
A Cross-Tissue Transcriptome-Wide Association Study Reveals Novel Susceptibility Genes for Diabetic Kidney Disease in the FinnGen Cohort

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. DKD GWAS Data Source
2.3. eQTL Files Source
2.4. Cross-Tissue TWAS Analysis Using sCCA
2.5. Single-Tissue TWAS Analysis Using FUSION
2.6. Conditional and Joint Analysis
2.7. Gene-Based Association Analysis
2.8. Single-Tissue TWAS Analysis Using Batch SMR
2.9. Single-Tissue TWAS Analysis Using FOCUS
2.10. Two-Sample MR
2.11. Over-Representation Analysis
2.12. GeneMANIA Analysis
2.13. Druggability Assessment and Tissue-Specific Expression Analysis
2.14. Ethic Approval
3. Results
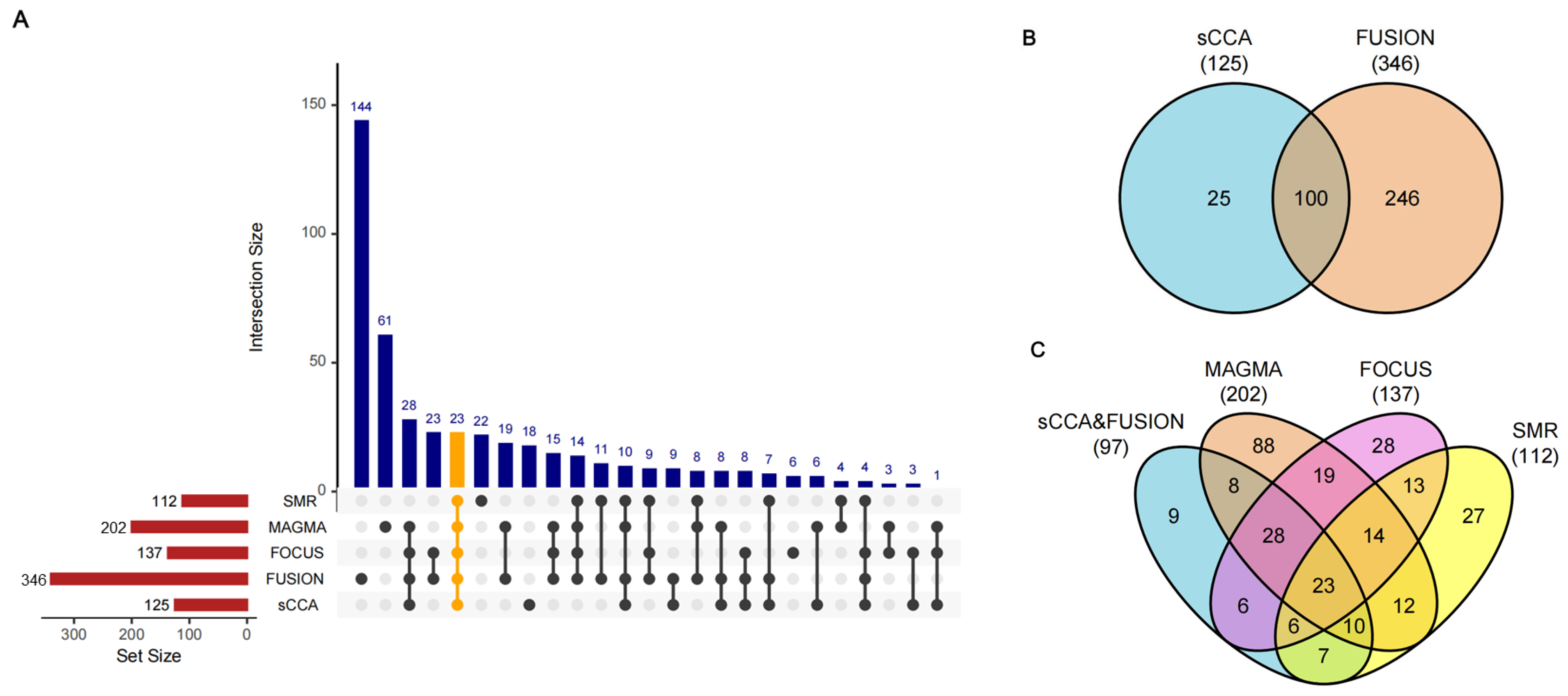
3.1. Discovery of DKD Causal Genes Through Integrative TWAS Analysis
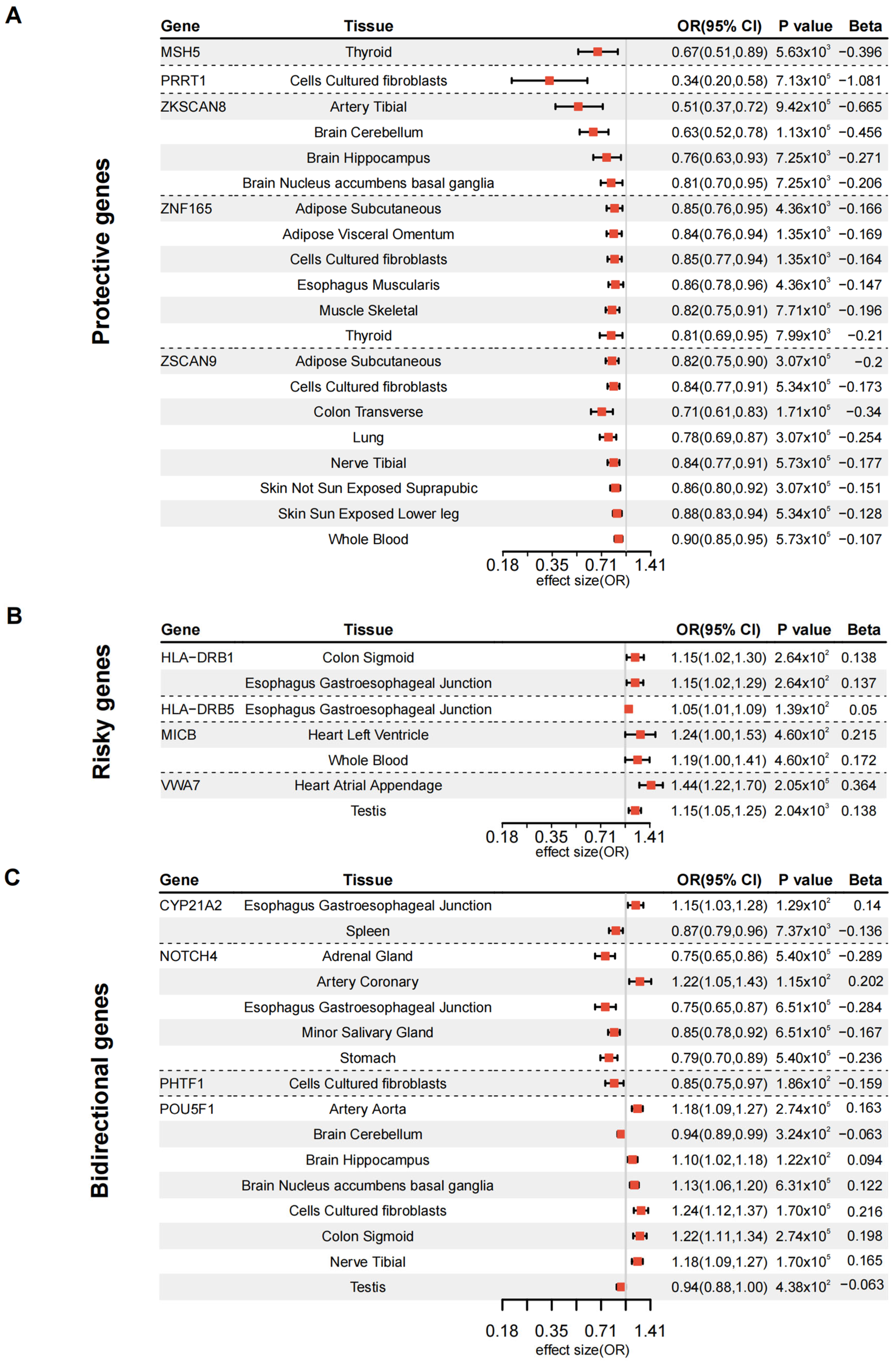
3.2. Validation of DKD Susceptibility Genes Through Tissue-Specific MR
3.3. Enrichment and Network Analysis Reveal Functional Gene Clusters in DKD Pathogenesis
3.4. MR-Validated Genes as Drug Targets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esposito, P.; Picciotto, D.; Cappadona, F.; Costigliolo, F.; Russo, E.; Macciò, L.; Viazzi, F. Multifaceted Relationship between Diabetes and Kidney Diseases: Beyond Diabetes. World J. Diabetes 2023, 14, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Costacou, T.; Orchard, T.J. Cumulative Kidney Complication Risk by 50 Years of Type 1 Diabetes: The Effects of Sex, Age, and Calendar Year at Onset. Diabetes Care 2018, 41, 426–433. [Google Scholar] [CrossRef]
- Casey, C.; Buckley, C.M.; Kearney, P.M.; Griffin, M.D.; Dinneen, S.F.; Griffin, T.P. Social Deprivation and Diabetic Kidney Disease: A European View. J. Diabetes Investig. 2024, 15, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Claessen, H.; Narres, M.; Kvitkina, T.; Wilk, A.; Friedel, H.; Günster, C.; Hoffmann, F.; Koch, M.; Jandeleit-Dahm, K.; Icks, A. Renal Replacement Therapy in People with and Without Diabetes in Germany, 2010–2016: An Analysis of More Than 25 Million Inhabitants. Diabetes Care 2021, 44, 1291–1299. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Agarwal, R.; Alpers, C.E.; Bakris, G.L.; Brosius, F.C.; Kolkhof, P.; Uribarri, J. Molecular Mechanisms and Therapeutic Targets for Diabetic Kidney Disease. Kidney Int. 2022, 102, 248–260. [Google Scholar] [CrossRef]
- Mancin, S.; Zarrella, A.; Petrelli, F.; Cosmai, S.; Cattani, D.; Lopane, D.; Scollo, S.; Morales Palomares, S.; Sguanci, M.; Amendola, A.; et al. Diabetes, Chronic Kidney Disease, and Vascular Ulcers: Prevention Strategies and Clinical Implications. Diabetology 2025, 6, 10. [Google Scholar] [CrossRef]
- Sandholm, N.; Dahlström, E.H.; Groop, P.-H. Genetic and Epigenetic Background of Diabetic Kidney Disease. Front. Endocrinol. 2023, 14, 1163001. [Google Scholar] [CrossRef]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-Wide Association Studies. Nat. Rev. Methods Primers 2021, 1, 59. [Google Scholar] [CrossRef]
- Hu, Y.; Li, M.; Lu, Q.; Weng, H.; Wang, J.; Zekavat, S.M.; Yu, Z.; Li, B.; Gu, J.; Muchnik, S.; et al. A Statistical Framework for Cross-Tissue Transcriptome-Wide Association Analysis. Nat. Genet. 2019, 51, 568–576. [Google Scholar] [CrossRef]
- Chen, Z.; Wen, W.; Beeghly-Fadiel, A.; Shu, X.-O.; Díez-Obrero, V.; Long, J.; Bao, J.; Wang, J.; Liu, Q.; Cai, Q.; et al. Identifying Putative Susceptibility Genes and Evaluating Their Associations with Somatic Mutations in Human Cancers. Am. J. Hum. Genet. 2019, 105, 477–492. [Google Scholar] [CrossRef]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; de Geus, E.J.C.; Boomsma, D.I.; Wright, F.A.; et al. Integrative Approaches for Large-Scale Transcriptome-Wide Association Studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, N.; Freund, M.K.; Johnson, R.; Shi, H.; Kichaev, G.; Gusev, A.; Pasaniuc, B. Probabilistic Fine-Mapping of Transcriptome-Wide Association Studies. Nat. Genet. 2019, 51, 675–682. [Google Scholar] [CrossRef]
- Barbeira, A.N.; Pividori, M.; Zheng, J.; Wheeler, H.E.; Nicolae, D.L.; Im, H.K. Integrating Predicted Transcriptome from Multiple Tissues Improves Association Detection. PLoS Genet. 2019, 15, e1007889. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, J.; Zhang, F.; Zhu, Z.; Qi, T.; Zheng, Z.; Lloyd-Jones, L.R.; Marioni, R.E.; Martin, N.G.; Montgomery, G.W.; et al. Integrative Analysis of Omics Summary Data Reveals Putative Mechanisms Underlying Complex Traits. Nat. Commun. 2018, 9, 918. [Google Scholar] [CrossRef]
- Feng, H.; Mancuso, N.; Gusev, A.; Majumdar, A.; Major, M.; Pasaniuc, B.; Kraft, P. Leveraging Expression from Multiple Tissues Using Sparse Canonical Correlation Analysis and Aggregate Tests Improves the Power of Transcriptome-Wide Association Studies. PLoS Genet. 2021, 17, e1008973. [Google Scholar] [CrossRef]
- Gui, J.; Yang, X.; Tan, C.; Wang, L.; Meng, L.; Han, Z.; Liu, J.; Jiang, L. A Cross-Tissue Transcriptome-Wide Association Study Reveals Novel Susceptibility Genes for Migraine. J. Headache Pain. 2024, 25, 94. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, R.; Chen, H.; Yang, X.; Dong, J.; Bai, M.; Lu, Y.; Leng, Y. Transcriptome-Wide Association Study Reveals Novel Susceptibility Genes for Coronary Atherosclerosis. Front. Cardiovasc. Med. 2023, 10, 1149113. [Google Scholar] [CrossRef]
- Ren, S.; Sun, C.; Zhai, W.; Wei, W.; Liu, J. Gaining New Insights into the Etiology of Ulcerative Colitis through a Cross-Tissue Transcriptome-Wide Association Study. Front. Genet. 2024, 15, 1425370. [Google Scholar] [CrossRef]
- Doke, T.; Huang, S.; Qiu, C.; Liu, H.; Guan, Y.; Hu, H.; Ma, Z.; Wu, J.; Miao, Z.; Sheng, X.; et al. Transcriptome-Wide Association Analysis Identifies DACH1 as a Kidney Disease Risk Gene That Contributes to Fibrosis. J. Clin. Investig. 2021, 131, 141801. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Qiu, M.; Tan, L.; Zhang, M.; Li, J.; Zhu, H.; Jiang, S.; Su, X.; Li, A. Renal and Cerebral RAS Interaction Contributes to Diabetic Kidney Disease. Am. J. Transl. Res. 2019, 11, 2925–2939. [Google Scholar]
- Miura, T.; Kuno, A.; Tanaka, M. Diabetes Modulation of the Myocardial Infarction-Acute Kidney Injury Axis. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H394–H405. [Google Scholar] [CrossRef] [PubMed]
- Folestad, E.; Mehlem, A.; Ning, F.C.; Oosterveld, T.; Palombo, I.; Singh, J.; Olauson, H.; Witasp, A.; Thorell, A.; Stenvinkel, P.; et al. Vascular Endothelial Growth Factor B-Mediated Fatty Acid Flux in the Adipose-Kidney Axis Contributes to Lipotoxicity in Diabetic Kidney Disease. Kidney Int. 2025, 107, 492–507. [Google Scholar] [CrossRef]
- Wu, I.-W.; Tsai, T.-H.; Lo, C.-J.; Chou, Y.-J.; Yeh, C.-H.; Cheng, M.-L.; Lai, C.-C.; Sytwu, H.-K.; Tsai, T.-F. Discovery of a Biomarker Signature That Reveals a Molecular Mechanism Underlying Diabetic Kidney Disease via Organ Cross Talk. Diabetes Care 2022, 45, e102–e104. [Google Scholar] [CrossRef]
- Li, S.-J.; Shi, J.-J.; Mao, C.-Y.; Zhang, C.; Xu, Y.-F.; Fan, Y.; Hu, Z.-W.; Yu, W.-K.; Hao, X.-Y.; Li, M.-J.; et al. Identifying Causal Genes for Migraine by Integrating the Proteome and Transcriptome. J. Headache Pain. 2023, 24, 111. [Google Scholar] [CrossRef]
- Liao, C.; Laporte, A.D.; Spiegelman, D.; Akçimen, F.; Joober, R.; Dion, P.A.; Rouleau, G.A. Transcriptome-Wide Association Study of Attention Deficit Hyperactivity Disorder Identifies Associated Genes and Phenotypes. Nat. Commun. 2019, 10, 4450. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of Summary Data from GWAS and eQTL Studies Predicts Complex Trait Gene Targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef]
- Zhang, B.; Huang, X.; Wang, X.; Chen, X.; Zheng, C.; Shao, W.; Wang, G.; Zhang, W. Using a Two-Sample Mendelian Randomization Analysis to Explore the Relationship between Physical Activity and Alzheimer’s Disease. Sci. Rep. 2022, 12, 12976. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor Package for Reactome Pathway Analysis and Visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The Druggable Genome and Support for Target Identification and Validation in Drug Development. Sci. Transl. Med. 2017, 9, eaag1166. [Google Scholar] [CrossRef]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and Deposition Resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef] [PubMed]
- Buniello, A.; Suveges, D.; Cruz-Castillo, C.; Llinares, M.B.; Cornu, H.; Lopez, I.; Tsukanov, K.; Roldán-Romero, J.M.; Mehta, C.; Fumis, L.; et al. Open Targets Platform: Facilitating Therapeutic Hypotheses Building in Drug Discovery. Nucleic Acids Res. 2025, 53, D1467–D1475. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liang, K.; Zhen, J.; Zhou, M.; Wang, X.; Wang, Z.; Wei, X.; Zhang, Y.; Sun, Y.; Zhou, Z.; et al. Sirt6 Deficiency Exacerbates Podocyte Injury and Proteinuria through Targeting Notch Signaling. Nat. Commun. 2017, 8, 413. [Google Scholar] [CrossRef]
- Ye, Q.; Ying, Q.; Chen, Y.; Liao, C.; Li, A. HLA-DRB5 Promotes Immune Thrombocytopenia via Activating CD8+ T Cells. Open Med. 2024, 19, 20240955. [Google Scholar] [CrossRef]
- Xu, J.; Chen, H.; Sun, C.; Wei, S.; Tao, J.; Jia, Z.; Chen, X.; Lv, W.; Lv, H.; Tang, G.; et al. Epigenome-Wide Methylation Haplotype Association Analysis Identified HLA-DRB1, HLA-DRB5 and HLA-DQB1 as Risk Factors for Rheumatoid Arthritis. Int. J. Immunogenet. 2023, 50, 291–298. [Google Scholar] [CrossRef]
- Ren, Y.; Ying, Q.; Chen, Y.; Liao, C.; Li, A.; Ye, Q. HLA-DRB5 Overexpression Promotes Platelet Reduction in Immune Thrombocytopenia Mice Model by Facilitating MHC-II-Mediated Antigen Presentation. Acta Haematol. 2025, 148, 68–76. [Google Scholar] [CrossRef]
- Jacobi, T.; Massier, L.; Klöting, N.; Horn, K.; Schuch, A.; Ahnert, P.; Engel, C.; Löffler, M.; Burkhardt, R.; Thiery, J.; et al. HLA Class II Allele Analyses Implicate Common Genetic Components in Type 1 and Non-Insulin-Treated Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2020, 105, dgaa027. [Google Scholar] [CrossRef]
- Li, L.; Tang, W.; Zhang, Y.; Jia, M.; Wang, L.; Li, Q.; Han, Q.; Peng, X.; Xie, Y.; Wu, J.; et al. Targeting Tissue-Resident Memory CD8+ T Cells in the Kidney Is a Potential Therapeutic Strategy to Ameliorate Podocyte Injury and Glomerulosclerosis. Mol. Ther. 2022, 30, 2746–2759. [Google Scholar] [CrossRef]
- Ackermann, D.; Vogt, B.; Bochud, M.; Burnier, M.; Martin, P.-Y.; Paccaud, F.; Ehret, G.; Guessous, I.; Ponte, B.; Pruijm, M.; et al. Increased Glucocorticoid Metabolism in Diabetic Kidney Disease. PLoS ONE 2022, 17, e0269920. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, X.; Zhou, J.; Qiu, K.; Deng, M.; Lin, Z.; Mosha, S.S.; Li, W. The Association Of Serum Cortisol Level with Microalbuminuria in Patients with Type 2 Diabetes and Prediabetes. Int. J. Med. Sci. 2020, 17, 2998–3004. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, Y. Relationship between Cortisol and Diabetic Microvascular Complications: A Retrospective Study. Eur. J. Med. Res. 2023, 28, 391. [Google Scholar] [CrossRef] [PubMed]
- Asao, T.; Oki, K.; Yoneda, M.; Tanaka, J.; Kohno, N. Hypothalamic-Pituitary-Adrenal Axis Activity Is Associated with the Prevalence of Chronic Kidney Disease in Diabetic Patients. Endocr. J. 2016, 63, 119–126. [Google Scholar] [CrossRef]
- Lao, Q.; Burkardt, D.D.; Kollender, S.; Faucz, F.R.; Merke, D.P. Congenital Adrenal Hyperplasia Due to Two Rare CYP21A2 Variant Alleles, Including a Novel Attenuated CYP21A1P/CYP21A2 Chimera. Mol. Genet. Genom. Med. 2023, 11, e2195. [Google Scholar] [CrossRef]
- Fracasso, P.M.; Goodner, S.A.; Wildi, J.D.; Naughton, M.J.; Linette, G.P.; Govindan, R.; Tan, B.R.; Blum, K.A.; Jones, G.J.; Pearce, T.E.; et al. A Phase I Study of Apolizumab, an Anti-HLA-DR ß-Chain Monoclonal Antibody, in Patients with Solid Tumor Malignancies. Am. J. Clin. Oncol. 2022, 45, 294–297. [Google Scholar] [CrossRef]
- Kamaliyan, Z.; Clarke, T.L. Zinc Finger Proteins: Guardians of Genome Stability. Front. Cell Dev. Biol. 2024, 12, 1448789. [Google Scholar] [CrossRef]
- Incorvaia, L.; Bazan Russo, T.D.; Gristina, V.; Perez, A.; Brando, C.; Mujacic, C.; Di Giovanni, E.; Bono, M.; Contino, S.; Ferrante Bannera, C.; et al. The Intersection of Homologous Recombination (HR) and Mismatch Repair (MMR) Pathways in DNA Repair-Defective Tumors. NPJ Precis. Oncol. 2024, 8, 190. [Google Scholar] [CrossRef]
- DeWitt, J.T.; Raghunathan, M.; Haricharan, S. Nonrepair Functions of DNA Mismatch Repair Proteins: New Avenues for Precision Oncology. Trends Cancer 2025, 11, 49–61. [Google Scholar] [CrossRef]
- Hess, D.L.; Kelly-Goss, M.R.; Cherepanova, O.A.; Nguyen, A.T.; Baylis, R.A.; Tkachenko, S.; Annex, B.H.; Peirce, S.M.; Owens, G.K. Perivascular Cell-Specific Knockout of the Stem Cell Pluripotency Gene Oct4 Inhibits Angiogenesis. Nat. Commun. 2019, 10, 967. [Google Scholar] [CrossRef]
- Mou, Y.; Yue, Z.; Wang, X.; Li, W.; Zhang, H.; Wang, Y.; Li, R.; Sun, X. OCT4 Remodels the Phenotype and Promotes Angiogenesis of HUVECs by Changing the Gene Expression Profile. Int. J. Med. Sci. 2016, 13, 386–394. [Google Scholar] [CrossRef]
- Huang, X.; Geng, S.; Weng, J.; Lu, Z.; Zeng, L.; Li, M.; Deng, C.; Wu, X.; Li, Y.; Du, X. Analysis of the Expression of PHTF1 and Related Genes in Acute Lymphoblastic Leukemia. Cancer Cell Int. 2015, 15, 93. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Liang, G.; Liu, Z.; Cai, H. Associations of GWAS-Supported Non-MHC Genes with Autoimmune Thyroiditis in Patients with Type 1 Diabetes. Diabetes Metab. Syndr. Obes. 2021, 14, 3017–3026. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, P.; Abu-Farha, M.; Alkayal, F.; Nizam, R.; Elkum, N.; Melhem, M.; John, S.E.; Channanath, A.; Abubaker, J.; Bennakhi, A.; et al. Genome-Wide Association Study Identifies Novel Risk Variants from RPS6KA1, CADPS, VARS, and DHX58 for Fasting Plasma Glucose in Arab Population. Sci. Rep. 2020, 10, 152. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene_id | Protein Names | Druggability Tier | Target Type | Drugs |
|---|---|---|---|---|---|
| CYP21A2 | ENSG00000231852 | Steroid 21-hydroxylase | 3B | Clinical trial | BBP-631 |
| HLA-DRB1 | ENSG00000196126 | HLA class II histocompatibility antigen, DRB1 beta chain | 1 | Successful | Glatiramer acetate; Apolizumab |
| HLA-DRB5 | ENSG00000198502 | HLA class II histocompatibility antigen, DR beta 5 chain | 1 | Clinical trial | Apolizumab |
| MICB | ENSG00000204516 | MHC class I polypeptide-related sequence B | / | / | / |
| MSH5 | ENSG00000204410 | MutS protein homolog 5 | / | / | / |
| NOTCH4 | ENSG00000204301 | Neurogenic locus notch homolog protein 4 | 3A | Clinical trial | Parsatuzumab (MEGF0444A); Crenigacestat (LY3039478) |
| PHTF1 | ENSG00000116793 | Protein PHTF1 | / | / | / |
| POU5F1 | ENSG00000204531 | POU domain, class 5, transcription factor 1 | / | / | / |
| PRRT1 | ENSG00000204314 | Proline-rich transmembrane protein 1 | / | / | / |
| VWA7 | ENSG00000204396 | von Willebrand factor A domain-containing protein 7 | / | / | / |
| ZKSCAN8 | ENSG00000198315 | Zinc finger protein with KRAB and SCAN domains 8 | / | / | / |
| ZNF165 | ENSG00000197279 | Zinc finger protein 165 | / | / | / |
| ZSCAN9 | ENSG00000137185 | Zinc finger and SCAN domain-containing protein 9 | / | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, M.; Li, Z.; Lu, Y.; Sun, P.; Chen, Y.; Yang, L. A Cross-Tissue Transcriptome-Wide Association Study Reveals Novel Susceptibility Genes for Diabetic Kidney Disease in the FinnGen Cohort. Biomedicines 2025, 13, 1231. https://doi.org/10.3390/biomedicines13051231
Liu M, Li Z, Lu Y, Sun P, Chen Y, Yang L. A Cross-Tissue Transcriptome-Wide Association Study Reveals Novel Susceptibility Genes for Diabetic Kidney Disease in the FinnGen Cohort. Biomedicines. 2025; 13(5):1231. https://doi.org/10.3390/biomedicines13051231
Chicago/Turabian StyleLiu, Menghan, Zehua Li, Yao Lu, Pingping Sun, Ying Chen, and Li Yang. 2025. "A Cross-Tissue Transcriptome-Wide Association Study Reveals Novel Susceptibility Genes for Diabetic Kidney Disease in the FinnGen Cohort" Biomedicines 13, no. 5: 1231. https://doi.org/10.3390/biomedicines13051231
APA StyleLiu, M., Li, Z., Lu, Y., Sun, P., Chen, Y., & Yang, L. (2025). A Cross-Tissue Transcriptome-Wide Association Study Reveals Novel Susceptibility Genes for Diabetic Kidney Disease in the FinnGen Cohort. Biomedicines, 13(5), 1231. https://doi.org/10.3390/biomedicines13051231