


A Narrative Review of Current and Investigational Therapies in Hypertrophic Cardiomyopathy
 ,
,
Abstract
1. Introduction
2. Methods
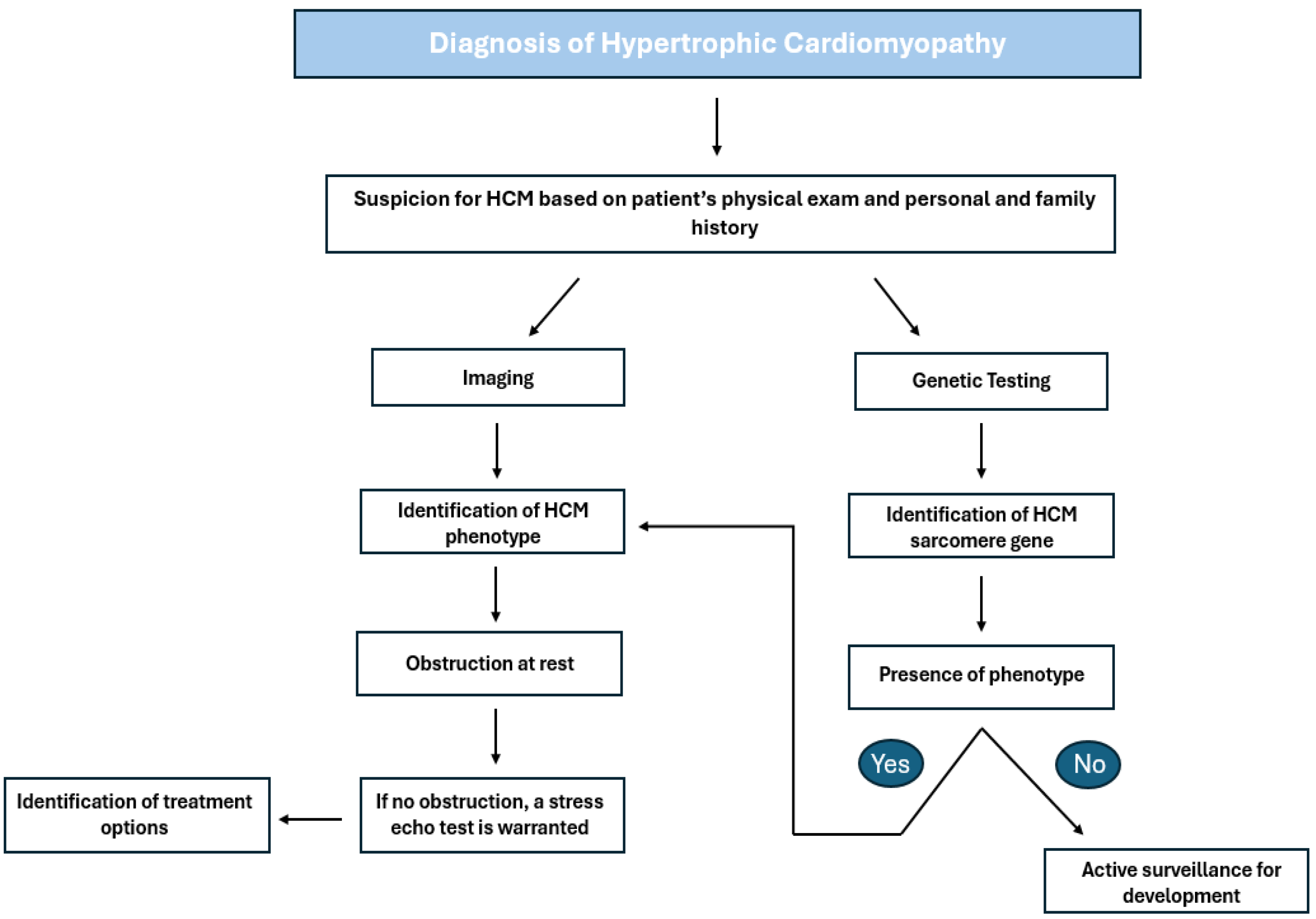
3. Indications for Treatment
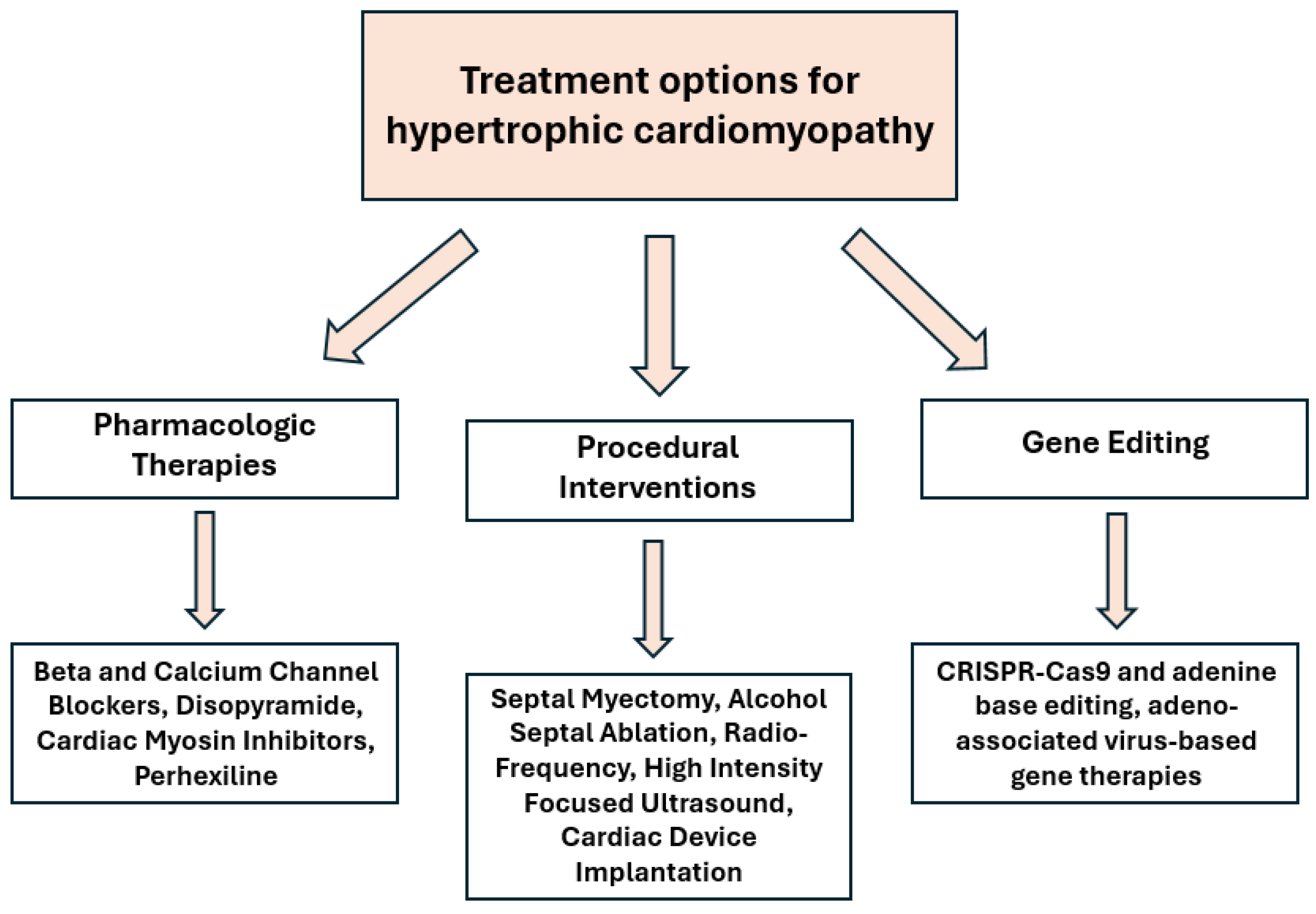
4. Current Pharmacological and Investigative Therapies
4.1. Beta Blockers and Non-Dihydropyridine Calcium Channel Blockers
4.2. Disopyramide
4.3. Cardiac Myosin Inhibitors
4.4. Ranolazine
4.5. Sacubitril/Valsartan in oHCM
4.6. Angiotensin II Receptor Blockers (ARBs) in oHCM
4.7. Perhexiline
5. Procedural Interventions
5.1. Septal Myectomy (SM)
5.2. Alcohol Septal Ablation
5.3. Radiofrequency Ablation
5.4. High-Intensity Focused Ultrasound
5.5. Cardiac Device Implantation
6. Gene Editing
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antunes, M.d.O.; Scudeler, T.L. Hypertrophic Cardiomyopathy. Int. J. Cardiol. Heart Vasc. 2020, 27, 100503. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Mealiffe, M.E.; Bach, R.G.; Bhattacharya, M.; Choudhury, L.; Edelberg, J.M.; Hegde, S.M.; Jacoby, D.; Lakdawala, N.K.; Lester, S.J.; et al. Evaluation of Mavacamten in Symptomatic Patients with Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Massera, D.; Sherrid, M.V.; Maron, M.S.; Rowin, E.J.; Maron, B.J. How Common Is Hypertrophic Cardiomyopathy… Really?: Disease Prevalence Revisited 27 Years After CARDIA. Int. J. Cardiol. 2023, 382, 64–67. [Google Scholar] [CrossRef]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef]
- Braunwald, E. Hypertrophic Cardiomyopathy: A Brief Overview. Am. J. Cardiol. 2024, 212S, S1–S3. [Google Scholar] [CrossRef]
- Ommen, S.R.; Ho, C.Y.; Asif, I.M.; Balaji, S.; Burke, M.A.; Day, S.M.; Dearani, J.A.; Epps, K.C.; Evanovich, L.; Ferrari, V.A.; et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1239–e1311. [Google Scholar] [CrossRef]
- Geske, J.B.; Ommen, S.R.; Gersh, B.J. Hypertrophic Cardiomyopathy. JACC Heart Fail. 2018, 6, 364–375. [Google Scholar] [CrossRef]
- Massera, D.; Sherrid, M.V.; Adlestein, E.; Bokhari, N.; Alvarez, I.C.; Wu, W.Y.; Reuter, M.C.; Maron, M.S.; Maron, B.J.; Rowin, E.J. Disopyramide Revisited for Treatment of Symptomatic Obstructive Hypertrophic Cardiomyopathy: Efficacy and Safety in Patients Treated for at Least 5 Years. J. Am. Heart Assoc. 2025, 14, e037639. [Google Scholar] [CrossRef]
- Ismayl, M.; Abbasi, M.A.; Marar, R.; Geske, J.B.; Gersh, B.J.; Anavekar, N.S. Mavacamten Treatment for Hypertrophic Cardiomyopathy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Curr. Probl. Cardiol. 2023, 48, 101429. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Coats, C.J.; Düngen, H.-D.; et al. Aficamten for Symptomatic Obstructive Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2024, 390, 1849–1861. [Google Scholar] [CrossRef]
- Masri, A.; Sherrid, M.V.; Abraham, T.P.; Choudhury, L.; Garcia-Pavia, P.; Kramer, C.M.; Barriales-Villa, R.; Owens, A.T.; Rader, F.; Nagueh, S.F.; et al. Efficacy and Safety of Aficamten in Symptomatic Nonobstructive Hypertrophic Cardiomyopathy: Results From the REDWOOD-HCM Trial, Cohort 4. J. Card. Fail. 2024, 30, 1439–1448. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Camici, P.G.; Merlini, P.A.; Rapezzi, C.; Patten, M.; Climent, V.; Sinagra, G.; Tomberli, B.; Marin, F.; Ehlermann, P.; et al. Efficacy of Ranolazine in Patients With Symptomatic Hypertrophic Cardiomyopathy: The RESTYLE-HCM Randomized, Double-Blind, Placebo-Controlled Study. Circ. Heart Fail. 2018, 11, e004124. [Google Scholar] [CrossRef] [PubMed]
- Groeneweg, J.A.; van Dalen, B.M.; Cox, M.P.G.J.; Heymans, S.; Braam, R.L.; Michels, M.; Asselbergs, F.W. 2023 European Society of Cardiology Guidelines on the Management of Cardiomyopathies: Statement of Endorsement by the NVVC. Neth. Heart J. 2025, 33, 148–156. [Google Scholar] [CrossRef]
- Velicki, L.; Popovic, D.; Okwose, N.C.; Preveden, A.; Tesic, M.; Tafelmeier, M.; Charman, S.J.; Barlocco, F.; MacGowan, G.A.; Seferovic, P.M.; et al. Sacubitril/Valsartan for the Treatment of Non-Obstructive Hypertrophic Cardiomyopathy: An Open-Label Randomized Controlled Trial (SILICOFCM). Eur. J. Heart Fail. 2024, 26, 1361–1368. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef]
- Penicka, M.; Gregor, P.; Kerekes, R.; Marek, D.; Curila, K.; Krupicka, J. Candesartan use in Hypertrophic and Non-obstructive Cardiomyopathy Estate (CHANCE) Study Investigators the Effects of Candesartan on Left Ventricular Hypertrophy and Function in Nonobstructive Hypertrophic Cardiomyopathy: A Pilot, Randomized Study. J. Mol. Diagn. 2009, 11, 35–41. [Google Scholar] [CrossRef]
- Axelsson, A.; Iversen, K.; Vejlstrup, N.; Ho, C.; Norsk, J.; Langhoff, L.; Ahtarovski, K.; Corell, P.; Havndrup, O.; Jensen, M.; et al. Efficacy and Safety of the Angiotensin II Receptor Blocker Losartan for Hypertrophic Cardiomyopathy: The INHERIT Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Diabetes Endocrinol. 2015, 3, 123–131. [Google Scholar] [CrossRef]
- Abdelazeem, B.; Abbas, K.S.; Ahmad, S.; Raslan, H.; Labieb, F.; Savarapu, P. The Effect of Angiotensin II Receptor Blockers in Patients with Hypertrophic Cardiomyopathy: An Updated Systematic Review and Meta-Analysis of Randomized Controlled Trials. Rev. Cardiovasc. Med. 2022, 23, 141. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Axelsson, A.; Russell, M.W.; Zahka, K.; Lever, H.M.; Pereira, A.C.; Colan, S.D.; Margossian, R.; Murphy, A.M.; et al. Valsartan in Early-Stage Hypertrophic Cardiomyopathy: A Randomized Phase 2 Trial. Nat. Med. 2021, 27, 1818–1824. [Google Scholar] [CrossRef]
- Ananthakrishna, R.; Lee, S.L.; Foote, J.; Sallustio, B.C.; Binda, G.; Mangoni, A.A.; Woodman, R.; Semsarian, C.; Horowitz, J.D.; Selvanayagam, J.B. Randomized Controlled Trial of Perhexiline on Regression of Left Ventricular Hypertrophy in Patients with Symptomatic Hypertrophic Cardiomyopathy (RESOLVE-HCM Trial). Am. Heart J. 2021, 240, 101–113. [Google Scholar] [CrossRef]
- Rastegar, H.; Boll, G.; Rowin, E.J.; Dolan, N.; Carroll, C.; Udelson, J.E.; Wang, W.; Carpino, P.; Maron, B.J.; Maron, M.S.; et al. Results of Surgical Septal Myectomy for Obstructive Hypertrophic Cardiomyopathy: The Tufts Experience. Ann. Cardiothorac. Surg. 2017, 6, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Sedehi, D.; Finocchiaro, G.; Tibayan, Y.; Chi, J.; Pavlovic, A.; Kim, Y.M.; Tibayan, F.A.; Reitz, B.A.; Robbins, R.C.; Woo, J.; et al. Long-Term Outcomes of Septal Reduction for Obstructive Hypertrophic Cardiomyopathy. J. Cardiol. 2015, 66, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Rastegar, H.; Dolan, N.; Carpino, P.; Koethe, B.; Maron, B.J.; Rowin, E.J. Outcomes Over Follow-up ≥10 Years After Surgical Myectomy for Symptomatic Obstructive Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2022, 163, 91–97. [Google Scholar] [CrossRef]
- Veselka, J.; Lawrenz, T.; Stellbrink, C.; Zemanek, D.; Branny, M.; Januska, J.; Sitar, J.; Dimitrow, P.; Krejci, J.; Dabrowski, M.; et al. Early Outcomes of Alcohol Septal Ablation for Hypertrophic Obstructive Cardiomyopathy: A European Multicenter and Multinational Study. Catheter. Cardiovasc. Interv. 2014, 84, 101–107. [Google Scholar] [CrossRef]
- Veselka, J.; Krejčí, J.; Tomašov, P.; Zemánek, D. Long-Term Survival after Alcohol Septal Ablation for Hypertrophic Obstructive Cardiomyopathy: A Comparison with General Population. Eur. Heart J. 2014, 35, 2040–2045. [Google Scholar] [CrossRef]
- Veselka, J.; Jensen, M.K.; Liebregts, M.; Januska, J.; Krejci, J.; Bartel, T.; Dabrowski, M.; Hansen, P.R.; Almaas, V.M.; Seggewiss, H.; et al. Long-Term Clinical Outcome After Alcohol Septal Ablation for Obstructive Hypertrophic Cardiomyopathy: Results from the Euro-ASA Registry. Eur. Heart J. 2016, 37, 1517–1523. [Google Scholar] [CrossRef]
- Matsuda, J.; Takano, H.; Imori, Y.; Ishihara, K.; Sangen, H.; Kubota, Y.; Nakata, J.; Miyachi, H.; Hosokawa, Y.; Tara, S.; et al. Long-Term Clinical Outcomes After Alcohol Septal Ablation for Hypertrophic Obstructive Cardiomyopathy in Japan: A Retrospective Study. Heart Vessel. 2025, 40, 496–508. [Google Scholar] [CrossRef]
- Sorajja, P.; Binder, J.; Nishimura, R.A.; Holmes, D.R.; Rihal, C.S.; Gersh, B.J.; Bresnahan, J.F.; Ommen, S.R. Predictors of an Optimal Clinical Outcome with Alcohol Septal Ablation for Obstructive Hypertrophic Cardiomyopathy. Catheter. Cardiovasc. Interv. 2013, 81, E58–E67. [Google Scholar] [CrossRef]
- Cui, H.; Schaff, H.V.; Wang, S.; Lahr, B.D.; Rowin, E.J.; Rastegar, H.; Hu, S.; Eleid, M.F.; Dearani, J.A.; Kimmelstiel, C.; et al. Survival Following Alcohol Septal Ablation or Septal Myectomy for Patients with Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 1647–1655. [Google Scholar] [CrossRef]
- Zhou, M.; Ta, S.; Hahn, R.T.; Hsi, D.H.; Leon, M.B.; Hu, R.; Zhang, J.; Zuo, L.; Li, J.; Wang, J.; et al. Percutaneous Intramyocardial Septal Radiofrequency Ablation in Patients with Drug-Refractory Hypertrophic Obstructive Cardiomyopathy. JAMA Cardiol. 2022, 7, 529–538. [Google Scholar] [CrossRef]
- Lawrenz, T.; Borchert, B.; Leuner, C.; Bartelsmeier, M.; Reinhardt, J.; Strunk-Mueller, C.; Meyer Zu Vilsendorf, D.; Schloesser, M.; Beer, G.; Lieder, F.; et al. Endocardial Radiofrequency Ablation for Hypertrophic Obstructive Cardiomyopathy: Acute Results and 6 Months’ Follow-up in 19 Patients. J. Am. Coll. Cardiol. 2011, 57, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Canzi, C.C.; do Prado Júnior, E.R.; da Silva Menezes Júnior, A.; Rezende, A.L.; Botelho, S.M.; da Ressurreiçao Santos, L. Radiofrequency Ablation in Patients with Obstructive Hypertrophic Cardiomyopathy: A Systematic Review and Meta-Analysis. Am. Heart J. Plus 2022, 24, 100229. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, Y.; Xue, Y.; Luo, S. Efficacy and Safety of Radiofrequency Ablation for Hypertrophic Obstructive Cardiomyopathy: A Systematic Review and Meta-Analysis. Clin. Cardiol. 2020, 43, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Chen, S.; Cui, Y.; Zhou, Z.; Lu, J.; Du, Z.; Ding, J.; Xing, K.; Zhang, Y.; Zhou, Y.; et al. Midterm Outcomes of Percutaneous Intramyocardial Septal Radiofrequency Ablation for Hypertrophic Cardiomyopathy: A Single-Center, Observational Study. J. Am. Heart Assoc. 2024, 13, e034080. [Google Scholar] [CrossRef]
- Ren, F.; Sui, Y.; Gong, X.; Xing, Q.; Wang, Z. High-Intensity Focused Ultrasound in Interventricular Septal Myocardial Ablation. Int. Heart J. 2022, 63, 1158–1165. [Google Scholar] [CrossRef]
- Engel, D.J.; Muratore, R.; Hirata, K.; Otsuka, R.; Fujikura, K.; Sugioka, K.; Marboe, C.; Lizzi, F.L.; Homma, S. Myocardial Lesion Formation Using High-Intensity Focused Ultrasound. J. Am. Soc. Echocardiogr. 2006, 19, 932–937. [Google Scholar] [CrossRef]
- Miller, D.L.; Lu, X.; Dou, C.; Zhu, Y.I.; Fuller, R.; Fields, K.; Fabiilli, M.L.; Owens, G.E.; Gordon, D.; Kripfgans, O.D. Ultrasonic Cavitation-Enabled Treatment for Therapy of Hypertrophic Cardiomyopathy: Proof of Principle. Ultrasound Med. Biol. 2018, 44, 1439–1450. [Google Scholar] [CrossRef]
- Miller, D.L.; Dou, C.; Owens, G.E.; Kripfgans, O.D. Timing of High-Intensity Pulses for Myocardial Cavitation-Enabled Therapy. J. Ther. Ultrasound 2014, 2, 20. [Google Scholar] [CrossRef]
- Otsuka, R.; Fujikura, K.; Abe, Y.; Okajima, K.; Pulerwitz, T.; Engel, D.J.; Muratore, R.; Ketterling, J.A.; Kalisz, A.; Sciacca, R.; et al. Extracardiac Ablation of the Left Ventricular Septum in Beating Canine Hearts Using High-Intensity Focused Ultrasound. J. Am. Soc. Echocardiogr. 2007, 20, 1400–1406. [Google Scholar] [CrossRef]
- Rong, S.; Woo, K.; Zhou, Q.; Zhu, Q.; Wu, Q.; Wang, Q.; Deng, C.; Liu, D.; Yang, G.; Jiang, Y.; et al. Septal Ablation Induced by Transthoracic High-Intensity Focused Ultrasound in Canines. J. Am. Soc. Echocardiogr. 2013, 26, 1228–1234. [Google Scholar] [CrossRef]
- Zhao, X.; Luo, W.; Hu, J.; Zuo, L.; Wang, J.; Hu, R.; Wang, B.; Xu, L.; Li, J.; Wu, M.; et al. Cardiomyocyte-Targeted and 17β-Estradiol-Loaded Acoustic Nanoprobes as a Theranostic Platform for Cardiac Hypertrophy. J. Nanobiotechnol. 2018, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Dou, C.; Lu, X.; Zhu, Y.I.; Fabiilli, M.L.; Owens, G.E.; Kripfgans, O.D. Use of Theranostic Strategies in Myocardial Cavitation-Enabled Therapy. Ultrasound Med. Biol. 2015, 41, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.I.; Miller, D.L.; Dou, C.; Kripfgans, O.D. Passive Microlesion Detection and Mapping for Treatment of Hypertrophic Cardiomyopathy. AIP Conf. Proc. 2017, 1816, 030002. [Google Scholar] [CrossRef]
- Khazanie, P.; Hammill, B.G.; Qualls, L.G.; Fonarow, G.C.; Hammill, S.C.; Heidenreich, P.A.; Al-Khatib, S.M.; Piccini, J.P.; Masoudi, F.A.; Peterson, P.N.; et al. Clinical Effectiveness of Cardiac Resynchronization Therapy versus Medical Therapy Alone among Patients with Heart Failure: Analysis of the ICD Registry and ADHERE. Circ. Heart Fail. 2014, 7, 926–934. [Google Scholar] [CrossRef]
- Solomou, E.; Gatzoulis, K.A.; Skiadas, I.; Doundoulakis, I.; Arsenos, P.; Dilaveris, P.; Sideris, S.; Tousoulis, D. Upgrade to Cardiac Resynchronization Therapy Difibrillator Device of a Pacemaker-Dependent Patient with End-Stage Hypertrophic Cardiomyopathy. Hell. J. Cardiol. 2020, 61, 65–67. [Google Scholar] [CrossRef]
- Radu, A.D.; Cojocaru, C.; Onciul, S.; Scarlatescu, A.; Zlibut, A.; Nastasa, A.; Dorobantu, M. Cardiac Resynchronization Therapy and Hypertrophic Cardiomyopathy: A Comprehensive Review. Biomedicines 2023, 11, 350. [Google Scholar] [CrossRef]
- Iskandarani, G.; Khamis, A.M.; Sabra, M.; Cai, M.; Akl, E.A.; Refaat, M. Transvenous versus Subcutaneous Implantable Cardiac Defibrillators for People at Risk of Sudden Cardiac Death. Cochrane Database Syst. Rev. 2020, 2020, CD013615. [Google Scholar] [CrossRef]
- Fong, K.Y.; Ng, C.J.R.; Wang, Y.; Yeo, C.; Tan, V.H. Subcutaneous Versus Transvenous Implantable Defibrillator Therapy: A Systematic Review and Meta-Analysis of Randomized Trials and Propensity Score–Matched Studies. J. Am. Heart Assoc. 2022, 11, e024756. [Google Scholar] [CrossRef]
- Rella, V.; Maurizi, N.; Bernardini, A.; Brasca, F.M.; Salerno, S.; Meda, M.; Mariani, D.; Torchio, M.; Ravaro, S.; Cerea, P.; et al. Candidacy and Long-Term Outcomes of Subcutaneous Implantable Cardioverter-Defibrillators in Current Practice in Patients with Hypertrophic Cardiomyopathy. Int. J. Cardiol. 2024, 409, 132202. [Google Scholar] [CrossRef]
- da Silva Menezes Júnior, A.; Oliveira, I.C.; de Sousa, A.M.; Paro Piai, R.F.; Oliveira, V.M.R. Subcutaneous versus Transvenous Implantable Cardioverter Defibrillator in Hypertrophic Cardiomyopathy: A Systematic Review and Meta-Analysis. Cardiovasc. Diagn. Ther. 2024, 14, 318–327. [Google Scholar] [CrossRef]
- Wynne, E.; Bergin, J.D.; Ailawadi, G.; Kern, J.A.; Kennedy, J.L.W. Use of a Left Ventricular Assist Device in Hypertrophic Cardiomyopathy. J. Card. Surg. 2011, 26, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Roesel, M.J.; Nersesian, G.; Neuber, S.; Thau, H.; Wolff von Gudenberg, R.; Lanmueller, P.; Hennig, F.; Falk, V.; Potapov, E.; Knosalla, C.; et al. LVAD as a Bridge to Transplantation-Current Status and Future Perspectives. Rev. Cardiovasc. Med. 2024, 25, 176. [Google Scholar] [CrossRef] [PubMed]
- Niamat, J.; Ramjankhan, F.; Van Der Kaaij, N.; Gianoli, M.; Van Laake, L.W.; Mokhles, M.M. Outcome After Left Ventricular Assist Device Exchange. Eur. J. Cardiothorac Surg. 2024, 66, ezae317. [Google Scholar] [CrossRef]
- Venturiello, D.; Tiberi, P.G.; Perulli, F.; Nardoianni, G.; Guida, L.; Barsali, C.; Terrone, C.; Cianca, A.; Lustri, C.; Sclafani, M.; et al. Unveiling the Future of Cardiac Care: A Review of Gene Therapy in Cardiomyopathies. Int. J. Mol. Sci. 2024, 25, 13147. [Google Scholar] [CrossRef]
- Park, F. The Heart Is Where AAV9 Lies. Physiol. Genomics 2022, 54, 316–318. [Google Scholar] [CrossRef]
- Reichart, D.; Newby, G.A.; Wakimoto, H.; Lun, M.; Gorham, J.M.; Curran, J.J.; Raguram, A.; DeLaughter, D.M.; Conner, D.A.; Marsiglia, J.D.C.; et al. Efficient in Vivo Genome Editing Prevents Hypertrophic Cardiomyopathy in Mice. Nat. Med. 2023, 29, 412–421. [Google Scholar] [CrossRef]
- Chai, A.C.; Cui, M.; Chemello, F.; Li, H.; Chen, K.; Tan, W.; Atmanli, A.; McAnally, J.R.; Zhang, Y.; Xu, L.; et al. Base Editing Correction of Hypertrophic Cardiomyopathy in Human Cardiomyocytes and Humanized Mice. Nat. Med. 2023, 29, 401–411. [Google Scholar] [CrossRef]
- Tenaya Therapeutics Reports Promising Early Data from MyPEAKTM-1 Phase 1b/2 Clinical Trial of TN-201 for Treatment of MYBPC3-Associated Hypertrophic Cardiomyopathy; Tenaya Therapeutics, Inc.: South San Francisco, CA, USA, 2024; Available online: https://investors.tenayatherapeutics.com/news-releases/news-release-details/tenaya-therapeutics-reports-promising-early-data-mypeaktm-1/ (accessed on 26 March 2025).
- Madaudo, C.; Parlati, A.L.M.; Di Lisi, D.; Carluccio, R.; Sucato, V.; Vadalà, G.; Nardi, E.; Macaione, F.; Cannata, A.; Manzullo, N.; et al. Artificial intelligence in cardiology: A peek at the future and the role of ChatGPT in cardiology practice. J. Cardiovasc. Med. 2024, 25, 766–771. [Google Scholar] [CrossRef]
- Desai, M.Y.; Jadam, S.; Abusafia, M.; Rutkowski, K.; Ospina, S.; Gaballa, A.; Sultana, S.; Thamilarasan, M.; Xu, B.; Popovic, Z.B. Real-World Artificial Intelligence-Based Electrocardiographic Analysis to Diagnose Hypertrophic Cardiomyopathy. JACC Clin. Electrophysiol. 2025. in press. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Diagnostic Criteria for Hypertrophic Cardiomyopathy in Adults Versus Children | |
|---|---|
| Adults | Identification of a maximal end-diastolic wall thickness of ≥15 mm in any part of the left ventricle (with no other cause of hypertrophy in adults). |
| An end-diastolic wall thickness ranging from 13 to 14 mm may serve as a diagnostic indicator, notably when correlated with a family history of hypertrophic cardiomyopathy (HCM) or a positive genetic test. | |
| Children | The diagnostic criteria exhibit variability; a z-score exceeding 2.5, adjusted for body surface area, may be suitable for identifying early hypertrophic cardiomyopathy (HCM) in asymptomatic children without a family history. Conversely, a z-score greater than two may be employed for children with a familial history of HCM or those who have received a positive genetic test result. |
| Sudden Cardiac Death Risk Factors in Adults with HCM | |
|---|---|
| Left Ventricular Hypertrophy | CMR or echocardiographic imaging demonstrating wall thickness > 30 mm in any segment. |
| Family History of SCD | First-degree relatives < 50 years of age who experienced SCD attributed to HCM. |
| Left Ventricular Apical Aneurysm | Apical aneurysm with transmural scar, regardless of size. |
| Unexplained Syncope | At least 1 episode of syncope that is deemed to be unrelated to vasovagal or LVOTO causes. |
| HCM in the Setting of Low Ejection Fraction | CMR or echocardiographic evidence of ejection fraction < 50%. |
| Non-Sustained Ventricular tachycardia upon Ambulatory Monitoring | NSVT defined as >3 beats at a rate of 120 beats per minute or higher occurring over 24 to 48 h of monitoring. |
| Late Gadolinium Enhancement on CMR | Fibrosis of >15% of the left ventricular mass detected by LGE. |
| Gene Editing Approaches | ||||
|---|---|---|---|---|
| Approach | Target Gene | Delivery Method | Model Studied | Key Findings |
| CRISPR-Cas9 [56] | MYH7 (R403Q) | AAV9 with cardiac-specific promoter | Mouse models with HCM | The mutated MYH7 (R403Q) was successfully silenced, leading to a decrease in hypertrophy and fibrosis. However, due to a restricted therapeutic window, dosage increases negatively affected cardiac contractility. |
| ABE8e (Adenine Base Editing) [56] | MYH7 (R403Q) | AAV9 with Tnnt2 promoter | Mouse models with HCM | ABE8e reduced hypertrophy and fibrosis in cardiac muscle within cardiomyocytes possessing mutated MYH7 (R403Q) alleles and was deemed to be safer and more effective than CRISPR. |
| ABEmax-VRQR [57] | MYH7 (R403Q) | AAV9 with sgRNA h403_sgRNA | iPSC-derived human cardiomyocytes and neonatal mice | Cardiomyocytes exhibiting mutated MYH7 (R403Q) alleles, targeted by ABEmax-VRQR, demonstrated restored contractility and ATP metabolism. An approximately 35% correction in mutated alleles was noted in vivo for heterozygous mice, coupled with prolonged survival in models with homozygous mutations. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogurek, I.; Gill, R.; Tasouli-Drakou, V.; Joseph, R.; Khalid, A.; Houshmand, N.; Tak, T. A Narrative Review of Current and Investigational Therapies in Hypertrophic Cardiomyopathy. Biomedicines 2025, 13, 1327. https://doi.org/10.3390/biomedicines13061327
Ogurek I, Gill R, Tasouli-Drakou V, Joseph R, Khalid A, Houshmand N, Tak T. A Narrative Review of Current and Investigational Therapies in Hypertrophic Cardiomyopathy. Biomedicines. 2025; 13(6):1327. https://doi.org/10.3390/biomedicines13061327
Chicago/Turabian StyleOgurek, Ian, Randeep Gill, Vasiliki Tasouli-Drakou, Ronnie Joseph, Arbab Khalid, Nazanin Houshmand, and Tahir Tak. 2025. "A Narrative Review of Current and Investigational Therapies in Hypertrophic Cardiomyopathy" Biomedicines 13, no. 6: 1327. https://doi.org/10.3390/biomedicines13061327
APA StyleOgurek, I., Gill, R., Tasouli-Drakou, V., Joseph, R., Khalid, A., Houshmand, N., & Tak, T. (2025). A Narrative Review of Current and Investigational Therapies in Hypertrophic Cardiomyopathy. Biomedicines, 13(6), 1327. https://doi.org/10.3390/biomedicines13061327