
Phospholipid-Rich DC-Vesicles with Preserved Immune Fingerprints: A Stable and Scalable Platform for Precision Immunotherapy

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
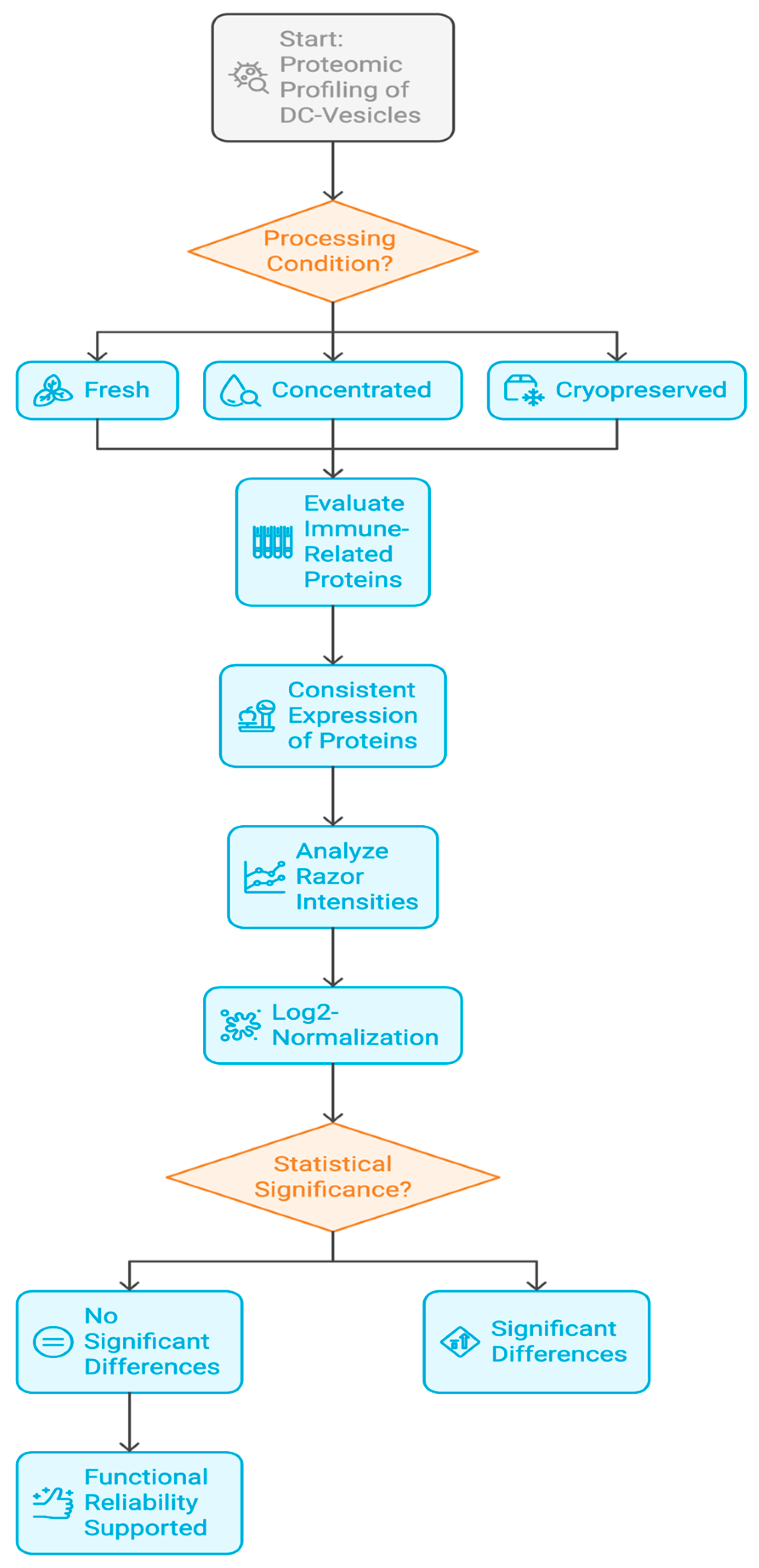
2.1. Experimental Design and Vesicle Preparation
2.2. Protein Extraction and Peptide Preparation
2.3. Mass Spectrometry and Data Acquisition
2.4. Protein Identification and Quantification
2.5. Functional Annotation and Protein Selection
2.6. Statistical Analysis and Visualization
2.7. Proteomic Fingerprinting and Batch Consistency
2.8. In Silico Modeling and Bioinformatic Validation
3. Results
3.1. Global Protein Yield and Identification Across Conditions
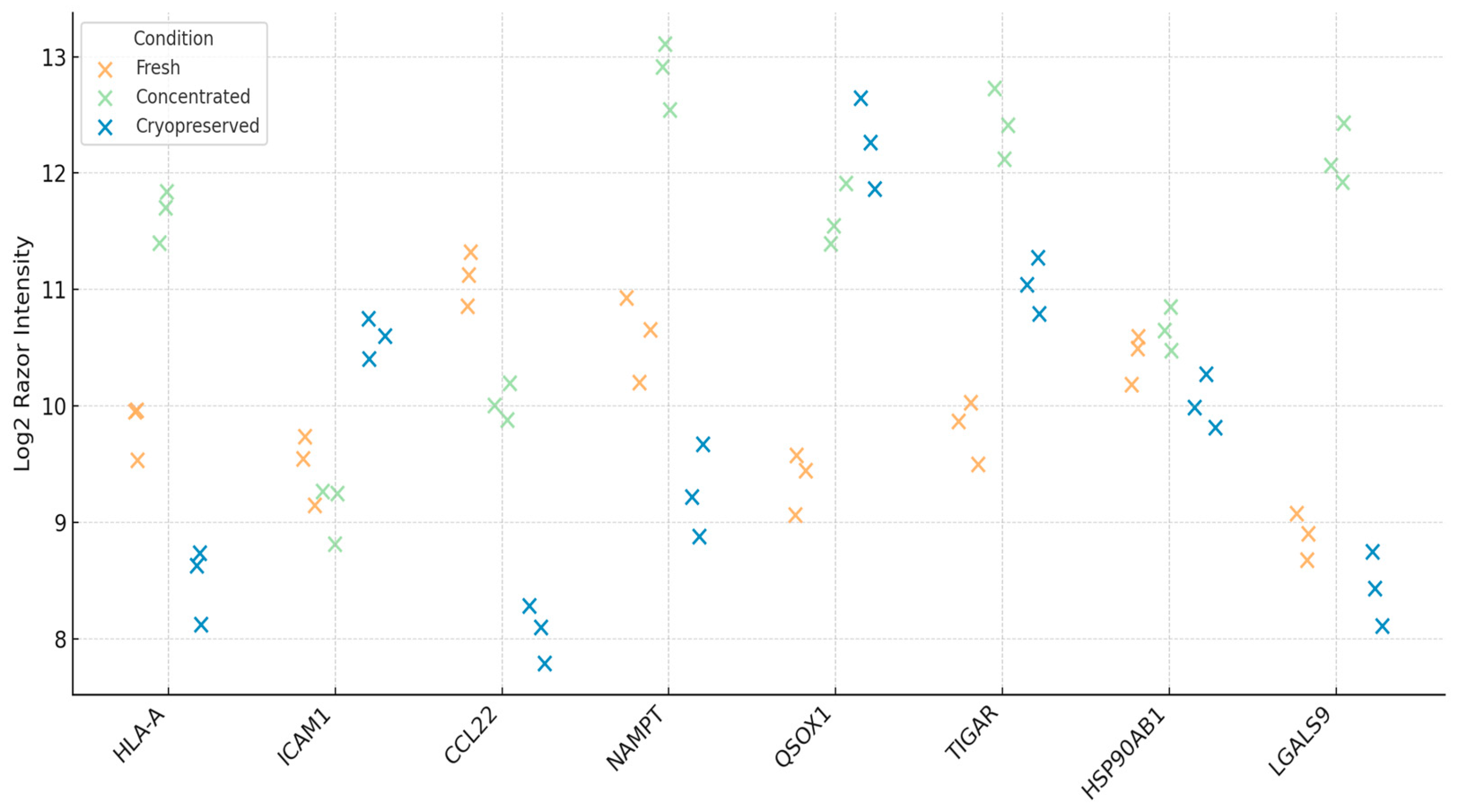
3.2. Functional Preservation of Immune-Relevant Proteins
3.3. Cytokine Modulation Profile Following DC-Vesicle Exposure
3.4. Proteomic Fingerprinting and Reproducibility Analysis
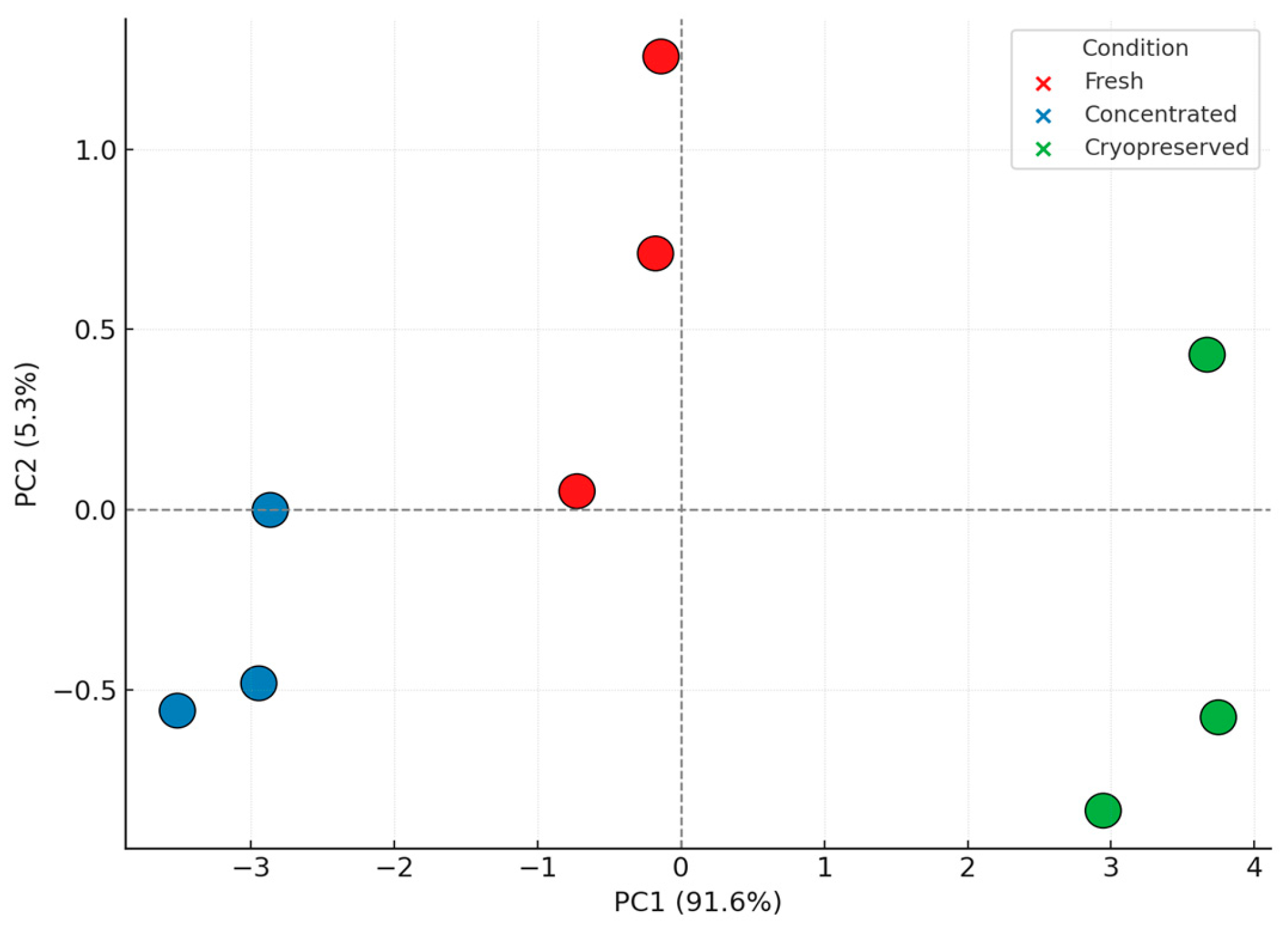
3.5. Principal Component Analysis and Structural Insights
4. Discussion
Limitations and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Abbreviations
| DC | Dendritic cell |
| DC-Vesicles | Dendritic cell-derived vesicles |
| DEX | Dendritic cell-derived exosomes |
| PBMC | Peripheral blood mononuclear cell |
| GMP | Good manufacturing practice |
| IL-4 | Interleukin 4 |
| IL-1β | Interleukin 1 beta |
| IL-10 | Interleukin 10 |
| IL-12 | Interleukin 12 |
| IL-15 | Interleukin 15 |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
| IFN-γ | Interferon gamma |
| CD | Cluster of differentiation |
| CAR-T | Chimeric antigen receptor T cell |
| CRS | Cytokine release syndrome |
| TME | Tumor microenvironment |
| Th1 | Type 1 helper T cell |
| Treg | Regulatory T cell |
| IDO | Indoleamine 2,3-dioxygenase |
| PGE2 | Prostaglandin E2 |
| HLA-A | Human leukocyte antigen A |
| ICAM1 | Intercellular adhesion molecule 1 |
| CCL22 | C-C motif chemokine ligand 22 |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| QSOX1 | Quiescin sulfhydryl oxidase 1 |
| TIGAR | TP53-induced glycolysis and apoptosis regulator |
| HSP90AB1 | Heat shock protein 90-beta |
| LGALS9 | Galectin-9 |
| MHC | Major histocompatibility complex |
| NCE | Non-New Chemical Entity |
| LFQ | Label-Free Quantification |
| PCA | Principal Component Analysis |
| ELISA | Enzyme-linked immunosorbent assay |
| MS/MS | Tandem mass spectrometry |
| nanoLC | Nanoflow liquid chromatography |
| ATMP | Advanced Therapy Medicinal Product |
References
- Petersen, J.D.; Zimmerberg, J. Endothelial cells release microvesicles harboring multivesicular bodies and secrete exosomes. J. Extracell. Biol. 2023, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Emerging targets for cancer therapy. Trends Mol. Med. 2014, 20, 385–393. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Patel, S.A.; Minn, A.J. Combination cancer immunotherapy strategies: Immune checkpoint blockade and beyond. Nat. Rev. Immunol. 2020, 20, 273–287. [Google Scholar]
- Whiteside, T.L. The role of immune cells in the tumor microenvironment. Cancer Treat. Res. 2006, 130, 103–124. [Google Scholar]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting: Implications for combination therapy. Lancet Oncol. 2022, 23, e731–e741. [Google Scholar]
- Finn, O.J. A believer’s overview of cancer immunosurveillance and immunotherapy: The journey to precision medicine. J. Immunol. 2023, 210, 323–332. [Google Scholar]
- Vader, P.; Kooijmans, S.A.; Stremersch, S.; Raemdonck, K. New considerations in the preparation of nucleic acid-loaded extracellular vesicles. Ther. Deliv. 2014, 5, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Geynisman, D.M.; Ajiboye, A.S.; Lorkowski, D.; Perera, S.; Reddy, P.S.; Wong, Y.N. An overview of immune checkpoint inhibitors in the management of metastatic non-small-cell lung cancer. J. Oncol. Pharm. Pract. 2018, 24, 389–399. [Google Scholar]
- Choi, C.; Bae, S.; Lee, J.; Cho, H.D.; Choi, S.Y.; Kim, H.R. Clinical significance of tumor-infiltrating lymphocytes in patients with non-small cell lung cancer. Cancers 2022, 14, 758. [Google Scholar]
- Benoit, A.; Vogin, G.; Duhem, C.; Berchem, G.; Janji, B. Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy. Cells 2023, 12, 1787. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, H.; Wang, Z.; Xu, X.; Jin, Y.; Wu, X.; Xie, Y.; Tang, X. Therapeutic potential of targeting cancer stem cells in cancer immunotherapy. Cancer Cell Int. 2024, 24, 315. [Google Scholar]
- Cao, Y.; Xu, P.; Shen, Y.; Wu, W.; Chen, M.; Wang, F.; Zhu, Y.; Yan, F.; Gu, W.; Lin, Y. Exosomes and cancer immunotherapy: A review of recent cancer research. Front. Oncol. 2023, 12, 1118101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lu, Q.; Kou, D.; Lou, S.; Ashrafizadeh, M.; Aref, A.R.; Canadas, I.; Tian, Y.; Niu, X.; Wang, Y.; Torabian, P.; et al. Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy. J. Hematol. Oncol. 2024, 17, 16. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nie, L.; Ma, J.; Yu, Y.; Tao, Y.; Song, Z.; Li, J. Exosomes as carriers to stimulate an anti-cancer immune response in immunotherapy and as predictive markers. Biochem. Pharmacol. 2025, 232, 116699. [Google Scholar] [CrossRef]
- Gutierrez-Sandoval, R.; Gutiérrez-Castro, F.; Muñoz-Godoy, N.; Rivadeneira, I.; Sobarzo, A.; Iturra, J.; Krakowiak, F.; Alarcón, L.; Dorado, W.; Lagos, A.; et al. Beyond Exosomes: An Ultrapurified Phospholipoproteic Complex (PLPC) as a Scalable Immunomodulatory Platform for Reprogramming Immune Suppression in Metastatic Cancer. Cancers 2025, 17, 1658. [Google Scholar] [CrossRef]
- Lyu, C.; Sun, H.; Sun, Z.; Liu, Y.; Wang, Q. Roles of exosomes in immunotherapy for solid cancers. Cell Death Dis. 2024, 15, 106. [Google Scholar] [CrossRef] [PubMed]
- Pi, Y.-N.; Xia, B.-R.; Jin, M.-Z.; Jin, W.-L.; Lou, G. Exosomes: Powerful weapon for cancer nano-immunoengineering. Biochem. Pharmacol. 2021, 186, 114487. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jiao, J.; Ke, H.; Ouyang, W.; Wang, L.; Pan, J.; Li, X. Role of exosomes in the development of the immune microenvironment in hepatocellular carcinoma. Front. Immunol. 2023, 14, 1200201. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Han, W.; Dong, H.; Liu, X.; Su, X. The rising roles of exosomes in the tumor microenvironment reprogramming and cancer immunotherapy. MedComm 2024, 5, e541. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, S.; Li, X.; Zhang, Y.; Cao, P.; Zhang, X. Advances in immune checkpoint inhibitors and their combination strategies in cancer therapy. J. Immunother. Cancer 2024, 12, e00582. [Google Scholar]
- Ho, T.; Msallam, R. Tissues and Tumor Microenvironment (TME) in 3D: Models to Shed Light on Immunosuppression in Cancer. Cells 2021, 10, 831. [Google Scholar] [CrossRef]
- Barrera, E.; Medina, R.; Mendoza, R.; Salinas, P.; Aguirre, J.; Paredes, M. Inmunoterapia en linfomas: Una nueva era en su tratamiento. Rev. Hematol. Argent. 2020, 46, 174–182. [Google Scholar]
- Yu, L.; Xu, L.; Chen, Y.; Rong, Y.; Zou, Y.; Ge, S.; Wu, T.; Lai, Y.; Xu, Q.; Guo, W.; et al. IDO1 inhibition promotes activation of tumor-intrinsic STAT3 pathway and induces adverse tumor-protective effects. J. Immunol. 2024, 212, 1232–1243. [Google Scholar] [CrossRef]
- Sosnowska, A.; Chlebowska-Tuz, J.; Matryba, P.; Pilch, Z.; Greig, A.; Wolny, A.; Grzywa, T.M.; Rydzynska, Z.; Sokolowska, O.; Rygiel, T.P.; et al. Inhibition of arginase modulates T-cell response in the tumor microenvironment of lung carcinoma. Oncoimmunology 2021, 10, 1956143. [Google Scholar] [CrossRef]
- Lorentzen, C.L.; Martinenaite, E.; Kjeldsen, J.W.; Holmstroem, R.B.; Mørk, S.K.; Pedersen, A.W.; Ehrnrooth, E.; Andersen, M.H.; Svane, I.M. Arginase-1 targeting peptide vaccine in patients with metastatic solid tumors—A phase I trial. Front. Immunol. 2022, 13, 1023023. [Google Scholar] [CrossRef]
- Martin-Orozco, E.; Sanchez-Fernandez, A.; Ortiz-Parra, I.; Ayala-San Nicolas, M. WNT signaling in tumors: The way to evade drugs and immunity. Front. Immunol. 2019, 10, 2854. [Google Scholar] [CrossRef] [PubMed]
- Dillen, A.; Bui, I.; Jung, M.; Agioti, S.; Zaravinos, A.; Bonavida, B. Regulation of PD-L1 expression by YY1 in cancer: Therapeutic efficacy of targeting YY1. Cancers 2024, 16, 1237. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Zhu, Y.; Zhang, H.; Gao, L.; Zhang, Z.; Xu, X.; Ying, L.; Zhang, L.; Li, Y.; Yun, Z.; et al. Open-source throttling of CD8+ T cells in brain with low-intensity focused ultrasound-guided sequential delivery of CXCL10, IL-2, and aPD-L1 for glioblastoma immunotherapy. Adv. Mater. 2024, 36, e2407235. [Google Scholar] [CrossRef] [PubMed]
- Bence, C.; Hofman, V.; Chamorey, E.; Long-Mira, E.; Lassalle, S.; Albertini, A.; Liolios, I.; Zahaf, K.; Picard, A.; Montaudié, H.; et al. Association of combined PD-L1 expression and tumour-infiltrating lymphocyte features with survival and treatment outcomes in patients with metastatic melanoma. J. Dermatol. Venereol. 2020, 34, 984–994. [Google Scholar] [CrossRef]
- Bacolod, M.D.; Barany, F.; Pilones, K.; Fisher, P.B.; de Castro, R.J. Pathways- and epigenetic-based assessment of relative immune infiltration in various types of solid tumors. Adv. Cancer Res. 2019, 142, 107–143. [Google Scholar]
- Dai, E.; Zhu, Z.; Wahed, S.; Qu, Z.; Storkus, W.J.; Guo, Z.S. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 2021, 20, 171. [Google Scholar] [CrossRef]
- Gomez, S.; Tabernacki, T.; Kobyra, J.; Roberts, P.; Chiappinelli, K.B. Combining epigenetic and immune therapy to overcome cancer resistance. Semin. Cancer Biol. 2020, 65, 99–113. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2020, 527, 249–253. [Google Scholar] [CrossRef]
- Qin, X.H.; Wang, H.-X.; Ma, L.; Shen, J.; Liu, Q.-H.; Xue, L. Knockout of the Placenta Specific 8 Gene affects the proliferation and migration of human embryonic kidney 293T cell. Cell Biochem. Biophys. 2020, 78, 55–64. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Z.; Yu, Q.; Chen, Y.; Sun, Y.; Zhu, Q.; Yang, J.; Jiang, R. PLAC8 is an innovative biomarker for immunotherapy participating in remodeling the immune microenvironment of renal clear cell carcinoma. Front. Oncol. 2023, 13, 1207551. [Google Scholar] [CrossRef]
- Singh, S.; Kachhawaha, K.; Singh, S.K. Comprehensive approaches to preclinical evaluation of monoclonal antibodies and their next-generation derivatives. Biochem. Pharmacol. 2024, 225, 116303. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed]
- Notarbartolo, S.; Abrignani, S. Human T lymphocytes at tumor sites. Scand. J. Immunol. 2022, 44, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Beumer-Chuwonpad, A.; Taggenbrock, R.L.R.E.; Ngo, T.A.; van Gisbergen, K.P.J.M. The potential of tissue-resident memory T cells for adoptive immunotherapy against cancer. Cells 2021, 10, 2234. [Google Scholar] [CrossRef]
- Micevic, G.; Thakral, D.; McGeary, M.; Bosenberg, M.W. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. 2019, 32, 435–440. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Micevic, G.; Bosenberg, M.W.; Yan, Q. The crossroads of cancer epigenetics and immune checkpoint therapy. Clin. Cancer Res. 2023, 29, 1173–1182. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Ferrone, C. Neoadjuvant or adjuvant therapy for resectable or borderline resectable pancreatic cancer: Which is preferred? J. Clin. Oncol. 2020, 38, 1757–1759. [Google Scholar] [CrossRef]
- Principe, D.R.; Korc, M.; Kamath, S.D.; Munshi, H.G.; Rana, A. Trials and tribulations of pancreatic cancer immunotherapy. Cancer Lett. 2021, 504, 1–14. [Google Scholar] [CrossRef]
- Wang, Y.; Duan, M.; Peng, Z.; Fan, R.; He, Y.; Zhang, H.; Xiong, W.; Jiang, W. Advances of DNA Damage Repair-Related Drugs and Combination With Immunotherapy in Tumor Treatment. Front. Immunol. 2022, 13, 854730. [Google Scholar] [CrossRef]
- Mohammadi, S.; Yousefi, F.; Shabaninejad, Z.; Movahedpour, A.; Tehran, M.M.; Shafiee, A.; Moradizarmehri, S.; Hajighadimi, S.; Savardashtaki, A.; Mirzaei, H. Exosomes and cancer: From oncogenic roles to therapeutic applications. IUBMB Life 2020, 72, 724–748. [Google Scholar] [CrossRef] [PubMed]
- Pourhanifeh, M.H.; Mahjoubin-Tehran, M.; Shafiee, A.; Hajighadimi, S.; Moradizarmehri, S.; Mirzaei, H.; Asemi, Z. MicroRNAs and exosomes: Small molecules with big actions in multiple myeloma pathogenesis. IUBMB Life 2020, 72, 314–333. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Xu, S. Advances in personalized neoantigen vaccines for cancer immunotherapy. Biosci. Trends 2020, 14, 349–353. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Singh, K.; Ritchie, T.M.; Aguiar, J.A.; Fan, I.Y.; Portillo, A.L.; Rojas, E.A.; Vahedi, F.; El-Sayes, A.; Xing, S.; et al. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 2021, 33, 1205–1220.e5. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Liu, Y.; Zuckier, L.S.; Ghesani, N.V. Lipid metabolism as a target for cancer drug resistance. Front. Pharmacol. 2023, 14, 1274335. [Google Scholar]
- An, Q.; Lin, R.; Wang, D.; Wang, C. Emerging roles of fatty acid metabolism in cancer and their targeted drug development. Eur. J. Med. Chem. 2022, 240, 114613. [Google Scholar] [CrossRef]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, H.; Song, P.; Zhang, M. Constructing a Glioblastoma Prognostic Model Related to Fatty Acid Metabolism Using Machine Learning and Identifying F13A1 as a Potential Target. Biomedicines 2025, 13, 256. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, L.; Che, X.; Wu, G. Discovery of paradoxical genes: Reevaluating the prognostic impact of overexpressed genes in cancer. Front. Cell Dev. Biol. 2025, 13, e1525345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, Z.; Liu, Y.; Pei, J.; Li, R.; Yang, Y. Targeting lipid metabolism: Novel insights and therapeutic advances in pancreatic cancer treatment. Lipids Health Dis. 2025, 24, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Chen, Y.; Lin, J.; Lin, Y.; Song, H.; Huang, W.; Shen, L.; Chen, F.; Liu, F.; Wang, J.; et al. Identification of novel serum lipid metabolism potential markers and metabolic pathways for oral cancer: A population-based study. BMC Cancer 2025, 25, 177. [Google Scholar] [CrossRef]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm 2020, 2, 27–59. [Google Scholar] [CrossRef]
- Fan, T.W.; Higashi, R.M.; Chernayavskaya, Y.; Lane, A.N. Resolving metabolic heterogeneity in experimental models of the tumor microenvironment from a stable isotope resolved metabolomics perspective. Metabolites 2020, 10, 249. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Poeller, P.; Komkova, D.; Röhrig, F.; Schlicker, L.; Winkelkotte, A.M.; Chaves-Filho, A.B.; Al-Shami, K.M.; Caballero, C.D.; Koltsaki, I.; et al. Targeting aldolase A in hepatocellular carcinoma leads to imbalanced glycolysis and energy stress due to uncontrolled FBP accumulation. Nat. Metab. 2025, 7, 348–366. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gašparović, A.Č.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Update 2020, 49, 100670. [Google Scholar] [CrossRef]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, M.; Su, J.; Li, Y.; Long, J.; Chu, J.; Wan, X.; Cao, Y.; Li, Q. Lipid Metabolism and Cancer. Life 2022, 12, 784. [Google Scholar] [CrossRef]
- Xu, D.; Shao, F.; Bian, X.; Meng, Y.; Liang, T.; Lu, Z. The Evolving Landscape of Noncanonical Functions of Metabolic Enzymes in Cancer and Other Pathologies. Cell Metab. 2021, 33, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Li, X.; Shao, F.; Lv, G.; Lv, H.; Lee, J.-H.; Qian, X.; Wang, Z.; Xia, Y.; Du, L.; et al. The protein kinase activity of fructokinase A specifies the antioxidant responses of tumor cells by phosphorylating p62. Sci. Adv. 2019, 5, eaav4570. [Google Scholar] [CrossRef] [PubMed]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells—Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xie, F.; Yang, Y.; Wang, S. Reprogramming of fatty acid metabolism in breast cancer: A narrative review. Transl. Breast Cancer Res. 2021, 2, 5. [Google Scholar] [CrossRef]
- Gong, Y.; Ji, P.; Yang, Y.-S.; Xie, S.; Yu, T.-J.; Xiao, Y.; Jin, M.-L.; Ma, D.; Guo, L.-W.; Pei, Y.-C.; et al. Metabolic-Pathway-Based Subtyping of Triple-Negative Breast Cancer Reveals Potential Therapeutic Targets. Cell Metab. 2021, 33, 51–64.e9. [Google Scholar] [CrossRef]
- Chen, Y.; Li, K.; Gong, D.; Zhang, J.; Li, Q.; Zhao, G.; Lin, P. ACLY: A biomarker of recurrence in breast cancer. Pathol. Res. Pract. 2020, 216, 153076. [Google Scholar] [CrossRef]
- Yang, J.; Wang, L.; Jia, R. Role of de novo cholesterol synthesis enzymes in cancer. J. Cancer 2020, 11, 1761–1767. [Google Scholar] [CrossRef]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef]
- Saito, R.F.; Andrade, L.N.d.S.; Bustos, S.O.; Chammas, R. Phosphatidylcholine-Derived Lipid Mediators: The Crosstalk Between Cancer Cells and Immune Cells. Front. Immunol. 2022, 13, 768606. [Google Scholar] [CrossRef]
- Saito, R.F.; Rangel, M.C.; Halman, J.R.; Chandler, M.; Andrade, L.N.d.S.; Odete-Bustos, S.; Furuya, T.K.; Carrasco, A.G.M.; Chaves-Filho, A.B.; Yoshinaga, M.Y.; et al. Simultaneous Silencing of Lysophosphatidylcholine Acyltransferases 1–4 by Nucleic Acid Nanoparticles (NANPs) Improves Radiation Response of Melanoma Cells. Nanomedicine 2021, 36, 102418. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function | Fold-Change (Cryo vs. Fresh) | Adjusted p-Value | Functional Note |
|---|---|---|---|---|
| HLA-A | Antigen presentation (MHC I) | −0.28 | 0.074 | Activates CD8+ T cells |
| ICAM1 | Co-stimulation/adhesion | −0.12 | 0.268 | Enhances immune synapse |
| CCL22 | Treg recruitment | +0.35 | 0.112 | Chemotactic signal for Tregs |
| NAMPT | NAD + metabolism | −0.42 | 0.056 | Redox-linked immunoregulator |
| QSOX1 | Protein folding/redox enzyme | −0.05 | 0.685 | Enhances disulfide bond formation |
| HSP90AB1 | Molecular chaperone | −0.09 | 0.324 | Stabilizes unfolded proteins under stress |
| TIGAR | p53-regulated glycolysis | −0.61 | 0.033 | Controls ROS and glycolytic flux |
| LGALS9 | Galectin-9 (Th1/Th2 modulator) | −1.25 | 0.027 | Involved in immune polarization |
| Combination Strategy | DC-Vesicle Role | Potentiated Immune Effects | Clinical Rationale |
|---|---|---|---|
| DC-Vesicles + PD-1/PD-L1 inhibitors | Immune priming/checkpoint sensitization | CD8+ activation, Treg suppression | Improves checkpoint efficacy in cold tumors |
| DC-Vesicles + CAR-T cells | TME remodeling/metabolic modulation | Enhanced CAR-T infiltration and persistence | Overcomes TME resistance and immunometabolic suppression |
| DC-Vesicles + IL-12 mimetics | Cytokine delivery vector | Amplified Th1 cytokine signature | Sustains pro-inflammatory tone during blockade therapy |
| DC-Vesicles + Tumor Vaccines | Antigen presentation/adjuvant | Increased APC-T cell cross-talk | Augments vaccine-induced immunogenicity |
| Conference | Year | Location | Code | Title of the Work |
|---|---|---|---|---|
| ESMO | 2024 | Geneva, Switzerland | FPN Code: 60P | Innovative applications of neoantigens in dendritic cell-derived exosome therapy |
| ESMO | 2025 | Geneva, Switzerland | FPN Code: 61P | Optimized protocol for accelerated production of DEX |
| SITC | 2025 | San Diego, CA, USA | FPN Code: 42 | PLPC: A multifunctional bioactive platform for TME reprogramming |
| SITC | 2025 | San Diego, CA, USA | FPN Code: 43 | Precision Engineered Dendritic Vesicles |
| SITC | 2025 | San Diego, CA, USA | FPN Code: 44 | Lyophilized Dendritic Exosomes |
| ASCO | 2025 | Chicago, IL, USA | Abstract #e14522 | Disruptive advances in exosome lyophilization |
| ASCO | 2025 | Chicago, IL, USA | Abstract #e14537 | Decoding NAMPT and TIGAR |
| ASCO | 2025 | Chicago, IL, USA | Abstract #e14511 | PLP-driven exosomal breakthroughs |
| ASCO | 2025 | Chicago, IL, USA | Abstract #e14512 | PLP-powered exosomal therapeutics |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutierrez-Sandoval, R.; Gutierrez-Castro, F.; Muñoz-Godoy, N.; Rivadeneira, I.; Sobarzo, A.; Alarcón, L.; Dorado, W.; Lagos, A.; Montenegro, D.; Muñoz, I.; et al. Phospholipid-Rich DC-Vesicles with Preserved Immune Fingerprints: A Stable and Scalable Platform for Precision Immunotherapy. Biomedicines 2025, 13, 1299. https://doi.org/10.3390/biomedicines13061299
Gutierrez-Sandoval R, Gutierrez-Castro F, Muñoz-Godoy N, Rivadeneira I, Sobarzo A, Alarcón L, Dorado W, Lagos A, Montenegro D, Muñoz I, et al. Phospholipid-Rich DC-Vesicles with Preserved Immune Fingerprints: A Stable and Scalable Platform for Precision Immunotherapy. Biomedicines. 2025; 13(6):1299. https://doi.org/10.3390/biomedicines13061299
Chicago/Turabian StyleGutierrez-Sandoval, Ramon, Francisco Gutierrez-Castro, Natalia Muñoz-Godoy, Ider Rivadeneira, Adolay Sobarzo, Luis Alarcón, Wilson Dorado, Andy Lagos, Diego Montenegro, Ignacio Muñoz, and et al. 2025. "Phospholipid-Rich DC-Vesicles with Preserved Immune Fingerprints: A Stable and Scalable Platform for Precision Immunotherapy" Biomedicines 13, no. 6: 1299. https://doi.org/10.3390/biomedicines13061299
APA StyleGutierrez-Sandoval, R., Gutierrez-Castro, F., Muñoz-Godoy, N., Rivadeneira, I., Sobarzo, A., Alarcón, L., Dorado, W., Lagos, A., Montenegro, D., Muñoz, I., Aguilera, R., Iturra, J., Krakowiak, F., Peña-Vargas, C., & Toledo, A. (2025). Phospholipid-Rich DC-Vesicles with Preserved Immune Fingerprints: A Stable and Scalable Platform for Precision Immunotherapy. Biomedicines, 13(6), 1299. https://doi.org/10.3390/biomedicines13061299