

Genetic Heterogeneity in Four Probands Reveals HGSNAT, KDM6B, LMNA and WFS1 Related Neurodevelopmental Disorders


Abstract
1. Introduction
2. Materials and Methods
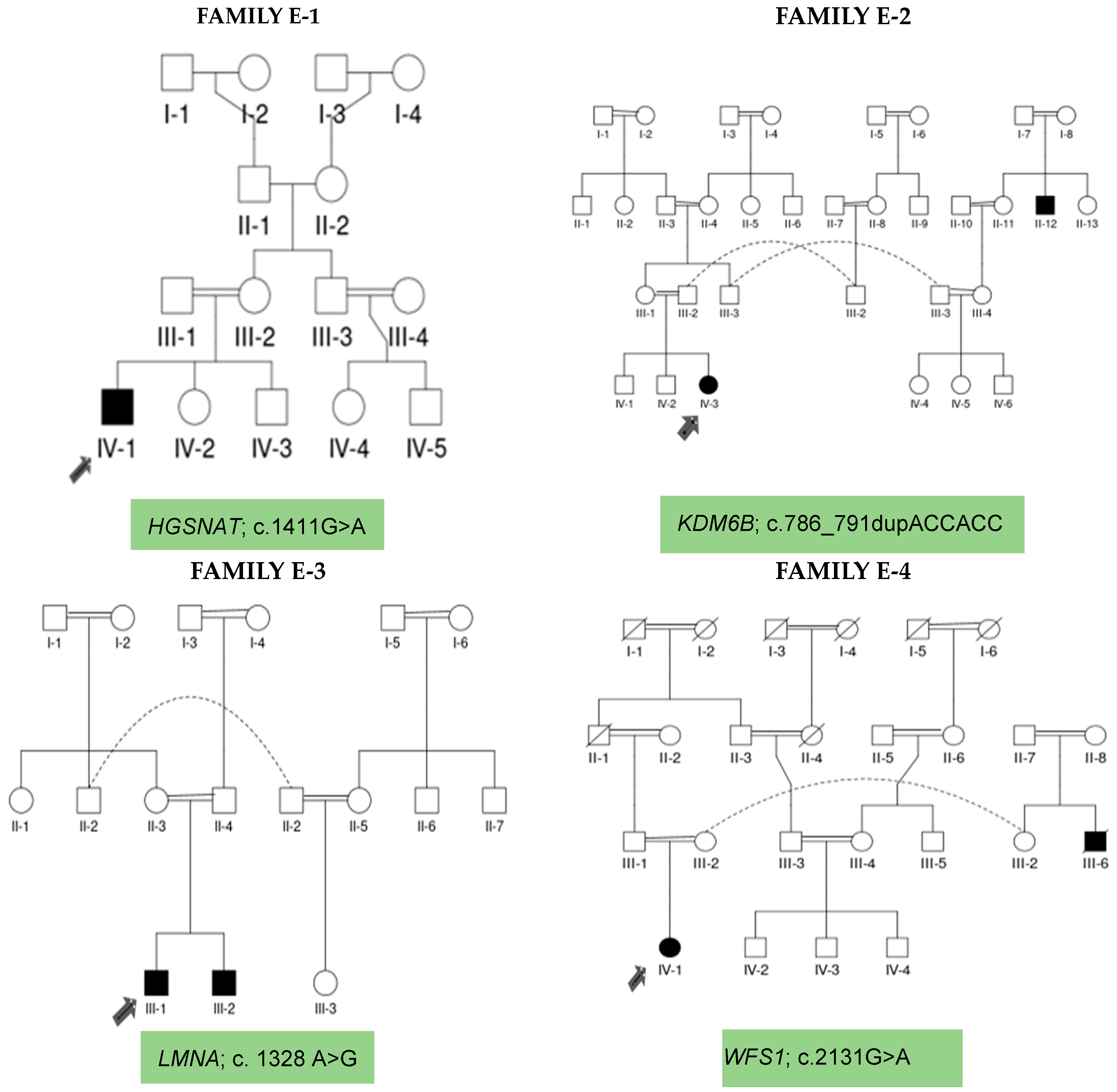
2.1. Enrolment of Probands in Four Families
2.2. Pedigree Analysis, Blood Sample Collection and DNA Extraction and Storage
2.3. Whole Exome Sequencing and Data Analysis
2.4. Validation of Variants and In Silico Protein Modelling
3. Results
3.1. Demographic/Clinical Characteristics of E-1, E-2, E-3, and E-4 Families
3.2. Whole Exome Sequencing Data Analysis and Variant Identification
3.3. Sanger Sequencing for Co-Segregation Analysis
3.4. In-Silico Protein Modelling
4. Discussion
5. Conclusions
Limitations
- Whole exome sequencing (WES) is an effective way of detecting mutations for monogenic hereditary illnesses. Although rapid and precise, WES fails to detect mutations in about 35% of instances due to diverse genetic variants of unknown significance, variants that are in deep intronic regions and hence ignored by exome capture techniques. This is a methodological constraint that precludes the efficient detection of sequence modifications.
- Complementary approaches, such as whole genome sequencing, may be used to aid in the discovery of disease-associated genetic changes.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NDD | neurodevelopmental disorder |
| ID | intellectual disability |
| DD | developmental delay |
| ER | endoplasmic reticulum |
| HS | heparan sulphate |
| CNS | central nervous system |
| BBH | Benazir Bhutto Hospital |
| RIC | Rawalpindi Institute of Cardiology |
References
- Khodosevich, K.; Sellgren, C.M. Neurodevelopmental disorders—High-resolution rethinking of disease modeling. Mol. Psychiatry 2023, 28, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Vasistha, N.A.; Pardo-Navarro, M.; Gasthaus, J.; Weijers, D.; Müller, M.K.; García-González, D.; Malwade, S.; Korshunova, I.; Pfisterer, U.; von Engelhardt, J.; et al. Maternal inflammation has a profound effect on cortical interneuron development in a stage and subtype-specific manner. Mol. Psychiatry 2020, 25, 2313–2329. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.E.; Martin, H.C.; Rice, D.L.; Gallone, G.; Gordon, S.; Kelemen, M.; McAloney, K.; McRae, J.; Radford, E.J.; Yu, S.; et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 2018, 562, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gao, X.; Liu, X.; Shen, L.; Wang, K.; Fan, Y.; Sun, Y.; Luo, X.; Liu, H.; Wang, L.; et al. Genetic Diagnostic Evaluation of Trio-Based Whole Exome Sequencing Among Children with Diagnosed or Suspected Autism Spectrum Disorder. Front. Genet. 2018, 9, 594. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.I.; Saarentaus, E.; Pietiläinen, O.; Gormley, P.; Lal, D.; Kerminen, S.; Torniainen-Holm, M.; Hämäläinen, E.; Rahikkala, E.; Keski-Filppula, R.; et al. Contribution of rare and common variants to intellectual disability in a sub-isolate of Northern Finland. Nat. Commun. 2019, 10, 410. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, L.; Jensen, M.; Polyak, A.; Rosenfeld, J.A.; Mannik, K.; Krishnan, A.; McCready, E.; Pichon, O.; Le Caignec, C.; Van Dijck, A.; et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet. Med. 2019, 21, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Iyer, J.; Singh, M.D.; Jensen, M.; Patel, P.; Pizzo, L.; Huber, E.; Koerselman, H.; Weiner, A.T.; Lepanto, P.; Vadodaria, K.; et al. Pervasive genetic interactions modulate neurodevelopmental defects of the autism-associated 16p11.2 deletion in Drosophila melanogaster. Nat. Commun. 2018, 9, 2548. [Google Scholar] [CrossRef]
- Wengert, E.R.; Tronhjem, C.E.; Wagnon, J.L.; Johannesen, K.M.; Petit, H.; Krey, I.; Saga, A.U.; Panchal, P.S.; Strohm, S.M.; Lange, J.; et al. Biallelic inherited SCN8A variants, a rare cause of SCN8A-related developmental and epileptic encephalopathy. Epilepsia 2019, 60, 2277–2285. [Google Scholar] [CrossRef]
- Guo, H.; Wang, T.; Wu, H.; Long, M.; Coe, B.P.; Li, H.; Xun, G.; Ou, J.; Chen, B.; Duan, G.; et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol. Autism 2018, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Akdemir, Z.H.C.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Liu, P.; Meng, L.; Normand, E.A.; Xia, F.; Song, X.; Ghazi, A.; Rosenfeld, J.; Magoulas, P.L.; Braxton, A.; Ward, P.; et al. Reanalysis of Clinical Exome Sequencing Data. N. Engl. J. Med. 2019, 380, 2478–2480. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Goyal, K.; Mathur, P.; Mathur, A. A Rare case of Sanfilippo syndrome type ‘C’. Indian J. Child Health 2020, 7, 236–238. [Google Scholar] [CrossRef]
- Rots, D.; Jakub, T.E.; Keung, C.; Jackson, A.; Banka, S.; Pfundt, R.; de Vries, B.B.; van Jaarsveld, R.H.; Hopman, S.M.; van Binsbergen, E.; et al. The clinical and molecular spectrum of the KDM6B-related neurodevelopmental disorder. Am. J. Hum. Genet. 2023, 110, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Transcriptional programs regulating neuronal differentiation are disrupted in DLG2 knockout human embryonic stem cells and enriched for schizophrenia and related disorders risk variants. Nat. Commun. 2022, 13, 27. [CrossRef]
- Yilmaz, S.; Beyazit, U.; Bütün Ayhan, A. Genetic Etiology of Neurodevelopmental Disorders. In The Palgrave Encyclopedia of Disability; Springer Nature: Berlin, Germany, 2024; Available online: https://link.springer.com/referenceworkentry/10.1007/978-3-031-40858-8_188-1 (accessed on 26 August 2024).
- Pineda, I.I.; González, C.L.G. The KDM6B mutation: Phenotype and clinical characteristics—Report of a case. Span. J. Psychiatry Ment. Health Engl. Ed. 2022, 15, 88–93. [Google Scholar] [CrossRef]
- Parry, D.A.; Martin, C.A.; Greene, P.; Marsh, J.A.; Blyth, M.; Cox, H.; Donnelly, D.; Greenhalgh, L.; Greville-Heygate, S.; Harrison, V. Heterozygous lamin B1 and lamin B2 variants cause primary microcephaly and define a novel laminopathy. Genet. Med. 2021, 23, 408–414. [Google Scholar] [CrossRef]
- Salih, N.N.M.; AL Azkwi, H.; AL Yahyai, M.; Almdhani, S. 60 Wolfram syndrome with variable presentation: Case series. BMJ Paediatr. Open 2024, 8 (Suppl. S4). [Google Scholar] [CrossRef]
- Liang, Y.; Gao, X.; Lu, D.; Zhang, H.; Zhang. Mucopolysaccharidosis type IIIC in chinese mainland: Clinical and molecular characteristics of ten patients and report of six novel variants in the HGSNAT gene. Metab. Brain Dis. 2023, 38, 2013–2023. [Google Scholar] [CrossRef]
- Cardoso, A.R.; Lopes-Marques, M.; Silva, R.M.; Serrano, C.; Amorim, A.; Prata, M.J.; Azevedo, L. Essential genetic findings in neurodevelopmental disorders. Hum. Genom. 2019, 13, 31. [Google Scholar] [CrossRef]
- Riazuddin, S.; Hussain, M.; Razzaq, A.; Iqbal, Z.; Shahzad, M.; Polla, D.L.; Song, Y.; Van Beusekom, E.; Khan, A.A.; Tomas-Roca, L.; et al. Exome sequencing of Pakistani consanguineous families identifies 30 novel candidate genes for recessive intellectual disability. Mol. Psychiatry 2017, 22, 1604–1614. [Google Scholar] [CrossRef]
- Velasco, H.M.; Sanchez, Y.; Martin, A.M.; Umaña, L.A. Natural History of Sanfilippo Syndrome Type C in Boyacá, Colombia: A Neurogenetic Description. J. Child Neurol. 2017, 32, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Mucopolysaccharidosis Type III—Abstract—Europe PMC. Available online: https://europepmc.org/article/NBK/nbk546574 (accessed on 28 August 2024).
- Kim, M.S.; Yang, A.; Noh, E.S.; Kim, C.; Bae, G.Y.; Lim, H.H.; Park, H.D.; Cho, S.Y.; Jin, D.K. Natural History and Molecular Characteristics of Korean Patients with Mucopolysaccharidosis Type III. J. Pers. Med. 2022, 12, 665. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.G.; Eisengart, J.B. The natural history of neurocognition in MPS disorders: A review. Mol. Genet. Metab. 2021, 133, 8–34. [Google Scholar] [CrossRef] [PubMed]
- Bruno, L. Pathophysiology of Synapse Function in Sanfilippo Syndrome. Master’s Thesis, McGill University, Montreal, QC, Canada, 2014. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwj6ivXG1fyJAxULgP0HHfx9PJwQFnoECBYQAQ&url=https%3A%2F%2Fescholarship.mcgill.ca%2Fconcern%2Ftheses%2Fwp988n90s&usg=AOvVaw3FPQirMBg8IC8Ghe-CxVCr&opi=89978449 (accessed on 28 August 2024).
- Schiff, E.R.; Varela, M.D.; Robson, A.G.; Pierpoint, K.; Ba-Abbad, R.; Nutan, S.; Zein, W.M.; Ullah, E.; Huryn, L.A.; Tuupanen, S.; et al. A genetic and clinical study of individuals with nonsyndromic retinopathy consequent upon sequence variants in HGSNAT, the gene associated with Sanfilippo C mucopolysaccharidosis. Am. J. Med Genet. Part C Semin. Med Genet. 2020, 184, 631–643. [Google Scholar] [CrossRef]
- Benetó, N.; Vilageliu, L.; Grinberg, D.; Canals, I. Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 7819. [Google Scholar] [CrossRef]
- Ludwig, J.; Sawant, O.B.; Wood, J.; Singamsetty, S.; Pan, X.; Bonilha, V.L.; Rao, S.; Pshezhetsky, A.V. Histological characterization of retinal degeneration in mucopolysaccharidosis type IIIC. Exp. Eye Res. 2023, 229, 109433. [Google Scholar] [CrossRef]
- Cyske, Z.; Anikiej-Wiczenbach, P.; Wisniewska, K.; Gaffke, L.; Pierzynowska, K.; Mański, A.; Wegrzyn, G. Sanfilippo Syndrome: Optimizing Care with a Multidisciplinary Approach. J. Multidiscip. Healthc. 2022, 15, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.N.; Isabel, C. Mucopolysaccharidosis Type IIIC: Molecular Defects and Pathophysiological Mechanism. 2022. Available online: https://papyrus.bib.umontreal.ca/xmlui/handle/1866/27635 (accessed on 28 August 2024).
- Mohammed, E.E.; Fayez, A.G.; Abdelfattah, N.M.; Fateen, E. Novel gene-specific Bayesian Gaussian mixture model to predict the missense variants pathogenicity of Sanfilippo syndrome. Sci. Rep. 2024, 14, 12148. [Google Scholar] [CrossRef]
- Zhang, C.; Ye, W.; Zhao, M.; Long, L.; Xia, D.; Fan, Z. KDM6B Negatively Regulates the Neurogenesis Potential of Apical Papilla Stem Cells via HES1. Int. J. Mol. Sci. 2023, 24, 10608. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A. KDM6B (JMJD3) and its dual role in cancer. Biochimie 2021, 184, 63–71. [Google Scholar] [CrossRef]
- Wright, H.; Aylwin, C.F.; Toro, C.A.; Ojeda, S.R.; Lomniczi, A. Polycomb represses a gene network controlling puberty via modulation of histone demethylase Kdm6b expression. Sci. Rep. 2021, 11, 1996. [Google Scholar] [CrossRef] [PubMed]
- Yalcintepe, S.; Gorker, I.; Bozatli, L.; Guler, H.S.; Zhuri, D.; Demir, S.; Atli, E.I.; Atli, E.; Eker, D.; Gurkan, H. Clinical exome sequencing reveals an important role for clinical diagnosis of intellectual disability with definition of seven novel variants. Neurol. Asia 2021, 26, 37–44. [Google Scholar] [CrossRef]
- Fichera, M.; Barone, R.; Grillo, L.; De Grandi, M.; Fiore, V.; Morana, I.; Maniscalchi, T.; Vinci, M.; Amata, S.; Spalletta, A.; et al. Familial 1q22 microduplication associated with psychiatric disorders, intellectual disability and late-onset autoimmune inflammatory response. Mol. Cytogenet. 2014, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Widyastuti, H.P.; Norden-Krichmar, T.M.; Grosberg, A.; Zaragoza, M.V. Gene expression profiling of fibroblasts in a family with LMNA-related cardiomyopathy reveals molecular pathways implicated in disease pathogenesis. BMC Med. Genet. 2021, 22, 152. [Google Scholar] [CrossRef] [PubMed]
- Storey, E.C.; Fuller, H.R. Genotype-Phenotype Correlations in Human Diseases Caused by Mutations of LINC Complex-Associated Genes: A Systematic Review and Meta-Summary. Cells 2024, 11, 4065. [Google Scholar] [CrossRef]
- Neri, I.; Ramazzotti, G.; Mongiorgi, S.; Rusciano, I.; Bugiani, M.; Conti, L.; Cousin, M.; Giorgio, E.; Padiath, Q.S.; Vaula, G.; et al. Understanding the Ultra-Rare Disease Autosomal Dominant Leukodystrophy: An Updated Review on Morpho-Functional Alterations Found in Experimental Models. Mol. Neurobiol. 2023, 60, 6362–6372. [Google Scholar] [CrossRef]
- Waszczykowska, A.; Zmysłowska, A.; Bartosiewicz, K.; Studzian, M.; Pułaski, Ł.; Braun, M.; Ivask, M.; Koks, S.; Jurowski, P.; Młynarski, W. Reduced Corneal Sensitivity with Neuronal Degeneration is a Novel Clinical Feature in Wolfram Syndrome. Am. J. Ophthalmol. 2022, 236, 63–68. [Google Scholar] [CrossRef]
- Kõks, S. Genomics of Wolfram Syndrome 1 (WFS1). Biomolecules 2023, 13, 1346. [Google Scholar] [CrossRef]
- Li, L.; Venkataraman, L.; Chen, S.; Fu, H. Function of WFS1 and WFS2 in the Central Nervous System: Implications for Wolfram Syndrome and Alzheimer’s disease. Neurosci. Biobehav. Rev. 2020, 118, 775–783. [Google Scholar] [CrossRef]
- Froukh, T.; Nafie, O.; Al Hait, S.A.S.; Laugwitz, L.; Sommerfeld, J.; Sturm, M.; Baraghiti, A.; Issa, T.; Al-Nazer, A.; Koch, P.A.; et al. Genetic basis of neurodevelopmental disorders in 103 Jordanian families. Clin. Genet. 2020, 97, 621–627. [Google Scholar] [CrossRef]
- Gavrilovici, C.; Jiang, Y.; Kiroski, I.; Sterley, T.L.; Vandal, M.; Bains, J.; Park, S.K.; Rho, J.M.; Teskey, G.C.; Nguyen, M.D. Behavioral Deficits in Mice with Postnatal Disruption of Ndel1 in Forebrain Excitatory Neurons: Implications for Epilepsy and Neuropsychiatric Disorders. Cereb. Cortex Commun. 2021, 2, tgaa096. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, J.; Morikawa, S.; Yagi, T.; Abreu, D.; Lu, S.; Kanekura, K.; Brown, C.M.; Urano, F. A soluble endoplasmic reticulum factor as regenerative therapy for Wolfram syndrome. Lab. Investig. 2020, 100, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Noormets, K.; Kõks, S.; Ivask, M.; Aunapuu, M.; Arend, A.; Vasar, E.; Tillmann, V. Energy Metabolism and Thyroid Function of Mice with Deleted Wolframin (Wfs1) Gene. Exp. Clin. Endocrinol. Diabetes 2014, 122, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Fauconnier, J.; Patergnani, S.; Rieusset, J.; Danese, A.; Affortit, C.A.; Jagodzinska, J.; Mégy, C.; Quiles, M.; Cazevieille, C.; et al. ER-mitochondria cross-talk is regulated by the Ca2+ sensor NCS1 and is impaired in Wolfram syndrome. Sci. Signal. 2018, 11, eaaq1380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, H.; Ming, G.-L. Genetics of human brain development. Nat. Rev. Genet. 2024, 25, 26–45. [Google Scholar] [CrossRef]
- Sultana, O.F.; Bandaru, M.; Islam, A.; Reddy, P.H. Unraveling the complexity of human brain: Structure, function in healthy and disease states. Ageing Res. Rev. 2024, 100, 102414. [Google Scholar] [CrossRef]
- Kumar, P.R.; Jha, R.K.; Kumar, P.A.; Raju, B.D. Improved neurological diagnoses and treatment strategies via automated human brain tissue segmentation from clinical magnetic resonance imaging. Intell. Med. 2024, 4, 161–169. [Google Scholar] [CrossRef]
- Khalaf, T.; Al Ojaimi, M.; Saleh, D.A.; Sulaiman, A.; Sohal, A.P.; Khan, A.; El-Hattab, A.W. The utility of exome sequencing in diagnosing pediatric neurodevelopmental disorders in a highly consanguineous population. Clin. Genet. 2024, 106, 82–89. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family ID | Ethnicity | Family Type | Proband Phenotype | Sibling Phenotype | Congenital Anomalies | Neurological Comorbidities | Family History for up to Five Previous Generations | Clinical Diagnosis | Genetic Identification |
|---|---|---|---|---|---|---|---|---|---|
| E-1 | Punjabi | Simplex | ID | Normal | Retinal degeneration | Seizures | Negative | NDD | Neuro degenerative Disorder |
| E-2 | Punjabi | Multiplex | ID | DD/Seizures | Tooth Decay Frontal Bossing | Seizures | Positive | NDD | NDD |
| E-3 | Punjabi | Simplex | ID | Normal | DD, DM, Cataract, Cardiac anomaly, Skin atrophy | Seizures | Positive | NDD | NDD |
| E-4 | Punjabi | Trio | ID | Microcephaly | Seizures | Positive | NDD | NDD |
| Members from Pedigrees | Family E-1 | Family E-2 | Family E-3 | Family E-4 | Tested by |
|---|---|---|---|---|---|
| Proband | IV-1 (+/+) | IV-3 (+/+) | III-1(+) | IV-1(+) | WES analysis |
| Parents | III-1, III-2 (+/−) | III-1, III-2 (+/−) | II-3, II-4 (−/−) | III-1, III-2 (−/−) | Co-segregation analyses by Sanger Sequencing |
| Siblings | IV-2, IV-3 (+/−) | IV-1, IV-2 (+/−) | III-2 (+) | Nil | |
| Close Relatives | IV-4, IV-5 (+/−) | III-3 (−/−) | II-1, II-2 (−/−) | IV-2, IV-3 (−/−) |
| Family ID | Proband Gender/Age | Gene | Exon | mRNA Variant | Protein Variant | Mutation Type | Mutation Effect |
|---|---|---|---|---|---|---|---|
| E-1 | M/7years | HGSNAT | 14 | NM_152419.3: c.1411G > A | p. Glu471Lys | Homozygous | Missense |
| E-2 | F/9 years | KDM6B | 10 | NM_001348716.2: c.786_791dupACCACC | p. Pro263_Pro264du | Homozygous | Non-frameshift |
| E-3 | M/16 years | LMNA | 8 | NM_170707.4: c. 1328 A > G | p. Glu443Gly | Heterozygous | Missense |
| E-4 | F/9years | WFS1 | 8 | NM_006005.3: c.2131G > A | p. Asp711Asn | Heterozygous | Missense |
| Gene | DECIPHER Values | Pathogenicity Prediction Values | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pHaplo | pTriplo | Missense Z-score | pLi | Polyphen2 | SIFT | MT | FATHMM | DANN | MetaLR | Fit Cons. | |
| HGSNAT | 0.79 | 0.56 | 2.85 | 0.92 | 0.9 (Damaging) | 0 (Del.) | 1 (Del.) | −4.21 (Del.) | 1 (Del.) | 0.53 (Del.) | 0.71 (Del.) |
| KDM6B | 0.89 | 0.93 | 2.51 | 1 | 0.8 | 0 | 1 | -4.5 | 1 | 0.56 | 0.8 |
| LMNA | 0.72 | 0.84 | 3.10 | 1 | 1 (Damaging) | 0 (Del). | 1 (Del.) | −4.87 (Del.) | 1 (Del.) | 0.94 (Del.) | 0.71 (Del.) |
| WFS1 | 0.27 | 0.13 | 2.43 | 0 | 0.92 (Damaging) | 0 (Del.) | 0.97 (Del.) | −4.24 (Del.) | 1 (Del.) | 0.85 (Del.) | 0.71 (Del.) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mudassir, B.U.; Mudassir, M.; Williams, J.B.; Agha, Z. Genetic Heterogeneity in Four Probands Reveals HGSNAT, KDM6B, LMNA and WFS1 Related Neurodevelopmental Disorders. Biomedicines 2024, 12, 2736. https://doi.org/10.3390/biomedicines12122736
Mudassir BU, Mudassir M, Williams JB, Agha Z. Genetic Heterogeneity in Four Probands Reveals HGSNAT, KDM6B, LMNA and WFS1 Related Neurodevelopmental Disorders. Biomedicines. 2024; 12(12):2736. https://doi.org/10.3390/biomedicines12122736
Chicago/Turabian StyleMudassir, Behjat Ul, Mujaddid Mudassir, Jamal B. Williams, and Zehra Agha. 2024. "Genetic Heterogeneity in Four Probands Reveals HGSNAT, KDM6B, LMNA and WFS1 Related Neurodevelopmental Disorders" Biomedicines 12, no. 12: 2736. https://doi.org/10.3390/biomedicines12122736
APA StyleMudassir, B. U., Mudassir, M., Williams, J. B., & Agha, Z. (2024). Genetic Heterogeneity in Four Probands Reveals HGSNAT, KDM6B, LMNA and WFS1 Related Neurodevelopmental Disorders. Biomedicines, 12(12), 2736. https://doi.org/10.3390/biomedicines12122736