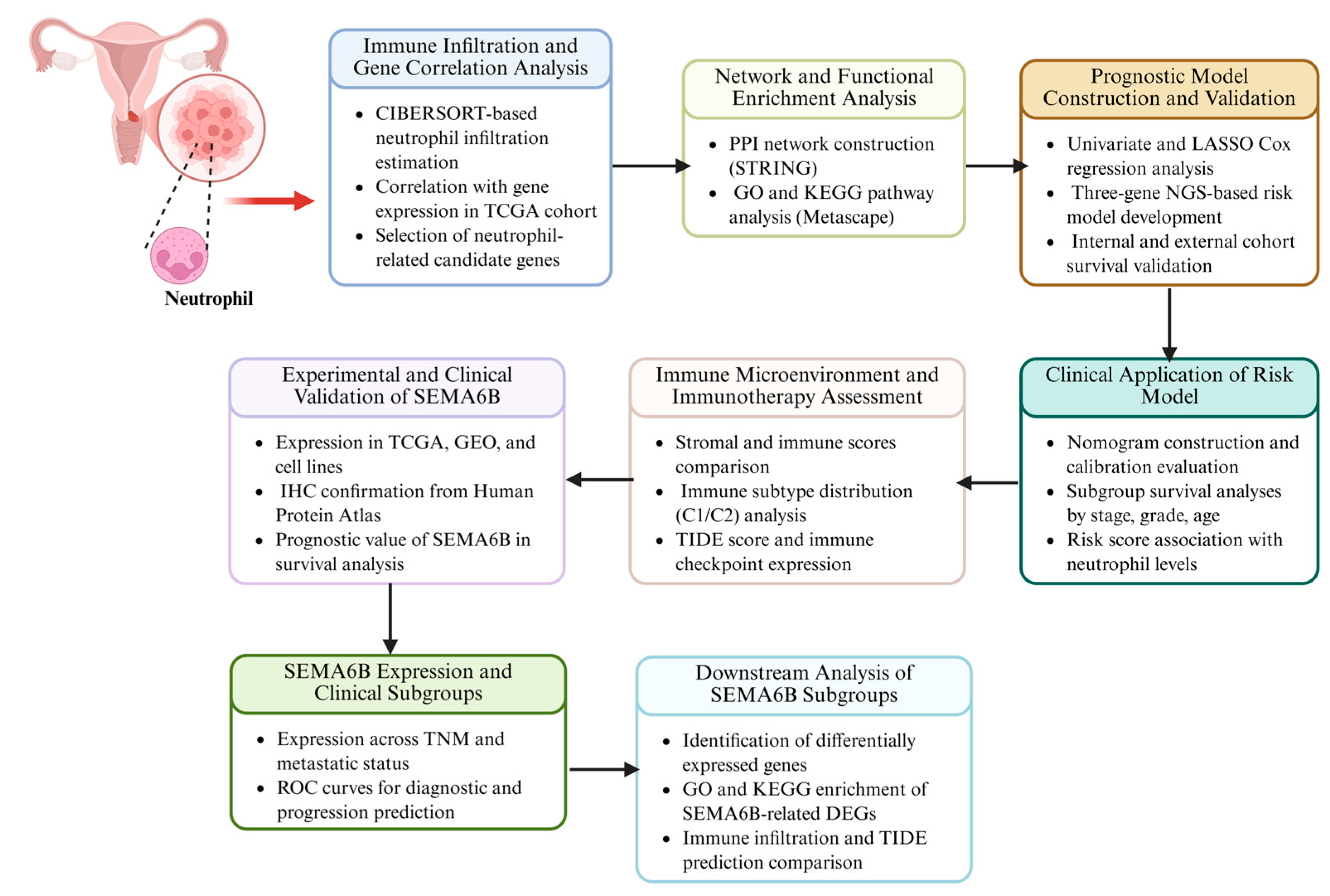
2. Materials and Methods
2.1. Data Collection and Preprocessing
2.2. Tumor Microenvironment Analysis
2.3. Identification of Neutrophil-Associated Genes
2.4. Construction and Validation of the Prognostic Model
2.5. Gene Expression Validation and Cell Experiments
2.6. Immunotherapy Data Analysis
2.7. Statistical Analysis
3. Results
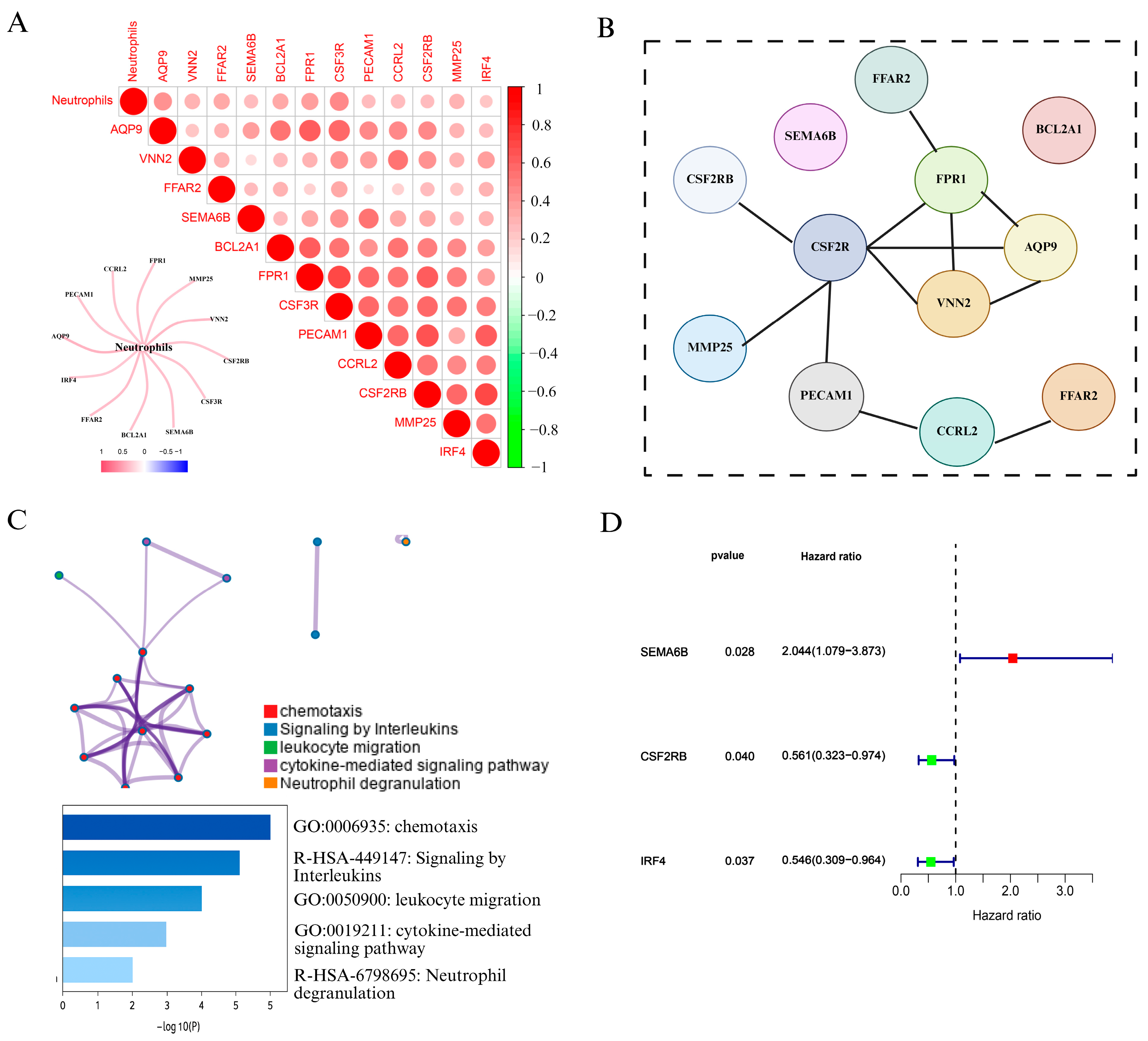
3.1. Identification of Neutrophil-Associated Genes and Prognostic Signatures in Cervical Cancer
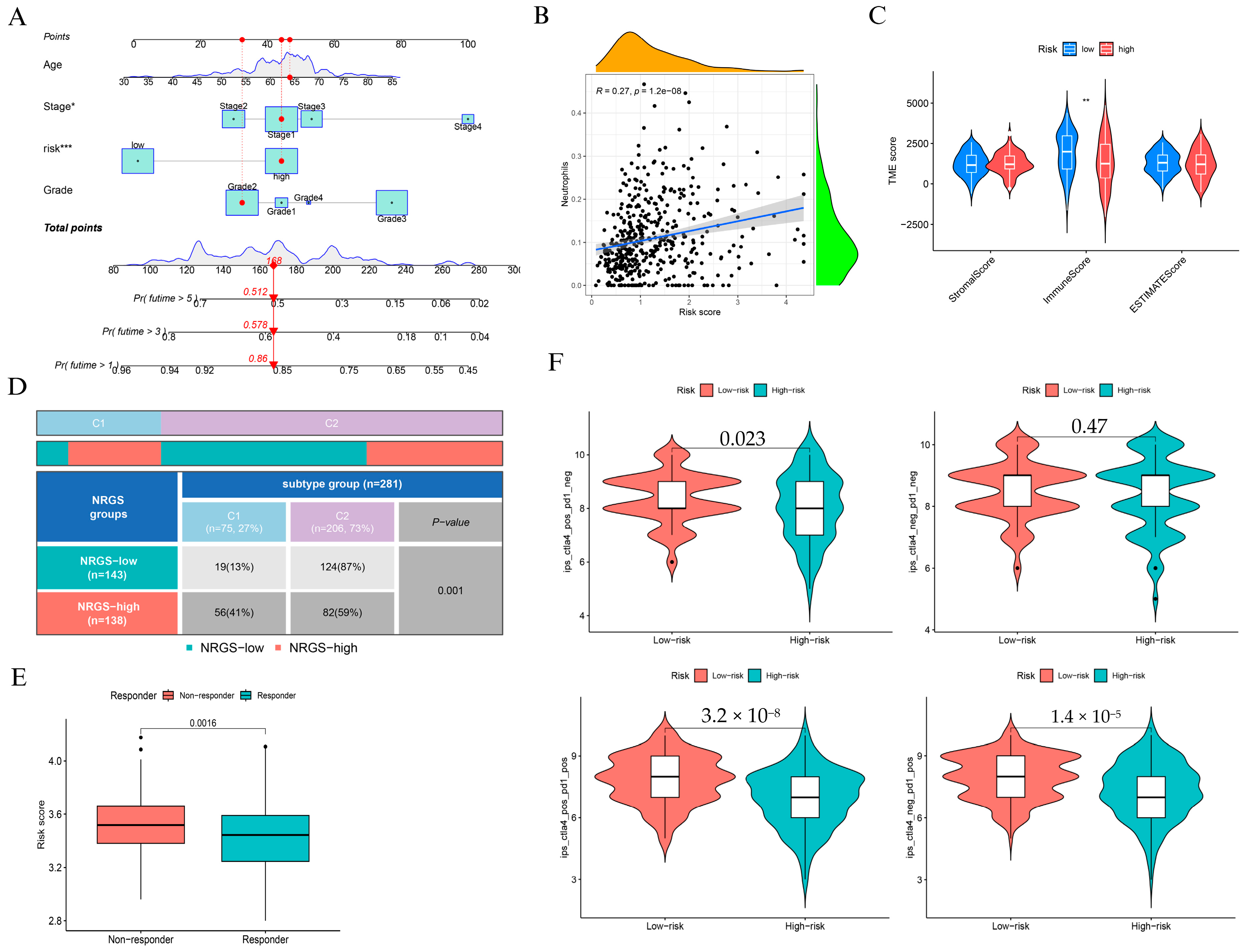
3.2. Construction and Validation of a Prognostic Risk Model Based on Neutrophil-Associated Genes
3.3. Clinical and Immunological Characterization of the NAGS-Based Risk Model
3.4. Expression Validation and Clinical Significance of SEMA6B in Cervical Cancer
3.5. Differential Expression and Functional Enrichment Analysis Between SEMA6B Expression Subgroups
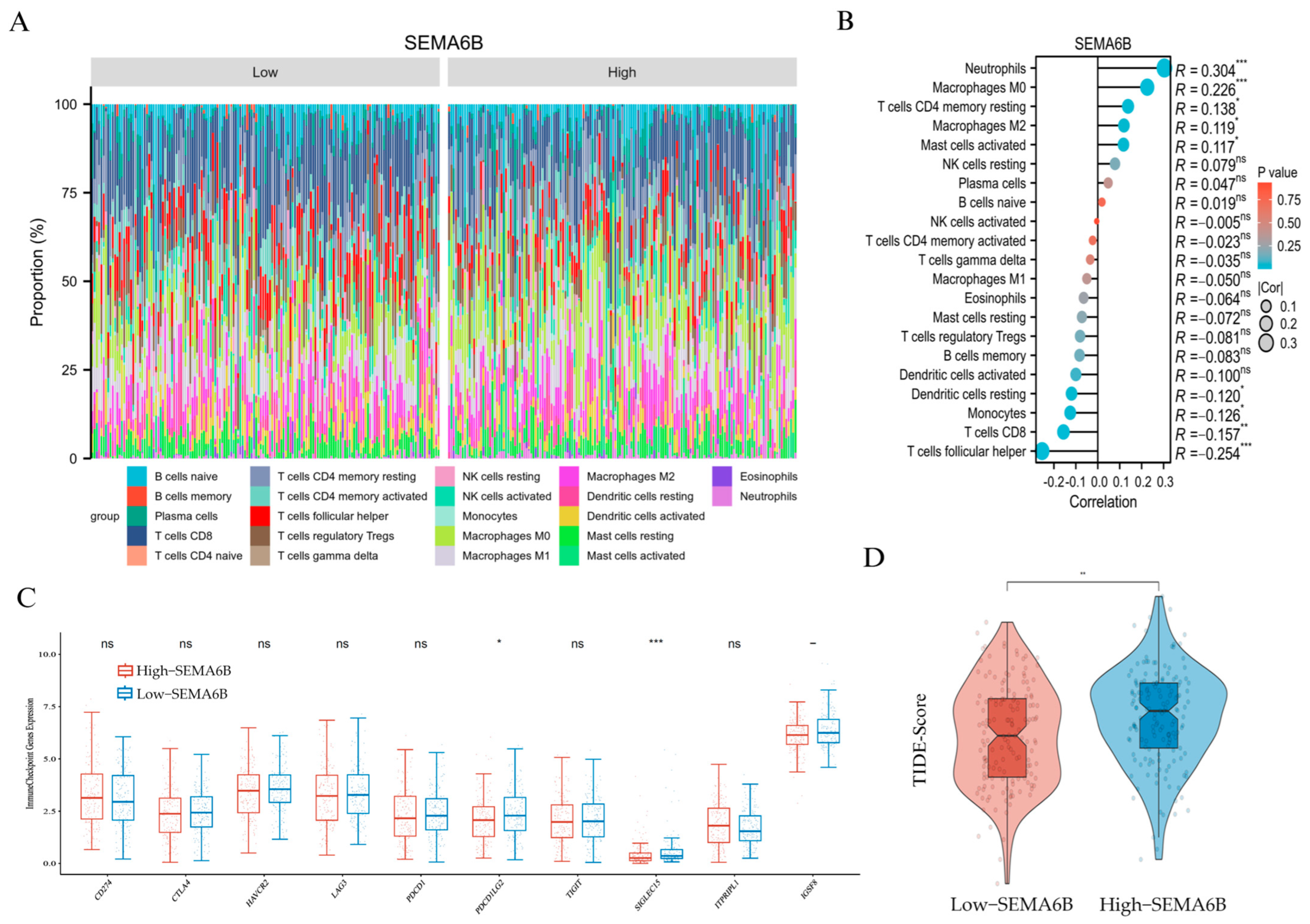
3.6. Association Between SEMA6B Expression and Tumor Immune Microenvironment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moore, D.H. Cervical cancer. Obstet. Gynecol. 2006, 107, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Deep, A.; Sharma, A.K. Current Treatment for Cervical Cancer: An Update. Anticancer. Agents Med. Chem. 2020, 20, 1768–1779. [Google Scholar] [CrossRef]
- Abu-Rustum, N.R.; Yashar, C.M.; Arend, R.; Barber, E.; Bradley, K.; Brooks, R.; Campos, S.M.; Chino, J.; Chon, H.S.; Crispens, M.A.; et al. NCCN Guidelines(R) Insights: Cervical Cancer, Version 1.2024. J. Natl. Compr. Canc Netw. 2023, 21, 1224–1233. [Google Scholar] [CrossRef]
- Perkins, R.B.; Wentzensen, N.; Guido, R.S.; Schiffman, M. Cervical Cancer Screening: A Review. JAMA 2023, 330, 547–558. [Google Scholar] [CrossRef]
- Yuan, Y.; Cai, X.; Shen, F.; Ma, F. HPV post-infection microenvironment and cervical cancer. Cancer Lett. 2021, 497, 243–254. [Google Scholar] [CrossRef]
- Bedell, S.L.; Goldstein, L.S.; Goldstein, A.R.; Goldstein, A.T. Cervical Cancer Screening: Past, Present, and Future. Sex. Med. Rev. 2020, 8, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, S.; Gupta, B. Screening for cervical cancer: Choices & dilemmas. Indian. J. Med. Res. 2021, 154, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ma, D. The precision prevention and therapy of HPV-related cervical cancer: New concepts and clinical implications. Cancer Med. 2018, 7, 5217–5236. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, B.; Feng, Y.; Ye, J.; Mao, Z.; Zhang, T.; Xu, M.; Zhang, W.; Jiao, X.; Zhang, Q.; et al. Targeting tumor-associated macrophage-derived CD74 improves efficacy of neoadjuvant chemotherapy in combination with PD-1 blockade for cervical cancer. J. Immunother. Cancer 2024, 12, e009024. [Google Scholar] [CrossRef]
- Colbert, L.E.; El Alam, M.B.; Wang, R.; Karpinets, T.; Lo, D.; Lynn, E.J.; Harris, T.A.; Elnaggar, J.H.; Yoshida-Court, K.; Tomasic, K.; et al. Tumor-resident Lactobacillus iners confer chemoradiation resistance through lactate-induced metabolic rewiring. Cancer Cell 2023, 41, 1945–1962.e1911. [Google Scholar] [CrossRef]
- Cao, G.; Yue, J.; Ruan, Y.; Han, Y.; Zhi, Y.; Lu, J.; Liu, M.; Xu, X.; Wang, J.; Gu, Q.; et al. Single-cell dissection of cervical cancer reveals key subsets of the tumor immune microenvironment. EMBO J. 2023, 42, e110757. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Hayatullah, G.; Zhou, H.; Chang, S.; Liu, L.; Qiu, H.; Duan, X.; Han, L. Tumor microenvironment characterization in cervical cancer identifies prognostic relevant gene signatures. PLoS ONE 2021, 16, e0249374. [Google Scholar] [CrossRef]
- Giese, M.A.; Hind, L.E.; Huttenlocher, A. Neutrophil plasticity in the tumor microenvironment. Blood 2019, 133, 2159–2167. [Google Scholar] [CrossRef]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef]
- Que, H.; Fu, Q.; Lan, T.; Tian, X.; Wei, X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188762. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Chen, Y.; Tian, T.; Gao, X.; Liu, X.; Wang, J.; Chu, H.; Zhao, C.; Yang, Y.; Lei, K.; et al. S100A7 orchestrates neutrophil chemotaxis and drives neutrophil extracellular traps (NETs) formation to facilitate lymph node metastasis in cervical cancer patients. Cancer Lett. 2024, 605, 217288. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Liu, Y.; Wang, S.; Shang, P.; Gao, Y.; Chen, X. Preoperative neutrophil-lymphocyte ratio before platelet-lymphocyte ratio predicts clinical outcome in patients with cervical cancer treated with initial radical surgery. Int. J. Gynecol. Cancer 2014, 24, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Lai, A.C.; Lin, Y.C.; Chi, P.Y.; Chen, Y.C.; Yang, Y.H.; Chen, C.H.; Shen, S.Y.; Hwang, T.L.; Su, M.W.; et al. Neutrophil extracellular trap production and CCL4L2 expression influence corticosteroid response in asthma. Sci. Transl. Med. 2023, 15, eadf3843. [Google Scholar] [CrossRef]
- Mu, C.; Wang, Y.; Han, C.; Song, H.; Wu, Q.; Yang, J.; Guo, N.; Ma, Y.; Zhang, C.; Zhang, J.; et al. Crosstalk between oxidative stress and neutrophil response in early ischemic stroke: A comprehensive transcriptome analysis. Front. Immunol. 2023, 14, 1134956. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.G.; Diao, L.; Karpinets, T.; Negrao, M.V.; Tran, H.T.; Parra, E.R.; Corsini, E.M.; Reuben, A.; Federico, L.; Bernatchez, C.; et al. Neutrophil expansion defines an immunoinhibitory peripheral and intratumoral inflammatory milieu in resected non-small cell lung cancer: A descriptive analysis of a prospectively immunoprofiled cohort. J. Immunother. Cancer 2020, 8, e000405. [Google Scholar] [CrossRef]
- Tang, G.; Song, Q.; Dou, J.; Chen, Z.; Hu, X.; Li, Z.; Li, X.; Wang, T.; Dong, S.; Zhang, H. Neutrophil-centric analysis of gastric cancer: Prognostic modeling and molecular insights. Cell Mol. Life Sci. 2024, 81, 452. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, C.; Li, L.; Zheng, C.; Wang, Y.; Chen, J.; Sun, B. Construction and multicohort validation of a colon cancer prognostic risk score system based on big data of neutrophil-associated differentially expressed genes. J. Cancer 2024, 15, 2866–2879. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.T.; Qiu, W.R.; Yu, W.K.; Xu, Z.C.; Zhang, S.H. Identifying TME signatures for cervical cancer prognosis based on GEO and TCGA databases. Heliyon 2023, 9, e15096. [Google Scholar] [CrossRef] [PubMed]
- Gui, Z.; Ye, Y.; Li, Y.; Ren, Z.; Wei, N.; Liu, L.; Wang, H.; Zhang, M. Construction of a novel cancer-associated fibroblast-related signature to predict clinical outcome and immune response in cervical cancer. Transl. Oncol. 2024, 46, 102001. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Jones, A.; Lee, S.H.; Ng, E.; Fiegl, H.; Zikan, M.; Cibula, D.; Sargent, A.; Salvesen, H.B.; Jacobs, I.J.; et al. The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women’s cancer. PLoS Genet. 2012, 8, e1002517. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Jones, A.; Fiegl, H.; Sargent, A.; Zhuang, J.J.; Kitchener, H.C.; Widschwendter, M. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 2012, 4, 24. [Google Scholar] [CrossRef]
- Shu, J.; Jiang, J.; Zhao, G. Identification of novel gene signature for lung adenocarcinoma by machine learning to predict immunotherapy and prognosis. Front. Immunol. 2023, 14, 1177847. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, J.; Zhang, Y.; Song, Z.; Bian, J.; Yi, H.; Ma, Z. Identifying neutrophil-associated subtypes in ulcerative colitis and confirming neutrophils promote colitis-associated colorectal cancer. Front. Immunol. 2023, 14, 1095098. [Google Scholar] [CrossRef] [PubMed]
- Passari, M.; Scutera, S.; Schioppa, T.; Tiberio, L.; Piantoni, S.; Tamassia, N.; Bugatti, M.; Vermi, W.; Angeli, F.; Caproli, A.; et al. Regulation of neutrophil associated RNASET2 expression in rheumatoid arthritis. Sci. Rep. 2024, 14, 26820. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Haji Mazdarani, M.; Jafarikia, M.; Nemati, F. Investigation of indolamine 2, 3 dioxygenase (IDO-1) gene expression by real-time PCR among patients with lung cancer. J. Cell Physiol. 2019, 234, 13781–13787. [Google Scholar] [CrossRef]
- Shih, H.C.; Choo, K.B.; Chang, T.J.; Yang, M.Y.; Shih, M.C.; Yeh, K.T.; Liu, T.C.; Lin, S.F.; Chang, J.G. Disturbance of circadian gene expression in endometrial cancer: Detection by real-time quantitative RT-PCR. Oncol. Rep. 2005, 14, 1533–1538. [Google Scholar] [CrossRef]
- Ning, S.; Wu, J.; Pan, Y.; Qiao, K.; Li, L.; Huang, Q. Identification of CD4(+) Conventional T Cells-Related lncRNA Signature to Improve the Prediction of Prognosis and Immunotherapy Response in Breast Cancer. Front. Immunol. 2022, 13, 880769. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lin, H.; Su, Q.; Li, C. Cuproptosis-related lncRNA predict prognosis and immune response of lung adenocarcinoma. World J. Surg. Oncol. 2022, 20, 275. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, B.; Zhang, W.; Zhang, T.; Liu, Q.; Jiao, X.; Ye, J.; Hao, Y.; Gao, Q.; Ma, G.; et al. APLN promotes the proliferation, migration, and glycolysis of cervical cancer through the PI3K/AKT/mTOR pathway. Arch. Biochem. Biophys. 2024, 755, 109983. [Google Scholar] [CrossRef]
- Huo, W.; Huang, Y.; Tian, B.; Chen, X.; Lu, J.; Huang, X.; Wu, M.; Yu, J.; Chen, D.; Wang, R. Unraveling the mechanisms of RECQL4-mediated cervical cancer progression through the PI3K/AKT pathway. Transl. Oncol. 2024, 50, 102146. [Google Scholar] [CrossRef]
- Bian, Z.; Wu, X.; Chen, Q.; Gao, Q.; Xue, X.; Wang, Y. Oct4 activates IL-17A to orchestrate M2 macrophage polarization and cervical cancer metastasis. Cancer Immunol. Immunother. 2024, 73, 73. [Google Scholar] [CrossRef]
- Bai, Y.; Li, H.; Lv, R. Interleukin-17 activates JAK2/STAT3, PI3K/Akt and nuclear factor-kappaB signaling pathway to promote the tumorigenesis of cervical cancer. Exp. Ther. Med. 2021, 22, 1291. [Google Scholar] [CrossRef]
- Huang, X.; Nepovimova, E.; Adam, V.; Sivak, L.; Heger, Z.; Valko, M.; Wu, Q.; Kuca, K. Neutrophils in Cancer immunotherapy: Friends or foes? Mol. Cancer 2024, 23, 107. [Google Scholar] [CrossRef]
- Xiong, S.; Dong, L.; Cheng, L. Neutrophils in cancer carcinogenesis and metastasis. J. Hematol. Oncol. 2021, 14, 173. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, C.C.; Malanchi, I. Neutrophils in cancer: Heterogeneous and multifaceted. Nat. Rev. Immunol. 2022, 22, 173–187. [Google Scholar] [CrossRef]
- Teijeira, A.; Garasa, S.; Ochoa, M.C.; Villalba, M.; Olivera, I.; Cirella, A.; Eguren-Santamaria, I.; Berraondo, P.; Schalper, K.A.; de Andrea, C.E.; et al. IL8, Neutrophils, and NETs in a Collusion against Cancer Immunity and Immunotherapy. Clin. Cancer Res. 2021, 27, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Franciosi, M.L.M.; do Carmo, T.I.T.; Zanini, D.; Cardoso, A.M. Inflammatory profile in cervical cancer: Influence of purinergic signaling and possible therapeutic targets. Inflamm. Res. 2022, 71, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Fullar, A.; Dudas, J.; Olah, L.; Hollosi, P.; Papp, Z.; Sobel, G.; Karaszi, K.; Paku, S.; Baghy, K.; Kovalszky, I. Remodeling of extracellular matrix by normal and tumor-associated fibroblasts promotes cervical cancer progression. BMC Cancer 2015, 15, 256. [Google Scholar] [CrossRef]
- Fernandes, P.C., Jr.; Garcia, C.B.; Micheli, D.C.; Cunha, F.Q.; Murta, E.F.; Tavares-Murta, B.M. Circulating neutrophils may play a role in the host response in cervical cancer. Int. J. Gynecol. Cancer 2007, 17, 1068–1074. [Google Scholar] [CrossRef]
- Mei, X.; Xiong, J.; Liu, J.; Huang, A.; Zhu, D.; Huang, Y.; Wang, H. DHCR7 promotes lymph node metastasis in cervical cancer through cholesterol reprogramming-mediated activation of the KANK4/PI3K/AKT axis and VEGF-C secretion. Cancer Lett. 2024, 584, 216609. [Google Scholar] [CrossRef]
- Yamamoto, N.; Kinoshita, T.; Nohata, N.; Itesako, T.; Yoshino, H.; Enokida, H.; Nakagawa, M.; Shozu, M.; Seki, N. Tumor suppressive microRNA-218 inhibits cancer cell migration and invasion by targeting focal adhesion pathways in cervical squamous cell carcinoma. Int. J. Oncol. 2013, 42, 1523–1532. [Google Scholar] [CrossRef]
- Dong, X.; Chen, X.; Xue, M.; Zhang, Y.; Jiang, P. EFNA1 promotes the tumorigenesis and metastasis of cervical cancer by phosphorylation pathway and epithelial-mesenchymal transition. Acta Histochem. 2025, 127, 152236. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Li, S.; Wang, L. Semaphorins in tumor microenvironment: Biological mechanisms and therapeutic progress. Int. Immunopharmacol. 2024, 132, 112035. [Google Scholar] [CrossRef] [PubMed]
- Bica, C.; Tirpe, A.; Nutu, A.; Ciocan, C.; Chira, S.; Gurzau, E.S.; Braicu, C.; Berindan-Neagoe, I. Emerging roles and mechanisms of semaphorins activity in cancer. Life Sci. 2023, 318, 121499. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Caprari, P.; Moretti, S.; Faronato, M.; Tamagnone, L.; Procopio, A. Semaphorin-3A is expressed by tumor cells and alters T-cell signal transduction and function. Blood 2006, 107, 3321–3329. [Google Scholar] [CrossRef]
- Qi, T.T.; Zhou, S.J.; Yu, Z.; Li, Y.; Chen, J.Q. Unveiling the heterogeneity and immunotherapy potency of tumor-associated neutrophils in the tumor microenvironment of gastric cancer. BMC Gastroenterol. 2025, 25, 303. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, T.; Wu, H.; Cheng, X.; Gao, H.; Yang, M. Integrative Analysis of Neutrophil-Associated Genes Reveals Prognostic Significance and Immune Microenvironment Modulation in Cervical Cancer. Biomedicines 2025, 13, 1348. https://doi.org/10.3390/biomedicines13061348
Hu T, Wu H, Cheng X, Gao H, Yang M. Integrative Analysis of Neutrophil-Associated Genes Reveals Prognostic Significance and Immune Microenvironment Modulation in Cervical Cancer. Biomedicines. 2025; 13(6):1348. https://doi.org/10.3390/biomedicines13061348
Chicago/Turabian StyleHu, Ting, Haijing Wu, Xinghan Cheng, Haoyue Gao, and Min Yang. 2025. "Integrative Analysis of Neutrophil-Associated Genes Reveals Prognostic Significance and Immune Microenvironment Modulation in Cervical Cancer" Biomedicines 13, no. 6: 1348. https://doi.org/10.3390/biomedicines13061348
APA StyleHu, T., Wu, H., Cheng, X., Gao, H., & Yang, M. (2025). Integrative Analysis of Neutrophil-Associated Genes Reveals Prognostic Significance and Immune Microenvironment Modulation in Cervical Cancer. Biomedicines, 13(6), 1348. https://doi.org/10.3390/biomedicines13061348