


Integrative Approaches in the Management of Hypertrophic Cardiomyopathy: A Comprehensive Review of Current Therapeutic Modalities

 , ,
, ,  , , , ,
, , , ,  and
and
Abstract
1. Introduction
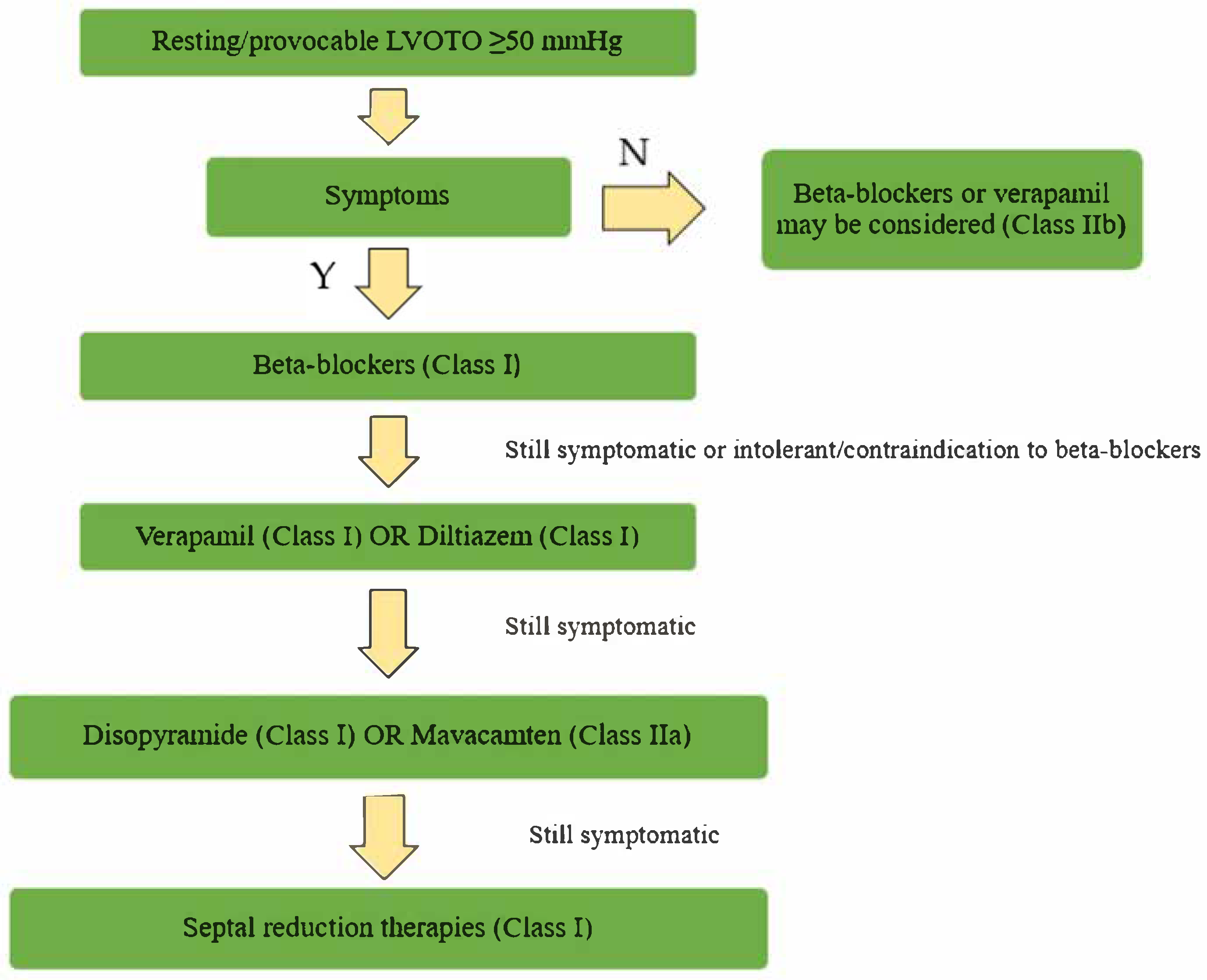
2. Pharmacological Therapy in Obstructive Forms
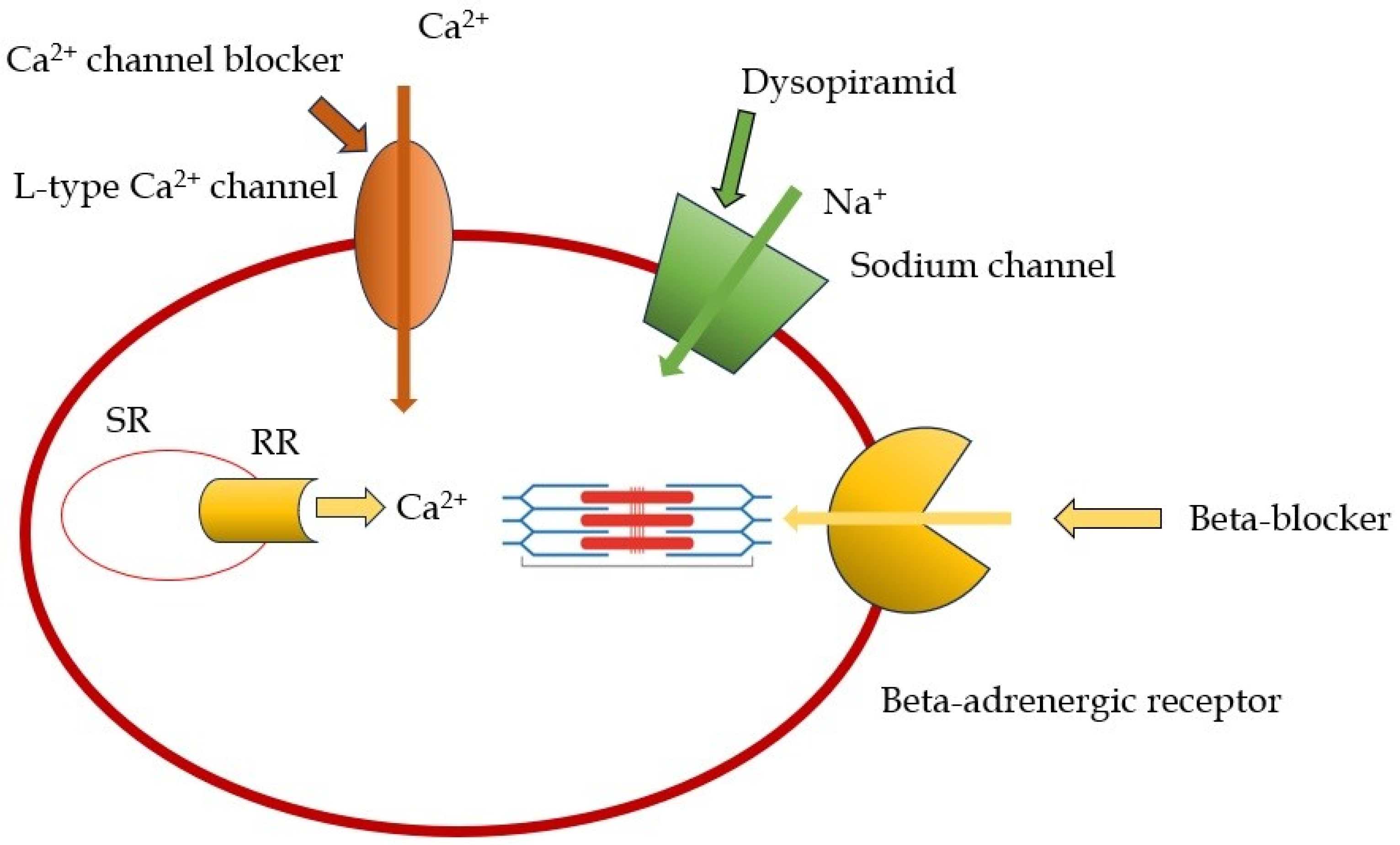
2.1. Beta-Blockers and Non-Dihydropiridine Calcium Channel Blockers
2.2. Disopyramid
2.3. Cardiac Myosin Modulators: Mavacamten and Aficamten
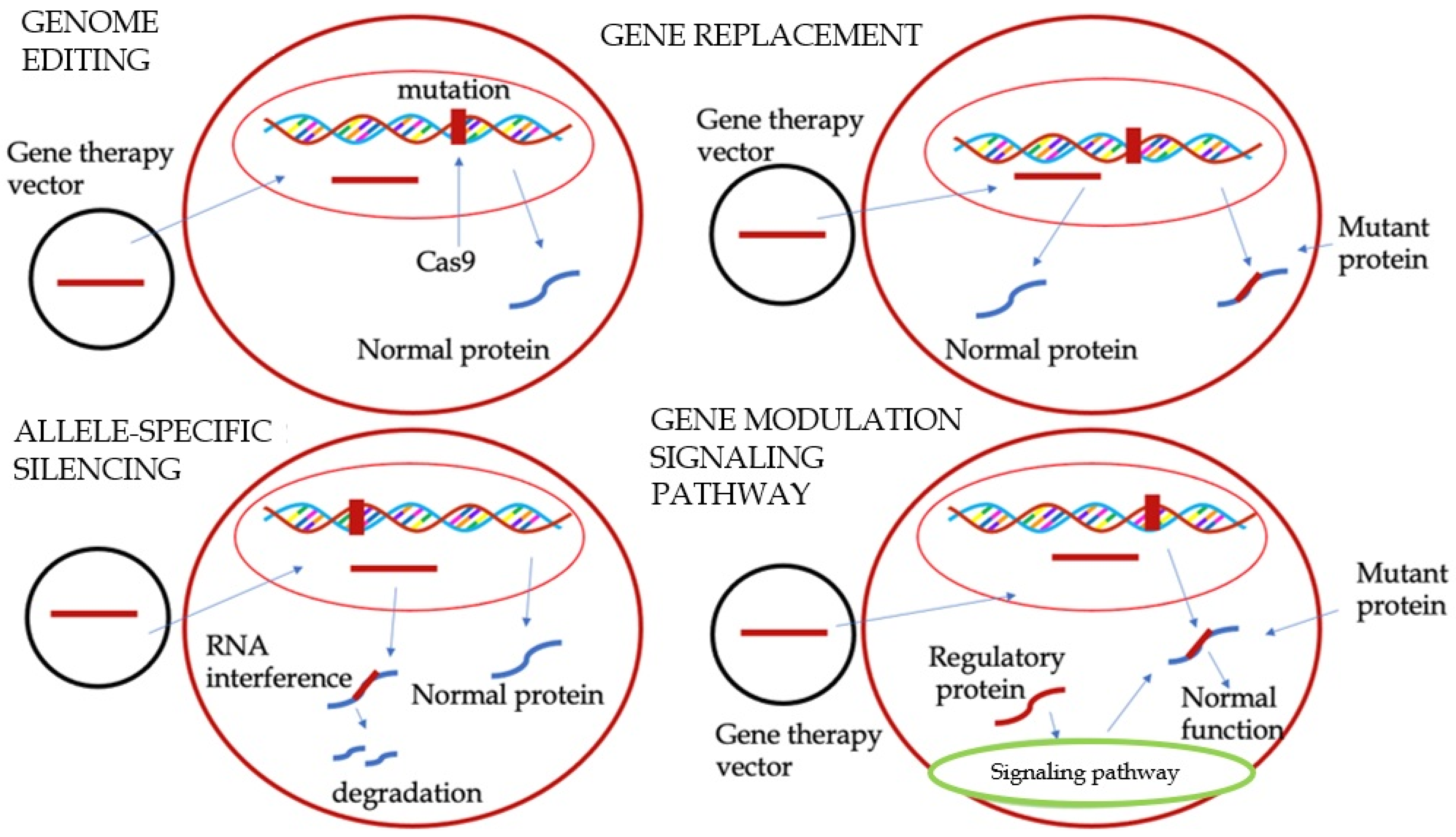
3. Gene Therapy
4. Invasive Strategies: Surgical Myectomy and Septal Ablation
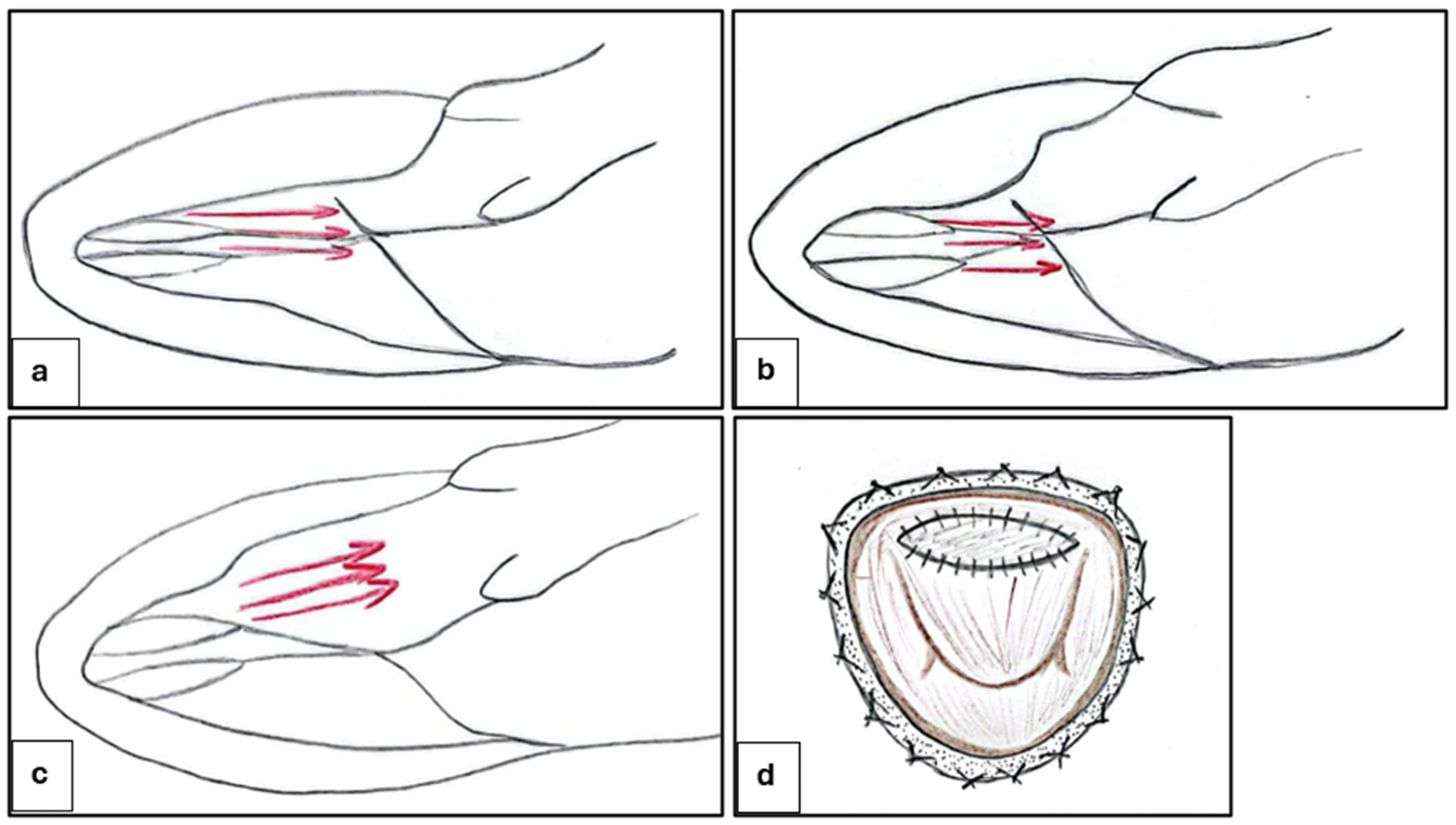
4.1. Ventricular Septal Myectomy
4.2. Alcohol Septal Ablation and Alternative Methods
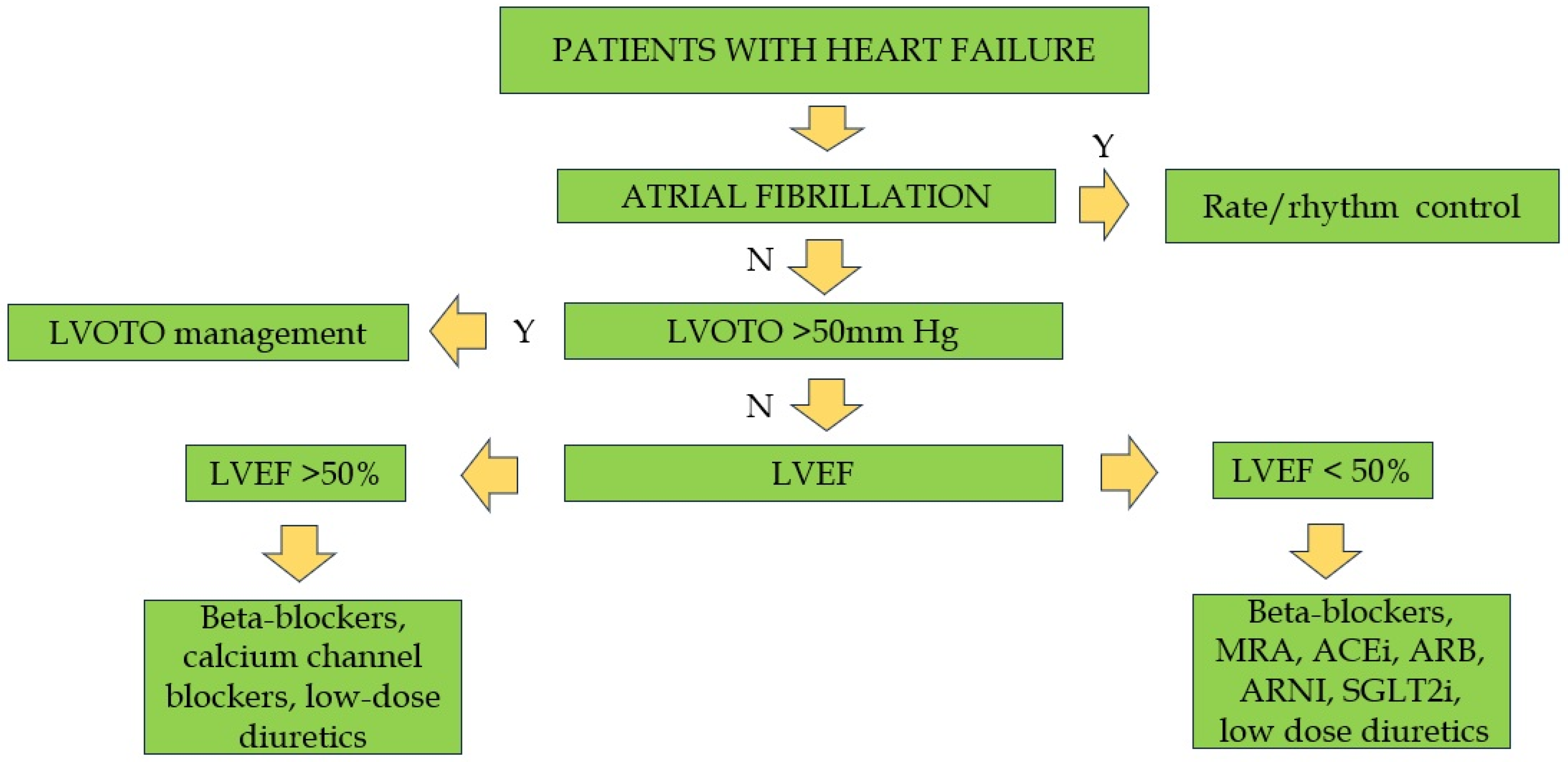
5. Management of Non-Obstructive Forms
6. Management of Atrial Fibrillation
7. Sudden Cardiac Death: Prognostic Factors and Prevention
8. Limitations and Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tuohy, C.V.; Kaul, S.; Song, H.K.; Nazer, B.; Heitner, S.B. Hypertrophic cardiomyopathy: The future of treatment. Eur. J. Heart Fail. 2020, 22, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Contri, R.; Coppini, R.; Cecchi, F.; Frigerio, M.; Olivotto, I. Pharmacological treatment of hypertrophic cardiomyopathy: Current practice and novel perspectives. Eur. J. Heart Fail. 2016, 18, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Merlo, M.; Gagno, G.; Baritussio, A.; Bauce, B.; Biagini, E.; Canepa, M.; Cipriani, A.; Castelletti, S.; Dellegrottaglie, S.; Guaricci, A.I.; et al. Clinical application of CMR in cardiomyopathies: Evolving concepts and techniques: A position paper of myocardial and pericardial diseases and cardiac magnetic resonance working groups of Italian society of cardiology. Heart Fail. Rev. 2023, 28, 77–95. [Google Scholar] [CrossRef]
- Forleo, C.; D’Erchia, A.M.; Sorrentino, S.; Manzari, C.; Chiara, M.; Iacoviello, M.; Guaricci, A.I.; De Santis, D.; Musci, R.L.; La Spada, A.; et al. Targeted next-generation sequencing detects novel gene-phenotype associations and expands the mutational spectrum in cardiomyopathies. PLoS ONE 2017, 12, e0181842. [Google Scholar] [CrossRef]
- Merlo, M.; Porcari, A.; Pagura, L.; Cameli, M.; Vergaro, G.; Musumeci, B.; Biagini, E.; Canepa, M.; Crotti, L.; Imazio, M.; et al. A national survey on prevalence of possible echocardiographic red flags of amyloid cardiomyopathy in consecutive patients undergoing routine echocardiography: Study design and patients characterization-the first insight from the AC-TIVE Study. Eur. J. Prev. Cardiol. 2021, 29, e173–e177. [Google Scholar] [CrossRef]
- Ghanbari, F.; Joyce, T.; Lorenzoni, V.; Guaricci, A.I.; Pavon, A.G.; Fusini, L.; Andreini, D.; Rabbat, M.G.; Aquaro, G.D.; Abete, R.; et al. AI Cardiac MRI Scar Analysis Aids Prediction of Major Arrhythmic Events in the Multicenter DERIVATE Registry. Radiology 2023, 307, e222239. [Google Scholar] [CrossRef]
- Baggiano, A.; Del Torto, A.; Guglielmo, M.; Muscogiuri, G.; Fusini, L.; Babbaro, M.; Collevecchio, A.; Mollace, R.; Scafuri, S.; Mushtaq, S.; et al. Role of CMR Mapping Techniques in Cardiac Hypertrophic Phenotype. Diagnostics 2020, 10, 770. [Google Scholar] [CrossRef]
- Mushtaq, S.; Chiesa, M.; Novelli, V.; Sommariva, E.; Biondi, M.L.; Manzoni, M.; Florio, A.; Lampus, M.L.; Avallone, C.; Zocchi, C.; et al. Role of advanced CMR features in identifying a positive genotype of hypertrophic cardiomyopathy. Int. J. Cardiol. 2024, 417, 132554. [Google Scholar] [CrossRef]
- Pagura, L.; Porcari, A.; Cameli, M.; Biagini, E.; Canepa, M.; Crotti, L.; Imazio, M.; Forleo, C.; Pavasini, R.; Limongelli, G.; et al. ECG/echo indexes in the diagnostic approach to amyloid cardiomyopathy: A head-to-head comparison from the AC-TIVE study. Eur. J. Intern. Med. 2023, 122, 68–77. [Google Scholar] [CrossRef]
- Carrabba, N.; Amico, M.A.; Guaricci, A.I.; Carella, M.C.; Maestrini, V.; Monosilio, S.; Pedrotti, P.; Ricci, F.; Monti, L.; Figliozzi, S.; et al. CMR Mapping: The 4th-Era Revolution in Cardiac Imaging. J. Clin. Med. 2024, 13, 337. [Google Scholar] [CrossRef] [PubMed]
- Dicorato, M.M.; Basile, P.; Muscogiuri, G.; Carella, M.C.; Naccarati, M.L.; Dentamaro, I.; Guglielmo, M.; Baggiano, A.; Mushtaq, S.; Fusini, L.; et al. Novel Insights into Non-Invasive Diagnostic Techniques for Cardiac Amyloidosis: A Critical Review. Diagnostics 2024, 14, 2249. [Google Scholar] [CrossRef] [PubMed]
- Carella, M.C.; Forleo, C.; Caretto, P.; Naccarati, M.L.; Dentamaro, I.; Dicorato, M.M.; Basile, P.; Carulli, E.; Latorre, M.D.; Baggiano, A.; et al. Overcoming Resistance in Anderson-Fabry Disease: Current Therapeutic Challenges and Future Perspectives. J. Clin. Med. 2024, 13, 7195. [Google Scholar] [CrossRef]
- Prondzynski, M.; Mearini, G.; Carrier, L. Gene therapy strategies in the treatment of hypertrophic cardiomyopathy. Pflugers Arch. 2019, 471, 807–815. [Google Scholar] [CrossRef]
- Ezzeddine, F.M.; Agboola, K.M.; Hassett, L.C.; Killu, A.M.; Del-Carpio Munoz, F.; DeSimone, C.V.; Kowlgi, G.N.; Deshmukh, A.J.; Siontis, K.C. Catheter ablation of atrial fibrillation in patients with and without hypertrophic cardiomyopathy: Systematic review and meta-analysis. EP Eur. 2023, 25, euad256. [Google Scholar] [CrossRef]
- Liang, L.W.; Lumish, H.S.; Sewanan, L.R.; Shimada, Y.J.; Maurer, M.S.; Weiner, S.D.; Clerkin, K.J. Evolving Strategies for the Management of Obstructive Hypertrophic Cardiomyopathy. J. Card. Fail. 2024, 30, 1136–1153. [Google Scholar] [CrossRef]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Dearani, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Management of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 390–414. [Google Scholar] [CrossRef]
- Colan, S.D. Hypertrophic cardiomyopathy in childhood. Heart Fail. Clin. 2010, 6, 433–444. [Google Scholar] [CrossRef]
- Yoerger, D.M.; Weyman, A.E. Hypertrophic obstructive cardiomyopathy: Mechanism of obstruction and response to therapy. Rev. Cardiovasc. Med. 2003, 4, 199–215. [Google Scholar]
- Dybro, A.M.; Rasmussen, T.B.; Nielsen, R.R.; Andersen, M.J.; Jensen, M.K.; Poulsen, S.H. Randomized Trial of Metoprolol in Patients with Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2021, 78, 2505–2517. [Google Scholar] [CrossRef]
- Borlaug, B.A.; Omote, K. Beta-Blockers and Exercise Hemodynamics in Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 1576–1578. [Google Scholar] [CrossRef] [PubMed]
- Rosing, D.R.; Idanpaan-Heikkila, U.; Maron, B.J.; Bonow, R.O.; Epstein, S.E. Use of calcium-channel blocking drugs in hypertrophic cardiomyopathy. Am. J. Cardiol. 1985, 55, 185B–195B. [Google Scholar] [CrossRef]
- Maltes, S.; Lopes, L.R. New perspectives in the pharmacological treatment of hypertrophic cardiomyopathy. Rev. Port. Cardiol. 2020, 39, 99–109. [Google Scholar] [CrossRef]
- Maron, M.S.; Rowin, E.J.; Olivotto, I.; Casey, S.A.; Arretini, A.; Tomberli, B.; Garberich, R.F.; Link, M.S.; Chan, R.H.M.; Lesser, J.R.; et al. Contemporary Natural History and Management of Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 1399–1409. [Google Scholar] [CrossRef]
- Epstein, S.E.; Rosing, D.R. Verapamil: Its potential for causing serious complications in patients with hypertrophic cardiomyopathy. Circulation 1981, 64, 437–441. [Google Scholar] [CrossRef]
- Pinto, G.; Chiarito, M.; Puscas, T.; Bacher, A.; Donal, E.; Reant, P.; Condorelli, G.; Hagege, A.; on behalf of the REMY working group of the French Society of Cardiology. Comparative Influences of Beta blockers and Verapamil on Cardiac Outcomes in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2025, 235, 9–15. [Google Scholar] [CrossRef]
- Sanchez-Nadales, A.; Anampa-Guzman, A.; Khan, A. Disopyramide for Hypertrophic Cardiomyopathy. Cureus 2019, 11, e4526. [Google Scholar] [CrossRef]
- Verlinden, N.J.; Coons, J.C. Disopyramide for Hypertrophic Cardiomyopathy: A Pragmatic Reappraisal of an Old Drug. Pharmacotherapy 2015, 35, 1164–1172. [Google Scholar] [CrossRef]
- Sherrid, M.V.; Massera, D. Disopyramide for symptomatic obstructive hypertrophic cardiomyopathy. Int. J. Cardiol. 2025, 423, 133030. [Google Scholar] [CrossRef]
- Sherrid, M.V.; Barac, I.; McKenna, W.J.; Elliott, P.M.; Dickie, S.; Chojnowska, L.; Casey, S.; Maron, B.J. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 1251–1258. [Google Scholar] [CrossRef]
- Sherrid, M.V.; Shetty, A.; Winson, G.; Kim, B.; Musat, D.; Alviar, C.L.; Homel, P.; Balaram, S.K.; Swistel, D.G. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with beta-blockade or verapamil. Circ. Heart Fail. 2013, 6, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Hamada, M.; Ikeda, S.; Shigematsu, Y. Advances in medical treatment of hypertrophic cardiomyopathy. J. Cardiol. 2014, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Maurizi, N.; Chiriatti, C.; Fumagalli, C.; Targetti, M.; Passantino, S.; Antiochos, P.; Skalidis, I.; Chiti, C.; Biagioni, G.; Tomberli, A.; et al. Real-World Use and Predictors of Response to Disopyramide in Patients with Obstructive Hypertrophic Cardiomyopathy. J. Clin. Med. 2023, 12, 2725. [Google Scholar] [CrossRef] [PubMed]
- Forleo, C.; Carella, M.C.; Basile, P.; Mandunzio, D.; Greco, G.; Napoli, G.; Carulli, E.; Dicorato, M.M.; Dentamaro, I.; Santobuono, V.E.; et al. The Role of Magnetic Resonance Imaging in Cardiomyopathies in the Light of New Guidelines: A Focus on Tissue Mapping. J. Clin. Med. 2024, 13, 2621. [Google Scholar] [CrossRef]
- Masri, A.; Lester, S.J.; Stendahl, J.C.; Hegde, S.M.; Sehnert, A.J.; Balaratnam, G.; Shah, A.; Fox, S.; Wang, A. Long-Term Safety and Efficacy of Mavacamten in Symptomatic Obstructive Hypertrophic Cardiomyopathy: Interim Results of the PIONEER-OLE Study. J. Am. Heart Assoc. 2024, 13, e030607. [Google Scholar] [CrossRef]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Desai, M.Y.; Owens, A.; Wolski, K.; Geske, J.B.; Saberi, S.; Wang, A.; Sherrid, M.; Cremer, P.C.; Lakdawala, N.K.; Tower-Rader, A.; et al. Mavacamten in Patients with Hypertrophic Cardiomyopathy Referred for Septal Reduction: Week 56 Results From the VALOR-HCM Randomized Clinical Trial. JAMA Cardiol. 2023, 8, 968–977. [Google Scholar] [CrossRef]
- Rader, F.; Oreziak, A.; Choudhury, L.; Saberi, S.; Fermin, D.; Wheeler, M.T.; Abraham, T.P.; Garcia-Pavia, P.; Zwas, D.R.; Masri, A.; et al. Mavacamten Treatment for Symptomatic Obstructive Hypertrophic Cardiomyopathy: Interim Results from the MAVA-LTE Study, EXPLORER-LTE Cohort. JACC Heart Fail. 2024, 12, 164–177. [Google Scholar] [CrossRef]
- Wheeler, M.T.; Jacoby, D.; Elliott, P.M.; Saberi, S.; Hegde, S.M.; Lakdawala, N.K.; Myers, J.; Sehnert, A.J.; Edelberg, J.M.; Li, W.; et al. Effect of beta-blocker therapy on the response to mavacamten in patients with symptomatic obstructive hypertrophic cardiomyopathy. Eur. J. Heart Fail. 2023, 25, 260–270. [Google Scholar] [CrossRef]
- Ismayl, M.; Abbasi, M.A.; Marar, R.; Geske, J.B.; Gersh, B.J.; Anavekar, N.S. Mavacamten Treatment for Hypertrophic Cardiomyopathy: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Curr. Probl. Cardiol. 2023, 48, 101429. [Google Scholar] [CrossRef]
- Bishev, D.; Fabara, S.; Loseke, I.; Alok, A.; Al-Ani, H.; Bazikian, Y. Efficacy and Safety of Mavacamten in the Treatment of Hypertrophic Cardiomyopathy: A Systematic Review. Heart Lung Circ. 2023, 32, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Mealiffe, M.E.; Bach, R.G.; Bhattacharya, M.; Choudhury, L.; Edelberg, J.M.; Hegde, S.M.; Jacoby, D.; Lakdawala, N.K.; Lester, S.J.; et al. Evaluation of Mavacamten in Symptomatic Patients with Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Masri, A.; Choudhury, L.; Olivotto, I.; Saberi, S.; Wang, A.; Garcia-Pavia, P.; Lakdawala, N.K.; Nagueh, S.F.; Rader, F.; et al. Phase 2 Study of Aficamten in Patients with Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.T.; Masri, A.; Abraham, T.P.; Choudhury, L.; Rader, F.; Symanski, J.D.; Turer, A.T.; Wong, T.C.; Tower-Rader, A.; Coats, C.J.; et al. Aficamten for Drug-Refractory Severe Obstructive Hypertrophic Cardiomyopathy in Patients Receiving Disopyramide: REDWOOD-HCM Cohort 3. J. Card. Fail. 2023, 29, 1576–1582. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Abraham, T.P.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Coats, C.J.; et al. Impact of Aficamten on Disease and Symptom Burden in Obstructive Hypertrophic Cardiomyopathy: Results from SEQUOIA-HCM. J. Am. Coll. Cardiol. 2024, 84, 1821–1831. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Coats, C.J.; Dungen, H.D.; et al. Aficamten for Symptomatic Obstructive Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2024, 390, 1849–1861. [Google Scholar] [CrossRef]
- Masri, A.; Cardoso, R.N.; Abraham, T.P.; Claggett, B.L.; Coats, C.J.; Hegde, S.M.; Kulac, I.J.; Lee, M.M.Y.; Maron, M.S.; Merkely, B.; et al. Effect of Aficamten on Cardiac Structure and Function in Obstructive Hypertrophic Cardiomyopathy: SEQUOIA-HCM CMR Substudy. J. Am. Coll. Cardiol. 2024, 84, 1806–1817. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Bilen, O.; Burroughs, M.; Costabel, J.P.; de Barros Correia, E.; Dybro, A.M.; Elliott, P.; Lakdawala, N.K.; Mann, A.; Nair, A.; et al. Aficamten vs Metoprolol for Obstructive Hypertrophic Cardiomyopathy: MAPLE-HCM Rationale, Study Design, and Baseline Characteristics. JACC Heart Fail. 2025, 13, 346–357. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 60, 705–715. [Google Scholar] [CrossRef]
- Forleo, C.; Carella, M.C.; Basile, P.; Carulli, E.; Dadamo, M.L.; Amati, F.; Loizzi, F.; Sorrentino, S.; Dentamaro, I.; Dicorato, M.M.; et al. Missense and Non-Missense Lamin A/C Gene Mutations Are Similarly Associated with Major Arrhythmic Cardiac Events: A 20-Year Single-Centre Experience. Biomedicines 2024, 12, 1293. [Google Scholar] [CrossRef]
- Paratz, E.D.; Mundisugih, J.; Rowe, S.J.; Kizana, E.; Semsarian, C. Gene Therapy in Cardiology: Is a Cure for Hypertrophic Cardiomyopathy on the Horizon? Can. J. Cardiol. 2024, 40, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Marti-Gutierrez, N.; Park, S.W.; Wu, J.; Lee, Y.; Suzuki, K.; Koski, A.; Ji, D.; Hayama, T.; Ahmed, R.; et al. Correction of a pathogenic gene mutation in human embryos. Nature 2017, 548, 413–419. [Google Scholar] [CrossRef]
- Chai, A.C.; Cui, M.; Chemello, F.; Li, H.; Chen, K.; Tan, W.; Atmanli, A.; McAnally, J.R.; Zhang, Y.; Xu, L.; et al. Base editing correction of hypertrophic cardiomyopathy in human cardiomyocytes and humanized mice. Nat. Med. 2023, 29, 401–411. [Google Scholar] [CrossRef]
- Reichart, D.; Newby, G.A.; Wakimoto, H.; Lun, M.; Gorham, J.M.; Curran, J.J.; Raguram, A.; DeLaughter, D.M.; Conner, D.A.; Marsiglia, J.D.C.; et al. Efficient in vivo genome editing prevents hypertrophic cardiomyopathy in mice. Nat. Med. 2023, 29, 412–421. [Google Scholar] [CrossRef]
- Mearini, G.; Stimpel, D.; Geertz, B.; Weinberger, F.; Kramer, E.; Schlossarek, S.; Mourot-Filiatre, J.; Stoehr, A.; Dutsch, A.; Wijnker, P.J.; et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat. Commun. 2014, 5, 5515. [Google Scholar] [CrossRef]
- Jiang, J.; Wakimoto, H.; Seidman, J.G.; Seidman, C.E. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 2013, 342, 111–114. [Google Scholar] [CrossRef]
- Helms, A.S.; Thompson, A.D.; Day, S.M. Translation of New and Emerging Therapies for Genetic Cardiomyopathies. JACC Basic. Transl. Sci. 2022, 7, 70–83. [Google Scholar] [CrossRef]
- Jaski, B.E.; Jessup, M.L.; Mancini, D.M.; Cappola, T.P.; Pauly, D.F.; Greenberg, B.; Borow, K.; Dittrich, H.; Zsebo, K.M.; Hajjar, R.J.; et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card. Fail. 2009, 15, 171–181. [Google Scholar] [CrossRef]
- Greenberg, B.; Butler, J.; Felker, G.M.; Ponikowski, P.; Voors, A.A.; Desai, A.S.; Barnard, D.; Bouchard, A.; Jaski, B.; Lyon, A.R.; et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): A randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 2016, 387, 1178–1186. [Google Scholar] [CrossRef]
- Menon, S.C.; Ackerman, M.J.; Ommen, S.R.; Cabalka, A.K.; Hagler, D.J.; O’Leary, P.W.; Dearani, J.A.; Cetta, F.; Eidem, B.W. Impact of septal myectomy on left atrial volume and left ventricular diastolic filling patterns: An echocardiographic study of young patients with obstructive hypertrophic cardiomyopathy. J. Am. Soc. Echocardiogr. 2008, 21, 684–688. [Google Scholar] [CrossRef]
- Bytyci, I.; Nistri, S.; Morner, S.; Henein, M.Y. Alcohol Septal Ablation versus Septal Myectomy Treatment of Obstructive Hypertrophic Cardiomyopathy: A Systematic Review and Meta-Analysis. J. Clin. Med. 2020, 9, 3062. [Google Scholar] [CrossRef] [PubMed]
- Karabulut, U.; Yilmaz Can, Y.; Duygu, E.; Karabulut, D.; Keskin, K.; Okay, T. Periprocedural, Short-Term, and Long-Term Outcomes of Alcohol Septal Ablation in Patients with Hypertrophic Obstructive Cardiomyopathy: A 20-Year Single-Center Experience. Anatol. J. Cardiol. 2022, 26, 316–324. [Google Scholar] [CrossRef]
- Kimmelstiel, C.; Rowin, E.J. Fixed, high-volume alcohol dose for septal ablation: High risk with no benefit. Catheter. Cardiovasc. Interv. 2020, 95, 1219–1220. [Google Scholar] [CrossRef]
- Lawin, D.; Lawrenz, T.; Marx, K.; Danielsmeier, N.B.; Poudel, M.R.; Stellbrink, C. Gender disparities in alcohol septal ablation for hypertrophic obstructive cardiomyopathy. Heart 2022, 108, 1623–1628. [Google Scholar] [CrossRef]
- Sasahira, Y.; Yamada, R.; Doi, N.; Uemura, S. Urgent percutaneous transluminal septal myocardial ablation for left ventricular outflow tract obstruction exacerbated after surgical aortic valve replacement. Clin. Case Rep. 2021, 9, e04789. [Google Scholar] [CrossRef]
- Buchel, J.; Leibundgut, G.; Badertscher, P.; Kuhne, M.; Krisai, P. Radiofrequency ablation in obstructive hypertrophic cardiomyopathy: A case report. Eur. Heart J. Case Rep. 2024, 8, ytae359. [Google Scholar] [CrossRef]
- Ommen, S.R.; Ho, C.Y.; Asif, I.M.; Balaji, S.; Burke, M.A.; Day, S.M.; Dearani, J.A.; Epps, K.C.; Evanovich, L.; Ferrari, V.A.; et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1239–e1311. [Google Scholar] [CrossRef]
- da Silva Menezes Junior, A.; de Oliveira, A.L.V.; Maia, T.A.; Botelho, S.M. A Narrative Review of Emerging Therapies for Hypertrophic Obstructive Cardiomyopathy. Curr. Cardiol. Rev. 2023, 19, e240323214927. [Google Scholar] [CrossRef]
- Maron, B.J.; Rowin, E.J.; Maron, M.S. Evolution of risk stratification and sudden death prevention in hypertrophic cardiomyopathy: Twenty years with the implantable cardioverter-defibrillator. Heart Rhythm. 2021, 18, 1012–1023. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Shimoda, T.; Shimada, Y.J.; Shimamura, J.; Akita, K.; Yasuda, R.; Takayama, H.; Kuno, T. Alcohol septal ablation versus surgical septal myectomy of obstructive hypertrophic cardiomyopathy: Systematic review and meta-analysis. Eur. J. Cardiothorac. Surg. 2023, 63, ezad043. [Google Scholar] [CrossRef]
- Afanasyev, A.V.; Bogachev-Prokophiev, A.V.; Zheleznev, S.I.; Zalesov, A.S.; Budagaev, S.A.; Shajahmetova, S.V.; Nazarov, V.M.; Demin, I.I.; Sharifulin, R.M.; Pivkin, A.N.; et al. Early post-septal myectomy outcomes for hypertrophic obstructive cardiomyopathy. Asian Cardiovasc. Thorac. Ann. 2022, 30, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Ullah, W.; Warner, E.; Khandait, H.; Sachdeva, S.; Abdalla, A.S.; Shafique, M.; Khan, M.A.; Roomi, S.; Khattak, F.; Alraies, M.C. Septal Myectomy or Alcohol Ablation for Hypertrophic Cardiomyopathy: A Nationwide Inpatient Sample (NIS) Database Analysis. Cardiovasc. Revasc. Med. 2023, 50, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Morrow, A.G.; Reitz, B.A.; Epstein, S.E.; Henry, W.L.; Conkle, D.M.; Itscoitz, S.B.; Redwood, D.R. Operative treatment in hypertrophic subaortic stenosis. Techniques, and the results of pre and postoperative assessments in 83 patients. Circulation 1975, 52, 88–102. [Google Scholar] [CrossRef]
- Tan, T.; Zhu, W.; Ma, J.; Fu, B.; Zeng, X.; Wang, R.; Li, X.; Liu, J.; Zhuang, J.; Chen, J.; et al. Clinical Effect of the Modified Morrow Septal Myectomy Procedure for Biventricular Hypertrophic Cardiomyopathy. Rev. Cardiovasc. Med. 2024, 25, 21. [Google Scholar] [CrossRef]
- Stefano, P.; Argiro, A.; Bacchi, B.; Iannone, L.; Bertini, A.; Zampieri, M.; Cerillo, A.; Olivotto, I. Does a standard myectomy exist for obstructive hypertrophic cardiomyopathy? From the Morrow variations to precision surgery. Int. J. Cardiol. 2023, 371, 278–286. [Google Scholar] [CrossRef]
- Lai, Y.; Guo, H.; Li, J.; Dai, J.; Ren, C.; Wang, Y. Comparison of surgical results in patients with hypertrophic obstructive cardiomyopathy after classic or modified morrow septal myectomy. Medicine 2017, 96, e9371. [Google Scholar] [CrossRef]
- Reis, R.L.; Hannah, H., 3rd; Carley, J.E.; Pugh, D.M. Surgical treatment of idiopathic hypertrophic subaortic stenosis (IHSS). Postoperative results in 30 patients following ventricular septal myotomy and myectomy (Morrow procedure). Circulation 1977, 56, II128–II132. [Google Scholar]
- Quinones, J.A.; DeLeon, S.Y.; Vitullo, D.A.; Hofstra, J.; Cziperle, D.J.; Shenoy, K.P.; Bell, T.J.; Fisher, E.A. Regression of hypertrophic cardiomyopathy after modified Konno procedure. Ann. Thorac. Surg. 1995, 60, 1250–1254. [Google Scholar] [CrossRef]
- Laredo, M.; Khraiche, D.; Raisky, O.; Gaudin, R.; Bajolle, F.; Maltret, A.; Chevret, S.; Bonnet, D.; Vouhe, P.R. Long-term results of the modified Konno procedure in high-risk children with obstructive hypertrophic cardiomyopathy. J. Thorac. Cardiovasc. Surg. 2018, 156, 2285–2294.e2. [Google Scholar] [CrossRef]
- Nguyen, S.N.; Chung, M.M.; Vinogradsky, A.V.; Richmond, M.E.; Zuckerman, W.A.; Goldstone, A.B.; Bacha, E.A. Long-term outcomes of surgery for obstructive hypertrophic cardiomyopathy in a pediatric cohort. JTCVS Open 2023, 16, 726–738. [Google Scholar] [CrossRef]
- Kunkala, M.R.; Schaff, H.V.; Nishimura, R.A.; Abel, M.D.; Sorajja, P.; Dearani, J.A.; Ommen, S.R. Transapical approach to myectomy for midventricular obstruction in hypertrophic cardiomyopathy. Ann. Thorac. Surg. 2013, 96, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Schaff, H.V.; Nishimura, R.A.; Geske, J.B.; Dearani, J.A.; Ommen, S.R. Transapical Septal Myectomy for Hypertrophic Cardiomyopathy with Midventricular Obstruction. Ann. Thorac. Surg. 2021, 111, 836–844. [Google Scholar] [CrossRef]
- Wehman, B.; Ghoreishi, M.; Foster, N.; Wang, L.; D’Ambra, M.N.; Maassel, N.; Maghami, S.; Quinn, R.; Dawood, M.; Fisher, S.; et al. Transmitral Septal Myectomy for Hypertrophic Obstructive Cardiomyopathy. Ann. Thorac. Surg. 2018, 105, 1102–1108. [Google Scholar] [CrossRef]
- Sakaguchi, T.; Totsugawa, T.; Tamura, K.; Hiraoka, A.; Chikazawa, G.; Yoshitaka, H. Minimally invasive trans-mitral septal myectomy for diffuse-type hypertrophic obstructive cardiomyopathy. Gen. Thorac. Cardiovasc. Surg. 2018, 66, 321–326. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, K.; Wang, F.; Yang, Z.; Yang, S.; Wang, C. Enlargement of left ventricular outflow tract using an autologous pericardial patch for anterior mitral valve leaflet and septal myectomy through trans-mitral approach for the treatment of hypertrophic obstructive cardiomyopathy. J. Card. Surg. 2021, 36, 4198–4202. [Google Scholar] [CrossRef]
- Massera, D.; Xia, Y.; Li, B.; Riedy, K.; Swistel, D.G.; Sherrid, M.V. Mitral annular calcification in hypertrophic cardiomyopathy. Int. J. Cardiol. 2022, 349, 83–89. [Google Scholar] [CrossRef]
- Wu, Z.; Nie, C.; Zhu, C.; Meng, Y.; Yang, Q.; Lu, T.; Lu, Z.; Liu, X.; Wang, S. Mitral annular calcification in obstructive hypertrophic cardiomyopathy: Incidence, risk factors, and prognostic value after myectomy. Int. J. Cardiol. 2023, 391, 131266. [Google Scholar] [CrossRef]
- Kuhn, H.; Seggewiss, H.; Gietzen, F.H.; Boekstegers, P.; Neuhaus, L.; Seipel, L. Catheter-based therapy for hypertrophic obstructive cardiomyopathy. First in-hospital outcome analysis of the German TASH Registry. Z. Kardiol. 2004, 93, 23–31. [Google Scholar] [CrossRef]
- Douglas, J.S., Jr. Current state of the roles of alcohol septal ablation and surgical myectomy in the treatment of hypertrophic obstructive cardiomyopathy. Cardiovasc. Diagn. Ther. 2020, 10, 36–44. [Google Scholar] [CrossRef]
- O’Mahony, C.; Mohiddin, S.A.; Knight, C. Alcohol Septal Ablation for the Treatment of Hypertrophic Obstructive Cardiomyopathy. Interv. Cardiol. 2014, 9, 108–114. [Google Scholar] [CrossRef]
- Gragnano, F.; Pelliccia, F.; Guarnaccia, N.; Niccoli, G.; De Rosa, S.; Piccolo, R.; Moscarella, E.; Fabris, E.; Montone, R.A.; Cesaro, A.; et al. Alcohol Septal Ablation in Patients with Hypertrophic Obstructive Cardiomyopathy: A Contemporary Perspective. J. Clin. Med. 2023, 12, 2810. [Google Scholar] [CrossRef] [PubMed]
- Bataiosu, D.R.; Rakowski, H. Septal Reduction Strategies in Hypertrophic Cardiomyopathy—The Scalpel, Catheter, or Wire? JAMA Cardiol. 2022, 7, 538–539. [Google Scholar] [CrossRef]
- Bode, M.F.; Ahmed, A.A.; Baron, S.J.; Labib, S.B.; Gadey, G. The use of MitraClip to prevent posttranscatheter aortic valve replacement left ventricular “suicide”. Catheter. Cardiovasc. Interv. 2021, 97, 369–372. [Google Scholar] [CrossRef]
- Veselka, J.; Faber, L.; Liebregts, M.; Cooper, R.; Januska, J.; Kashtanov, M.; Dabrowski, M.; Hansen, P.R.; Seggewiss, H.; Bonaventura, J.; et al. Alcohol dose in septal ablation for hypertrophic obstructive cardiomyopathy. Int. J. Cardiol. 2021, 333, 127–132. [Google Scholar] [CrossRef]
- Raimondo, C.; Balsam, P. Alternative Management Options for Hypertrophic Cardiomyopathy: Feasible? JACC Case Rep. 2020, 2, 389–391. [Google Scholar] [CrossRef]
- Okutucu, S.; Aytemir, K.; Oto, A. Glue septal ablation: A promising alternative to alcohol septal ablation. JRSM Cardiovasc. Dis. 2016, 5, 2048004016636313. [Google Scholar] [CrossRef]
- Iacob, M.; Pinte, F.; Tintoiu, I.; Cotuna, L.; Caroescu, M.; Popa, A.; Cristian, G.; Goleanu, V.; Greere, V.; Moscaliuc, I.; et al. Microcoil embolisation for ablation of septal hypertrophy in hypertrophic obstructive cardiomyopathy. Kardiol. Pol. 2004, 61, 350–355. [Google Scholar]
- Iacob, M.; Pinte, F.; Tintoiu, I.; Cotuna, L.; Coroescu, M.; Filip, S.; Popa, A.; Cristian, G.; Goleanu, V.; Greere, V.; et al. Microcoil embolization for ablation of septal hypertrophy in hypertrophic obstructive cardiomyopathy. EuroIntervention 2005, 1, 93–97. [Google Scholar] [CrossRef]
- Ates, A.H.; Sener, Y.Z.; Sahiner, M.L.; Kaya, E.B.; Aytemir, K. Single Center Experience of Percutaneous Septal Ablation in Patients with Hypertrophic Cardiomyopathy with A Novel Agent: Polidocanol. Am. J. Cardiol. 2023, 190, 1–7. [Google Scholar] [CrossRef]
- Ates, A.H.; Kivrak, A.; Canpolat, U.; Menemencioglu, C.; Dogan, M.; Coteli, C.; Sahiner, M.L.; Kaya, E.B.; Ozer, N.; Aytemir, K. Polidocanol ablation in midventricular obstructive cardiomyopathy: Novel approach and early outcomes. J. Invasive Cardiol. 2025. [Google Scholar] [CrossRef]
- Banthiya, S.; Check, L.; Atkins, J. Hypertrophic Cardiomyopathy as a Form of Heart Failure with Preserved Ejection Fraction: Diagnosis, Drugs, and Procedures. US Cardiol. 2024, 18, e17. [Google Scholar] [CrossRef] [PubMed]
- Udelson, J.E.; Bonow, R.O.; O’Gara, P.T.; Maron, B.J.; Van Lingen, A.; Bacharach, S.L.; Epstein, S.E. Verapamil prevents silent myocardial perfusion abnormalities during exercise in asymptomatic patients with hypertrophic cardiomyopathy. Circulation 1989, 79, 1052–1060. [Google Scholar] [CrossRef]
- Ommen, S.R.; Mital, S.; Burke, M.A.; Day, S.M.; Deswal, A.; Elliott, P.; Evanovich, L.L.; Hung, J.; Joglar, J.A.; Kantor, P.; et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2020, 142, e558–e631. [Google Scholar] [CrossRef]
- Basile, P.; Monitillo, F.; Santoro, D.; Falco, G.; Carella, M.C.; Khan, Y.; Moretti, A.; Santobuono, V.E.; Memeo, R.; Pontone, G.; et al. Impact on ventricular arrhythmic burden of SGLT2 inhibitors in patients with chronic heart failure evaluated with cardiac implantable electronic device monitoring. J. Cardiol. 2024, 85, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Velicki, L.; Popovic, D.; Okwose, N.C.; Preveden, A.; Tesic, M.; Tafelmeier, M.; Charman, S.J.; Barlocco, F.; MacGowan, G.A.; Seferovic, P.M.; et al. Sacubitril/valsartan for the treatment of non-obstructive hypertrophic cardiomyopathy: An open label randomized controlled trial (SILICOFCM). Eur. J. Heart Fail. 2024, 26, 1361–1368. [Google Scholar] [CrossRef]
- Shimada, Y.J.; Passeri, J.J.; Baggish, A.L.; O’Callaghan, C.; Lowry, P.A.; Yannekis, G.; Abbara, S.; Ghoshhajra, B.B.; Rothman, R.D.; Ho, C.Y.; et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 2013, 1, 480–487. [Google Scholar] [CrossRef]
- Olivotto, I.; Ashley, E.A. INHERIT (INHibition of the renin angiotensin system in hypertrophic cardiomyopathy and the Effect on hypertrophy—A Randomised Intervention Trial with losartan). Glob. Cardiol. Sci. Pract. 2015, 2015, 7. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Axelsson, A.; Russell, M.W.; Zahka, K.; Lever, H.M.; Pereira, A.C.; Colan, S.D.; Margossian, R.; Murphy, A.M.; et al. Valsartan in early-stage hypertrophic cardiomyopathy: A randomized phase 2 trial. Nat. Med. 2021, 27, 1818–1824. [Google Scholar] [CrossRef]
- Coats, C.J.; Pavlou, M.; Watkinson, O.T.; Protonotarios, A.; Moss, L.; Hyland, R.; Rantell, K.; Pantazis, A.A.; Tome, M.; McKenna, W.J.; et al. Effect of Trimetazidine Dihydrochloride Therapy on Exercise Capacity in Patients with Nonobstructive Hypertrophic Cardiomyopathy: A Randomized Clinical Trial. JAMA Cardiol. 2019, 4, 230–235. [Google Scholar] [CrossRef]
- Olivotto, I.; Camici, P.G.; Merlini, P.A.; Rapezzi, C.; Patten, M.; Climent, V.; Sinagra, G.; Tomberli, B.; Marin, F.; Ehlermann, P.; et al. Efficacy of Ranolazine in Patients with Symptomatic Hypertrophic Cardiomyopathy: The RESTYLE-HCM Randomized, Double-Blind, Placebo-Controlled Study. Circ. Heart Fail. 2018, 11, e004124. [Google Scholar] [CrossRef]
- Sherrid, M.V. Drug Therapy for Hypertrophic Cardiomypathy: Physiology and Practice. Curr. Cardiol. Rev. 2016, 12, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, M.; Monda, E.; Vritz, O.; Calila Albert, D.; Rubino, M.; Verrillo, F.; Caiazza, M.; Lioncino, M.; Amodio, F.; Guarnaccia, N.; et al. Medical treatment of patients with hypertrophic cardiomyopathy: An overview of current and emerging therapy. Arch. Cardiovasc. Dis. 2022, 115, 529–537. [Google Scholar] [CrossRef]
- Dicorato, M.M.; Basile, P.; Naccarati, M.L.; Carella, M.C.; Dentamaro, I.; Falagario, A.; Cicco, S.; Forleo, C.; Guaricci, A.I.; Ciccone, M.M.; et al. Predicting New-Onset Atrial Fibrillation in Hypertrophic Cardiomyopathy: A Review. J. Clin. Med. 2025, 14, 2018. [Google Scholar] [CrossRef]
- Garg, L.; Gupta, M.; Sabzwari, S.R.A.; Agrawal, S.; Agarwal, M.; Nazir, T.; Gordon, J.; Bozorgnia, B.; Martinez, M.W. Atrial fibrillation in hypertrophic cardiomyopathy: Prevalence, clinical impact, and management. Heart Fail. Rev. 2019, 24, 189–197. [Google Scholar] [CrossRef]
- McKenna, W.J.; Harris, L.; Rowland, E.; Kleinebenne, A.; Krikler, D.M.; Oakley, C.M.; Goodwin, J.F. Amiodarone for long-term management of patients with hypertrophic cardiomyopathy. Am. J. Cardiol. 1984, 54, 802–810. [Google Scholar] [CrossRef]
- Tendera, M.; Wycisk, A.; Schneeweiss, A.; Polonski, L.; Wodniecki, J. Effect of sotalol on arrhythmias and exercise tolerance in patients with hypertrophic cardiomyopathy. Cardiology 1993, 82, 335–342. [Google Scholar] [CrossRef]
- Miller, C.A.S.; Maron, M.S.; Estes, N.A.M., III; Price, L.L.; Rowin, E.J.; Maron, B.J.; Link, M.S. Safety, Side Effects and Relative Efficacy of Medications for Rhythm Control of Atrial Fibrillation in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2019, 123, 1859–1862. [Google Scholar] [CrossRef]
- Oliveri, F.; Pepe, A.; Bongiorno, A.; Fasolino, A.; Gentile, F.R.; Schirinzi, S.; Colombo, D.; Breviario, F.; Greco, A.; Turco, A.; et al. Hypertrophic Cardiomyopathy and Atrial Fibrillation: A Systematic Review and Meta-analysis of Anticoagulation Strategy. Am. J. Cardiovasc. Drugs 2023, 23, 269–276. [Google Scholar] [CrossRef]
- Castagno, D.; Di Donna, P.; Olivotto, I.; Frontera, A.; Calo, L.; Scaglione, M.; Arretini, A.; Anselmino, M.; Giustetto, C.; De Ferrari, G.M.; et al. Transcatheter ablation for atrial fibrillation in patients with hypertrophic cardiomyopathy: Long-term results and clinical outcomes. J. Cardiovasc. Electrophysiol. 2021, 32, 657–666. [Google Scholar] [CrossRef]
- Rowin, E.J.; Link, M.S.; Maron, M.S.; Maron, B.J. Evolving Contemporary Management of Atrial Fibrillation in Hypertrophic Cardiomyopathy. Circulation 2023, 148, 1797–1811. [Google Scholar] [CrossRef]
- Santangeli, P.; Di Biase, L.; Themistoclakis, S.; Raviele, A.; Schweikert, R.A.; Lakkireddy, D.; Mohanty, P.; Bai, R.; Mohanty, S.; Pump, A.; et al. Catheter ablation of atrial fibrillation in hypertrophic cardiomyopathy: Long-term outcomes and mechanisms of arrhythmia recurrence. Circ. Arrhythm. Electrophysiol. 2013, 6, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.Y.; Dukkipati, S.R.; Neuzil, P.; Anic, A.; Petru, J.; Funasako, M.; Cochet, H.; Minami, K.; Breskovic, T.; Sikiric, I.; et al. Pulsed Field Ablation of Paroxysmal Atrial Fibrillation: 1-Year Outcomes of IMPULSE, PEFCAT, and PEFCAT II. JACC Clin. Electrophysiol. 2021, 7, 614–627. [Google Scholar] [CrossRef]
- Lapenna, E.; Pozzoli, A.; De Bonis, M.; La Canna, G.; Nisi, T.; Nascimbene, S.; Vicentini, L.; Di Sanzo, S.; Del Forno, B.; Schiavi, D.; et al. Mid-term outcomes of concomitant surgical ablation of atrial fibrillation in patients undergoing cardiac surgery for hypertrophic cardiomyopathydagger. Eur. J. Cardiothorac. Surg. 2017, 51, 1112–1118. [Google Scholar] [CrossRef]
- Elliott, P.M.; Gimeno, J.R.; Thaman, R.; Shah, J.; Ward, D.; Dickie, S.; Tome Esteban, M.T.; McKenna, W.J. Historical trends in reported survival rates in patients with hypertrophic cardiomyopathy. Heart 2006, 92, 785–791. [Google Scholar] [CrossRef]
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur. Heart J. 2014, 35, 2010–2020. [Google Scholar] [CrossRef]
- Spirito, P.; Autore, C.; Rapezzi, C.; Bernabo, P.; Badagliacca, R.; Maron, M.S.; Bongioanni, S.; Coccolo, F.; Estes, N.A.; Barilla, C.S.; et al. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation 2009, 119, 1703–1710. [Google Scholar] [CrossRef]
- Ostman-Smith, I.; Wettrell, G.; Keeton, B.; Holmgren, D.; Ergander, U.; Gould, S.; Bowker, C.; Verdicchio, M. Age- and gender-specific mortality rates in childhood hypertrophic cardiomyopathy. Eur. Heart J. 2008, 29, 1160–1167. [Google Scholar] [CrossRef]
- Gimeno, J.R.; Tome-Esteban, M.; Lofiego, C.; Hurtado, J.; Pantazis, A.; Mist, B.; Lambiase, P.; McKenna, W.J.; Elliott, P.M. Exercise-induced ventricular arrhythmias and risk of sudden cardiac death in patients with hypertrophic cardiomyopathy. Eur. Heart J. 2009, 30, 2599–2605. [Google Scholar] [CrossRef]
- Spirito, P.; Bellone, P.; Harris, K.M.; Bernabo, P.; Bruzzi, P.; Maron, B.J. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N. Engl. J. Med. 2000, 342, 1778–1785. [Google Scholar] [CrossRef]
- Nistri, S.; Olivotto, I.; Betocchi, S.; Losi, M.A.; Valsecchi, G.; Pinamonti, B.; Conte, M.R.; Casazza, F.; Galderisi, M.; Maron, B.J.; et al. Prognostic significance of left atrial size in patients with hypertrophic cardiomyopathy (from the Italian Registry for Hypertrophic Cardiomyopathy). Am. J. Cardiol. 2006, 98, 960–965. [Google Scholar] [CrossRef]
- Autore, C.; Bernabo, P.; Barilla, C.S.; Bruzzi, P.; Spirito, P. The prognostic importance of left ventricular outflow obstruction in hypertrophic cardiomyopathy varies in relation to the severity of symptoms. J. Am. Coll. Cardiol. 2005, 45, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Freitas, P.; Ferreira, A.M.; Arteaga-Fernandez, E.; de Oliveira Antunes, M.; Mesquita, J.; Abecasis, J.; Marques, H.; Saraiva, C.; Matos, D.N.; Rodrigues, R.; et al. The amount of late gadolinium enhancement outperforms current guideline-recommended criteria in the identification of patients with hypertrophic cardiomyopathy at risk of sudden cardiac death. J. Cardiovasc. Magn. Reson. 2019, 21, 50. [Google Scholar] [CrossRef]
- Park, Y.M. Updated risk assessments for sudden cardiac death in hypertrophic cardiomyopathy patients with implantable cardioverter-defibrillator. Korean J. Intern. Med. 2023, 38, 7–15. [Google Scholar] [CrossRef]
- Lee, J.M.; Park, H.B.; Song, J.E.; Kim, I.C.; Song, J.H.; Kim, H.; Oh, J.; Youn, J.C.; Hong, G.R.; Kang, S.M. The impact of cardiopulmonary exercise-derived scoring on prediction of cardio-cerebral outcome in hypertrophic cardiomyopathy. PLoS ONE 2022, 17, e0259638. [Google Scholar] [CrossRef]
- Liao, M.T.; Wu, C.K.; Juang, J.J.; Lin, T.T.; Wu, C.C.; Lin, L.Y. Atrial fibrillation and the risk of sudden cardiac arrest in patients with hypertrophic cardiomyopathy—A nationwide cohort study. EClinicalMedicine 2021, 34, 100802. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, H.K.; Lee, S.C.; Kim, J.; Park, J.B.; Hwang, I.C.; Choi, Y.J.; Lee, S.P.; Chang, S.A.; Lee, W.; et al. Supplementary role of left ventricular global longitudinal strain for predicting sudden cardiac death in hypertrophic cardiomyopathy. Eur. Heart J. Cardiovasc. Imaging 2022, 23, 1108–1116. [Google Scholar] [CrossRef]
- Negri, F.; Muser, D.; Driussi, M.; Sanna, G.D.; Mase, M.; Cittar, M.; Poli, S.; De Bellis, A.; Fabris, E.; Puppato, M.; et al. Prognostic role of global longitudinal strain by feature tracking in patients with hypertrophic cardiomyopathy: The STRAIN-HCM study. Int. J. Cardiol. 2021, 345, 61–67. [Google Scholar] [CrossRef]
- Wu, G.; Liu, J.; Wang, S.; Yu, S.; Zhang, C.; Wang, D.; Zhang, M.; Yang, Y.; Kang, L.; Zhao, S.; et al. N-terminal pro-brain natriuretic peptide and sudden cardiac death in hypertrophic cardiomyopathy. Heart 2021, 107, 1576–1583. [Google Scholar] [CrossRef]
- Makavos, G.; Kappaairis, C.; Tselegkidi, M.E.; Karamitsos, T.; Rigopoulos, A.G.; Noutsias, M.; Ikonomidis, I. Hypertrophic cardiomyopathy: An updated review on diagnosis, prognosis, and treatment. Heart Fail. Rev. 2019, 24, 439–459. [Google Scholar] [CrossRef]
- Ramchand, J.; Fava, A.M.; Chetrit, M.; Desai, M.Y. Advanced imaging for risk stratification of sudden death in hypertrophic cardiomyopathy. Heart 2020, 106, 793–801. [Google Scholar] [CrossRef]
- Cardim, N.; Galderisi, M.; Edvardsen, T.; Plein, S.; Popescu, B.A.; D’Andrea, A.; Bruder, O.; Cosyns, B.; Davin, L.; Donal, E.; et al. Role of multimodality cardiac imaging in the management of patients with hypertrophic cardiomyopathy: An expert consensus of the European Association of Cardiovascular Imaging Endorsed by the Saudi Heart Association. Eur. Heart J. Cardiovasc. Imaging 2015, 16, 280. [Google Scholar] [CrossRef] [PubMed]
- Chatur, S.; Hegde, S.M. Monitoring treatment with cardiac myosin inhibitors in symptomatic obstructive hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2023, 38, 424–432. [Google Scholar] [CrossRef]
- Litt, M.J.; Ali, A.; Reza, N. Familial Hypertrophic Cardiomyopathy: Diagnosis and Management. Vasc. Health Risk Manag. 2023, 19, 211–221. [Google Scholar] [CrossRef]
- Mestres, C.A.; Bartel, T.; Sorgente, A.; Muller, S.; Gruner, C.; Dearani, J.; Quintana, E. Hypertrophic obstructive cardiomyopathy: What, when, why, for whom? Eur. J. Cardiothorac. Surg. 2018, 53, 700–707. [Google Scholar] [CrossRef]
- Doliner, B.; Gaddar, H.; Kalil, R.; Postalian, A. Modern Perspectives on Hypertrophic Cardiomyopathy—No One Size Fits All. Tex. Heart Inst. J. 2024, 51, e248423. [Google Scholar] [CrossRef]
- Li, S.; Feng, Z.; Xiao, C.; Wu, Y.; Ye, W. The Establishment of Hypertrophic Cardiomyopathy Diagnosis Model via Artificial Neural Network and Random Decision Forest Method. Mediat. Inflamm. 2022, 2022, 2024974. [Google Scholar] [CrossRef]
- Siontis, K.C.; Abreau, S.; Attia, Z.I.; Barrios, J.P.; Dewland, T.A.; Agarwal, P.; Balasubramanyam, A.; Li, Y.; Lester, S.J.; Masri, A.; et al. Patient-Level Artificial Intelligence-Enhanced Electrocardiography in Hypertrophic Cardiomyopathy: Longitudinal Treatment and Clinical Biomarker Correlations. JACC Adv. 2023, 2, 100582. [Google Scholar] [CrossRef]
- Tison, G.H.; Siontis, K.C.; Abreau, S.; Attia, Z.; Agarwal, P.; Balasubramanyam, A.; Li, Y.; Sehnert, A.J.; Edelberg, J.M.; Friedman, P.A.; et al. Assessment of Disease Status and Treatment Response with Artificial Intelligence-Enhanced Electrocardiography in Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 1032–1034. [Google Scholar] [CrossRef]
- Zargarzadeh, A.; Javanshir, E.; Ghaffari, A.; Mosharkesh, E.; Anari, B. Artificial intelligence in cardiovascular medicine: An updated review of the literature. J. Cardiovasc. Thorac. Res. 2023, 15, 204–209. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Indication | Starting Dose | Maximum Dose | Notes | Side Effects |
|---|---|---|---|---|---|
| Propranolol | Angina and dyspnea in patients with or without LVOTO; rate control, ectopic beats | 40 mg bid | 80 mg bid | Short half time; drug of choice in children | Asthma, bradycardia |
| Metoprolol [20] | Same as propranolol | 50 mg qd | 100 mg bid | Short half time, Not useful in OHCM | bradycardia |
| Bisoprolol [21] | Systolic dysfunction, HF | 1.25 mg qd | 15 mg qd | Not useful in OHCO | Asthma, bradycardia |
| Atenolol | Same as propranolol | 25 mg qd | 150 mg qd | Drug of choice in HCM and hypertension | Hypotension, bradycardia |
| Nadolol | Same as propanolol, reduction in NSVT and SCD when associated with amiodarone | 40 mg qd | 80 mg bid | Reduction of obstruction | Bradycardia, asthma |
| Verapamil [22,25] | Control of ventricular rate, improvement of diastolic filling | 40 mg bid | 240 mg bid | AV conduction decrease, peripheral edema | |
| Diltiazem [22] | Same as Verapamil | 60 mg bid | 180 mg bid | Same as Verapamil | |
| Felodipine | Refractory angina in HCM | 5 mg qd | Useful in microvascular disease | Same as Verapamil | |
| Disopyramid [27,28,29,30] | Reduce LVOTO at rest | 125 mg bid | 250 mg time | QTc prolongation, Anticholinergic effects |
| Drug | Indication | Starting Dose | Maximum Dose | Side Effects |
|---|---|---|---|---|
| Disopyramid [117] | Reduce LVOTO at rest | 125 mg bid | 250 mg time | QTc prolongation, Anticholinergic effects |
| Amiodarone [115] | AF prevention, control of recurrence SVT/VT, ectopic beats | 200 mg qd | 200 mg bid | QTc prolongation, thyroid disease, Pulmonary interstitial disease |
| Sotalol [116] | AF prevention, reduction of ectopic beats | 40 mg bid | 80 mg time |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dicorato, M.M.; Citarelli, G.; Mangini, F.; Alemanni, R.; Albanese, M.; Cicco, S.; Greco, C.A.; Forleo, C.; Basile, P.; Carella, M.C.; et al. Integrative Approaches in the Management of Hypertrophic Cardiomyopathy: A Comprehensive Review of Current Therapeutic Modalities. Biomedicines 2025, 13, 1256. https://doi.org/10.3390/biomedicines13051256
Dicorato MM, Citarelli G, Mangini F, Alemanni R, Albanese M, Cicco S, Greco CA, Forleo C, Basile P, Carella MC, et al. Integrative Approaches in the Management of Hypertrophic Cardiomyopathy: A Comprehensive Review of Current Therapeutic Modalities. Biomedicines. 2025; 13(5):1256. https://doi.org/10.3390/biomedicines13051256
Chicago/Turabian StyleDicorato, Marco Maria, Gaetano Citarelli, Francesco Mangini, Rossella Alemanni, Miriam Albanese, Sebastiano Cicco, Cosimo Angelo Greco, Cinzia Forleo, Paolo Basile, Maria Cristina Carella, and et al. 2025. "Integrative Approaches in the Management of Hypertrophic Cardiomyopathy: A Comprehensive Review of Current Therapeutic Modalities" Biomedicines 13, no. 5: 1256. https://doi.org/10.3390/biomedicines13051256
APA StyleDicorato, M. M., Citarelli, G., Mangini, F., Alemanni, R., Albanese, M., Cicco, S., Greco, C. A., Forleo, C., Basile, P., Carella, M. C., Ciccone, M. M., Guaricci, A. I., & Dentamaro, I. (2025). Integrative Approaches in the Management of Hypertrophic Cardiomyopathy: A Comprehensive Review of Current Therapeutic Modalities. Biomedicines, 13(5), 1256. https://doi.org/10.3390/biomedicines13051256