
Unlocking the Role of OCT4 in Cancer Lineage Plasticity: A Cross-Cancer Perspective with an Emphasis on Prostate Cancer
 , ,
, ,
Abstract
1. Introduction
2. OCT4 in Stemness and Plasticity
2.1. OCT4′s Function in Embryonic Stem Cells and Normal Tissue Development
2.2. OCT4 in Cancer Stem-like Cells and Lineage Plasticity
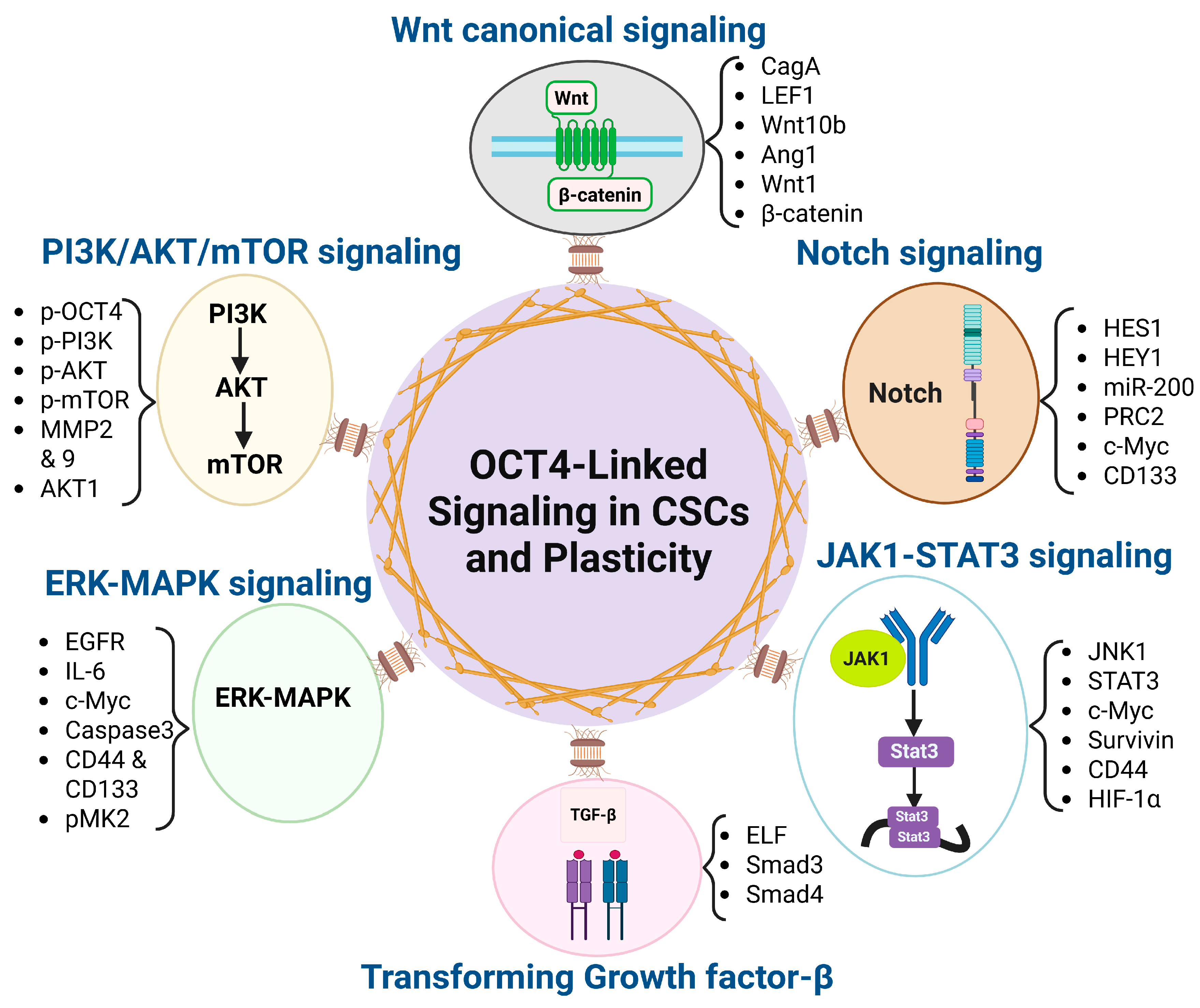
2.3. OCT4-Associated Signaling Pathways in CSC and Plasticity
2.3.1. Wnt/β-Catenin
2.3.2. TGF-β
2.3.3. PI3K/AKT/mTOR
2.3.4. Notch
2.3.5. JAK1-STAT3
2.3.6. ERK-MAPK
3. OCT4 in Prostate Cancer Progression and Lineage Plasticity
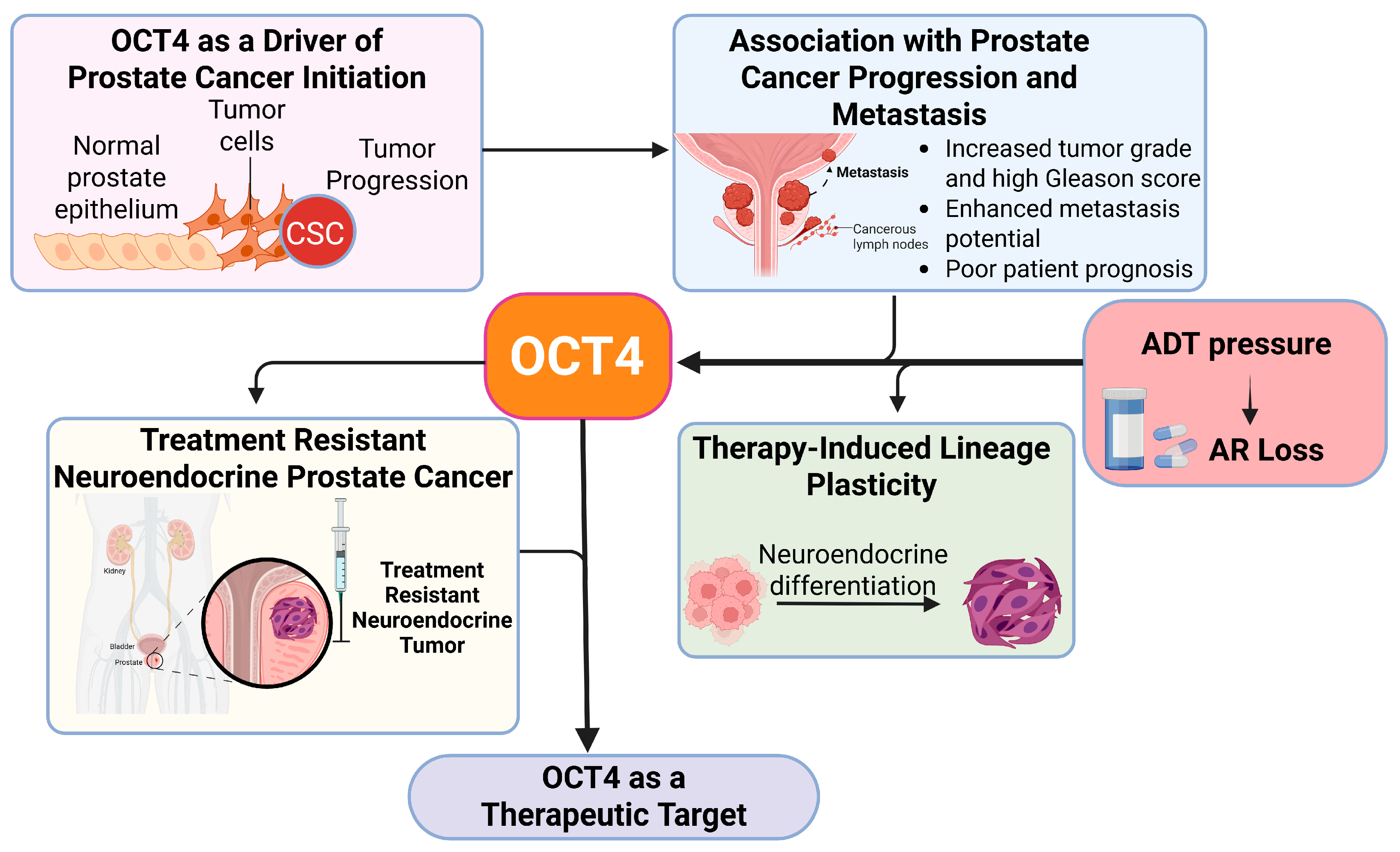
3.1. OCT4 as a Driver of Prostate Cancer Initiation
3.2. Association of OCT4 with Prostate Cancer Progression and Metastasis
- Increased Tumor Grade and High Gleason Score
- Enhanced Metastatic Potential
- Poor Patient Prognosis
3.3. OCT4 and Therapy-Induced Lineage Plasticity in Prostate Cancer
3.3.1. The Role of AR-Targeted Therapy in Driving Stemness and Plasticity
3.3.2. Chromatin Modifications in Driving Stemness and Plasticity in Prostate Cancer Contributing to Drug Resistance
3.4. OCT4 as a Therapeutic Target in Prostate Cancer
4. Glimpse into the Future
- 1.
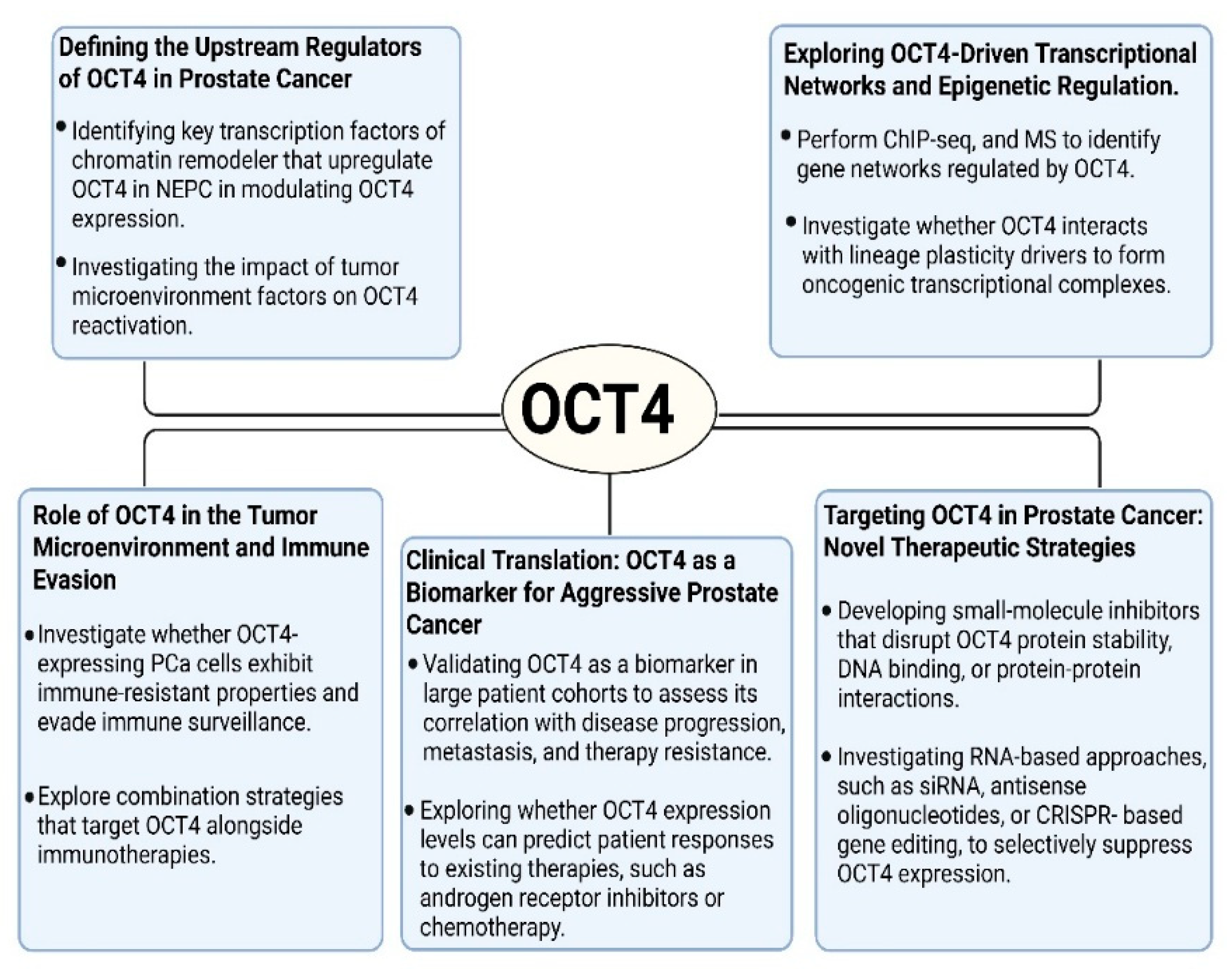
- Defining the Upstream Regulators of OCT4 in Prostate Cancer
- Identifying key transcription factors or chromatin remodelers that upregulate OCT4 in CRPC and NEPC.
- Investigating the role of long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) in modulating the OCT4 expression.
- Investigating the impact of tumor microenvironment factors, including hypoxia, inflammatory cytokines, and stromal interactions, on the OCT4 reactivation.
- 2.
- Exploring OCT4-Driven Transcriptional Networks and Epigenetic Regulation
- Perform chromatin immunoprecipitation sequencing (ChIP-seq), RNA sequencing (RNA-seq), and mass spectrometry (MS) to identify the gene networks regulated by OCT4 in PCa.
- Investigate whether OCT4 interacts with other lineage plasticity drivers, such as SOX2, NANOG, EZH2, or AURKA, to form oncogenic transcriptional complexes.
- Examine the role of super-enhancers (SEs) in sustaining OCT4 expression and whether disrupting SEs could be a viable therapeutic approach.
- 3.
- Role of OCT4 in the Tumor Microenvironment and Immune Evasion
- Investigate whether OCT4-expressing PCa cells exhibit immune-resistant properties and evade immune surveillance.
- Determine whether OCT4 influences immune checkpoint expression (e.g., PD-L1) or modulates tumor-associated macrophages, myeloid-derived suppressor cells (MDSCs), or T cells in the tumor microenvironment.
- Explore combination strategies that target OCT4 and other key master regulators—such as SOX2, MYC, EZH2, and BRN2—alongside immunotherapies, to more effectively disrupt the stemness and immune-evasive phenotypes associated with lineage plasticity in prostate cancer.
- 4.
- Clinical Translation: OCT4 as a Biomarker for Aggressive Prostate Cancer
- Validating OCT4 as a biomarker in large patient cohorts to assess its correlation with disease progression, metastasis, and therapy resistance.
- Developing non-invasive diagnostic tools (e.g., CTCs, exosomal OCT4 detection) to monitor disease progression in CRPC and NEPC patients.
- Exploring whether OCT4 expression levels can predict patient responses to existing therapies, such as androgen receptor inhibitors or chemotherapy.
- 5.
- Targeting OCT4 in Prostate Cancer: Novel Therapeutic Strategies
- Developing small-molecule inhibitors that disrupt OCT4 protein stability, DNA binding, or protein–protein interactions.
- Investigating RNA-based approaches, such as siRNA, antisense oligonucleotides, or CRISPR-based gene editing, to selectively suppress OCT4 expression.
- Identifying upstream regulatory pathways (e.g., NFκB, FGFR, or Wnt/β-catenin) that indirectly modulate OCT4 and could be targeted with existing inhibitors.
- Exploring the potential of targeted protein degradation strategies, such as PROTACs (proteolysis-targeting chimeras), to selectively degrade OCT4 in PCa cells.
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA 2025, 75, 10. [Google Scholar] [CrossRef]
- Shih, H.J.; Fang, S.C.; An, L.; Shao, Y.H.J. Early-onset prostate cancer is associated with increased risks of disease progression and cancer-specific mortality. Prostate 2021, 81, 118–126. [Google Scholar] [CrossRef]
- Teo, M.Y.; Rathkopf, D.E.; Kantoff, P. Treatment of advanced prostate cancer. Annu. Rev. Med. 2019, 70, 479–499. [Google Scholar] [CrossRef]
- Swami, U.; McFarland, T.R.; Nussenzveig, R.; Agarwal, N. Advanced prostate cancer: Treatment advances and future directions. Trends Cancer 2020, 6, 702–715. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, Z.; Yu, W.; Huang, H.; Wang, Y.; Niu, Y. Investigating High-risk Factors, Precise Diagnosis, and Treatment of Castration-Resistant Prostate Cancer (CRPC). Comb. Chem. High Throughput Screen. 2024, 27, 2598–2608. [Google Scholar] [CrossRef]
- Tashiro, K.; Kimura, S.; Tsuzuki, S.; Urabe, F.; Fukuokaya, W.; Mori, K.; Aikawa, K.; Murakami, M.; Sasaki, H.; Miki, K. Radiographic Progression at Castration-Resistant Prostate Cancer Diagnosis: A Prognostic Indicator of Metastatic Hormone-Sensitive Prostate Cancer. Clin. Genitourin. Cancer 2024, 22, 102075. [Google Scholar] [CrossRef]
- Yamada, Y.; Beltran, H. Clinical and biological features of neuroendocrine prostate cancer. Curr. Oncol. Rep. 2021, 23, 15. [Google Scholar] [CrossRef]
- Liu, S.; Alabi, B.R.; Yin, Q.; Stoyanova, T. Molecular mechanisms underlying the development of neuroendocrine prostate cancer. Semin. Cancer Biol. 2022, 86, 57–68. [Google Scholar] [CrossRef]
- Storck, W.K.; May, A.M.; Westbrook, T.C.; Duan, Z.; Morrissey, C.; Yates, J.A.; Alumkal, J.J. The role of epigenetic change in therapy-induced neuroendocrine prostate cancer lineage plasticity. Front. Endocrinol. 2022, 13, 926585. [Google Scholar] [CrossRef]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J. The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef]
- Ferguson, A.M.; Rubin, M.A. Lineage plasticity in prostate cancer: Looking beyond intrinsic alterations. Cancer Lett. 2022, 548, 215901. [Google Scholar] [CrossRef]
- Zhu, J.; Liang, X.; Wu, D.; Chen, S.; Yang, B.; Mao, W.; Shen, D. Clinicopathological characteristics and survival outcomes in neuroendocrine prostate cancer: A population-based study. Medicine 2021, 100, e25237. [Google Scholar] [CrossRef]
- Yao, J.; Liu, Y.; Liang, X.; Shao, J.; Zhang, Y.; Yang, J.; Zheng, M. Neuroendocrine carcinoma as an independent prognostic factor for patients with prostate cancer: A population-based study. Front. Endocrinol. 2021, 12, 778758. [Google Scholar] [CrossRef]
- Rubin, M.A.; Bristow, R.G.; Thienger, P.D.; Dive, C.; Imielinski, M. Impact of lineage plasticity to and from a neuroendocrine phenotype on progression and response in prostate and lung cancers. Mol. Cell 2020, 80, 562–577. [Google Scholar] [CrossRef]
- Takayama, K.-i.; Kosaka, T.; Suzuki, T.; Hongo, H.; Oya, M.; Fujimura, T.; Suzuki, Y.; Inoue, S. Subtype-specific collaborative transcription factor networks are promoted by OCT4 in the progression of prostate cancer. Nat. Commun. 2021, 12, 3766. [Google Scholar] [CrossRef]
- Imamura, J.; Ganguly, S.; Muskara, A.; Liao, R.S.; Nguyen, J.K.; Weight, C.; Wee, C.E.; Gupta, S.; Mian, O.Y. Lineage plasticity and treatment resistance in prostate cancer: The intersection of genetics, epigenetics, and evolution. Front. Endocrinol. 2023, 14, 1191311. [Google Scholar] [CrossRef]
- Swain, N.; Thakur, M.; Pathak, J.; Swain, B. SOX2, OCT4 and NANOG: The core embryonic stem cell pluripotency regulators in oral carcinogenesis. J. Oral Maxillofac. Pathol. 2020, 24, 368–373. [Google Scholar] [CrossRef]
- Esch, D.; Vahokoski, J.; Groves, M.R.; Pogenberg, V.; Cojocaru, V.; Vom Bruch, H.; Han, D.; Drexler, H.C.; Arauzo-Bravo, M.J.; Ng, C.K. A unique Oct4 interface is crucial for reprogramming to pluripotency. Nat. Cell Biol. 2013, 15, 295–301. [Google Scholar] [CrossRef]
- Robinson, M.; Gilbert, S.F.; Waters, J.A.; Lujano-Olazaba, O.; Lara, J.; Alexander, L.J.; Green, S.E.; Burkeen, G.A.; Patrus, O.; Sarwar, Z. Characterization of SOX2, OCT4 and NANOG in ovarian cancer tumor-initiating cells. Cancers 2021, 13, 262. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Wei, S.-J.; Kang, M.H. Role of OCT4 in cancer stem-like cells and chemotherapy resistance. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165432. [Google Scholar] [CrossRef]
- Kosaka, T.; Nagamatsu, G.; Saito, S.; Oya, M.; Suda, T.; Horimoto, K. Identification of drug candidate against prostate cancer from the aspect of somatic cell reprogramming. Cancer Sci. 2013, 104, 1017–1026. [Google Scholar] [CrossRef]
- Ma, Y. OCT4-positive circulating tumor cells may predict a poor prognosis in patients with metastatic castration-resistant prostate cancer treated with abiraterone plus prednisone therapy. Oncol. Lett. 2023, 26, 452. [Google Scholar] [CrossRef]
- Xie, W.; Yu, J.; Yin, Y.; Zhang, X.; Zheng, X.; Wang, X. OCT4 induces EMT and promotes ovarian cancer progression by regulating the PI3K/AKT/mTOR pathway. Front. Oncol. 2022, 12, 876257. [Google Scholar] [CrossRef]
- Bu, X.; Liu, Y.; Wang, L.; Yan, Z.; Xin, G.; Su, W. Oct4 promoted proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) in colon cancer cells by activating the SCF/c-Kit signaling pathway. Cell Cycle 2023, 22, 291–302. [Google Scholar] [CrossRef]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.-P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef]
- Nouruzi, S.; Ganguli, D.; Tabrizian, N.; Kobelev, M.; Sivak, O.; Namekawa, T.; Thaper, D.; Baca, S.C.; Freedman, M.L.; Aguda, A. ASCL1 activates neuronal stem cell-like lineage programming through remodeling of the chromatin landscape in prostate cancer. Nat. Commun. 2022, 13, 2282. [Google Scholar] [CrossRef]
- Kaarijärvi, R.; Kaljunen, H.; Ketola, K. Molecular and functional links between neurodevelopmental processes and treatment-induced neuroendocrine plasticity in prostate cancer progression. Cancers 2021, 13, 692. [Google Scholar] [CrossRef]
- Ko, J.; Meyer, A.N.; Haas, M.; Donoghue, D.J. Characterization of FGFR signaling in prostate cancer stem cells and inhibition via TKI treatment. Oncotarget 2021, 12, 22. [Google Scholar] [CrossRef]
- Guo, C.; Kadier, A.; Zhang, Z.; Mao, S.; Yang, B.; Zheng, J.; Yao, X. ADT increases prostate cancer cell invasion via altering AR/SALL4/SOX2-OCT4 stem cell signaling. Cell Biol. Toxicol. 2025, 41, 107. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, C.; Huang, L.; Niu, S.; Lu, Q.; Gong, D.; Huang, S.; Yuan, Y.; Chen, H. Prognostic value of association of OCT4 with LEF1 expression in esophageal squamous cell carcinoma and their impact on epithelial-mesenchymal transition, invasion, and migration. Cancer Med. 2018, 7, 3977–3987. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Shi, S.; Xu, Y.; Dai, X.; Li, H.; Wang, J.; Zhang, Q.; Wang, Y.; Sun, S. The prognostic and clinicopathologic characteristics of OCT4 and lung cancer: A meta-analysis. Curr. Mol. Med. 2019, 19, 54–75. [Google Scholar] [CrossRef]
- Kosaka, T.; Mikami, S.; Yoshimine, S.; Miyazaki, Y.; Daimon, T.; Kikuchi, E.; Miyajima, A.; Oya, M. The prognostic significance of OCT4 expression in patients with prostate cancer. Hum. Pathol. 2016, 51, 1–8. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, Z.; Zhu, Y.; Chen, J.; Li, W. The role and specific mechanism of OCT4 in cancer stem cells: A review. Int. J. Stem Cells 2020, 13, 312–325. [Google Scholar] [CrossRef]
- Cui, Y.; Niu, Y.; Zhou, J.; Chen, Y.; Cheng, Y.; Li, S.; Ai, Z.; Chu, C.; Wang, H.; Zheng, B. Generation of a precise Oct4-hrGFP knockin cynomolgus monkey model via CRISPR/Cas9-assisted homologous recombination. Cell Res. 2018, 28, 383–386. [Google Scholar] [CrossRef]
- Gao, L.; Yang, Y.; Xu, H.; Liu, R.; Li, D.; Hong, H.; Qin, M.; Wang, Y. MiR-335 functions as a tumor suppressor in pancreatic cancer by targeting OCT4. Tumor Biol. 2014, 35, 8309–8318. [Google Scholar] [CrossRef]
- Vaddi, P.K.; Stamnes, M.A.; Cao, H.; Chen, S. Elimination of SOX2/OCT4-associated prostate cancer stem cells blocks tumor development and enhances therapeutic response. Cancers 2019, 11, 1331. [Google Scholar] [CrossRef]
- Fogarty, N.M.; McCarthy, A.; Snijders, K.E.; Powell, B.E.; Kubikova, N.; Blakeley, P.; Lea, R.; Elder, K.; Wamaitha, S.E.; Kim, D. Genome editing reveals a role for OCT4 in human embryogenesis. Nature 2017, 550, 67–73. [Google Scholar] [CrossRef]
- Hisey, E.; Ross, P.J.; Meyers, S.A. A review of OCT4 functions and applications to equine embryos. J. Equine Vet. Sci. 2021, 98, 103364. [Google Scholar] [CrossRef]
- Niwa, H.; Miyazaki, J.-i.; Smith, A.G. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 2000, 24, 372–376. [Google Scholar] [CrossRef]
- Cai, W.; Wang, Z.; Wei, C.; Wu, M.; Zheng, W.; Zhang, H.; Liu, C.; Liu, L. Prognostic evaluation of NANOG and OCT4 expression for posttransplantation hepatocellular carcinoma recurrence. J. Cell. Biochem. 2019, 120, 8419–8429. [Google Scholar] [CrossRef]
- Noel, K.; Ibraheem, M.M.; Ahmed, B.S.; Hameed, A.F.; Khamees, N.H.; Akkila, S.S. Expression of OCT4 stem cell marker in benign prostatic hyperplasia and normal tissue around the prostatic carcinoma in a sample of Iraqi patients. Egypt. J. Histol. 2020, 43, 245–254. [Google Scholar]
- Su, B.-H.; Wang, C.-T.; Chang, J.-M.; Chen, H.-Y.; Huang, T.-H.; Yen, Y.-T.; Tseng, Y.-L.; Chang, M.-Y.; Lee, C.-H.; Cheng, L.-H. OCT4 promotes lung cancer progression through upregulation of VEGF-correlated chemokine-1. Int. J. Med. Sci. 2025, 22, 680. [Google Scholar] [CrossRef]
- Lambis-Anaya, L.; Fernández-Ruiz, M.; Liscano, Y.; Suarez-Causado, A. High OCT4 expression might be associated with an aggressive phenotype in rectal cancer. Cancers 2023, 15, 3740. [Google Scholar] [CrossRef]
- Pandian, J.; Panneerpandian, P.; Sekar, B.T.; Selvarasu, K.; Ganesan, K. OCT4-mediated transcription confers oncogenic advantage for a subset of gastric tumors with poor clinical outcome. Funct. Integr. Genom. 2022, 22, 1345–1360. [Google Scholar] [CrossRef]
- Ding, J.; Xu, H.; Faiola, F.; Ma’ayan, A.; Wang, J. Oct4 links multiple epigenetic pathways to the pluripotency network. Cell Res. 2012, 22, 155–167. [Google Scholar] [CrossRef]
- Ambrosetti, D.-C.; Basilico, C.; Dailey, L. Synergistic activation of the fibroblast growth factor 4 enhancer by Sox2 and Oct-3 depends on protein-protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol. Cell. Biol. 1997, 17, 6321–6329. [Google Scholar] [CrossRef]
- Cho, Y.; Kang, H.G.; Kim, S.-J.; Lee, S.; Jee, S.; Ahn, S.G.; Kang, M.J.; Song, J.S.; Chung, J.-Y.; Yi, E.C. Post-translational modification of OCT4 in breast cancer tumorigenesis. Cell Death Differ. 2018, 25, 1781–1795. [Google Scholar] [CrossRef]
- Sohn, E.J.; Moon, H.J.; Lim, J.K.; Kim, D.S.; Kim, J.H. Regulation of the protein stability and transcriptional activity of OCT4 in stem cells. Adv. Biol. Regul. 2021, 79, 100777. [Google Scholar] [CrossRef]
- Dan, S.; Kang, B.; Duan, X.; Wang, Y.-J. A cell-free system toward deciphering the post-translational modification barcodes of Oct4 in different cellular contexts. Biochem. Biophys. Res. Commun. 2015, 456, 714–720. [Google Scholar] [CrossRef]
- MacLean, M.R.; Walker, O.L.; Arun, R.P.; Fernando, W.; Marcato, P. Informed by cancer stem cells of solid tumors: Advances in treatments targeting tumor-promoting factors and pathways. Int. J. Mol. Sci. 2024, 25, 4102. [Google Scholar]
- Weng, Z.; Lin, J.; He, J.; Gao, L.; Lin, S.; Tsang, L.L.; Zhang, H.; He, X.; Wang, G.; Yang, X. Human embryonic stem cell-derived neural crest model unveils CD55 as a cancer stem cell regulator for therapeutic targeting in MYCN-amplified neuroblastoma. Neuro-Oncology 2022, 24, 872–885. [Google Scholar] [CrossRef]
- Liu, H.-L.; Tang, H.-t.; Yang, H.-l.; Deng, T.-T.; Xu, Y.-P.; Xu, S.-Q.; Peng, L.; Wang, Z.; Fang, Q.; Kuang, X.-Y. Oct4 regulates the transition of cancer stem-like cells to tumor endothelial-like cells in human liver cancer. Front. Cell Dev. Biol. 2020, 8, 563316. [Google Scholar] [CrossRef]
- Smith, B.A.; Sokolov, A.; Uzunangelov, V.; Baertsch, R.; Newton, Y.; Graim, K.; Mathis, C.; Cheng, D.; Stuart, J.M.; Witte, O.N. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. USA 2015, 112, E6544–E6552. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Song, P.; Gao, Z.; Bao, Y.; Chen, L.; Huang, Y.; Liu, Y.; Dong, Q.; Wei, X. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J. Hematol. Oncol. 2024, 17, 46. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Pandey, P.; Khan, F.; Seifeldin, S.A.; Alshaghdali, K.; Siddiqui, S.; Abdelwadoud, M.E.; Vyas, M.; Saeed, M.; Mazumder, A.; Saeed, A. Targeting Wnt/β-catenin pathway by flavonoids: Implication for cancer therapeutics. Nutrients 2023, 15, 2088. [Google Scholar] [CrossRef]
- Leonardo-Sousa, C.; Barriga, R.; Florindo, H.F.; Acúrcio, R.C.; Guedes, R.C. Structural Insights and Development of Small Molecule Inhibitors Targeting TGF-β Receptor I: A Comprehensive Review of Clinical Advances. Mol. Ther. Oncol. 2025, 33, 200945. [Google Scholar] [CrossRef]
- Du, L.; Zhu, W. Research Progress on TGF-Β Gene Family. Octa J. Environ. Res. 2024, 12, 1–10. [Google Scholar]
- Ebrahimi, M.; Nourbakhsh, E.; Hazara, A.Z.; Mirzaei, A.; Shafieyari, S.; Salehi, A.; Hoseinzadeh, M.; Payandeh, Z.; Barati, G. PI3K/Akt/mTOR signaling pathway in cancer stem cells. Pathol.-Res. Pract. 2022, 237, 154010. [Google Scholar]
- Song, J.-x.; Dong, Y.-q.; Han, R.-l.; Xie, J.; Zhu, A.-y.; Chen, X.; Yang, Y.-y.; Sheng, C.-x.; Jiang, T.; Zhao, H.-y. PI3K/AKT/mTOR Activation is Associated with Malignant Severity and Poorer Prognosis in Parathyroid Carcinomas. J. Clin. Endocrinol. Metab. 2025, dgaf042. [Google Scholar] [CrossRef]
- Versari, I.; Salucci, S.; Bavelloni, A.; Battistelli, M.; Traversari, M.; Wang, A.; Sampaolesi, M.; Faenza, I. The Emerging Role and Clinical Significance of PI3K-Akt-mTOR in Rhabdomyosarcoma. Biomolecules 2025, 15, 334. [Google Scholar] [CrossRef]
- Iluta, S.; Nistor, M.; Buruiana, S.; Dima, D. Notch and Hedgehog Signaling Unveiled: Crosstalk, Roles, and Breakthroughs in Cancer Stem Cell Research. Life 2025, 15, 228. [Google Scholar] [CrossRef]
- Lou, L.; Peng, K.; Ouyang, S.; Ding, W.; Mo, J.; Yan, J.; Gong, X.; Liu, G.; Lu, J.; Yue, P. Periostin-mediated NOTCH1 activation between tumor cells and HSCs crosstalk promotes liver metastasis of small cell lung cancer. J. Exp. Clin. Cancer Res. 2025, 44, 6. [Google Scholar] [CrossRef]
- Chen, C.; Du, Y.; Nie, R.; Wang, S.; Wang, H.; Li, P. Notch signaling in cancers: Mechanism and potential therapy. Front. Cell Dev. Biol. 2025, 13, 1542967. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Y.; Wu, H.; Wang, Y.; Deng, Y.; Chang, Y.; Su, K.; Yang, L.; Tao, W.; Liu, W. Exploring the mechanism of Jianpi Lishi Jiedu Granules against postoperative recurrence of colorectal adenoma based on IL-6/JAK/STAT3 signaling pathway. Cell. Signal. 2025, 127, 111535. [Google Scholar] [CrossRef]
- Ma, L.; Liu, X.; Roopashree, R.; Kazmi, S.W.; Jasim, S.A.; Phaninder Vinay, K.; Fateh, A.; Yang, F.; Rajabivahid, M.; Dehghani-Ghorbi, M. Long non-coding RNAs (lncRNAs) in cancer development: New insight from STAT3 signaling pathway to immune evasion. Clin. Exp. Med. 2025, 25, 53. [Google Scholar] [CrossRef]
- Gao, Y.; Lan, L.; Wang, C.; Wang, Y.; Shi, L.; Sun, L. Selective JAK1 inhibitors and the therapeutic applications thereof: A patent review (2016–2023). Expert Opin. Ther. Pat. 2025, 35, 181–195. [Google Scholar] [CrossRef]
- Duan, J.; Wang, Y.; Chen, Y.; Wang, Y.; Li, Q.; Liu, J.; Fu, C.; Cao, C.; Cong, Z.; Su, M. Silencing LY6D expression inhibits colon cancer in xenograft mice and regulates colon cancer stem cells’ proliferation, stemness, invasion, and apoptosis via the MAPK pathway. Molecules 2023, 28, 7776. [Google Scholar] [CrossRef]
- Emelyanova, A.; Zolotovskaia, M.; Poddubskaya, E.; Modestov, A.; Buzdin, A.; Kuzmin, D. Activation of P38 MAPK Signaling Cascade is Linked with Clinical Outcomes and Therapeutic Responses in Human Cancers. Biochemistry 2024, 89, 2155–2173. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Lin, T.-J.; Chong, K.-Y.; Chen, G.-Y.; Kuo, C.-Y.; Lin, Y.-Y.; Chang, C.-W.; Hsiao, T.-F.; Wang, C.-L.; Shih, Y.-C. Targeting the ERK1/2 and p38 MAPK pathways attenuates Golgi tethering factor golgin-97 depletion-induced cancer progression in breast cancer. Cell Commun. Signal. 2025, 23, 22. [Google Scholar] [CrossRef]
- Yong, X.; Tang, B.; Xiao, Y.-F.; Xie, R.; Qin, Y.; Luo, G.; Hu, C.-J.; Dong, H.; Yang, S.-M. Helicobacter pylori upregulates Nanog and Oct4 via Wnt/β-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett. 2016, 374, 292–303. [Google Scholar] [CrossRef]
- Sun, L.; Liu, T.; Zhang, S.; Guo, K.; Liu, Y. Oct4 induces EMT through LEF1/β-catenin dependent WNT signaling pathway in hepatocellular carcinoma. Oncol. Lett. 2017, 13, 2599–2606. [Google Scholar] [CrossRef]
- Yuan, F.; Zhou, W.; Zou, C.; Zhang, Z.; Hu, H.; Dai, Z.; Zhang, Y. Expression of Oct4 in HCC and modulation to wnt/β-catenin and TGF-β signal pathways. Mol. Cell. Biochem. 2010, 343, 155–162. [Google Scholar] [CrossRef]
- Wang, P.; Deng, Z.; Li, A.; Li, R.; Huang, W.; Cui, J.; Chen, S.; Li, B.; Zhang, S. β-Catenin promotes long-term survival and angiogenesis of peripheral blood mesenchymal stem cells via the Oct4 signaling pathway. Exp. Mol. Med. 2022, 54, 1434–1449. [Google Scholar] [CrossRef]
- Bassiouny, A. Regulation of Oct4 signaling on tumorigenesis and modulation of wnt/β-catenin and TGF-β signal pathways in hepatocellular carcinoma cells. J. Clin. Oncol. 2011, 29 (Suppl. 15), e13551. [Google Scholar]
- Kelly, K.F.; Ng, D.Y.; Jayakumaran, G.; Wood, G.A.; Koide, H.; Doble, B.W. β-catenin enhances Oct-4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell 2011, 8, 214–227. [Google Scholar]
- Guo, Y.; Li, B.; Yan, X.; Shen, X.; Ma, J.; Liu, S.; Zhang, D. Bisphenol A and polychlorinated biphenyls enhance the cancer stem cell properties of human ovarian cancer cells by activating the WNT signaling pathway. Chemosphere 2020, 246, 125775. [Google Scholar]
- Shen, W.; Zhang, X.; Tang, J.; Zhang, Z.; Du, R.; Luo, D.; Liu, X.; Xia, Y.; Li, Y.; Wang, S. CCL16 maintains stem cell-like properties in breast cancer by activating CCR2/GSK3β/β-catenin/OCT4 axis. Theranostics 2021, 11, 2297. [Google Scholar]
- Huang, H.; Wang, C.; Liu, F.; Li, H.-Z.; Peng, G.; Gao, X.; Dong, K.-Q.; Wang, H.-R.; Kong, D.-P.; Qu, M. Reciprocal network between cancer stem-like cells and macrophages facilitates the progression and androgen deprivation therapy resistance of prostate cancer. Clin. Cancer Res. 2018, 24, 4612–4626. [Google Scholar]
- Zhao, Q.W.; Zhou, Y.W.; Li, W.X.; Kang, B.; Zhang, X.Q.; Yang, Y.; ChENG, J.; Yin, S.Y.; Tong, Y.; He, J.Q. Akt-mediated phosphorylation of Oct4 is associated with the proliferation of stem-like cancer cells. Oncol. Rep. 2015, 33, 1621–1629. [Google Scholar] [CrossRef]
- Lin, Y.; Yang, Y.; Li, W.; Chen, Q.; Li, J.; Pan, X.; Zhou, L.; Liu, C.; Chen, C.; He, J. Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol. Cell 2012, 48, 627–640. [Google Scholar] [CrossRef]
- Li, W.; Zhou, Y.; Zhang, X.; Yang, Y.; Dan, S.; Su, T.; She, S.; Dong, W.; Zhao, Q.; Jia, J. Dual inhibiting OCT4 and AKT potently suppresses the propagation of human cancer cells. Sci. Rep. 2017, 7, 46246. [Google Scholar] [CrossRef]
- Park, G.B.; Kim, D. TLR5/7-mediated PI3K activation triggers epithelial-mesenchymal transition of ovarian cancer cells through WAVE3-dependent mesothelin or OCT4/SOX2 expression. Oncol. Rep. 2017, 38, 3167–3176. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, Y.; Zhang, Y.; Zhang, Z.; Peng, J.; Li, Z.; Han, L.; You, Q.; Chen, X.; Rao, X. Downregulation of cancer stem cell properties via mTOR signaling pathway inhibition by rapamycin in nasopharyngeal carcinoma. Int. J. Oncol. 2015, 47, 909–917. [Google Scholar] [CrossRef]
- Johnson, A.; Korleski, J.; Laterra, J.; Lopez-Bertoni, H. Abstract PR013: Oct4 and Sox2 induce cellular transition of glioma stem cells to an immune suppressive, regulatory T cell-like state. Cancer Res. 2022, 82 (Suppl. 10), PR013. [Google Scholar] [CrossRef]
- Hagiwara, M.; Yasumizu, Y.; Yamashita, N.; Rajabi, H.; Fushimi, A.; Long, M.D.; Li, W.; Bhattacharya, A.; Ahmad, R.; Oya, M. MUC1-C activates the BAF (mSWI/SNF) complex in prostate cancer stem cells. Cancer Res. 2021, 81, 1111–1122. [Google Scholar] [CrossRef]
- Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Wang, Z.; Sethi, S.; Sarkar, F.H. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE 2010, 5, e12445. [Google Scholar] [CrossRef]
- Zhang, L.; Sha, J.; Yang, G.; Huang, X.; Bo, J.; Huang, Y. Activation of Notch pathway is linked with epithelial-mesenchymal transition in prostate cancer cells. Cell Cycle 2017, 16, 999–1007. [Google Scholar] [CrossRef]
- Ibrahim, D.A.; Elsebai, E.A.; Fayed, A.; Abdelrahman, A.E. Prognostic value of NOTCH1 and OCT4 in gastric carcinoma. Indian J. Pathol. Microbiol. 2022, 65, 328–335. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, G.; Wang, Z.; Ding, X.; Qian, L.; Li, Y. Reck-notch1 signaling mediates miR-221/222 regulation of lung cancer stem cells in NSCLC. Front. Cell Dev. Biol. 2021, 9, 663279. [Google Scholar] [CrossRef]
- Wang, Y.; Han, Y.; Xu, S.; Zhang, L.; Zhang, X.; Deng, J.; Ye, W.; Liu, S. Targeting EGFR enriches stem cell-like properties in salivary adenoid cystic carcinoma by activating the Notch1 pathway. Cancer Manag. Res. 2020, 12, 6655–6663. [Google Scholar] [CrossRef]
- Cheng, J.-w.; Duan, L.-x.; Yu, Y.; Wang, P.; Feng, J.-l.; Feng, G.-z.; Liu, Y. Bone marrow mesenchymal stem cells promote prostate cancer cell stemness via cell–cell contact to activate the Jagged1/Notch1 pathway. Cell Biosci. 2021, 11, 87. [Google Scholar] [CrossRef]
- Bai, S.; Zhao, Y.; Chen, W.; Peng, W.; Wang, Y.; Xiong, S.; Aruna; Li, Y.; Yang, Y.; Chen, S. The stromal-tumor amplifying STC1-Notch1 feedforward signal promotes the stemness of hepatocellular carcinoma. J. Transl. Med. 2023, 21, 236. [Google Scholar] [CrossRef]
- Zhao, L.; Lei, J.; Gu, S.; Zhang, Y.; Jing, X.; Wang, L.; Zhang, L.; Ning, Q.; Luo, M.; Qi, Y. A yes-associated protein 1-Notch1 receptor positive feedback loop promotes breast cancer lung metastasis by attenuating the bone morphogenetic protein 4-SMAD family member 1/5 signaling. Carcinogenesis 2022, 43, 1162–1175. [Google Scholar] [CrossRef]
- Wang, G.; Zhou, H.; Gu, Z.; Gao, Q.; Shen, G. Oct4 promotes cancer cell proliferation and migration and leads to poor prognosis associated with the survivin/STAT3 pathway in hepatocellular carcinoma. Oncol. Rep. 2018, 40, 979–987. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell. Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef]
- Cheng, C.-C.; Shi, L.-H.; Wang, X.-J.; Wang, S.-X.; Wan, X.-Q.; Liu, S.-R.; Wang, Y.-F.; Lu, Z.; Wang, L.-H.; Ding, Y. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int. J. Oncol. 2018, 53, 339–348. [Google Scholar] [CrossRef]
- Sun, S.; Yang, H.; Wang, F.; Zhao, S. Oct4 downregulation-induced inflammation increases the migration and invasion rate of oral squamous cell carcinoma. Acta Biochim. Biophys. Sin. 2021, 53, 1440–1449. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, Q.; Dai, X.; Wen, X.; Luo, X.; Duan, Y.; Yang, Z.; Dai, Q. Alantolactone enhances the sensitivity of melanoma to MAPK pathway inhibitors by targeting inhibition of STAT3 activation and down-regulating stem cell markers. Cancer Cell Int. 2024, 24, 191. [Google Scholar] [CrossRef]
- Chen, M.; Ye, A.; Wei, J.; Wang, R.; Poon, K. Deoxycholic acid upregulates the reprogramming factors KFL4 and OCT4 through the IL-6/STAT3 pathway in esophageal adenocarcinoma cells. Technol. Cancer Res. Treat. 2020, 19, 1533033820945302. [Google Scholar] [CrossRef]
- Pandian, J.; Ganesan, K. Delineation of gastric tumors with activated ERK/MAPK signaling cascades for the development of targeted therapeutics. Exp. Cell Res. 2022, 410, 112956. [Google Scholar] [CrossRef]
- Jiang, P.; Li, F.; Liu, Z.; Hao, S.; Gao, J.; Li, S. BTB and CNC homology 1 (Bach1) induces lung cancer stem cell phenotypes by stimulating CD44 expression. Respir. Res. 2021, 22, 320. [Google Scholar] [CrossRef]
- Xu, H.; Du, Z.; Li, Z.; Liu, X.; Li, X.; Zhang, X.; Ma, J. MUC1-EGFR crosstalk with IL-6 by activating NF-κB and MAPK pathways to regulate the stemness and paclitaxel-resistance of lung adenocarcinoma. Ann. Med. 2024, 56, 2313671. [Google Scholar] [CrossRef]
- Wei, S.-J.; Nguyen, T.H.; Yang, I.-H.; Mook, D.G.; Makena, M.R.; Verlekar, D.; Hindle, A.; Martinez, G.M.; Yang, S.; Shimada, H. MYC transcription activation mediated by OCT4 as a mechanism of resistance to 13-cis RA-mediated differentiation in neuroblastoma. Cell Death Dis. 2020, 11, 368. [Google Scholar] [CrossRef]
- Emhemmed, F.; Azouaou, S.A.; Thuaud, F.; Schini-Kerth, V.; Désaubry, L.; Muller, C.D.; Fuhrmann, G. Selective anticancer effects of a synthetic flavagline on human Oct4-expressing cancer stem-like cells via a p38 MAPK-dependent caspase-3-dependent pathway. Biochem. Pharmacol. 2014, 89, 185–196. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar]
- Roy, A.; Mishra, J.; Chakraborty, S.; Singh, S.P.; Patra, S.K. Epigenetic regulation of pluripotency inducer genes NANOG and SOX2 in human prostate cancer. Prog. Mol. Biol. Transl. Sci. 2023, 197, 241–260. [Google Scholar]
- Costa, C.D.; Justo, A.A.; Kobayashi, P.E.; Story, M.M.; Palmieri, C.; Amorim, R.L.; Fonseca-Alves, C.E. Characterization of OCT3/4, Nestin, NANOG, CD44 and CD24 as stem cell markers in canine prostate cancer. Int. J. Biochem. Cell Biol. 2019, 108, 21–28. [Google Scholar] [CrossRef]
- Qi, Y.-F.; Wu, L.; Li, Z.-Q.; Wu, M.-L.; Wang, H.-F.; Chan, K.-Y.; Lu, L.-L.; Cai, S.-H.; Wang, H.-S.; Du, J. Nodal signaling modulates the expression of Oct-4 via nuclear translocation of β-catenin in lung and prostate cancer cells. Arch. Biochem. Biophys. 2016, 608, 34–41. [Google Scholar] [CrossRef]
- De Resende, M.F.; Chinen, L.T.D.; Vieira, S.; Jampietro, J.; Da Fonseca, F.P.; Vassallo, J.; Campos, L.C.; Guimarães, G.C.; Soares, F.A.; Rocha, R.M. Prognostication of OCT4 isoform expression in prostate cancer. Tumor Biol. 2013, 34, 2665–2673. [Google Scholar] [CrossRef]
- Nong, S.; Guan, Y.; Wang, Z.; Wei, Z.; Zhang, Y.; Ni, J.; He, C.; Ma, L.; Zhou, S.; Li, W. Significant association between IL-18 and OCT4 gene polymorphisms in susceptibility and clinical characteristics of prostate cancer. Oncol. Transl. Med. 2019, 5, 123–130. [Google Scholar] [CrossRef]
- Caputo, S.; Grioni, M.; Brambillasca, C.S.; Monno, A.; Brevi, A.; Freschi, M.; Piras, I.S.; Elia, A.R.; Pieri, V.; Baccega, T. Galectin-3 in prostate cancer stem-like cells is immunosuppressive and drives early metastasis. Front. Immunol. 2020, 11, 540641. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, J.; Wang, W. Expression and significance of S100P, CD147, and OCT4 in different prostate cancer tissue TNM stages. Genet. Mol. Res. 2015, 14, 6844–6851. [Google Scholar] [CrossRef]
- Hepburn, A.; Steele, R.; Veeratterapillay, R.; Wilson, L.; Kounatidou, E.; Barnard, A.; Berry, P.; Cassidy, J.; Moad, M.; El-Sherif, A. The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene 2019, 38, 4412–4424. [Google Scholar] [CrossRef]
- Miyashita, M.; Tomogane, M.; Nakamura, Y.; Shimizu, T.; Fujihara, A.; Ukimura, O.; Ashihara, E. Sphere-derived prostate cancer stem cells are resistant to γδ T cell cytotoxicity. Anticancer Res. 2020, 40, 5481–5487. [Google Scholar] [CrossRef]
- Federer-Gsponer, J.R.; Müller, D.C.; Zellweger, T.; Eggimann, M.; Marston, K.; Ruiz, C.; Seifert, H.H.; Rentsch, C.A.; Bubendorf, L.; Le Magnen, C. Patterns of stemness-associated markers in the development of castration-resistant prostate cancer. Prostate 2020, 80, 1108–1117. [Google Scholar] [CrossRef]
- Liu, B.; Jiang, H.-Y.; Yuan, T.; Luo, J.; Zhou, W.-D.; Jiang, Q.-Q.; Wu, D. Enzalutamide-induced upregulation of PCAT6 promotes prostate cancer neuroendocrine differentiation by regulating miR-326/HNRNPA2B1 axis. Front. Oncol. 2021, 11, 650054. [Google Scholar] [CrossRef]
- Fujimoto, N.; Tsubonuma, Y.; Nagata, Y.; Minato, A.; Tomisaki, I.; Harada, K.; Miyamoto, H. Second-line systemic therapy for highly aggressive neuroendocrine prostate cancer. Anticancer Res. 2023, 43, 3841–3847. [Google Scholar] [CrossRef]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F. The master neural transcription factor BRN2 is an androgen receptor–suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef]
- Sotomayor, P.; Godoy, A.; Smith, G.J.; Huss, W.J. Oct4A is expressed by a subpopulation of prostate neuroendocrine cells. Prostate 2009, 69, 401–410. [Google Scholar] [CrossRef]
- Gillessen, S.; Turco, F.; Davis, I.D.; Efstathiou, J.A.; Fizazi, K.; James, N.D.; Shore, N.; Small, E.; Smith, M.; Sweeney, C.J. Management of patients with advanced prostate cancer. Report from the 2024 Advanced Prostate Cancer Consensus Conference (APCCC). Eur. Urol. 2025, 87, 157–216. [Google Scholar]
- Thomson, A.; Al Saffar, H.; Tempo, J.; Lawrentschuk, N.; Murphy, D.G.; Perera, M. Time to castrate the cost? the rising expense of chemical castration for the management of prostate cancer. Prostate Int. 2025. [Google Scholar] [CrossRef]
- Liu, X.; Li, W.; Puzanov, I.; Goodrich, D.W.; Chatta, G.; Tang, D.G. Prostate cancer as a dedifferentiated organ: Androgen receptor, cancer stem cells, and cancer stemness. Essays Biochem. 2022, 66, 291–303. [Google Scholar]
- Sanchez, B.G.; Bort, A.; Vara-Ciruelos, D.; Diaz-Laviada, I. Androgen deprivation induces reprogramming of prostate cancer cells to stem-like cells. Cells 2020, 9, 1441. [Google Scholar] [CrossRef]
- Quintero, J.C.; Díaz, N.F.; Rodríguez-Dorantes, M.; Camacho-Arroyo, I. Cancer stem cells and androgen receptor signaling: Partners in disease progression. Int. J. Mol. Sci. 2023, 24, 15085. [Google Scholar] [CrossRef]
- Tiwari, R.; Manzar, N.; Ateeq, B. Dynamics of cellular plasticity in prostate cancer progression. Front. Mol. Biosci. 2020, 7, 130. [Google Scholar] [CrossRef]
- Ge, R.; Wang, Z.; Montironi, R.; Jiang, Z.; Cheng, M.; Santoni, M.; Huang, K.; Massari, F.; Lu, X.; Cimadamore, A. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann. Oncol. 2020, 31, 470–479. [Google Scholar] [CrossRef]
- Al Salhi, Y.; Sequi, M.B.; Valenzi, F.M.; Fuschi, A.; Martoccia, A.; Suraci, P.P.; Carbone, A.; Tema, G.; Lombardo, R.; Cicione, A. Cancer stem cells and prostate cancer: A narrative review. Int. J. Mol. Sci. 2023, 24, 7746. [Google Scholar] [CrossRef]
- Verma, S.; Shankar, E.; Kalayci, F.N.C.; Mukunda, A.; Alassfar, M.; Singh, V.; Chan, E.R.; MacLennan, G.T.; Gupta, S. Androgen deprivation induces transcriptional reprogramming in prostate cancer cells to develop stem cell-like characteristics. Int. J. Mol. Sci. 2020, 21, 9568. [Google Scholar] [CrossRef]
- Banerjee, P.; Kapse, P.; Siddique, S.; Kundu, M.; Choudhari, J.; Mohanty, V.; Malhotra, D.; Gosavi, S.W.; Gacche, R.N.; Kundu, G.C. Therapeutic implications of cancer stem cells in prostate cancer. Cancer Biol. Med. 2023, 20, 401–420. [Google Scholar] [CrossRef]
- Fatma, H.; Siddique, H.R. Cancer cell plasticity, stem cell factors, and therapy resistance: How are they linked? Cancer Metastasis Rev. 2024, 43, 423–440. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53-and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef]
- Kainulainen, K.; Niskanen, E.A.; Kinnunen, J.; Mäki-Mantila, K.; Hartikainen, K.; Paakinaho, V.; Malinen, M.; Ketola, K.; Pasonen-Seppänen, S. Secreted factors from M1 macrophages drive prostate cancer stem cell plasticity by upregulating NANOG, SOX2, and CD44 through NFκB-signaling. Oncoimmunology 2024, 13, 2393442. [Google Scholar] [CrossRef]
- Linn, D.E.; Yang, X.; Sun, F.; Xie, Y.; Chen, H.; Jiang, R.; Chen, H.; Chumsri, S.; Burger, A.M.; Qiu, Y. A role for OCT4 in tumor initiation of drug-resistant prostate cancer cells. Genes Cancer 2010, 1, 908–916. [Google Scholar] [CrossRef]
- Chen, X. Super-enhancer in prostate cancer: Transcriptional disorders and therapeutic targets. npj Precis. Oncol. 2020, 4, 31. [Google Scholar] [CrossRef]
- AlAbdi, L.; Saha, D.; He, M.; Dar, M.S.; Utturkar, S.M.; Sudyanti, P.A.; McCune, S.; Spears, B.H.; Breedlove, J.A.; Lanman, N.A. Oct4-mediated inhibition of Lsd1 activity promotes the active and primed state of pluripotency enhancers. Cell Rep. 2020, 30, 1478–1490.e6. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, X.; Pauklin, S. 3D chromatin architecture and epigenetic regulation in cancer stem cells. Protein Cell 2021, 12, 440–454. [Google Scholar] [CrossRef]
- Kar, S.; Niharika, N.; Roy, A.; Patra, S.K. Overexpression of SOX2 gene by histone modifications: SOX2 enhances human prostate and breast cancer progression by prevention of apoptosis and enhancing cell proliferation. Oncology 2023, 101, 591–608. [Google Scholar] [CrossRef]
- Davies, A.; Nouruzi, S.; Ganguli, D.; Namekawa, T.; Thaper, D.; Linder, S.; Karaoğlanoğlu, F.; Omur, M.E.; Kim, S.; Kobelev, M. An androgen receptor switch underlies lineage infidelity in treatment-resistant prostate cancer. Nat. Cell Biol. 2021, 23, 1023–1034. [Google Scholar] [CrossRef]
- Logotheti, S.; Papadaki, E.; Zolota, V.; Logothetis, C.; Vrahatis, A.G.; Soundararajan, R.; Tzelepi, V. Lineage Plasticity and Stemness Phenotypes in Prostate Cancer: Harnessing the Power of Integrated “Omics” Approaches to Explore Measurable Metrics. Cancers 2023, 15, 4357. [Google Scholar] [CrossRef]
- Yasumizu, Y.; Rajabi, H.; Jin, C.; Hata, T.; Pitroda, S.; Long, M.D.; Hagiwara, M.; Li, W.; Hu, Q.; Liu, S. MUC1-C regulates lineage plasticity driving progression to neuroendocrine prostate cancer. Nat. Commun. 2020, 11, 338. [Google Scholar] [CrossRef]
- Shokraii, F.; Moharrami, M.; Motamed, N.; Shahhoseini, M.; Totonchi, M.; Ezzatizadeh, V.; Firouzi, J.; Khosravani, P.; Ebrahimi, M. Histone modification marks strongly regulate Cdh1 promoter in prostospheres as a model of prostate cancer stem like cells. Cell J. 2019, 21, 124. [Google Scholar]
- Saha, S.K.; Jeong, Y.; Cho, S.; Cho, S.-G. Systematic expression alteration analysis of master reprogramming factor OCT4 and its three pseudogenes in human cancer and their prognostic outcomes. Sci. Rep. 2018, 8, 14806. [Google Scholar] [CrossRef]
- Pacheco, M.B.; Camilo, V.; Henrique, R.; Jerónimo, C. Epigenetic editing in prostate cancer: Challenges and opportunities. Epigenetics 2022, 17, 564–588. [Google Scholar] [CrossRef]
- Grillo, G.; Keshavarzian, T.; Linder, S.; Arlidge, C.; Mout, L.; Nand, A.; Teng, M.; Qamra, A.; Zhou, S.; Kron, K.J. Transposable elements are co-opted as oncogenic regulatory elements by lineage-specific transcription factors in prostate cancer. Cancer Discov. 2023, 13, 2470–2487. [Google Scholar] [CrossRef]
- Kushwaha, P.P.; Verma, S.; Kumar, S.; Gupta, S. Role of prostate cancer stem-like cells in the development of antiandrogen resistance. Cancer Drug Resist. 2022, 5, 459. [Google Scholar] [CrossRef]
- Escudero-Lourdes, C.; Alvarado-Morales, I.; Tokar, E.J. Stem cells as target for prostate cancer therapy: Opportunities and challenges. Stem Cell Rev. Rep. 2022, 18, 2833–2851. [Google Scholar] [CrossRef]
- Verma, P.; Shukla, N.; Kumari, S.; Ansari, M.; Gautam, N.K.; Patel, G.K. Cancer stem cell in prostate cancer progression, metastasis and therapy resistance. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2023, 1878, 188887. [Google Scholar]
- Wolf, I.; Gratzke, C.; Wolf, P. Prostate cancer stem cells: Clinical aspects and targeted therapies. Front. Oncol. 2022, 12, 935715. [Google Scholar] [CrossRef]
- Murakami, S.; Ninomiya, W.; Sakamoto, E.; Shibata, T.; Akiyama, H.; Tashiro, F. SRY and OCT4 are required for the acquisition of cancer stem cell-like properties and are potential differentiation therapy targets. Stem Cells 2015, 33, 2652–2663. [Google Scholar] [CrossRef]
- Zhu, X.; Ding, C.-K.C.; Aggarwal, R.R. Emerging Therapeutic Targets of Neuroendocrine Prostate Cancer. Curr. Oncol. Rep. 2025, 27, 362–374. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.; Carles, J.; Fay, A.P.; Matsubara, N.; Szczylik, C.; De Giorgi, U.; Joung, J.Y.; Fong, P.C.; Voog, E. Final overall survival (OS) with talazoparib (TALA)+ enzalutamide (ENZA) as first-line treatment in unselected patients with metastatic castration-resistant prostate cancer (mCRPC) in the phase 3 TALAPRO-2 trial. Am. Soc. Clin. Oncol. 2025, 43, 5. [Google Scholar] [CrossRef]
- Ramesh, S.; Selvakumar, P.; Ameer, M.Y.; Lian, S.; Abdullah Alzarooni, A.I.M.; Ojha, S.; Mishra, A.; Tiwari, A.; Kaushik, A.; Jung, Y.D. State-of-the-art therapeutic strategies for targeting cancer stem cells in prostate cancer. Front. Oncol. 2023, 13, 1059441. [Google Scholar] [CrossRef]
- Zhu, M.; Yu, X.; Zheng, Z.; Huang, J.; Yang, X.; Shi, H. Capsaicin suppressed activity of prostate cancer stem cells by inhibition of Wnt/β-catenin pathway. Phytother. Res. 2020, 34, 817–824. [Google Scholar] [CrossRef]
- Zhu, J.; Qin, P.; Cao, C.; Dai, G.; Xu, L.; Yang, D. Use of miR-145 and testicular nuclear receptor 4 inhibition to reduce chemoresistance to docetaxel in prostate cancer. Oncol. Rep. 2021, 45, 963–974. [Google Scholar] [CrossRef]
- Zhang, S. The mechanism of TR4 participate in prostate cancer metastasis and therapy. Theor. Nat. Sci. 2024, 61, 21–26. [Google Scholar]
- Mehravar, M.; Ghaemimanesh, F.; Poursani, E.M. An overview on the complexity of OCT4: At the level of DNA, RNA and protein. Stem Cell Rev. Rep. 2021, 17, 1121–1136. [Google Scholar] [CrossRef]
- Panayiotou, T.; Eftychiou, M.; Patera, E.; Promponas, V.J.; Strati, K. A paradigm for post-embryonic Oct4 re-expression: E7-induced hydroxymethylation regulates Oct4 expression in cervical cancer. J. Med. Virol. 2023, 95, e29264. [Google Scholar] [CrossRef]
- Lei, M.M.L.; Lee, T.K.W. Cancer stem cells: Emerging key players in immune evasion of cancers. Front. Cell Dev. Biol. 2021, 9, 692940. [Google Scholar] [CrossRef]
- Skvortsov, S.; Skvortsova, I.-I.; Tang, D.G.; Dubrovska, A. Concise review: Prostate cancer stem cells: Current understanding. Stem Cells 2018, 36, 1457–1474. [Google Scholar] [CrossRef]
- Su, H.; Huang, L.; Zhou, J.; Yang, G. Prostate cancer stem cells and their targeted therapies. Front. Cell Dev. Biol. 2024, 12, 1410102. [Google Scholar] [CrossRef]
- Deng, X.; Jiao, Y.; Hao, H.; Guo, Z.; An, G.; Zhang, W.; Xue, D.; Han, S. Dandelion extract suppresses the stem-like properties of triple-negative breast cancer cells by regulating CUEDC2/β-catenin/OCT4 signaling axis. J. Ethnopharmacol. 2025, 342, 119408. [Google Scholar] [CrossRef]
- Lin, T.-C.; Wang, K.-H.; Chuang, K.-H.; Kao, A.-P.; Kuo, T.-C. Oct-4 induces cisplatin resistance and tumor stem cell-like properties in endometrial carcinoma cells. Taiwan. J. Obstet. Gynecol. 2023, 62, 16–21. [Google Scholar] [CrossRef]
- Kasaju, M.; Mihailescu, M.-R. BPS2025-Investigation of miR-145-5p and FMRP binding to the 3′-UTR Oct4 mRNA G-quadruplex forming sequence. Biophys. J. 2025, 124, 87a. [Google Scholar] [CrossRef]
- Zou, J.; Chen, J.; Deng, L.; Xu, B.; Yu, T.; Wang, J.; He, C. Mechanistic Insights into SENP1 and OCT4 Interaction in Promoting Drug Resistance and Stem Cell Features in Colon Cancer. Am. J. Physiol.-Cell Physiol. 2025, 328, C1260–C1278. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signaling Pathways | Key Role in Lineage Plasticity | Interaction with OCT4 | Downstream Effector | Cancer Type | Therapeutic Implication | Clinical Evidence Level | References |
|---|---|---|---|---|---|---|---|
| Wnt/β-catenin | Elevation in CSC-like properties, promotes dedifferentiation, induces transcription, and enhances epithelial–mesenchymal transition, Angiogenesis, and Antiapoptotic effect, drug resistance |
|
|
|
|
| [72,73,74,75,76,77,78,79,80] |
| PI3K/AKT/mTOR | Enhance proliferation, EMT, and plasticity |
|
|
|
|
| [23,81,82,83,84,85] |
| TGF-β | Induces EMT and plasticity |
|
|
|
|
| [74,76,86] |
| Notch1 | Drives CSC maintenance, tumor aggressiveness promotes therapy resistance, and integrates EMT with CSC self-renewal |
|
|
|
|
| [87,88,89,90,91,92,93,94,95] |
| JAK1-STAT3 | Promotes CSC plasticity and enhances tumor progression, viability, migration, and invasion. Contributes to chemoresistance and poor prognosis. |
|
|
|
|
| [96,97,98,99,100,101] |
| ERK/MAPK | Regulates tumor progression, differentiation, apoptosis, and EMT. Promotes CSC-like properties |
|
|
|
|
| [102,103,104,105,106] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esfini Farahani, M.; Zhang, Y.; Akinyemi, A.O.; Seilani, F.; Alam, M.R.; Liu, X. Unlocking the Role of OCT4 in Cancer Lineage Plasticity: A Cross-Cancer Perspective with an Emphasis on Prostate Cancer. Biomedicines 2025, 13, 1642. https://doi.org/10.3390/biomedicines13071642
Esfini Farahani M, Zhang Y, Akinyemi AO, Seilani F, Alam MR, Liu X. Unlocking the Role of OCT4 in Cancer Lineage Plasticity: A Cross-Cancer Perspective with an Emphasis on Prostate Cancer. Biomedicines. 2025; 13(7):1642. https://doi.org/10.3390/biomedicines13071642
Chicago/Turabian StyleEsfini Farahani, Mohammad, Yanquan Zhang, Amos Olalekan Akinyemi, Fatemeh Seilani, Md Rakibul Alam, and Xiaoqi Liu. 2025. "Unlocking the Role of OCT4 in Cancer Lineage Plasticity: A Cross-Cancer Perspective with an Emphasis on Prostate Cancer" Biomedicines 13, no. 7: 1642. https://doi.org/10.3390/biomedicines13071642
APA StyleEsfini Farahani, M., Zhang, Y., Akinyemi, A. O., Seilani, F., Alam, M. R., & Liu, X. (2025). Unlocking the Role of OCT4 in Cancer Lineage Plasticity: A Cross-Cancer Perspective with an Emphasis on Prostate Cancer. Biomedicines, 13(7), 1642. https://doi.org/10.3390/biomedicines13071642