

Molecular Biomarkers of Glioma


 , , , , ,
, , , , , 
Abstract

1. Introduction
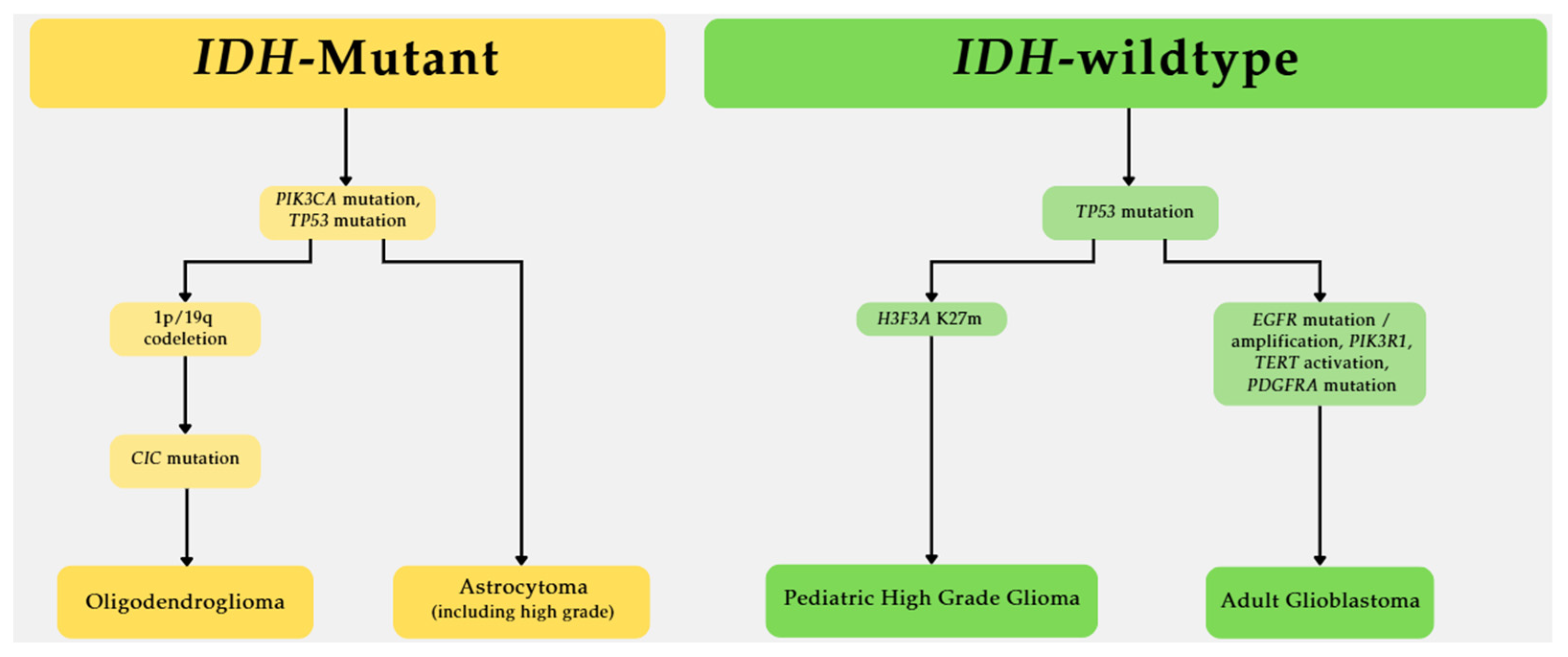
2. IDH1 and IDH2 Mutations Are Crucial Biomarkers for Glioma
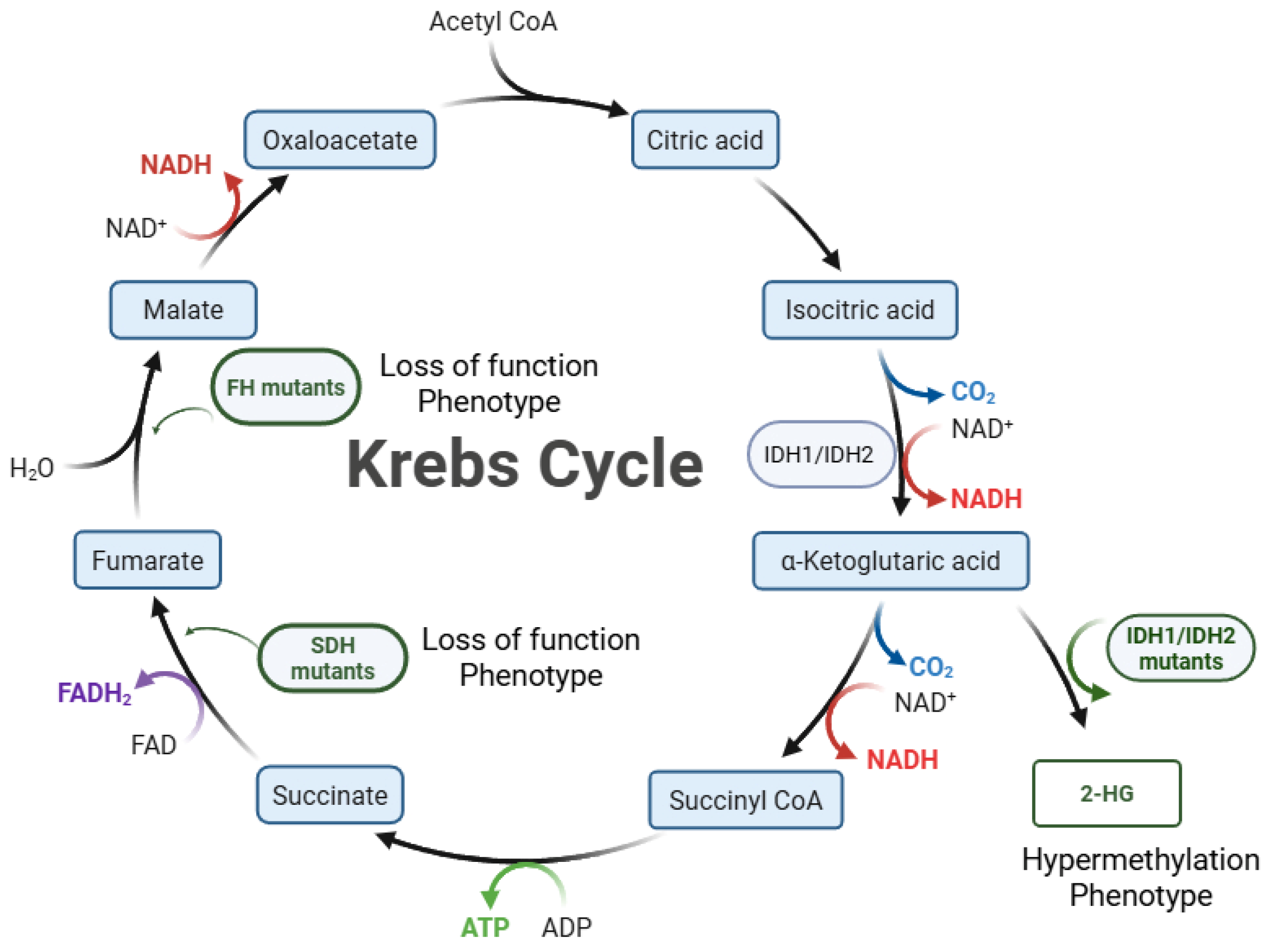
2.1. The Frequency of IDH Mutations in Glioma
2.2. IDH Mutations Associated with Immature Glial Differentiation Programs
2.3. IDH Mutation and Prognosis in Glioma
2.4. Association Between Drug Efficacy and IDH Mutation
3. Molecular Markers on the PI3K Pathway in Glioma
3.1. PTEN (Phosphatase and TENsin) Loss of Function Mutations in Glioblastoma
3.2. PIK3CA-Activating Mutations in Glioma
3.3. PIK3R1 Mutations in Glioblastoma
4. Biomarkers on the RAS Pathway in Glioma
The Capicua Transcriptional Repressor as a Glioma Biomarker on the Ras Pathway
5. TP53 as a Glioma Biomarker
6. LRP1B Is a Key Tumor Suppressor in Glioma
7. The SMARCB1 Gene as a Glioma Biomarker
8. TERT Mutations Drive Telomerase in Glioma
9. KMT2C Gene Mutation and Glioma
10. H3F3A Is a Biomarker for Pediatric Glioma
11. Wnt Pathway as Glioma Biomarkers
11.1. The Wnt Pathway in Glioblastoma
11.2. CTNNB1 Mutations Are Rare in Glioblastoma
11.3. Prognosis
11.4. The Impact of the Wnt Pathway on Drug Efficacy in Glioblastoma
12. The NOTCH Pathway Is Commonly Activated in Glioma
13. Future Directions
14. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro Oncol. 2023, 25, iv1–iv99. [Google Scholar] [CrossRef] [PubMed]
- Price, M.; Neff, C.; Nagarajan, N.; Kruchko, C.; Waite, K.A.; Cioffi, G.; Cordeiro, B.B.; Willmarth, N.; Penas-Prado, M.; Gilbert, M.R.; et al. CBTRUS Statistical Report: American Brain Tumor Association & NCI Neuro-Oncology Branch Adolescent and Young Adult Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro Oncol. 2024, 26, iii1–iii53. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Goncalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I., Jr.; Jahromi, M.S.; Xekouki, P.; et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef]
- Cho, U.; Yang, S.H.; Yoo, C. Estimation of the occurrence rates of IDH1 and IDH2 mutations in gliomas and the reconsideration of IDH-wildtype anaplastic astrocytomas: An institutional experience. J. Int. Med. Res. 2021, 49, 3000605211019258. [Google Scholar] [CrossRef]
- Stancheva, G.; Goranova, T.; Laleva, M.; Kamenova, M.; Mitkova, A.; Velinov, N.; Poptodorov, G.; Mitev, V.; Kaneva, R.; Gabrovsky, N. IDH1/IDH2 but not TP53 mutations predict prognosis in Bulgarian glioblastoma patients. Biomed. Res. Int. 2014, 2014, 654727. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef]
- Krell, D.; Assoku, M.; Galloway, M.; Mulholland, P.; Tomlinson, I.; Bardella, C. Screen for IDH1, IDH2, IDH3, D2HGDH and L2HGDH mutations in glioblastoma. PLoS ONE 2011, 6, e19868. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef]
- Glasgow, S.M.; Carlson, J.C.; Zhu, W.; Chaboub, L.S.; Kang, P.; Lee, H.K.; Clovis, Y.M.; Lozzi, B.E.; McEvilly, R.J.; Rosenfeld, M.G.; et al. Glia-specific enhancers and chromatin structure regulate NFIA expression and glioma tumorigenesis. Nat. Neurosci. 2017, 20, 1520–1528. [Google Scholar] [CrossRef]
- Yamauchi, T.; Ohno, M.; Matsushita, Y.; Takahashi, M.; Miyakita, Y.; Kitagawa, Y.; Kondo, E.; Tsushita, N.; Satomi, K.; Yoshida, A.; et al. Radiological characteristics based on isocitrate dehydrogenase mutations and 1p/19q codeletion in grade II and III gliomas. Brain Tumor Pathol. 2018, 35, 148–158. [Google Scholar] [CrossRef]
- Metellus, P.; Coulibaly, B.; Colin, C.; de Paula, A.M.; Vasiljevic, A.; Taieb, D.; Barlier, A.; Boisselier, B.; Mokhtari, K.; Wang, X.W.; et al. Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol. 2010, 120, 719–729. [Google Scholar] [CrossRef]
- Cabezas-Camarero, S.; Garcia-Barberan, V.; Perez-Alfayate, R.; Casado-Farinas, I.; Sloane, H.; Jones, F.S.; Perez-Segura, P. Detection of IDH1 Mutations in Plasma Using BEAMing Technology in Patients with Gliomas. Cancers 2022, 14, 2891. [Google Scholar] [CrossRef]
- Gatto, L.; Franceschi, E.; Di Nunno, V.; Tosoni, A.; Lodi, R.; Brandes, A.A. Liquid Biopsy in Glioblastoma Management: From Current Research to Future Perspectives. Oncologist 2021, 26, 865–878. [Google Scholar] [CrossRef]
- Choate, K.A.; Pratt, E.P.S.; Jennings, M.J.; Winn, R.J.; Mann, P.B. IDH Mutations in Glioma: Molecular, Cellular, Diagnostic, and Clinical Implications. Biology 2024, 13, 885. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.I.; Brander, T.; Gold, J.; Paul, D.; Klein, P.; Cheng, K.; Liu, J.M.; Marcellino, B.K. Diagnostic and Practical Challenges in Applying National Comprehensive Cancer Network Guidelines for Suspected Pathogenic TP53 Mosaicism. JCO Precis. Oncol. 2024, 8, e2400006. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, X.; Xu, H.; Teschendorff, A.E.; Xu, L.; Li, J.; Fu, M.; Liu, J.; Zhou, H.; Wang, Y.; et al. Integrative analysis of genomic and epigenomic regulation reveals miRNA mediated tumor heterogeneity and immune evasion in lower grade glioma. Commun. Biol. 2024, 7, 824. [Google Scholar] [CrossRef]
- Sharma, N.; Mallela, A.N.; Shi, D.D.; Tang, L.W.; Abou-Al-Shaar, H.; Gersey, Z.C.; Zhang, X.; McBrayer, S.K.; Abdullah, K.G. Isocitrate dehydrogenase mutations in gliomas: A review of current understanding and trials. Neurooncol. Adv. 2023, 5, vdad053. [Google Scholar] [CrossRef]
- Sun, X.; Turcan, S. From Laboratory Studies to Clinical Trials: Temozolomide Use in IDH-Mutant Gliomas. Cells 2021, 10, 1225. [Google Scholar] [CrossRef]
- Turkalp, Z.; Karamchandani, J.; Das, S. IDH mutation in glioma: New insights and promises for the future. JAMA Neurol. 2014, 71, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Cetintas, V.B.; Batada, N.N. Is there a causal link between PTEN deficient tumors and immunosuppressive tumor microenvironment? J. Transl. Med. 2020, 18, 45. [Google Scholar] [CrossRef]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef]
- Koul, D.; Takada, Y.; Shen, R.; Aggarwal, B.B.; Yung, W.K. PTEN enhances TNF-induced apoptosis through modulation of nuclear factor-kappaB signaling pathway in human glioma cells. Biochem. Biophys. Res. Commun. 2006, 350, 463–471. [Google Scholar] [CrossRef]
- Brito, C.; Tomas, A.; Azevedo, A.; Esteves, S.; Mafra, M.; Roque, L.; Pojo, M. PIK3CA Mutations in Diffuse Gliomas: An Update on Molecular Stratification, Prognosis, Recurrence, and Aggressiveness. Clin. Med. Insights Oncol. 2022, 16, 11795549211068804. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Wu, C.; Zhang, J.; Yu, J.; Li, G.; Jiang, H.; Zhang, X.; Yu, R.; Liu, X. Dual blockade of EGFR and PI3K signaling pathways offers a therapeutic strategy for glioblastoma. Cell Commun. Signal. 2023, 21, 363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-F.; Wang, J.; Shao, W.; Wu, C.-P.; Chen, Z.-P.; To, S.-S.T.; Li, W.-P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef]
- Choo, E.F.; Ng, C.M.; Berry, L.; Belvin, M.; Lewin-Koh, N.; Merchant, M.; Salphati, L. PK-PD modeling of combination efficacy effect from administration of the MEK inhibitor GDC-0973 and PI3K inhibitor GDC-0941 in A2058 xenografts. Cancer Chemother. Pharmacol. 2013, 71, 133–143. [Google Scholar] [CrossRef] [PubMed]
- El Meskini, R.; Iacovelli, A.J.; Kulaga, A.; Gumprecht, M.; Martin, P.L.; Baran, M.; Householder, D.B.; Van Dyke, T.; Weaver Ohler, Z. A preclinical orthotopic model for glioblastoma recapitulates key features of human tumors and demonstrates sensitivity to a combination of MEK and PI3K pathway inhibitors. Dis. Model Mech. 2015, 8, 45–56. [Google Scholar] [CrossRef]
- Wen, P.Y.; Lee, E.Q.; Reardon, D.A.; Ligon, K.L.; Alfred Yung, W.K. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro Oncol. 2012, 14, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.W.; Mills, G.B. Targeting therapeutic liabilities engendered by PIK3R1 mutations for cancer treatment. Pharmacogenomics 2016, 17, 297–307. [Google Scholar] [CrossRef]
- Nonoguchi, N.; Ohta, T.; Oh, J.E.; Kim, Y.H.; Kleihues, P.; Ohgaki, H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol. 2013, 126, 931–937. [Google Scholar] [CrossRef]
- Cheung, L.W.; Yu, S.; Zhang, D.; Li, J.; Ng, P.K.; Panupinthu, N.; Mitra, S.; Ju, Z.; Yu, Q.; Liang, H.; et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer Cell 2014, 26, 479–494. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B.; Kamel, A.; Ciubotaru, G.V.; Onose, G.; Sevastre, A.S.; Sfredel, V.; Danoiu, S.; Dricu, A.; Tataranu, L.G. An Overview of EGFR Mechanisms and Their Implications in Targeted Therapies for Glioblastoma. Int. J. Mol. Sci. 2023, 24, 11110. [Google Scholar] [CrossRef] [PubMed]
- Eskilsson, E.; Rosland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 2018, 20, 743–752. [Google Scholar] [CrossRef]
- Rios, S.A.; Oyervides, S.; Uribe, D.; Reyes, A.M.; Fanniel, V.; Vazquez, J.; Keniry, M. Emerging Therapies for Glioblastoma. Cancers 2024, 16, 1485. [Google Scholar] [CrossRef]
- Zou, Y.; Cao, Y.; Yue, Z.; Liu, J. Gamma-secretase inhibitor DAPT suppresses glioblastoma growth via uncoupling of tumor vessel density from vessel function. Clin. Exp. Med. 2013, 13, 271–278. [Google Scholar] [CrossRef]
- Tanaka, S.; Nakada, M.; Yamada, D.; Nakano, I.; Todo, T.; Ino, Y.; Hoshii, T.; Tadokoro, Y.; Ohta, K.; Ali, M.A.; et al. Strong therapeutic potential of gamma-secretase inhibitor MRK003 for CD44-high and CD133-low glioblastoma initiating cells. J. Neurooncol. 2015, 121, 239–250. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar] [CrossRef] [PubMed]
- Tohma, Y.; Gratas, C.; Biernat, W.; Peraud, A.; Fukuda, M.; Yonekawa, Y.; Kleihues, P.; Ohgaki, H. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J. Neuropathol. Exp. Neurol. 1998, 57, 684–689. [Google Scholar] [CrossRef]
- Wang, S.I.; Puc, J.; Li, J.; Bruce, J.N.; Cairns, P.; Sidransky, D.; Parsons, R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997, 57, 4183–4186. [Google Scholar]
- Li, Y.; Liang, Y.; Sun, Z.; Xu, K.; Fan, X.; Li, S.; Zhang, Z.; Jiang, T.; Liu, X.; Wang, Y. Radiogenomic analysis of PTEN mutation in glioblastoma using preoperative multi-parametric magnetic resonance imaging. Neuroradiology 2019, 61, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Hu, R.; Yang, H.; Liu, J.; Sui, J.; Xiang, X.; Wang, F.; Chu, L.; Song, S. PTEN gene mutations correlate to poor prognosis in glioma patients: A meta-analysis. Onco Targets Ther. 2016, 9, 3485–3492. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.B.; Wang, S.; Yang, B.; Jiang, Z.; Lenahan, C.; Wang, J.; Zhang, J.; Shao, A. Transcriptome analyses reveal molecular mechanisms underlying phenotypic differences among transcriptional subtypes of glioblastoma. J. Cell. Mol. Med. 2020, 24, 3901–3916. [Google Scholar] [CrossRef]
- Yang, Y.; Shan, N.; Luo, G.; Li, L.; Zheng, L.; Nilsson-Ehle, P.; Xu, N. Mutations of PTEN Gene in Gliomas Correlate to Tumor Differentiation and Short-term Survival Rate. Anticancer Res. 2010, 30, 981–985. [Google Scholar]
- Scheer, M.; Leisz, S.; Sorge, E.; Storozhuk, O.; Prell, J.; Ho, I.; Harder, A. Neurofibromatosis Type 1 Gene Alterations Define Specific Features of a Subset of Glioblastomas. Int. J. Mol. Sci. 2021, 23, 352. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Butterfield, Y.S.; Morozova, O.; Chittaranjan, S.; Blough, M.D.; An, J.; Birol, I.; Chesnelong, C.; Chiu, R.; Chuah, E.; et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J. Pathol. 2012, 226, 7–16. [Google Scholar] [CrossRef]
- Chen, H.; Lin, F.; Zhang, J.; Lv, X.; Zhou, J.; Li, Z.C.; Chen, Y. Deep Learning Radiomics to Predict PTEN Mutation Status From Magnetic Resonance Imaging in Patients with Glioma. Front. Oncol. 2021, 11, 734433. [Google Scholar] [CrossRef]
- Palande, V.; Siegal, T.; Detroja, R.; Gorohovski, A.; Glass, R.; Flueh, C.; Kanner, A.A.; Laviv, Y.; Har-Nof, S.; Levy-Barda, A.; et al. Detection of gene mutations and gene-gene fusions in circulating cell-free DNA of glioblastoma patients: An avenue for clinically relevant diagnostic analysis. Mol. Oncol. 2022, 16, 2098–2114. [Google Scholar] [CrossRef]
- Lee, D.Y.; Oh, J.S.; Kim, J.W.; Oh, M.; Oh, S.J.; Lee, S.; Kim, Y.H.; Kim, J.H.; Nam, S.J.; Song, S.W.; et al. Pre-operative dual-time-point [(18)F]FET PET differentiates CDKN2A/B loss and PIK3CA mutation status in adult-type diffuse glioma: A single-center prospective study. Eur. J. Nucl. Med. Mol. Imaging 2025, 52, 669–682. [Google Scholar] [CrossRef]
- Weber, G.L.; Parat, M.O.; Binder, Z.A.; Gallia, G.L.; Riggins, G.J. Abrogation of PIK3CA or PIK3R1 reduces proliferation, migration, and invasion in glioblastoma multiforme cells. Oncotarget 2011, 2, 833–849. [Google Scholar] [CrossRef]
- Quayle, S.N.; Lee, J.Y.; Cheung, L.W.; Ding, L.; Wiedemeyer, R.; Dewan, R.W.; Huang-Hobbs, E.; Zhuang, L.; Wilson, R.K.; Ligon, K.L.; et al. Somatic mutations of PIK3R1 promote gliomagenesis. PLoS ONE 2012, 7, e49466. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kang, M.R.; Eom, H.S.; Han, J.Y.; Ahn, C.H.; Kim, S.S.; Lee, S.H.; Yoo, N.J. Somatic mutation of PIK3R1 gene is rare in common human cancers. Acta Oncol. 2010, 49, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Thapa, N.; Chen, M.; Cryns, V.L.; Anderson, R. A p85 isoform switch enhances PI3K activation on endosomes by a MAP4- and PI3P-dependent mechanism. Cell Rep. 2024, 43, 114119. [Google Scholar] [CrossRef] [PubMed]
- Park, A.K.; Kim, P.; Ballester, L.Y.; Esquenazi, Y.; Zhao, Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro Oncol. 2019, 21, 59–70. [Google Scholar] [CrossRef]
- Muench, A.; Teichmann, D.; Spille, D.; Kuzman, P.; Perez, E.; May, S.A.; Mueller, W.C.; Kombos, T.; Nazari-Dehkordi, S.; Onken, J.; et al. A Novel Type of IDH-wildtype Glioma Characterized by Gliomatosis Cerebri-like Growth Pattern, TERT Promoter Mutation, and Distinct Epigenetic Profile. Am. J. Surg. Pathol. 2023, 47, 1364–1375. [Google Scholar] [CrossRef]
- Draaisma, K.; Wijnenga, M.M.; Weenink, B.; Gao, Y.; Smid, M.; Robe, P.; van den Bent, M.J.; French, P.J. PI3 kinase mutations and mutational load as poor prognostic markers in diffuse glioma patients. Acta Neuropathol. Commun. 2015, 3, 88. [Google Scholar] [CrossRef]
- Chen, L.; Yang, L.; Yao, L.; Kuang, X.Y.; Zuo, W.J.; Li, S.; Qiao, F.; Liu, Y.R.; Cao, Z.G.; Zhou, S.L.; et al. Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat. Commun. 2018, 9, 1357. [Google Scholar] [CrossRef]
- Punekar, S.R.; Velcheti, V.; Neel, B.G.; Wong, K.K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Soriano, O.; Alcon-Perez, M.; Vicente-Manzanares, M.; Castellano, E. The Crossroads between RAS and RHO Signaling Pathways in Cellular Transformation, Motility and Contraction. Genes 2021, 12, 819. [Google Scholar] [CrossRef]
- Knobbe, C.B.; Reifenberger, J.; Reifenberger, G. Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta Neuropathol. 2004, 108, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Sherief, M.; Ioannou, M.; Chinnasamy, V.; Chen, L.; Frost, M.; Mattson-Hoss, M.; Sarnoff, H.; Kamson, D.O.; Holdhoff, M.; et al. NF1 expression profiling in IDH-wildtype glioblastoma: Genomic associations and survival outcomes. Acta Neuropathol. Commun. 2024, 12, 172. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Parada, L.F.; Silva, A.J.; Ratner, N. Neurofibromatosis type 1: Modeling CNS dysfunction. J. Neurosci. 2012, 32, 14087–14093. [Google Scholar] [CrossRef]
- Francis, J.M.; Zhang, C.Z.; Maire, C.L.; Jung, J.; Manzo, V.E.; Adalsteinsson, V.A.; Homer, H.; Haidar, S.; Blumenstiel, B.; Pedamallu, C.S.; et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 2014, 4, 956–971. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Suki, D.; Yang, D.; Shi, W.; Aldape, K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 2005, 3, 38. [Google Scholar] [CrossRef]
- Ozawa, T.; Brennan, C.W.; Wang, L.; Squatrito, M.; Sasayama, T.; Nakada, M.; Huse, J.T.; Pedraza, A.; Utsuki, S.; Yasui, Y.; et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010, 24, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Huang, K.; He, X.; Zhang, J.; Ma, Y.; Liu, H. ATRX mutation modifies the DNA damage response in glioblastoma multiforme tumor cells and enhances patient prognosis. Medicine 2025, 104, e41180. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, M.A.; Shah, S.; Eberhart, C.G.; Rodriguez, F.J. Clinicopathologic implications of NF1 gene alterations in diffuse gliomas. Hum. Pathol. 2015, 46, 1323–1330. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef]
- McNulty, S.N.; Schwetye, K.E.; Ferguson, C.; Storer, C.E.; Ansstas, G.; Kim, A.H.; Gutmann, D.H.; Rubin, J.B.; Head, R.D.; Dahiya, S. BRAF mutations may identify a clinically distinct subset of glioblastoma. Sci. Rep. 2021, 11, 19999. [Google Scholar] [CrossRef]
- Prieto, R.; Barrios, L.; Ebrat-Mancilla, E.; Martin, P.; Tejerina, E. The Significance of BRAF Mutation in the Epithelioid Glioblastoma Subtype: A Systematic Literature Review and a Case Report with a Unique Intraventricular Topography. Int. J. Surg. Pathol. 2024, 32, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, Y.; Nagaishi, M. Clinical relevance of BRAF status in glial and glioneuronal tumors: A systematic review. J. Clin. Neurosci. 2019, 66, 196–201. [Google Scholar] [CrossRef]
- Lopez-Gines, C.; Gil-Benso, R.; Ferrer-Luna, R.; Benito, R.; Serna, E.; Gonzalez-Darder, J.; Quilis, V.; Monleon, D.; Celda, B.; Cerda-Nicolas, M. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod. Pathol. 2010, 23, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Higa, N.; Akahane, T.; Yokoyama, S.; Yonezawa, H.; Uchida, H.; Takajo, T.; Otsuji, R.; Hamada, T.; Matsuo, K.; Kirishima, M.; et al. Prognostic impact of PDGFRA gain/amplification and MGMT promoter methylation status in patients with IDH wild-type glioblastoma. Neurooncol. Adv. 2022, 4, vdac097. [Google Scholar] [CrossRef]
- Lo, H.W. EGFR-targeted therapy in malignant glioma: Novel aspects and mechanisms of drug resistance. Curr. Mol. Pharmacol. 2010, 3, 37–52. [Google Scholar] [CrossRef]
- Pan, P.C.; Magge, R.S. Mechanisms of EGFR Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 8471. [Google Scholar] [CrossRef]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Wang, Y.; Ge, P.; Bailey, C.; Zhang, P.; Zhang, D.; Meng, Z.; Qi, C.; Chen, Q.; Chen, J.; et al. The HIF1alpha-PDGFD-PDGFRalpha axis controls glioblastoma growth at normoxia/mild-hypoxia and confers sensitivity to targeted therapy by echinomycin. J. Exp. Clin. Cancer Res. 2021, 40, 278. [Google Scholar] [CrossRef]
- Vuong, H.G.; Altibi, A.M.A.; Duong, U.N.P.; Ngo, H.T.T.; Pham, T.Q.; Fung, K.M.; Hassell, L. BRAF Mutation is Associated with an Improved Survival in Glioma-a Systematic Review and Meta-analysis. Mol. Neurobiol. 2018, 55, 3718–3724. [Google Scholar] [CrossRef]
- Zhu, J.; Ye, J.; Zhang, L.; Xia, L.; Hu, H.; Jiang, H.; Wan, Z.; Sheng, F.; Ma, Y.; Li, W.; et al. Differential Expression of Circular RNAs in Glioblastoma Multiforme and Its Correlation with Prognosis. Transl. Oncol. 2017, 10, 271–279. [Google Scholar] [CrossRef]
- Basindwah, S.; Alkhalidi, H.; Abdelwarith, A.; Elwatidy, S. Ten-year survival in glioblastoma patient with neurofibromatosis type 1: Illustrative case. J. Neurosurg. Case Lessons 2022, 3, CASE21630. [Google Scholar] [CrossRef]
- Gleize, V.; Alentorn, A.; Connen de Kerillis, L.; Labussiere, M.; Nadaradjane, A.A.; Mundwiller, E.; Ottolenghi, C.; Mangesius, S.; Rahimian, A.; Ducray, F.; et al. CIC inactivating mutations identify aggressive subset of 1p19q codeleted gliomas. Ann. Neurol. 2015, 78, 355–374. [Google Scholar] [CrossRef]
- Bunda, S.; Heir, P.; Metcalf, J.; Li, A.S.C.; Agnihotri, S.; Pusch, S.; Yasin, M.; Li, M.; Burrell, K.; Mansouri, S.; et al. CIC protein instability contributes to tumorigenesis in glioblastoma. Nat. Commun. 2019, 10, 661. [Google Scholar] [CrossRef]
- Darabi, S.; Xiu, J.; Samec, T.; Kesari, S.; Carrillo, J.; Aulakh, S.; Walsh, K.M.; Sengupta, S.; Sumrall, A.; Spetzler, D.; et al. Capicua (CIC) mutations in gliomas in association with MAPK activation for exposing a potential therapeutic target. Med. Oncol. 2023, 40, 197. [Google Scholar] [CrossRef]
- Sondka, Z.; Dhir, N.B.; Carvalho-Silva, D.; Jupe, S.; Madhumita, N.; McLaren, K.; Starkey, M.; Ward, S.; Wilding, J.; Ahmed, M.; et al. COSMIC: A curated database of somatic variants and clinical data for cancer. Nucleic Acids Res. 2024, 52, D1210–D1217. [Google Scholar] [CrossRef]
- Zhang, L.; Giuste, F.; Vizcarra, J.C.; Li, X.; Gutman, D. Radiomics Features Predict CIC Mutation Status in Lower Grade Glioma. Front. Oncol. 2020, 10, 937. [Google Scholar] [CrossRef]
- Balana, C.; Castaner, S.; Carrato, C.; Moran, T.; Lopez-Paradis, A.; Domenech, M.; Hernandez, A.; Puig, J. Preoperative Diagnosis and Molecular Characterization of Gliomas with Liquid Biopsy and Radiogenomics. Front. Neurol. 2022, 13, 865171. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Levine, A.J. Spontaneous and inherited TP53 genetic alterations. Oncogene 2021, 40, 5975–5983. [Google Scholar] [CrossRef]
- Lee, Y.J.; Seo, H.W.; Baek, J.H.; Lim, S.H.; Hwang, S.G.; Kim, E.H. Gene expression profiling of glioblastoma cell lines depending on TP53 status after tumor-treating fields (TTFields) treatment. Sci. Rep. 2020, 10, 12272. [Google Scholar] [CrossRef]
- Jha, P.; Suri, V.; Singh, G.; Jha, P.; Purkait, S.; Pathak, P.; Sharma, V.; Sharma, M.C.; Suri, A.; Gupta, D.; et al. Characterization of molecular genetic alterations in GBMs highlights a distinctive molecular profile in young adults. Diagn. Mol. Pathol. 2011, 20, 225–232. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Yang, W.J.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.X.; Liu, Y.H.; You, C.; Mao, Q. Mutant TP53 enhances the resistance of glioblastoma cells to temozolomide by up-regulating O6-methylguanine DNA-methyltransferase. Neurol. Sci. 2013, 34, 1421–1428. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Li, S.; Zhang, W.; Chen, B.; Jiang, T.; Wang, Z. Prognostic and predictive value of p53 in low MGMT expressing glioblastoma treated with surgery, radiation and adjuvant temozolomide chemotherapy. Neurol. Res. 2010, 32, 690–694. [Google Scholar] [CrossRef]
- Garrido-Navas, M.C.; Garcia-Diaz, A.; Molina-Vallejo, M.P.; Gonzalez-Martinez, C.; Alcaide Lucena, M.; Canas-Garcia, I.; Bayarri, C.; Delgado, J.R.; Gonzalez, E.; Lorente, J.A.; et al. The Polemic Diagnostic Role of TP53 Mutations in Liquid Biopsies from Breast, Colon and Lung Cancers. Cancers 2020, 12, 3343. [Google Scholar] [CrossRef]
- Principe, C.; Dionisio de Sousa, I.J.; Prazeres, H.; Soares, P.; Lima, R.T. LRP1B: A Giant Lost in Cancer Translation. Pharmaceuticals 2021, 14, 836. [Google Scholar] [CrossRef]
- Tabouret, E.; Labussiere, M.; Alentorn, A.; Schmitt, Y.; Marie, Y.; Sanson, M. LRP1B deletion is associated with poor outcome for glioblastoma patients. J. Neurol. Sci. 2015, 358, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Ogawa, S.; Kawamata, N.; Tunici, P.; Finocchiaro, G.; Eoli, M.; Ruckert, C.; Huynh, T.; Liu, G.; Kato, M.; et al. High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol. Cancer Res. 2009, 7, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Sergi, C.M. Commentary on: SMARCB1 as a novel diagnostic and prognostic biomarker for osteosarcoma. Biosci. Rep. 2022, 42, BSR20220040. [Google Scholar] [CrossRef]
- Zhang, Z.K.; Davies, K.P.; Allen, J.; Zhu, L.; Pestell, R.G.; Zagzag, D.; Kalpana, G.V. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol. Cell. Biol. 2002, 22, 5975–5988. [Google Scholar] [CrossRef]
- Mukherjee, S.; Stroberg, E.; Wang, F.; Morales, L.; Shan, Y.; Rao, A.; Huang, J.H.; Wu, E.; Fonkem, E. SMARCB1 Gene Mutation Predisposes to Earlier Development of Glioblastoma: A Case Report of Familial GBM. J. Neuropathol. Exp. Neurol. 2020, 79, 562–565. [Google Scholar] [CrossRef]
- Cooper, G.W.; Hong, A.L. SMARCB1-Deficient Cancers: Novel Molecular Insights and Therapeutic Vulnerabilities. Cancers 2022, 14, 3645. [Google Scholar] [CrossRef]
- Stockman, D.L.; Curry, J.L.; Torres-Cabala, C.A.; Watson, I.R.; Siroy, A.E.; Bassett, R.L.; Zou, L.; Patel, K.P.; Luthra, R.; Davies, M.A.; et al. Use of clinical next-generation sequencing to identify melanomas harboring SMARCB1 mutations. J. Cutan. Pathol. 2015, 42, 308–317. [Google Scholar] [CrossRef]
- Choi, S.K.; Kim, M.J.; You, J.S. SMARCB1 Acts as a Quiescent Gatekeeper for Cell Cycle and Immune Response in Human Cells. Int. J. Mol. Sci. 2020, 21, 3969. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, Q.; Gou, M.; Wang, X.; Liu, Y.; Liang, R.; Mao, Y.; Luo, J.; Mao, Q. TERT mutation in glioma: Frequency, prognosis and risk. J. Clin. Neurosci. 2016, 26, 57–62. [Google Scholar] [CrossRef]
- Olympios, N.; Gilard, V.; Marguet, F.; Clatot, F.; Di Fiore, F.; Fontanilles, M. TERT Promoter Alterations in Glioblastoma: A Systematic Review. Cancers 2021, 13, 1147. [Google Scholar] [CrossRef]
- Johanns, T.M.; Fu, Y.; Kobayashi, D.K.; Mei, Y.; Dunn, I.F.; Mao, D.D.; Kim, A.H.; Dunn, G.P. High incidence of TERT mutation in brain tumor cell lines. Brain Tumor Pathol. 2016, 33, 222–227. [Google Scholar] [CrossRef]
- El Zarif, T.; Machaalani, M.; Nawfal, R.; Nassar, A.H.; Xie, W.; Choueiri, T.K.; Pomerantz, M. TERT Promoter Mutations Frequency Across Race, Sex, and Cancer Type. Oncologist 2024, 29, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Labussiere, M.; Boisselier, B.; Mokhtari, K.; Di Stefano, A.L.; Rahimian, A.; Rossetto, M.; Ciccarino, P.; Saulnier, O.; Paterra, R.; Marie, Y.; et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology 2014, 83, 1200–1206. [Google Scholar] [CrossRef]
- Jones, J.J.; Nguyen, H.; Wong, S.Q.; Whittle, J.; Iaria, J.; Stylli, S.; Towner, J.; Pieters, T.; Gaillard, F.; Kaye, A.H.; et al. Plasma ctDNA liquid biopsy of IDH1, TERTp, and EGFRvIII mutations in glioma. Neurooncol. Adv. 2024, 6, vdae027. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Wu, L.J.; Trinh, B.; Tsai, R.Y.L. IMPDH Inhibition Decreases TERT Expression and Synergizes the Cytotoxic Effect of Chemotherapeutic Agents in Glioblastoma Cells. Int. J. Mol. Sci. 2024, 25, 5992. [Google Scholar] [CrossRef]
- Jiao, Y.; Lv, Y.; Liu, M.; Liu, Y.; Han, M.; Xiong, X.; Zhou, H.; Zhong, J.; Kang, X.; Su, W. The modification role and tumor association with a methyltransferase: KMT2C. Front. Immunol. 2024, 15, 1444923. [Google Scholar] [CrossRef]
- Hu, X.; Biswas, A.; De, S. KMT2C-deficient tumors have elevated APOBEC mutagenesis and genomic instability in multiple cancers. NAR Cancer 2022, 4, zcac023. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y.; Wang, M.; Wu, Z.; Li, N.; Zhang, J.; Yang, C. EZH2-, CHD4-, and IDH-linked epigenetic perturbation and its association with survival in glioma patients. J. Mol. Cell Biol. 2017, 9, 477–488. [Google Scholar] [CrossRef]
- Lian, J.; Xu, C.; Chen, X.; Huang, S.; Wu, D. Histone methyltransferase KMT2C plays an oncogenic role in prostate cancer. J. Cancer Res. Clin. Oncol. 2022, 148, 1627–1640. [Google Scholar] [CrossRef]
- Liu, X.; Qiu, R.; Xu, M.; Meng, M.; Zhao, S.; Ji, J.; Yang, Y. KMT2C is a potential biomarker of prognosis and chemotherapy sensitivity in breast cancer. Breast Cancer Res. Treat. 2021, 189, 347–361. [Google Scholar] [CrossRef]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Felsani, A.; Tringali, G.; Cifola, I.; Pozzoli, G.; Cenciarelli, C. Exome sequencing of glioblastoma-derived cancer stem cells reveals rare clinically relevant frameshift deletion in MLLT1 gene. Cancer Cell Int. 2022, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Sarker, A.; Uddin, B.; Ahmmed, R.; Mahmud, S.; Ajadee, A.; Pappu, M.A.A.; Aziz, M.A.; Mollah, M.N.H. Discovery of mutated oncodriver genes associated with glioblastoma originated from stem cells of subventricular zone through whole exome sequence profile analysis, and drug repurposing. Heliyon 2025, 11, e42052. [Google Scholar] [CrossRef] [PubMed]
- Rampias, T.; Karagiannis, D.; Avgeris, M.; Polyzos, A.; Kokkalis, A.; Kanaki, Z.; Kousidou, E.; Tzetis, M.; Kanavakis, E.; Stravodimos, K.; et al. The lysine-specific methyltransferase KMT2C/MLL3 regulates DNA repair components in cancer. EMBO Rep. 2019, 20, e46821. [Google Scholar] [CrossRef]
- Wang, N.; Pachai, M.R.; Li, D.; Lee, C.J.; Warda, S.; Khudoynazarova, M.N.; Cho, W.H.; Xie, G.; Shah, S.R.; Yao, L.; et al. Loss of Kmt2c or Kmt2d primes urothelium for tumorigenesis and redistributes KMT2A-menin to bivalent promoters. Nat. Genet. 2025, 57, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Saratsis, A.M.; Knowles, T.; Petrovic, A.; Nazarian, J. H3K27M mutant glioma: Disease definition and biological underpinnings. Neuro Oncol. 2024, 26, S92–S100. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Aggarwal, P.; Luo, W.; Pehlivan, K.C.; Hoang, H.; Rajappa, P.; Cripe, T.P.; Cassady, K.A.; Lee, D.A.; Cairo, M.S. Pediatric versus adult high grade glioma: Immunotherapeutic and genomic considerations. Front. Immunol. 2022, 13, 1038096. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Santi, M.; Felicella, M.M.; Yarilin, D.; Phillips, J.J.; Sullivan, L.M.; Martinez, D.; Perry, A.; Lewis, P.W.; Thompson, C.B.; et al. A sensitive and specific histopathologic prognostic marker for H3F3A K27M mutant pediatric glioblastomas. Acta Neuropathol. 2014, 128, 743–753. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Hojfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Williams, M.J.; Singleton, W.G.; Lowis, S.P.; Malik, K.; Kurian, K.M. Therapeutic Targeting of Histone Modifications in Adult and Pediatric High-Grade Glioma. Front. Oncol. 2017, 7, 45. [Google Scholar] [CrossRef]
- Chi, A.S.; Tarapore, R.S.; Hall, M.D.; Shonka, N.; Gardner, S.; Umemura, Y.; Sumrall, A.; Khatib, Z.; Mueller, S.; Kline, C.; et al. Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J. Neurooncol. 2019, 145, 97–105. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/beta-catenin signaling in cancers and targeted therapies. Signal Transduct. Target. Ther. 2021, 6, 307. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Ye, T.; Wang, W.; Song, W.; Tan, T. CTNNB1 in neurodevelopmental disorders. Front. Psychiatry 2023, 14, 1143328. [Google Scholar] [CrossRef]
- Nager, M.; Sallan, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macia, A.; Yeramian, A.; Canti, C.; Herreros, J. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef]
- Denysenko, T.; Annovazzi, L.; Cassoni, P.; Melcarne, A.; Mellai, M.; Schiffer, D. WNT/beta-catenin Signaling Pathway and Downstream Modulators in Low- and High-grade Glioma. Cancer Genom. Proteom. 2016, 13, 31–45. [Google Scholar]
- Moreno, D.A.; Bonatelli, M.; Antoniazzi, A.P.; de Paula, F.E.; Leal, L.F.; Garcia, F.A.O.; de Paula, A.E.; Teixeira, G.R.; Santana, I.V.V.; Saggioro, F.; et al. High frequency of WNT-activated medulloblastomas with CTNNB1 wild type suggests a higher proportion of hereditary cases in a Latin-Iberian population. Front. Oncol. 2023, 13, 1237170. [Google Scholar] [CrossRef]
- Coelho, B.P.; Fernandes, C.F.L.; Boccacino, J.M.; Souza, M.; Melo-Escobar, M.I.; Alves, R.N.; Prado, M.B.; Iglesia, R.P.; Cangiano, G.; Mazzaro, G.R.; et al. Multifaceted WNT Signaling at the Crossroads Between Epithelial-Mesenchymal Transition and Autophagy in Glioblastoma. Front. Oncol. 2020, 10, 597743. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/beta-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef]
- Zechner, D.; Fujita, Y.; Hulsken, J.; Muller, T.; Walther, I.; Taketo, M.M.; Crenshaw, E.B., 3rd; Birchmeier, W.; Birchmeier, C. beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev. Biol. 2003, 258, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Chenn, A.; Walsh, C.A. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 2002, 297, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, J.K.; Ahn, S.H.; Lee, J.; Nam, D.H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. 2016, 96, 137–150. [Google Scholar] [CrossRef]
- Liu, C.; Tu, Y.; Sun, X.; Jiang, J.; Jin, X.; Bo, X.; Li, Z.; Bian, A.; Wang, X.; Liu, D.; et al. Wnt/beta-Catenin pathway in human glioma: Expression pattern and clinical/prognostic correlations. Clin. Exp. Med. 2011, 11, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Nager, M.; Bhardwaj, D.; Canti, C.; Medina, L.; Nogues, P.; Herreros, J. beta-Catenin Signalling in Glioblastoma Multiforme and Glioma-Initiating Cells. Chemother. Res. Pract. 2012, 2012, 192362. [Google Scholar] [CrossRef]
- Latour, M.; Her, N.G.; Kesari, S.; Nurmemmedov, E. WNT Signaling as a Therapeutic Target for Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8428. [Google Scholar] [CrossRef]
- Yun, E.J.; Kim, S.; Hsieh, J.T.; Baek, S.T. Wnt/beta-catenin signaling pathway induces autophagy-mediated temozolomide-resistance in human glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef]
- Tyagi, A.; Sharma, A.K.; Damodaran, C. A Review on Notch Signaling and Colorectal Cancer. Cells 2020, 9, 1549. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, Q.; Geng, R.; Liu, H.; Yuan, F.E.; Xu, Y.; Qi, Y.; Jiang, H.; Chen, Q.; Liu, B. Notch intracellular domain regulates glioblastoma proliferation through the Notch1 signaling pathway. Oncol. Lett. 2021, 21, 303. [Google Scholar] [CrossRef]
- Anusewicz, D.; Orzechowska, M.; Bednarek, A.K. Notch Signaling Pathway in Cancer-Review with Bioinformatic Analysis. Cancers 2021, 13, 768. [Google Scholar] [CrossRef]
- Zheng, Z.Q.; Zhang, G.G.; Yuan, G.Q.; Hao, J.H.; Nie, Q.Q.; Zheng, M.C.; Wang, Z. Development and validation of an immune infiltration/tumor proliferation-related Notch3 nomogram for predicting survival in patients with primary glioblastoma. Front. Genet. 2023, 14, 1148126. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Nakamura, H.; Suzuki, H.; Matsuo, K.; Kataoka, K.; Shimamura, T.; Motomura, K.; Ohka, F.; Shiina, S.; Yamamoto, T.; et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2018, 20, 66–77. [Google Scholar] [CrossRef]
- Hai, L.; Zhang, C.; Li, T.; Zhou, X.; Liu, B.; Li, S.; Zhu, M.; Lin, Y.; Yu, S.; Zhang, K.; et al. Notch1 is a prognostic factor that is distinctly activated in the classical and proneural subtype of glioblastoma and that promotes glioma cell survival via the NF-kappaB(p65) pathway. Cell Death Dis. 2018, 9, 158. [Google Scholar] [CrossRef]
- Liu, G.; Bu, C.; Guo, G.; Zhang, Z.; Sheng, Z.; Deng, K.; Wu, S.; Xu, S.; Bu, Y.; Gao, Y.; et al. Molecular and clonal evolution in vivo reveal a common pathway of distant relapse gliomas. iScience 2023, 26, 107528. [Google Scholar] [CrossRef]
- Greenwald, A.C.; Darnell, N.G.; Hoefflin, R.; Simkin, D.; Mount, C.W.; Gonzalez Castro, L.N.; Harnik, Y.; Dumont, S.; Hirsch, D.; Nomura, M.; et al. Integrative spatial analysis reveals a multi-layered organization of glioblastoma. Cell 2024, 187, 2485–2501.E26. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Ren, Y.; Huang, Z.; Zhou, L.; Xiao, P.; Song, J.; He, P.; Xie, C.; Zhou, R.; Li, M.; Dong, X.; et al. Spatial transcriptomics reveals niche-specific enrichment and vulnerabilities of radial glial stem-like cells in malignant gliomas. Nat. Commun. 2023, 14, 1028. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, H.K. Current Understanding of Hypoxia in Glioblastoma Multiforme and Its Response to Immunotherapy. Cancers 2022, 14, 1176. [Google Scholar] [CrossRef] [PubMed]
- Lino, M.M.; Merlo, A.; Boulay, J.L. Notch signaling in glioblastoma: A developmental drug target? BMC Med. 2010, 8, 72. [Google Scholar] [CrossRef]
- Gersey, Z.; Osiason, A.D.; Bloom, L.; Shah, S.; Thompson, J.W.; Bregy, A.; Agarwal, N.; Komotar, R.J. Therapeutic Targeting of the Notch Pathway in Glioblastoma Multiforme. World Neurosurg. 2019, 131, 252–263.e252. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Biomarker | Drug Treatment |
|---|---|
| IDH |
|
| |
| |
| PTEN |
|
| |
| |
| |
| |
| PIK3CA |
|
| |
| |
| |
| |
| PIK3R1 | |
| |
| EGFR | |
| NOTCH1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajakaruna, P.; Rios, S.; Elnahas, H.; Villanueva, A.; Uribe, D.; Leslie, S.; Abbas, W.A.; Barroso, L.; Oyervides, S.; Persans, M.; et al. Molecular Biomarkers of Glioma. Biomedicines 2025, 13, 1298. https://doi.org/10.3390/biomedicines13061298
Rajakaruna P, Rios S, Elnahas H, Villanueva A, Uribe D, Leslie S, Abbas WA, Barroso L, Oyervides S, Persans M, et al. Molecular Biomarkers of Glioma. Biomedicines. 2025; 13(6):1298. https://doi.org/10.3390/biomedicines13061298
Chicago/Turabian StyleRajakaruna, Punsasi, Stella Rios, Hana Elnahas, Ashley Villanueva, David Uribe, Sophia Leslie, Walaa A. Abbas, Larissa Barroso, Stephanie Oyervides, Michael Persans, and et al. 2025. "Molecular Biomarkers of Glioma" Biomedicines 13, no. 6: 1298. https://doi.org/10.3390/biomedicines13061298
APA StyleRajakaruna, P., Rios, S., Elnahas, H., Villanueva, A., Uribe, D., Leslie, S., Abbas, W. A., Barroso, L., Oyervides, S., Persans, M., Innis-Whitehouse, W., & Keniry, M. (2025). Molecular Biomarkers of Glioma. Biomedicines, 13(6), 1298. https://doi.org/10.3390/biomedicines13061298