

DBI as a Novel Immunotherapeutic Candidate in Colorectal Cancer: Dissecting Genetic Risk and the Immune Landscape via GWAS, eQTL, and pQTL


Abstract
1. Introduction
2. Materials and Methods
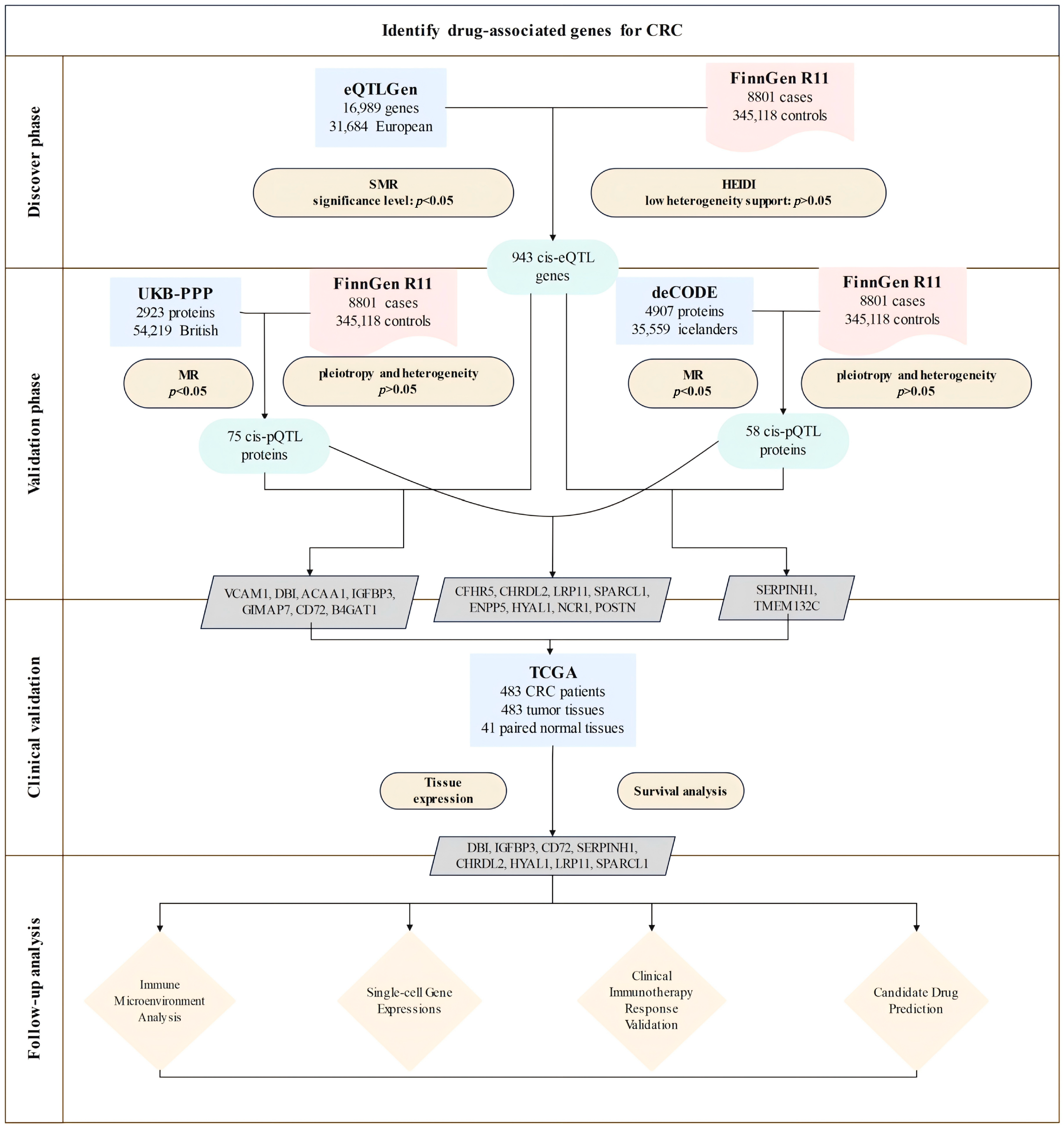
2.1. Research Design
2.2. Exposure Data
2.3. Outcome Data
2.4. SMR Analysis and HEIDI Test
2.5. Mendelian Randomization Analysis
2.6. Transcriptomic Profiling and Prognostic Assessment
2.7. Immune Microenvironment Analysis
2.8. Analysis of CRC Single-Cell Transcriptomes
2.9. Clinical Drug Resistance Analysis
2.10. Drug Target Prediction
3. Results
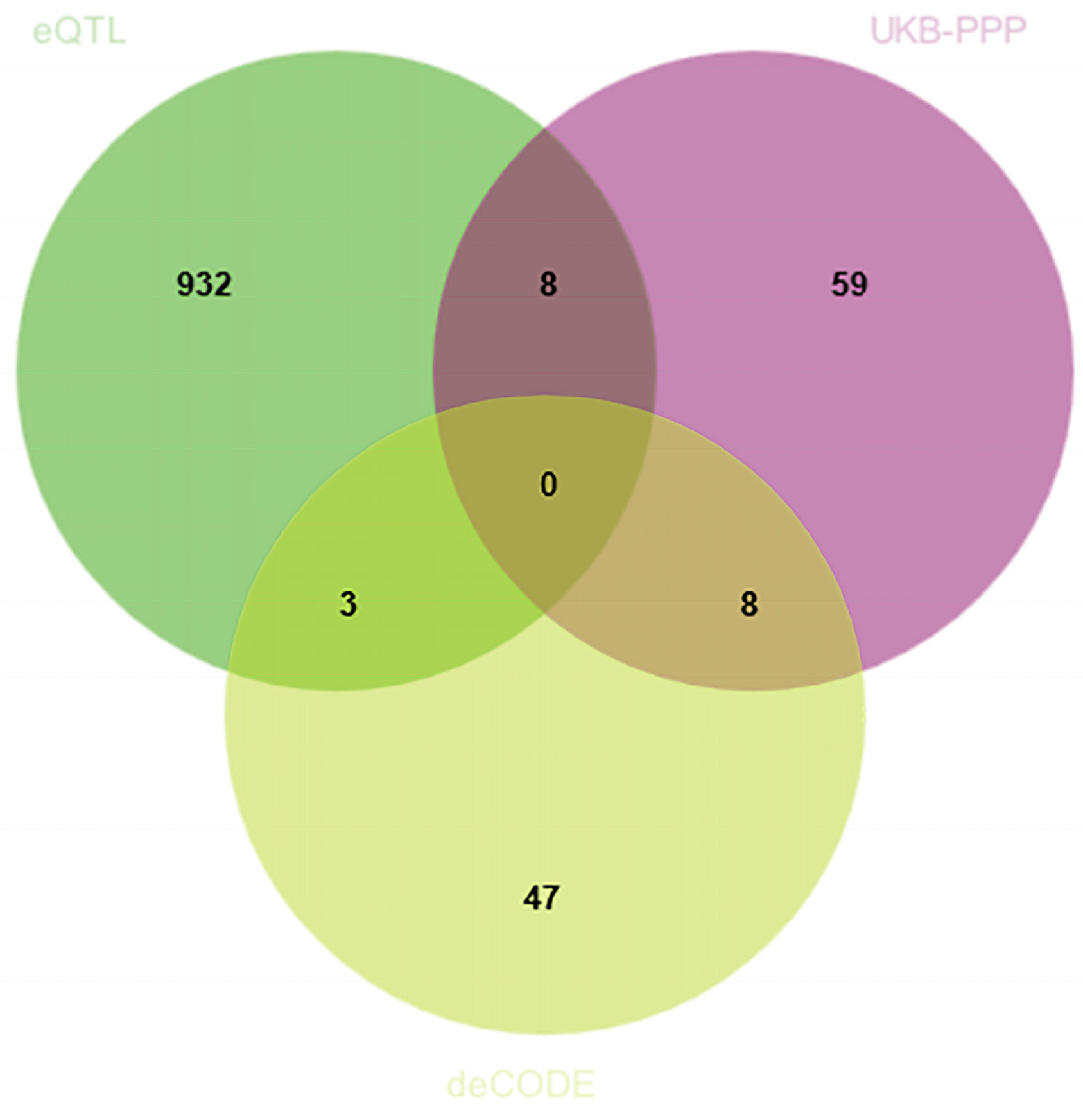
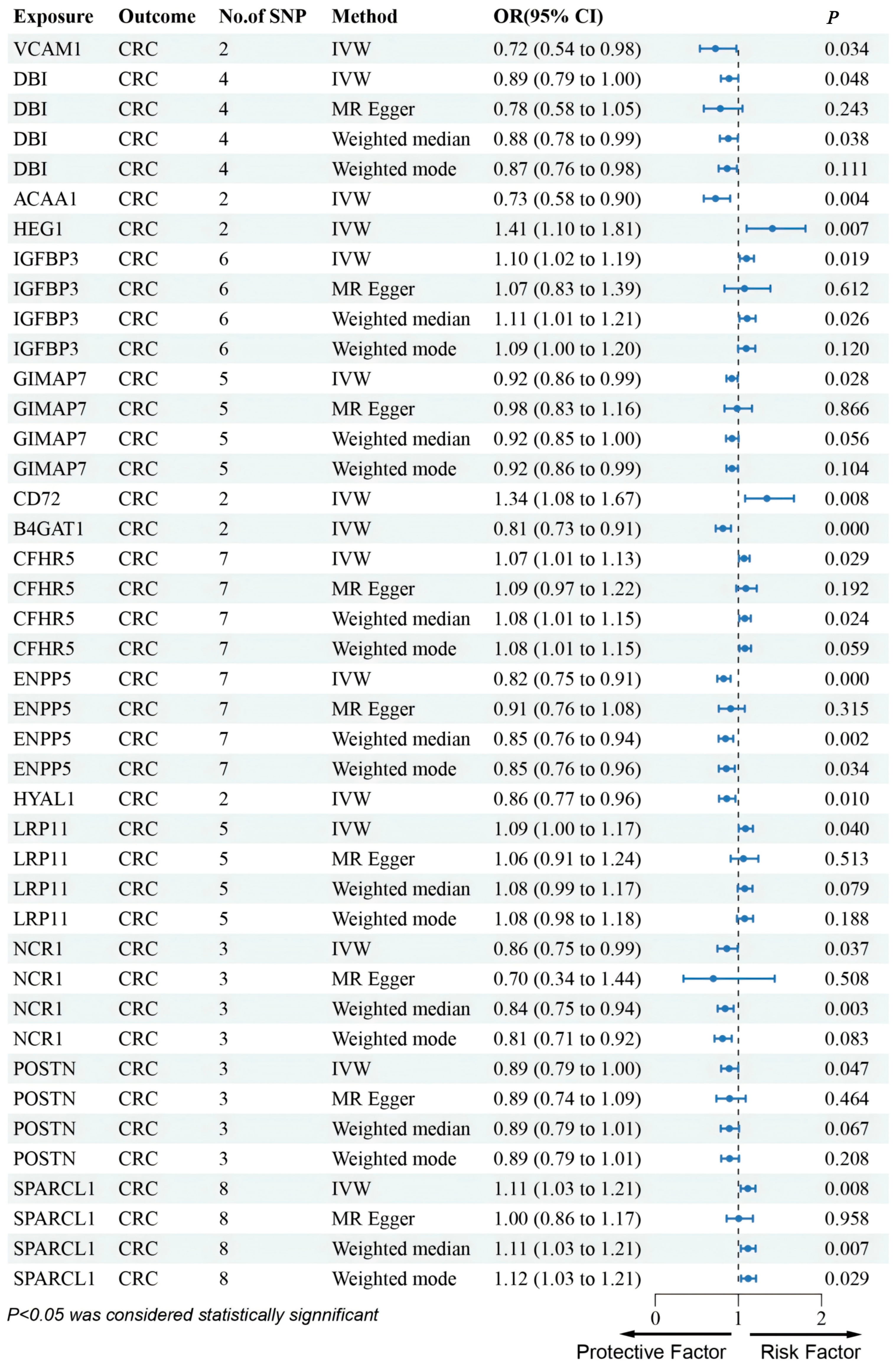
3.1. Discovery of Potential Cis-eQTL Genes and CRC
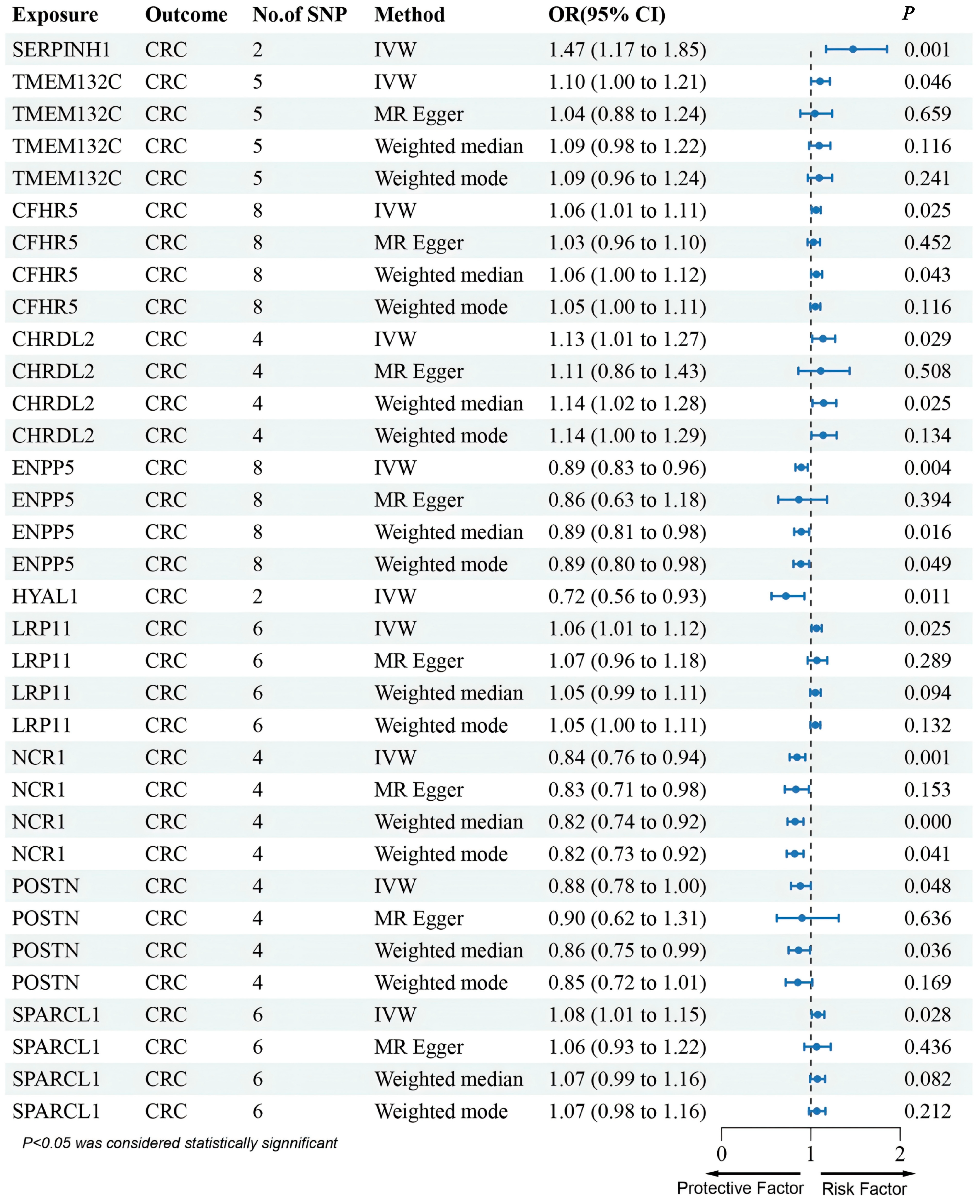
3.2. MR Analysis of pQTLs in Validation Phase
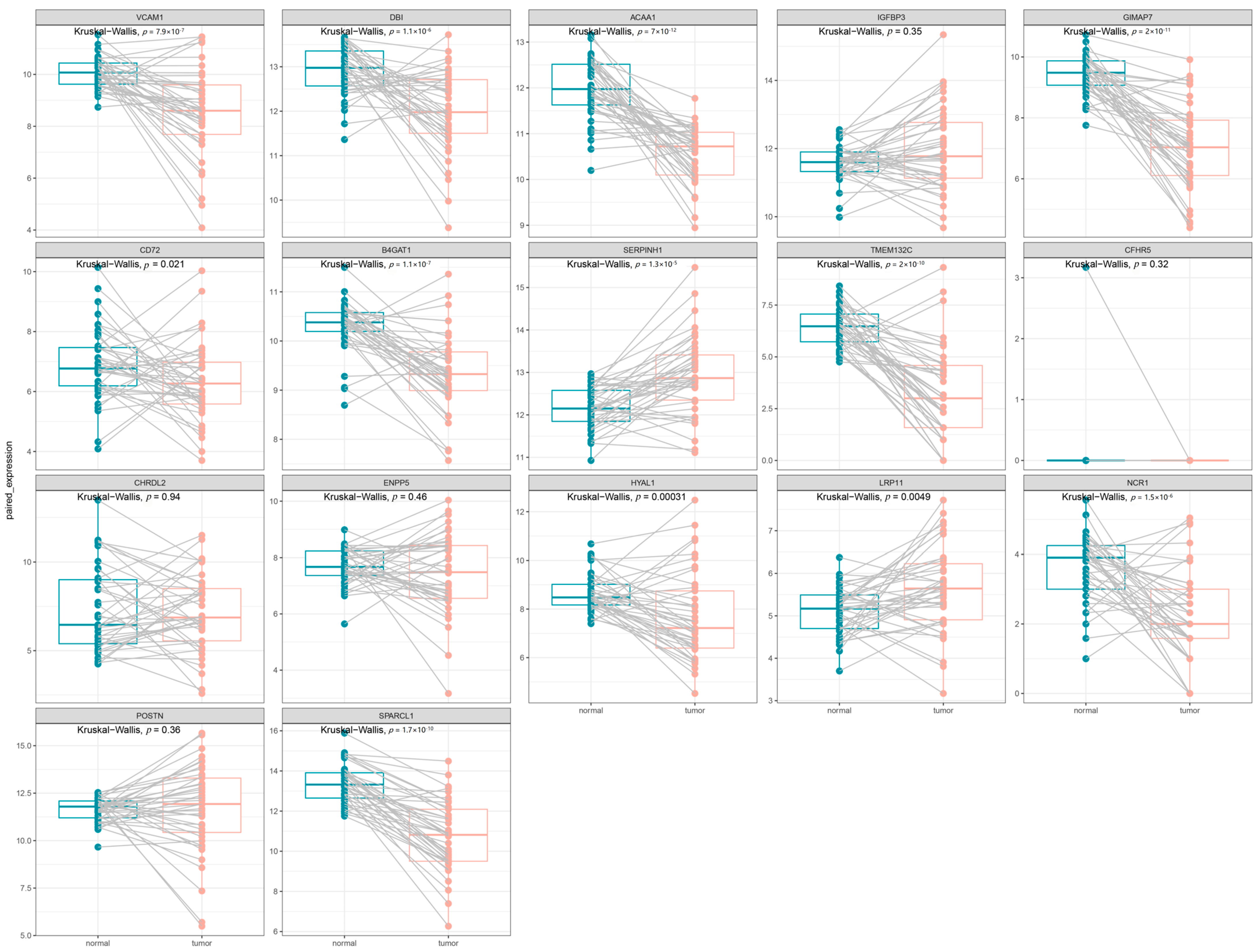
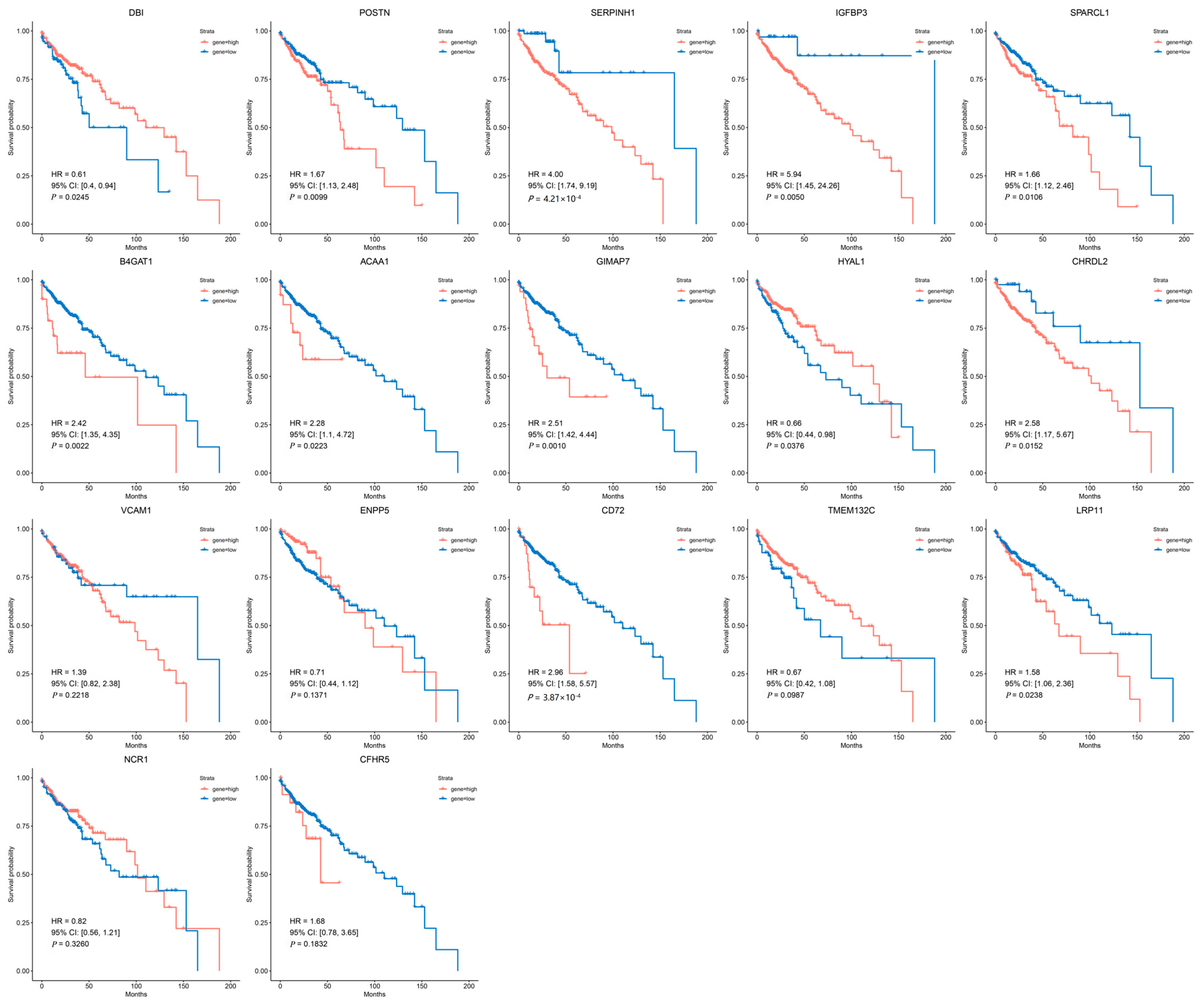
3.3. Clinical Expression and Survival Analysis
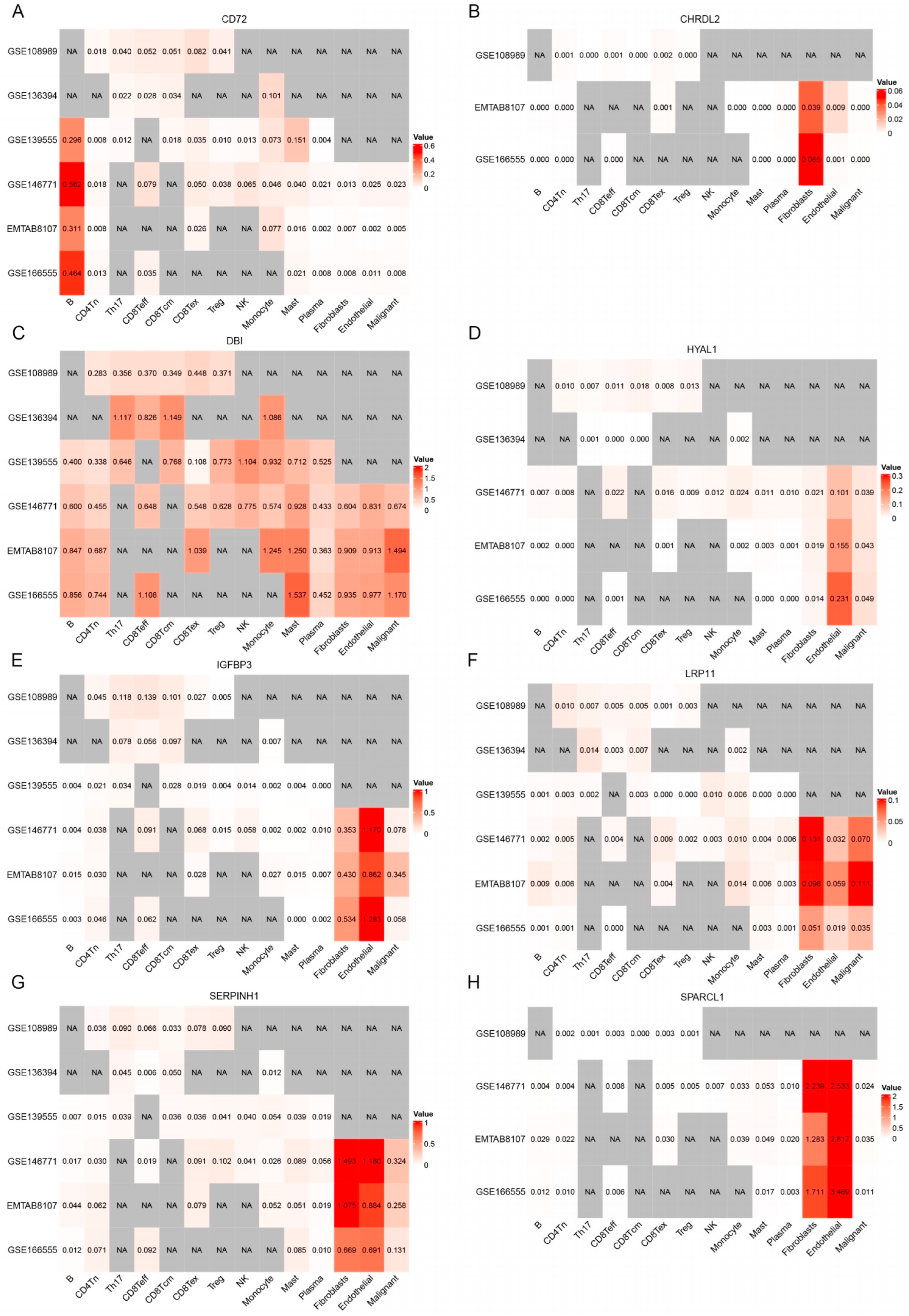
3.4. Immune Microenvironment Analysis of Candidate Genes in CRC
3.5. Analysis of Single-Cell Gene Expressions in CRC
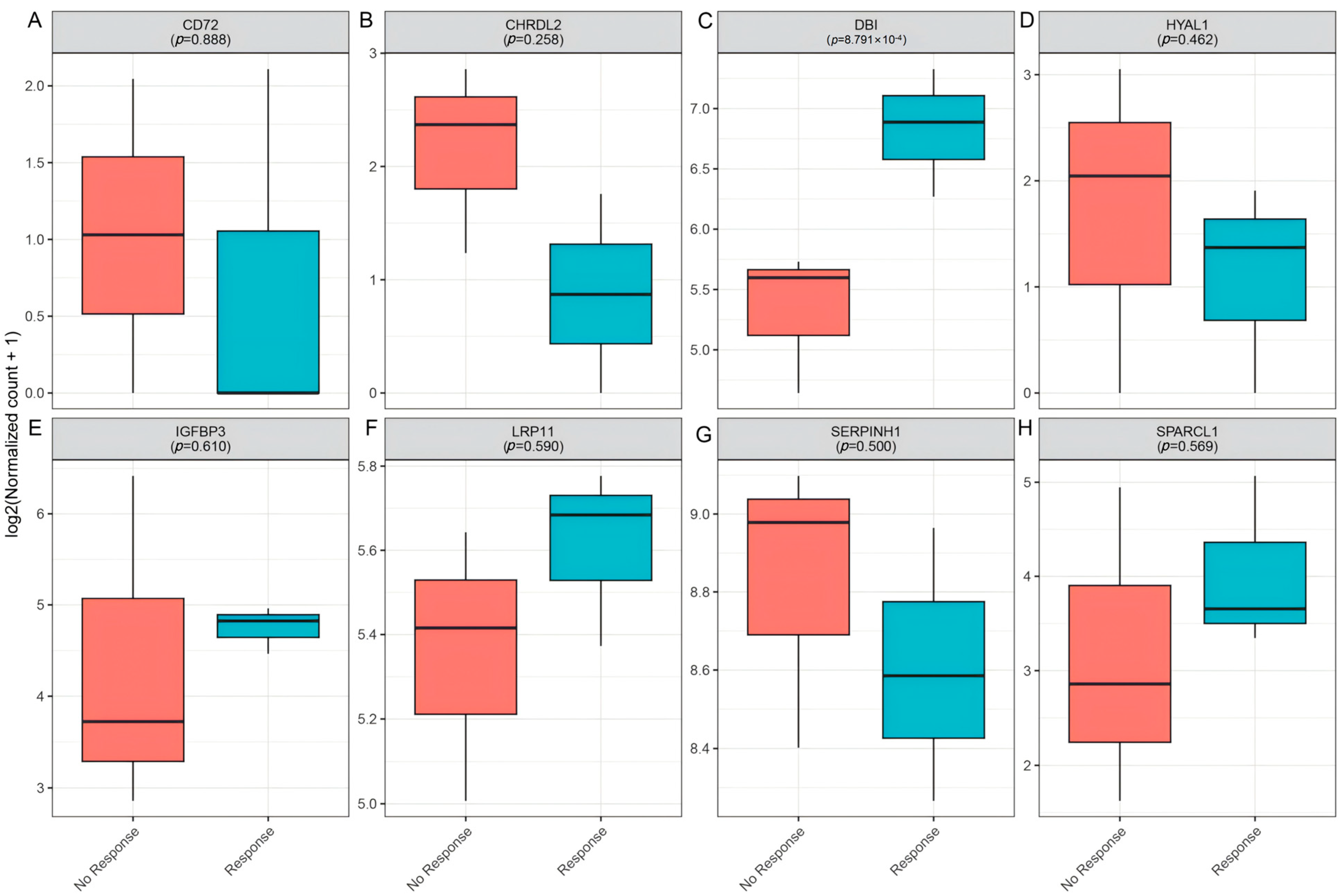
3.6. Clinical Immunotherapy Response Validation
3.7. Drug Target Prediction of DBI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, M.; Zhai, Z.; Wang, Y.; Chen, F.; Liu, R.; Yang, X.; Zhao, G. Advancements in the Application of Artificial Intelligence in the Field of Colorectal Cancer. Front. Oncol. 2025, 15, 1499223. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.E.; Giancotti, F.G.; Rustgi, A.K. Metastatic Colorectal Cancer: Mechanisms and Emerging Therapeutics. Trends Pharmacol. Sci. 2023, 44, 222–236. [Google Scholar] [CrossRef]
- Kanani, A.; Veen, T.; Søreide, K. Neoadjuvant Immunotherapy in Primary and Metastatic Colorectal Cancer. Br. J. Surg. 2021, 108, 1417–1425. [Google Scholar] [CrossRef]
- Kumar, A.; Gautam, V.; Sandhu, A.; Rawat, K.; Sharma, A.; Saha, L. Current and Emerging Therapeutic Approaches for Colorectal Cancer: A Comprehensive Review. World J. Gastrointest. Surg. 2023, 15, 495–519. [Google Scholar] [CrossRef]
- Franke, A.J.; Skelton, W.P.; Starr, J.S.; Parekh, H.; Lee, J.J.; Overman, M.J.; Allegra, C.; George, T.J. Immunotherapy for Colorectal Cancer: A Review of Current and Novel Therapeutic Approaches. J. Natl. Cancer Inst. 2019, 111, 1131–1141. [Google Scholar] [CrossRef]
- Atreya, C.E.; Yaeger, R.; Chu, E. Systemic Therapy for Metastatic Colorectal Cancer: From Current Standards to Future Molecular Targeted Approaches. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 246–256. [Google Scholar] [CrossRef]
- Kasprzak, A. The Role of Tumor Microenvironment Cells in Colorectal Cancer (CRC) Cachexia. Int. J. Mol. Sci. 2021, 22, 1565. [Google Scholar] [CrossRef] [PubMed]
- Nenkov, M.; Ma, Y.; Gaßler, N.; Chen, Y. Metabolic Reprogramming of Colorectal Cancer Cells and the Microenvironment: Implication for Therapy. Int. J. Mol. Sci. 2021, 22, 6262. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P. Involvement of Tumor Immune Microenvironment Metabolic Reprogramming in Colorectal Cancer Progression, Immune Escape, and Response to Immunotherapy. Front. Immunol. 2024, 15, 1353787. [Google Scholar] [CrossRef]
- AlMusawi, S.; Ahmed, M.; Nateri, A.S. Understanding Cell-Cell Communication and Signaling in the Colorectal Cancer Microenvironment. Clin. Transl. Med. 2021, 11, e308. [Google Scholar] [CrossRef]
- Lin, K.X.; Istl, A.C.; Quan, D.; Skaro, A.; Tang, E.; Zheng, X. PD-1 and PD-L1 Inhibitors in Cold Colorectal Cancer: Challenges and Strategies. Cancer Immunol. Immun. 2023, 72, 3875–3893. [Google Scholar] [CrossRef]
- Jin, C.; Zhu, X.; Huang, X.; Gong, T.; Wei, Z.; You, J. Efficacy and Safety of PD-1/PD-L1 and CTLA-4 Immune Checkpoint Inhibitors in Colorectal Cancer: A Meta-Analysis. J. Comp. Eff. Res. 2022, 11, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially Organized Multicellular Immune Hubs in Human Colorectal Cancer. Cell 2021, 184, 4734–4752. [Google Scholar] [CrossRef]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Yarmolinsky, J.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Higgins, J.P.T.; Timpson, N.J.; Dimou, N.; et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA 2021, 326, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Shen, X.; Pan, W. Constrained Maximum Likelihood-Based Mendelian Randomization Robust to Both Correlated and Uncorrelated Pleiotropic Effects. Am. J. Hum. Genet. 2021, 108, 1251–1269. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Tang, L.; Liu, Y.; Hu, H.; Liu, J. Causal Relationship between Immune Cell Signatures and Colorectal Cancer: A Bi-Directional, Two-Sample Mendelian Randomization Study. BMC Cancer 2025, 25, 387. [Google Scholar] [CrossRef]
- Yun, Z.; Guo, Z.; Li, X.; Shen, Y.; Nan, M.; Dong, Q.; Hou, L. Genetically Predicted 486 Blood Metabolites in Relation to Risk of Colorectal Cancer: A Mendelian Randomization Study. Cancer Med. 2023, 12, 13784–13799. [Google Scholar] [CrossRef]
- Fernandez-Rozadilla, C.; Timofeeva, M.; Chen, Z.; Law, P.; Thomas, M.; Schmit, S.; Díez-Obrero, V.; Hsu, L.; Fernandez-Tajes, J.; Palles, C.; et al. Deciphering Colorectal Cancer Genetics through Multi-Omic Analysis of 100,204 Cases and 154,587 Controls of European and East Asian Ancestries. Nat. Genet. 2023, 55, 89–99. [Google Scholar] [CrossRef]
- Sun, X.; Chen, B.; Qi, Y.; Wei, M.; Chen, W.; Wu, X.; Wang, Q.; Li, J.; Lei, X.; Luo, G. Multi-Omics Mendelian Randomization Integrating GWAS, eQTL and pQTL Data Revealed GSTM4 as a Potential Drug Target for Migraine. J. Headache Pain 2024, 25, 117. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, J.; Jiang, F.; Wang, L.; Xiao, Q.; Han, F.; Chen, J.; Yuan, S.; Wei, J.; Larsson, S.C.; et al. Identification of Novel Protein Biomarkers and Drug Targets for Colorectal Cancer by Integrating Human Plasma Proteome with Genome. Genome Med. 2023, 15, 75. [Google Scholar] [CrossRef]
- King, E.A.; Davis, J.W.; Degner, J.F. Are Drug Targets with Genetic Support Twice as Likely to Be Approved? Revised Estimates of the Impact of Genetic Support for Drug Mechanisms on the Probability of Drug Approval. PLoS Genet. 2019, 15, e1008489. [Google Scholar] [CrossRef]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The Support of Human Genetic Evidence for Approved Drug Indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef]
- Võsa, U.; Claringbould, A.; Westra, H.J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-Scale Cis- and Trans-eQTL Analyses Identify Thousands of Genetic Loci and Polygenic Scores That Regulate Blood Gene Expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef]
- Sun, B.B.; Chiou, J.; Traylor, M.; Benner, C.; Hsu, Y.H.; Richardson, T.G.; Surendran, P.; Mahajan, A.; Robins, C.; Vasquez-Grinnell, S.G.; et al. Plasma Proteomic Associations with Genetics and Health in the UK Biobank. Nature 2023, 622, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Ferkingstad, E.; Sulem, P.; Atlason, B.A.; Sveinbjornsson, G.; Magnusson, M.I.; Styrmisdottir, E.L.; Gunnarsdottir, K.; Helgason, A.; Oddsson, A.; Halldorsson, B.V.; et al. Large-Scale Integration of the Plasma Proteome with Genetics and Disease. Nat. Genet. 2021, 53, 1712–1721. [Google Scholar] [CrossRef]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen Provides Genetic Insights from a Well-Phenotyped Isolated Population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of Summary Data from GWAS and eQTL Studies Predicts Complex Trait Gene Targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, J.; Zhang, F.; Zhu, Z.; Qi, T.; Zheng, Z.; Lloyd-Jones, L.R.; Marioni, R.E.; Martin, N.G.; Montgomery, G.W.; et al. Integrative Analysis of Omics Summary Data Reveals Putative Mechanisms Underlying Complex Traits. Nat. Commun. 2018, 9, 918. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An Integrated Map of Genetic Variation from 1092 Human Genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef]
- Davey Smith, G.; Hemani, G. Mendelian Randomization: Genetic Anchors for Causal Inference in Epidemiological Studies. Hum. Mol. Genet. 2014, 23, R89–R98. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian Randomization Analysis with Multiple Genetic Variants Using Summarized Data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian Randomization with Invalid Instruments: Effect Estimation and Bias Detection through Egger Regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Chen, C.Y.; Neale, B.; Do, R. Detection of Widespread Horizontal Pleiotropy in Causal Relationships Inferred from Mendelian Randomization between Complex Traits and Diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Fang, Y.; Qiu, W.; Luo, P.; Wang, S.; Shen, R.; Gu, W.; Huang, X.; Mao, Q.; Wang, G.; et al. Enhancing Immuno-Oncology Investigations through Multidimensional Decoding of Tumor Microenvironment with IOBR 2.0. Cell Rep. Methods 2024, 4, 100910. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-Cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A Pan-Cancer Blueprint of the Heterogeneous Tumor Microenvironment Revealed by Single-Cell Profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage Tracking Reveals Dynamic Relationships of T Cells in Colorectal Cancer. Nature 2018, 564, 268–272. [Google Scholar] [CrossRef]
- Uhlitz, F.; Bischoff, P.; Peidli, S.; Sieber, A.; Trinks, A.; Lüthen, M.; Obermayer, B.; Blanc, E.; Ruchiy, Y.; Sell, T.; et al. Mitogen-Activated Protein Kinase Activity Drives Cell Trajectories in Colorectal Cancer. EMBO Mol. Med. 2021, 13, e14123. [Google Scholar] [CrossRef]
- Wu, T.D.; Madireddi, S.; de Almeida, P.E.; Banchereau, R.; Chen, Y.J.J.; Chitre, A.S.; Chiang, E.Y.; Iftikhar, H.; O’Gorman, W.E.; Au-Yeung, A.; et al. Peripheral T Cell Expansion Predicts Tumour Infiltration and Clinical Response. Nature 2020, 579, 274–278. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef]
- Lu, Y.C.; Jia, L.; Zheng, Z.; Tran, E.; Robbins, P.F.; Rosenberg, S.A. Single-Cell Transcriptome Analysis Reveals Gene Signatures Associated with T-Cell Persistence Following Adoptive Cell Therapy. Cancer Immunol. Res. 2019, 7, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ma, Y.; Yang, L.; Ma, S.; Yu, Z.; Ren, X.; Kong, X.; Zhang, X.; Li, D.; Liu, Z. CTR-DB 2.0: An Updated Cancer Clinical Transcriptome Resource, Expanding Primary Drug Resistance and Newly Adding Acquired Resistance Datasets and Enhancing the Discovery and Validation of Predictive Biomarkers. Nucleic Acids Res. 2025, 53, D1335–D1347. [Google Scholar] [CrossRef]
- Yoo, M.; Shin, J.; Kim, J.; Ryall, K.A.; Lee, K.; Lee, S.; Jeon, M.; Kang, J.; Tan, A.C. DSigDB: Drug Signatures Database for Gene Set Analysis. Bioinformatics 2015, 31, 3069–3071. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Naschberger, E.; Liebl, A.; Schellerer, V.S.; Schütz, M.; Britzen-Laurent, N.; Kölbel, P.; Schaal, U.; Haep, L.; Regensburger, D.; Wittmann, T.; et al. Matricellular Protein SPARCL1 Regulates Tumor Microenvironment-Dependent Endothelial Cell Heterogeneity in Colorectal Carcinoma. J. Clin. Investig. 2016, 126, 4187–4204. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Li, C.; Yuan, H.; Jiang, K.; Deng, G. Immunological Role and Prognostic Value of SPARCL1 in Pan-Cancer Analysis. Pathol. Oncol. Res. 2022, 28, 1610687. [Google Scholar] [CrossRef]
- Eiza, N.; Sabag, A.; Kessler, O.; Toubi, E.; Vadasz, Z. Soluble CD72, Is a T-Cell Activator Probably via Binding to CD6 in Homeostasis and Autoimmunity. Front. Immunol. 2024, 15, 1367120. [Google Scholar] [CrossRef]
- Tian, L.; Wang, Y.; Zhang, Z.; Feng, X.; Xiao, F.; Zong, M. CD72, a New Immune Checkpoint Molecule, Is a Novel Prognostic Biomarker for Kidney Renal Clear Cell Carcinoma. Eur. J. Med. Res. 2023, 28, 531. [Google Scholar] [CrossRef]
- Sica, V.; Martins, I.; Pietrocola, F.; Bravo-San Pedro, J.M. Quantification of Intracellular ACBP/DBI Levels. Methods Cell Biol. 2021, 165, 111–122. [Google Scholar] [CrossRef]
- Knudsen, J.; Mandrup, S.; Rasmussen, J.T.; Andreasen, P.H.; Poulsen, F.; Kristiansen, K. The Function of Acyl-CoA-Binding Protein (ACBP)/Diazepam Binding Inhibitor (DBI). Mol. Cell Biochem. 1993, 123, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Montégut, L.; Abdellatif, M.; Motiño, O.; Madeo, F.; Martins, I.; Quesada, V.; López-Otín, C.; Kroemer, G. Acyl Coenzyme A Binding Protein (ACBP): An Aging- and Disease-Relevant “Autophagy Checkpoint”. Aging Cell 2023, 22, e13910. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mingoia, S.; Montégut, L.; Lambertucci, F.; Chen, H.; Dong, Y.; De Palma, F.D.E.; Scuderi, S.A.; Rong, Y.; Carbonnier, V.; et al. Atlas of Expression of Acyl CoA Binding Protein/Diazepam Binding Inhibitor (ACBP/DBI) in Human and Mouse. Cell Death Dis. 2025, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Motiño, O.; Lambertucci, F.; Anagnostopoulos, G.; Li, S.; Martins, I.; Kroemer, G. Cardio-, Hepato- and Pneumoprotective Effects of Autophagy Checkpoint Inhibition by Targeting DBI/ACBP. Autophagy 2023, 19, 1604–1606. [Google Scholar] [CrossRef]
- Yao, J.; Liu, Y.; Yang, J.; Li, M.; Li, S.; Zhang, B.; Yang, R.; Zhang, Y.; Cui, X.; Feng, C. Single-Cell Sequencing Reveals That DBI Is the Key Gene and Potential Therapeutic Target in Quiescent Bladder Cancer Stem Cells. Front. Genet. 2022, 13, 904536. [Google Scholar] [CrossRef]
- Montégut, L.; Martins, I.; Kroemer, G. Neutralization of the Autophagy-Repressive Tissue Hormone DBI/ACBP (Diazepam Binding Inhibitor, Acyl-CoA Binding Protein) Enhances Anticancer Immunosurveillance. Autophagy 2024, 20, 2836–2838. [Google Scholar] [CrossRef]
- Alquier, T.; Christian-Hinman, C.A.; Alfonso, J.; Færgeman, N.J. From Benzodiazepines to Fatty Acids and beyond: Revisiting the Role of ACBP/DBI. Trends Endocrinol. Metab. 2021, 32, 890–903. [Google Scholar] [CrossRef]
- Aristin Revilla, S.; Kranenburg, O.; Coffer, P.J. Colorectal Cancer-Infiltrating Regulatory T Cells: Functional Heterogeneity, Metabolic Adaptation, and Therapeutic Targeting. Front. Immunol. 2022, 13, 903564. [Google Scholar] [CrossRef]
- Hicks, K.C.; Tyurina, Y.Y.; Kagan, V.E.; Gabrilovich, D.I. Myeloid Cell-Derived Oxidized Lipids and Regulation of the Tumor Microenvironment. Cancer Res. 2022, 82, 187–194. [Google Scholar] [CrossRef]
- Hunt, E.G.; Hurst, K.E.; Riesenberg, B.P.; Kennedy, A.S.; Gandy, E.J.; Andrews, A.M.; Pauneto, C.D.M.A.; Ball, L.E.; Wallace, E.D.; Gao, P.; et al. Acetyl-CoA Carboxylase Obstructs CD8+ T Cell Lipid Utilization in the Tumor Microenvironment. Cell Metab. 2024, 36, 969–983.e10. [Google Scholar] [CrossRef]
- Al Zein, M.; Boukhdoud, M.; Shammaa, H.; Mouslem, H.; El Ayoubi, L.M.; Iratni, R.; Issa, K.; Khachab, M.; Assi, H.I.; Sahebkar, A.; et al. Immunotherapy and Immunoevasion of Colorectal Cancer. Drug Discov. Today 2023, 28, 103669. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A. Immunotherapy in Colorectal Cancer: Rationale, Challenges and Potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Zhao, Z.; Yang, M.; Ji, J.; Zhu, D. T-Cell-Based Immunotherapy in Colorectal Cancer. Cancer Lett. 2021, 498, 201–209. [Google Scholar] [CrossRef]
- Juat, D.J.; Hachey, S.J.; Billimek, J.; Del Rosario, M.P.; Nelson, E.L.; Hughes, C.C.W.; Zell, J.A. Adoptive T-Cell Therapy in Advanced Colorectal Cancer: A Systematic Review. Oncologist 2022, 27, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Wieder, T.; Mauerer, B.; Schäfer, L.; Kesselring, R.; Braumüller, H. T Cells in Colorectal Cancer: Unravelling the Function of Different T Cell Subsets in the Tumor Microenvironment. Int. J. Mol. Sci. 2023, 24, 11673. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Status | p-Value | Adjusted p-Value | Odds Ratio |
|---|---|---|---|---|
| Coenzyme A | Approved | N.A. | N.A. | N.A. |
| Hexadecanal | Experimental | N.A. | N.A. | N.A. |
| Benzo[b]fluoranthene | Candidate | 3.65 × 10−3 | 4.29 × 10−2 | 19,927 |
| Dibenz[a,h]anthracene | Candidate | 3.90 × 10−3 | 4.29 × 10−2 | 19,922 |
| baclofen | Candidate | 1.17 × 10−2 | 8.58 × 10−2 | 19,766 |
| diltiazem | Candidate | 1.82 × 10−2 | 9.17 × 10−2 | 19,636 |
| paclitaxel | Candidate | 2.08 × 10−2 | 9.17 × 10−2 | 19,583 |
| ambroxol | Candidate | 3.11 × 10−2 | 9.53 × 10−2 | 19,378 |
| puromycin | Candidate | 3.61 × 10−2 | 9.53 × 10−2 | 19,278 |
| hydralazine | Candidate | 3.88 × 10−2 | 9.53 × 10−2 | 19,224 |
| Vitinoin | Candidate | 3.90 × 10−2 | 9.53 × 10−2 | 19,220 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, T.; Han, H.; Huang, J.; Ma, J.; Ran, R. DBI as a Novel Immunotherapeutic Candidate in Colorectal Cancer: Dissecting Genetic Risk and the Immune Landscape via GWAS, eQTL, and pQTL. Biomedicines 2025, 13, 1115. https://doi.org/10.3390/biomedicines13051115
Tian T, Han H, Huang J, Ma J, Ran R. DBI as a Novel Immunotherapeutic Candidate in Colorectal Cancer: Dissecting Genetic Risk and the Immune Landscape via GWAS, eQTL, and pQTL. Biomedicines. 2025; 13(5):1115. https://doi.org/10.3390/biomedicines13051115
Chicago/Turabian StyleTian, Ting, Huan Han, Jingtao Huang, Jun’e Ma, and Ruoxi Ran. 2025. "DBI as a Novel Immunotherapeutic Candidate in Colorectal Cancer: Dissecting Genetic Risk and the Immune Landscape via GWAS, eQTL, and pQTL" Biomedicines 13, no. 5: 1115. https://doi.org/10.3390/biomedicines13051115
APA StyleTian, T., Han, H., Huang, J., Ma, J., & Ran, R. (2025). DBI as a Novel Immunotherapeutic Candidate in Colorectal Cancer: Dissecting Genetic Risk and the Immune Landscape via GWAS, eQTL, and pQTL. Biomedicines, 13(5), 1115. https://doi.org/10.3390/biomedicines13051115