Abstract
These days, in vitro functional analysis of gene variants is becoming increasingly important for risk stratification of cardiac ion channelopathies. So far, such risk stratification has been applied to SCN5A, KCNQ1, and KCNH2 gene variants associated with Brugada syndrome and long QT syndrome types 1 and 2, respectively, but risk stratification of HCN4 gene variants related to sick sinus syndrome has not yet been performed. HCN4 is the gene responsible for the hyperpolarization-activated ‘funny’ current If, which is an important modulator of the spontaneous diastolic depolarization underlying the sinus node pacemaker activity. In the present study, we carried out a risk classification assay on those loss-of-function mutations in HCN4 for which in vivo as well as in vitro data have been published. We used the in vitro data to compute the charge carried by If (Qf) during the diastolic depolarization phase of a prerecorded human sinus node action potential waveform and assessed the extent to which this Qf predicts (1) the beating rate of the comprehensive Fabbri–Severi model of a human sinus node cell with mutation-induced changes in If and (2) the heart rate observed in patients carrying the associated mutation in HCN4. The beating rate of the model cell showed a very strong correlation with Qf from the simulated action potential clamp experiments (R2 = 0.95 under vagal tone). The clinically observed minimum or resting heart rates showed a strong correlation with Qf (R2 = 0.73 and R2 = 0.71, respectively). While a translational perspective remains to be seen, we conclude that action potential clamp on transfected cells, without the need for further voltage clamp experiments and data analysis to determine individual biophysical parameters of If, is a promising tool for risk stratification of sinus bradycardia due to loss-of-function mutations in HCN4. In combination with an If blocker, this tool may also prove useful when applied to human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) obtained from mutation carriers and non-carriers.
1. Introduction
Over the past decade, risk stratification has become common practice for a large variety of diseases [1,2,3,4,5], including cardiac rhythm abnormalities [6,7,8,9,10,11,12]. Attempts at arrhythmic risk stratification are frequently based on patient clinical parameters, including electrical history and basic and advanced electrocardiographic indices [9,13,14,15,16,17,18,19,20]. However, the stratification of arrhythmic risk in patients can be difficult and controversial [15,17,21,22,23]. These days, genetic testing for the presence of gene variants is increasingly becoming part of the clinical management and risk stratification of cardiac ion channelopathies [6,15,24,25,26]. Variants of genes underlying the major ion channels involved in cardiac depolarization and repolarization can be classified as ‘pathogenic’, ‘likely pathogenic’, ‘variant of uncertain significance’, ‘likely benign’, and ‘benign’, according to a classification scheme developed by the American College of Medical Genetics and Genomics (ACMG) [27]. The ACMG criteria for the classification of pathogenic and benign variants include in vitro functional data, which for arrhythmia syndromes are mainly based on patch clamp experiments.
The patch clamp technique is a >40-year-old electrophysiological tool [28] and is considered the gold standard in electrophysiology because of its ability to measure both action potentials (APs) and specific membrane currents, and even single-channel currents, in detail [29]. Functional consequences of variations in genes encoding cardiac ion channels (for reviews, see Marbán [30], Schwartz et al. [10], and Wilde et al. [15]) can be studied in a large variety of models, such as isolated cells from genetically modified animals [31,32,33] and human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) [34,35,36]. However, mammalian cell lines in which the wild-type and the variant cDNA of a given ion channel can be expressed are still widely used to study the functional properties of ion currents [37,38]. Initially, patch clamp experiments were performed manually, but in recent years, the pathogenicity of SCN5A, KCNQ1, and KCNH2 variants involved in the inherited cardiac channelopathies Brugada syndrome and long QT syndrome types 1 and 2, respectively, have also been determined using high-throughput reclassification assays based on automated patch clamp devices [25,39,40,41,42,43,44]. Various biophysical parameters of the membrane currents have been used to classify the risk of genetic variants, but current density is the most reliable marker for predicting the risk of SCN5A [45], KCNQ1 [39], and KCNH2 [46] ion channel variants, followed by their voltage dependence of activation (through their half-activation voltage, V½).
Here, we performed a risk classification assay for sick sinus syndrome, a group of disorders with the common feature that the heart cannot adequately perform its normal pacemaking function [47,48,49]. Impaired cardiac pacemaker function may be due to reduced impulse formation in the sinoatrial node (SAN), defects in impulse conduction from SAN to atria, or widespread atrial electrophysiological abnormalities [49,50,51,52], but in the present study, we limited ourselves to familial sinus bradycardia due to loss-of-function mutations in HCN4. The HCN4 gene encodes the HCN4 protein, which is the major HCN isoform of the ion channels in the human SAN that mediate the hyperpolarization-activated ‘funny’ current If (also called ‘pacemaker current’) [53,54]. If is a depolarizing inward current during the diastolic depolarization phase of human SAN APs and fulfills an important modulatory role [55]. To date, several HCN4 variants have been identified, as reviewed by us and others [56,57,58,59,60], and they all affect pacemaker function by one or more alterations in the unique set of biophysical parameters of If, including activation upon hyperpolarization, time constants of activation and deactivation, reversal potential, modulation by cAMP, and density of channels. In the present study, we tested whether the affected biophysical parameters of HCN4 variants can explain the severity of sinus bradycardia. Therefore, we first selected from the literature those loss-of-function mutations in HCN4 that have been associated with familial sinus bradycardia and for which both clinical and in vitro data are available, with the condition that the clinical data include quantitative heart rate data from at least two mutation carriers. The in vitro data were then used to compute the charge carried by If (Qf) during the diastolic depolarization of a prerecorded human SAN AP waveform [61] as a measure of the physiological impact of If that can be readily determined in AP clamp experiments without the need for further voltage clamp experiments and data analysis to determine individual biophysical parameters of If [55,60,61]. We assessed the extent to which this Qf predicts (1) the beating rate of the comprehensive Fabbri–Severi model of a human SAN pacemaker cell [62] with mutation-induced changes in If, and (2) the heart rate observed in patients carrying the associated mutation in HCN4. We demonstrate that the beating rate of the model cell, as well as the clinically observed minimum or resting heart rate, show a strong correlation with Qf and conclude that risk stratification by AP clamp is a promising tool for risk stratification of sinus bradycardia due to loss-of-function mutations in HCN4.
2. Materials and Methods
2.1. Simulations of Action Potential Clamp Experiments
A prerecorded AP waveform from a single isolated human SAN cell [61], with a cycle length of 813 ms, was used to construct a train of 100 APs that could be employed as a command signal of ≈82 s duration under voltage clamp conditions, which was long enough to achieve stable behavior of the HCN4 current during the simulated AP clamp experiments. This typical AP waveform had been recorded from a pacemaker cell isolated from a human SAN in the whole cell configuration of the patch clamp technique [61]. Recordings were made at 36 ± 0.2 °C, and the extracellular Na+, K+, and Ca2+ concentrations amounted to 140, 5.4, and 1.8 mmol/L, respectively, whereas the recording pipette solution contained 145 mmol/L K+ and 5.0 mmol/L Na+. Of note, these ion concentrations are identical or almost identical to those in the Fabbri–Severi human SAN cell model [62] (Section 2.2). Only the intracellular K+ concentration of the model cell (140 mmol/L) is slightly different from the K+ concentration in the pipette solution (145 mmol/L).
The custom software to simulate such AP clamp experiments was compiled as a 32-bit Windows application using Intel Visual Fortran Composer XE 2013 and run on an Intel Core i7 processor-based workstation. For the numerical reconstruction of the HCN4 current, we used equations based on our experimental data on If acquired from the same set of single isolated human SAN cells [61], as described in detail by Verkerk et al. [63] and also employed by Fabbri et al. [62] in their Fabbri–Severi model of a human SAN pacemaker cell. We applied a simple and efficient Euler-type integration scheme with a time step of 10 μs for the numerical integration of the differential equations [64].
2.2. Simulations of the Electrical Activity of Human Sinoatrial Node Pacemaker Cells
The electrical activity of a single human SAN pacemaker cell was simulated using the comprehensive model of such a cell developed by Fabbri et al. [62], known as the Fabbri–Severi model, with updated equations for the slow delayed rectifier potassium current (IKs) [65]. Vagal tone was simulated by setting the model concentration of acetylcholine (ACh) to 20 nmol/L, whereas β-adrenergic tone was simulated by adopting our ‘High Iso’ settings [65] that are intermediate between the model settings used by Fabbri et al. [62] to simulate the administration of 1 µmol/L of isoprenaline and the model settings that they used to arrive at a pacemaking rate near 180 beats/min.
The CellML code [66] of the Fabbri–Severi model, available from the CellML Model Repository [67] at https://www.cellml.org/ (accessed on 9 June 2023), was edited and run in version 0.9.31.1409 of the Windows-based Cellular Open Resource (COR) environment [68]. All simulations were run for a period of 100 s, which was long enough to achieve stable behavior. The data analyzed are from the final five seconds of this 100 s period.
3. Results
First, we reviewed the scientific literature for publications on sinus bradycardia due to a loss-of-function mutation in HCN4, in which the clinical data on heart rate were preferably accompanied by in vitro data on the functional effects of the mutation of interest on the HCN4 current. If the clinical data were not accompanied by such experimental data, we reviewed the scientific literature for separate studies on the functional effects of the specific mutation, resulting in a set of loss-of-function mutations in HCN4 for which both clinical and in vitro data were available. These mutations are summarized in Figure 1 and include the R375C [69], R378C [70], A414G [71], G480R [72], Y481H [71,73], G482R [71,74,75], A485V [76], K530N [77], R550C [78], R666Q [79], and S672R [80] missense mutations, and the 695X truncating mutation [81].
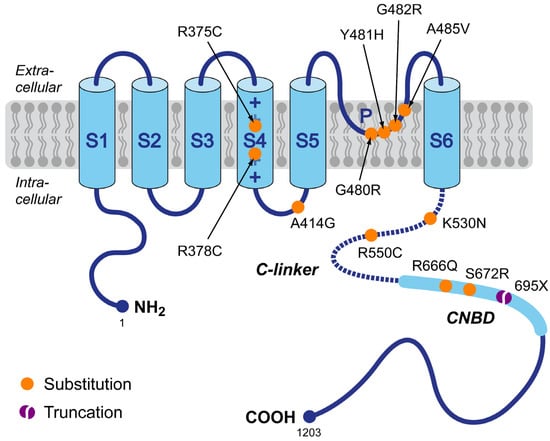
Figure 1.
Schematic topology of the HCN4 protein and the set of loss-of-function mutations in HCN4 associated with familial sinus bradycardia for which both clinical and in vitro data were available, requiring that these clinical data include quantitative heart rate data from at least two mutation carriers. Tetramers of HCN4 α-subunits form the cardiac ion channels that conduct the hyperpolarization-activated ‘funny’ current (If). The HCN4 protein has six transmembrane segments (S1–S6), a pore-forming loop (P), and intracellular N- and C-termini. The voltage sensor of the channel is formed by the positively charged S4 helix. The C-terminus contains the C-linker (dotted line) and the cyclic nucleotide-binding domain (CNBD), which is known to mediate cyclic AMP (cAMP)-dependent changes in HCN channel gating. Colored dots indicate the location of the loss-of-function mutations in the HCN4 protein of the present study. This set of mutations includes eleven substitutions (R375C, R378C, A414G, G480R, Y481H, G482R, A485V, K530N, R550C, R666Q, and S672R) and one truncation (695X).
The clinical and in vitro data resulting from our review are presented in Section 3.1 and Section 3.2, respectively. In these sections, we have only included mutations with quantitative data on heart rate in at least two mutation carriers, thus, for example, ignoring the initial studies by Schulze-Bahr et al. [82] and Ueda et al. [83] on the 573X and D553N mutations, respectively, and the parts of the studies by Schweizer et al. [74] and Möller et al. [70] dealing with the P883R mutation and the R550H and E1193Q mutations, respectively.
In Section 3.3, we use the in vitro data in Section 3.2 to compute the charge carried by If (Qf) during the diastolic depolarization of a prerecorded human SAN AP waveform for each of the collected mutations. In Section 3.4 and Section 3.5, we demonstrate the extent to which Qf can predict the beating rate of a single human sinus node pacemaker cell and the heart rate of mutation carriers, respectively.
3.1. Loss-of-Function Mutations in HCN4: Clinical Observations
Clinical data on heart rate in the presence of (all heterozygous) loss-of-function mutations in HCN4 are listed in Table 1 and Table 2, ordered by the location of the mutation sites on the HCN4 protein (Figure 1). In some studies, data are reported as minimum, average, and maximum heart rates from 24 h Holter recordings. Data from these studies are presented in Table 1. In a similar number of studies, data are reported as resting heart rates and maximum heart rates during exercise testing. Data from the latter studies are listed in Table 2. In several studies, on the G480R mutation by Nof et al. [72], the G482R mutation by Schweizer et al. [74] and Brunet-Garcia et al. [75], the A485 mutation by Laish-Farkash et al. [76], the K530N mutation by Duhme et al. [77], and the 695X mutation by Schweizer et al. [81], both types of data were reported. This explains why these studies appear in both Table 1 and Table 2.

Table 1.
Minimum, average, and maximum heart rates from 24 h Holter recordings.
Table 2.
Resting heart rates and maximum heart rates during exercise testing.
In Table 2, we have omitted the heart rate during exercise testing reported by Alonso-Fernández-Gatta et al. [69] for carriers of the R375C mutation. This is because this heart rate of only 81.5 ± 2.8 beats/min (n = 11) was not obtained after completion of the full Bruce protocol for exercise testing but rather after completion of the third stage of this six-stage protocol.
The R375C, G482R, and A485V mutations were also related to sinus bradycardia (and ventricular non-compaction) by Chanavat et al. [84], but these were all single cases, and no quantitative data, either in vivo or in vitro, were provided. In a study of left ventricular non-compaction, Richard et al. [85] presented a family with four heterozygous carriers of the G480C (p.Gly480Cys) mutation, all of whom had sinus bradycardia. However, no quantitative data were provided, neither on the bradycardia itself nor the functional effects of the mutation. The G480C mutation was also identified by Cambon-Viala et al. [86] in a single patient out of a group of 19 HCN4 mutation carriers with heart rates between 41 and 50 beats/min. Unfortunately, no further clinical data were reported and no functional studies were performed.
3.2. Loss-of-Function Mutations in HCN4: In Vitro Data
In this section, we present the functional effects of the loss-of-function mutations in HCN4 collected in Section 3.1, focusing on the functional differences between wild type (WT) and heteromeric mutant HCN4 channels, again ordered by the location of the mutation sites on the HCN4 protein (Figure 1). Also, we explain how we translated these experimentally observed differences in HCN4 current characteristics into changes in the parameter settings of If in our reconstructions of If during diastolic depolarization (Section 3.3) as well as in our simulations with the Fabbri–Severi model of a human SAN pacemaker cell [62] (Section 3.4). These changes in parameter settings are summarized in Table 3. The changes are limited to a decrease in the fully activated conductance of If (gf), representing an experimentally observed decrease in the fully activated HCN4 current, and/or a hyperpolarizing shift in the voltage dependence of the steady-state activation (y∞) curve and the bell-shaped (de)activation time constant (τy) curve. The shape of these curves and the effects of hyperpolarizing shifts are illustrated for the R375C mutation in Section 3.2.1 below.
Table 3.
Heteromeric mutation-induced changes in If characteristics derived from HCN4-transfected cells.
Only a few of the functional studies presented data on the sensitivity of heteromeric HCN4 mutant channels to cAMP. Such data are limited to the K530N [77] and 695X [81] mutations and are described in the corresponding subsections below. In other cases, data on the sensitivity to cAMP were only gathered for homomeric mutant channels or attempts to obtain data on sensitivity to cAMP failed. For the sake of completeness, these cases are also briefly documented below.
3.2.1. R375C (p.Arg375Cys)
In their recent study of the R375C mutation in HCN4, Alonso-Fernández-Gatta et al. [69] carried out whole cell patch clamp experiments at room temperature on Chinese hamster ovary (CHO) cells expressing WT, homomeric R375C mutant, or heteromeric R375C mutant (WT + R375C) HCN4 channels. Estimated from the graphical representation of their patch clamp data, the fully activated WT + R375C HCN4 current density at −140 mV was ≈50% of the current density of the WT current, whereas the steady-state activation curve was shifted by ≈−14 mV, as illustrated in Figure 2A, with no change in the reversal potential of the HCN4 current. The mutation-induced ≈50% decrease in fully activated HCN4 current was incorporated into the model as a 50% decrease in fully activated conductance of If (gf), whereas the shift in the steady-state activation curve was incorporated as a −14 mV shift in the voltage dependence of the If activation gate, thus also applying this hyperpolarizing shift to the bell-shaped (de)activation time constant curve, as illustrated in Figure 2B. This latter shift results in an increase in the time constant of activation at highly negative membrane potentials, which would explain the experimentally observed mutation-induced slowing of the HCN4 current activation at −130 mV.
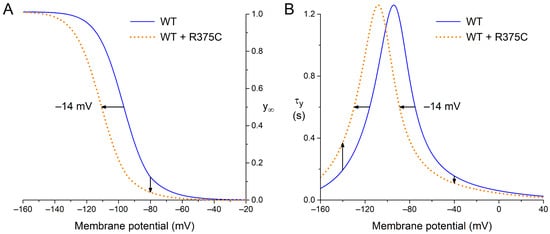
Figure 2.
Voltage dependence of wild type (WT; solid blue lines) and heteromeric R375C mutant (WT + R375C; orange dotted lines) HCN4 current (de)activation. (A) Steady-state activation (y∞). Horizontal arrow: the mutation-induced hyperpolarizing shift in half-maximum activation voltage. Vertical arrow: the mutation-induced decrease in maximally available HCN4 current. (B) Time constant of (de)activation (τy). Horizontal arrow: the mutation-induced hyperpolarizing shift in the bell-shaped curve. Upward vertical arrow: the mutation-induced decrease in the rate of (de)activation at highly negative membrane potentials. Downward vertical arrow: the mutation-induced increase in the rate of (de)activation at less negative membrane potentials.
3.2.2. R378C (p.Arg378Cys)
Möller et al. [70] recorded whole cell currents from Xenopus oocytes at room temperature in their study of the R378C mutation. These oocytes were injected with cRNAs to make them express WT, R378C, or WT + R378C HCN4 channels. Somewhat similar to R375 (Section 3.2.1), the fully activated WT + R378C HCN4 current was reduced by 57%, the steady-state activation curve was shifted by −7.9 mV, and activation was slowed at −140 mV, which were incorporated into the Fabbri–Severi model as a 57% decrease in the fully activated conductance of If and a −7.9 mV shift in the voltage dependence of the If activation gate (Table 3). Consistent with the associated −7.9 mV shift in the bell-shaped time constant curve, Möller et al. [70] observed a decrease in the deactivation time constant of the HCN4 current at +20 mV. Möller et al. [70] did not observe any ‘dramatic impairment of cAMP activation’ of the mutant channels.
3.2.3. A414G (p.Ala414Gly)
We have recently extended the voltage clamp experiments presented by Milano et al. [71] on CHO cells expressing WT or WT + A414G heterozygous mutant HCN4 channels, which were also carried out in our laboratory. For details on materials and methods, we, therefore, refer to the study by Milano et al. [71]. Using the amphotericin-perforated patch clamp technique at 36 ± 0.2 °C, we found that the half-maximal activation voltage (V½) of the WT + A414G current showed a shift of −19.9 mV relative to WT and the voltage dependence of its (de)activation time constant showed a shift of −11.9 mV, whereas no differences were observed in the slope factor (k) of the steady-state activation curve, the fully activated current density, and the reversal potential. Accordingly, a −19.9 mV shift in the steady-state activation curve and a −11.9 mV shift in the voltage dependence of the time constant of (de)activation were applied to the model If.
3.2.4. G480R (p.Gly480Arg)
In their study of the G480R mutation, Nof et al. [72] injected Xenopus oocytes with mRNAs to make them express WT, G480R, or WT + G480R HCN4 channels. In whole cell voltage clamp experiments at room temperature, they observed that the WT + G480R current activated more slowly and at more negative potentials than the WT current, with no change in the reversal potential. Also, the fully activated WT + G480R current density was substantially smaller than the WT current density. Unfortunately, their data on the heterozygous mutant current are limited. Roughly estimated, the WT + G480R kinetics showed a −10 mV shift in voltage dependence relative to WT, whereas the fully activated current was reduced by 54%. These estimated changes were applied to the If of the model cell as a combined −10 mV shift of the steady-state activation curve and the bell-shaped time constant curve and a 54% reduction in the fully activated If conductance. Nof et al. [72] were unable to test the β-adrenergic regulation of either WT or mutant channels in response to epinephrine. It is likely, as suggested by Nof et al. [72], that their attempts failed due to the high levels of endogenous cAMP in their oocytes.
3.2.5. Y481H (p.Tyr481His)
In the study by Milano et al. [71], HCN4 channels were expressed in CHO cells, and HCN4 currents were recorded at 37 ± 0.2 °C using the amphotericin-perforated patch clamp technique. The steady-state activation curve of the WT + Y481H mutant current showed a hyperpolarizing shift of as much as 44 mV compared to WT, whereas no difference in its slope factor k was observed. The apparent decrease in fully activated current density at −160 mV did not reach statistical significance. The effects of the mutation were implemented by a −44 mV shift in the steady-state activation curve of If. Given the common observation that a mutation-induced hyperpolarizing shift in the steady-state activation curve of If is accompanied by a similar shift in the bell-shaped time constant curve, we also applied this shift to the latter curve. Of note, the more recent study by Vermeer et al. [73], which identified a novel family with the Y481H mutation, focused on the structural effects of the mutation and did not include additional patch clamp data.
3.2.6. G482R (p.Gly482Arg)
Clinical data on the G482R mutation were presented by Milano et al. [71], Schweizer et al. [74], Brunet-Garcia et al. [75], and, with a strict focus on ventricular non-compaction, Cambon-Viala et al. [86]. Patch clamp data were only presented by Milano et al. [71] and Schweizer et al. [74]. Using the same experimental approach as for the Y481H mutation (see Section 3.2.5), Milano et al. [71] observed a −39 mV shift in the steady-state activation curve of the WT + G482R mutant current compared to WT, without a change in k. As also observed for the Y481H mutation, there was an apparent decrease in fully activated current density at −160 mV that did not reach statistical significance. In contrast to the findings by Milano et al. [71], Schweizer et al. [74], who expressed HCN4 channels in human embryonic kidney (HEK-293) cells for their whole cell patch clamp recordings at room temperature, did not observe any change in the steady-state activation curve or kinetic properties of the WT + G482R mutant current compared to WT, whereas the fully activated current density at −120 mV was reduced by 65%. We incorporated the experimental findings of Milano et al. [71] into the Fabbri–Severi model as a −39 mV shift in the steady-state activation curve of If (applying the same shift to the bell-shaped time constant curve) and those in Schweizer et al. [74] as a 65% decrease in its fully activated conductance.
3.2.7. A485V (p.Ala485Val)
Laish-Farkash et al. [76] injected Xenopus oocytes with WT and/or A485V mutant mRNA to let them express WT, A485V, or WT + A485V HCN4 channels. In whole cell voltage clamp experiments at room temperature, the WT + A485V HCN4 current activated more slowly and at more hyperpolarized potentials (below −80 vs. −65 mV) than the WT current, with no significant difference in the reversal potential. Also, the fully activated WT + A485V current was substantially smaller than the WT current. As a rough estimate, the HCN4 current kinetics showed a −15 mV shift in their voltage dependence, whereas the fully activated conductance was reduced by 68%. These estimated changes were applied to If of the model cell.
3.2.8. K530N (p.Lys530Asn)
In their study of the K530N mutation, Duhme et al. [77] performed whole cell patch clamp experiments at room temperature on transfected HEK-293 cells. Compared to WT, the heteromeric WT + K530N current activated more slowly at −120 mV and showed a −14 mV shift in V½, without a change in k, reversal potential, and fully activated current density at −120 mV. These experimental findings were incorporated into the cell model as a −14 mV shift in the voltage dependence of the If kinetics. Interestingly, the heteromeric WT + K530N channels showed a significantly higher sensitivity to cAMP, with a +7.5 mV larger cAMP-induced depolarizing shift in the steady-state activation curve and a significantly more accelerated activation at −120 mV. Surprisingly, the electrophysiological properties of the homomeric K530N channels were almost indistinguishable from WT channels. In our simulations, we accounted for the larger cAMP-induced shift through a +5 mV larger shift in our ‘High Iso’ settings of the voltage dependence of the If kinetics, which also resulted in a more accelerated activation at −120 mV.
3.2.9. R550C (p.Arg550Cys)
Campostrini et al. [78] transfected both CHO cells and neonatal rat ventricular cardiomyocytes (NRVCs) with WT, R550C, or WT + R550C hHCN4 and carried out whole cell patch clamp experiments at room temperature and 36 ± 1 °C, respectively, to assess the functional effects of the R550C mutation. The V½ of the WT + R550C current in CHO cells showed a small but statistically significant shift of −4.6 mV compared to WT. No differences were observed in other electrophysiological properties, including k, current density, and time constants of activation and deactivation. Highly similar results were obtained in NRVCs, with a small but statistically significant shift of −3.7 mV in V½ and no significant differences in k, current density, and time constants of activation and deactivation at multiple membrane potentials, except for a significantly smaller time constant of deactivation at −65 mV. The effects of the mutation were implemented by a −4 mV shift in the voltage dependence of the If kinetics, keeping in mind that a −4 mV shift in the membrane potential sensitivity of the time constant of (de)activation would be barely detectable. Homomeric WT and R550C channels showed a very similar sensitivity to cAMP.
3.2.10. R666Q (p.Arg666Gln)
Recently, Wang et al. [79] carried out whole cell patch clamp experiments at room temperature on HEK-293T cells expressing WT, homomeric R666Q mutant, or heteromeric R666Q mutant HCN4 channels. The V½ and k of the WT and mutant HCN4 currents were highly similar. However, the current density of WT + R666Q at −130 mV was significantly lower than WT. In 24 to 36 h and 36 to 48 h after transfection, it was 50% and 42% of WT, respectively. We incorporated these experimental results into the Fabbri–Severi model as a 54% decrease in the fully activated conductance of If. The sensitivity of homomeric WT and R666Q channels to cAMP was highly similar.
3.2.11. S672R (p.Ser672Arg)
The effects of the S672R mutation were determined by Milanesi et al. [80] through whole cell patch clamp experiments at room temperature on HEK-293 cells that were transfected to express WT or S672R homomeric or heteromeric mutant HCN4 channels. For heteromeric mutant channels, the changes in electrophysiological properties relative to WT were limited to a −4.9 mV shift in V½ and a slight decrease in the time constants of deactivation at multiple membrane potentials. These experimental findings were incorporated into the cell model as a −4.9 mV shift in the voltage dependence of the If kinetics (i.e., a combined −4.9 mV shift in the steady-state activation curve and the bell-shaped time constant curve). Homomeric WT and S672R channels showed a highly similar sensitivity to cAMP.
3.2.12. 695X (p.695X)
In their study of the 695X mutation, Schweizer et al. [81] performed whole cell patch clamp experiments at room temperature on transfected HEK-293 cells. Under cAMP-free conditions, the V½, k, and time constant of activation at −120 mV of the heteromeric WT + 695X channels were highly similar to the WT channels. Changes in electrophysiological properties were observed only in the presence of cAMP (10 µmol/L), as a result of the truncation of the cyclic nucleotide-binding domain (CNBD; Figure 1) due to an insertion of 13 nucleotides in exon 6 of the HCN4 gene that leads to a premature stop codon [81], which exerted a dominant-negative effect on the cAMP-induced increase in HCN4 current. These experimental findings were incorporated into the cell model as a fixed −10.9 mV shift in the voltage dependence of the If kinetics (which is the maximum shift at high ACh concentrations in the model equations).
3.3. Qf: Charge Carried by If during Diastolic Depolarization
Using the in vitro data on the biophysical effects of the loss-of-function mutations in HCN4 in Section 3.2, we computed the charge that is carried by the associated heteromeric mutant If during diastolic depolarization of a human SAN pacemaker cell (Qf). To this end, we simulated AP clamp experiments using a prerecorded human SAN AP waveform [61] and If equations based on our patch clamp data on If in human SAN cells [63]. As already set out in Section 2.1, these equations are also part of the Fabbri–Severi model [62].
Figure 3, A and B, show the prerecorded AP with its diastolic depolarization and the associated reconstructed WT If, which contributes to the diastolic depolarization as an inward current that carries a charge of 1.00 pC (Figure 3B, filled area). Reconstructing If with its gf halved and its voltage dependence shifted by −14 mV, as listed in Table 3 and set out in Section 3.2.1, we obtained the WT + R375C If, which is much smaller in amplitude and carries a charge of only 0.20 pC during diastolic depolarization (Figure 3C). Similarly, we reconstructed If for each of the other (heterozygous) mutations listed in Table 3 (and described in detail in Section 3.2) and computed Qf. The obtained data on Qf are summarized in Table 4.
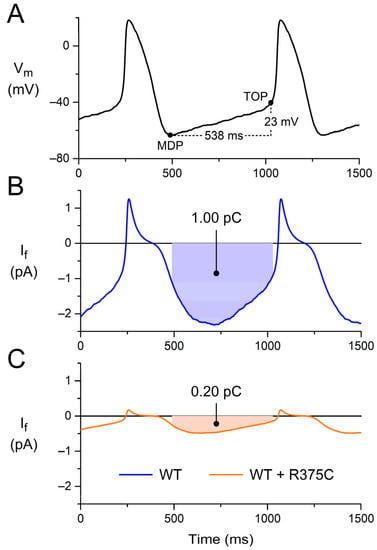
Figure 3.
Charge carried by If during diastolic depolarization. (A) Prerecorded AP waveform of an isolated human sinus node pacemaker cell. During the diastolic depolarization from the maximum diastolic potential (MDP) to the take-off potential (TOP), which takes 538 ms, the membrane potential (Vm) depolarizes by 23 mV. (B) Associated reconstructed WT If, which carries a charge of 1.00 pC (filled area) as an inward current during diastolic depolarization. (C) Associated reconstructed WT + R375C If, which carries a charge of 0.20 pC during diastolic depolarization. The AP waveform of panel A is a typical waveform obtained from a set of single isolated human SAN pacemaker cells [61], and the If curve of panel B is reconstructed from this typical AP waveform and the If equations of the Fabbri–Severi model [62], which are based on the If data obtained in voltage clamp experiments on the same set of single-isolated human SAN pacemaker cells [61,63].
Table 4.
Charge carried by If during diastolic depolarization of a human SAN pacemaker cell.
3.4. Can Qf Predict the Beating Rate of a Single Human Sinus Node Pacemaker Cell?
We questioned to what extent the data on Qf obtained from (simulated) AP clamp experiments can predict the beating rate of a single human sinus node pacemaker cell. Therefore, we performed computer simulations with the Fabbri–Severi model under control conditions (default ‘wild-type’ If) and for each of the (heterozygous) mutations listed in Table 3 at different levels of autonomic tone. Figure 4 shows the results that we obtained for the WT + R375C mutation compared to WT. Under vagal tone (20 nmol/L ACh; Figure 4A), the modulatory If is already small. Yet, the cycle length is substantially increased due to the mutation-induced decrease in If, and the beating rate is reduced from 42.4 to 29.3 beats/min. With the default model (no rate modulation; Figure 4B), where If is larger than under vagal tone, the mutation-induced decrease in If results in a decrease in the beating rate from 70.2 to 56.9 beats/min. Under β-adrenergic tone (‘High Iso’; Figure 4C), If exerts its modulatory role and is again larger. The mutation-induced decrease in If now results in a decrease in the beating rate from 109.5 to 81.2 beats/min.
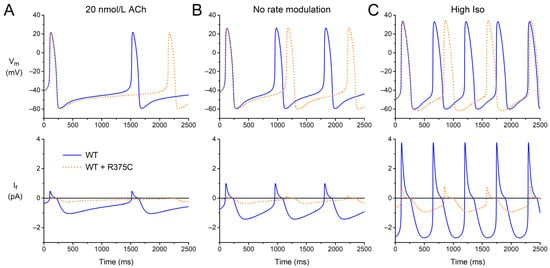
Figure 4.
Electrical activity of the Fabbri–Severi model of a human SAN pacemaker cell with its default ‘wild-type’ If (WT; solid blue lines) and heteromeric R375C mutant If (WT + R375C; orange dotted lines) at different levels of autonomic tone. (A) Vagal tone (simulated ACh concentration of 20 nmol/L). (B) No rate modulation (default model). (C) β-Adrenergic tone (‘High Iso’ settings of the model).
Having obtained data on the beating rate, as illustrated in Figure 4, for each of the mutations, we plotted these beating rates against the associated Qf from the simulated AP clamp experiments (Table 4), resulting in Figure 5. At each level of autonomic tone, there is a very strong correlation between the beating rate and Qf, suggesting that data on Qf obtained in AP clamp experiments on cells expressing the (heteromeric) HCN4 channels of interest, compared to Qf for wild type channels, may predict the amount of sinus bradycardia in mutation carriers.
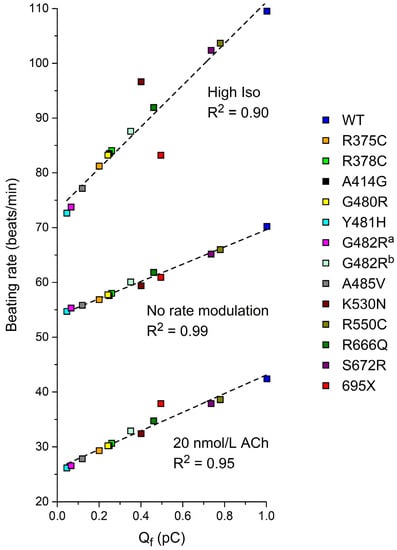
Figure 5.
The beating rate of the Fabbri–Severi model of a human SAN pacemaker cell with its default ‘wild-type’ If (WT) and heteromeric mutant If, simulated with the settings presented in Section 3.2 as a function of Qf (Section 3.3) at different levels of autonomic tone. a If parameters are based on Milano et al. [71]. b If parameters are based on Schweizer et al. [74]. Dashed lines are linear fits.
3.5. Can Qf Predict the Heart Rate of Mutation Carriers?
Keeping in mind that the promising results of Section 3.4 are based on the ideal case of an If in a (simulated) human SAN cell with biophysical parameters that are completely identical to those of the HCN4 current in an expression system, we assessed to what extent the data on Qf obtained from (simulated) AP clamp experiments can predict the clinically observed heart rates of mutation carriers. To this end, we first plotted the minimum, average, and maximum heart rates from 24 h Holter recordings (Table 1) against the associated Qf (Table 4), resulting in Figure 6. The clinically observed minimum heart rate shows a strong correlation with Qf (R2 = 0.73; p < 0.001, ANOVA). The average heart rate shows a less clear relationship (R2 = 0.56) but is still statistically significant (p = 0.002). For the maximum heart rate, the relationship is not statistically significant (p = 0.07).
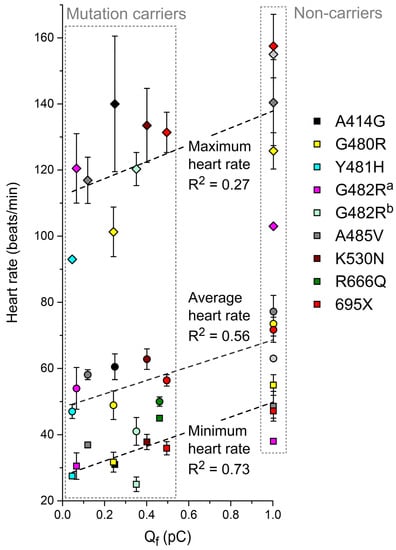
Figure 6.
Minimum, average, and maximum heart rates obtained during 24 h Holter recordings from heterozygous carriers of the mutations in HCN4 as indicated or from non-carriers of the same family (Table 1) as a function of Qf (Section 3.3). a If parameters are based on Milano et al. [71]. b If parameters are based on Schweizer et al. [74]. Dashed lines are linear fits.
We also plotted the clinically observed resting heart rates and maximum heart rates during exercise testing in Table 2 against the associated Qf. As shown in Figure 7A, the resting heart rate shows a strong correlation with Qf (R2 = 0.71; p = 0.001). Data on the maximum heart rate during exercise testing are limited, and no clear correlation can be discerned (Figure 7B).
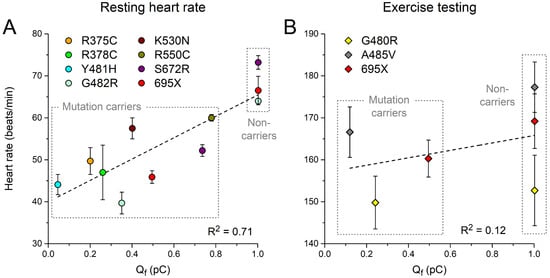
Figure 7.
(A) Resting heart rates and (B) maximum heart rates during exercise testing from heterozygous carriers of the mutations in HCN4 as indicated or from non-carriers of the same family (Table 2) as a function of Qf (Section 3.3). Dashed lines are linear fits.
4. Discussion
These days, risk stratification of ion channelopathies is largely based on changes in the electrophysiological properties of ion channel variants. So far, such risk stratification has been performed not only for a selection of cardiac ion channelopathies, as already outlined in the Introduction section, but also for neuromuscular diseases with NaV1.4 variants [87], hearing loss related to KCNQ4 variants [88], and encephalopathies, including schizophrenia, with CACNA1I variants [89], and epilepsy with SCN1A [90], KCNB1 [91], HCN1 [92], KCNQ2 [93], and SCN2A [94] variants. In the present study, we tested whether risk stratification for sinus bradycardia can be based on AP clamp experiments on transfected cells to compute the charge carried by (mutant) If during the diastolic depolarization phase of a prerecorded human SAN AP, using this AP as command potential. The mutation-induced changes in the biophysical parameters of an HCN4 variant of interest are summarized in the mutation-induced change in this charge, without the need to characterize each of these parameters separately.
In our study, we first reviewed the scientific literature for publications on loss-of-function mutations in HCN4 with both clinical and in vitro data on the effects of the mutation. This resulted in clinical data on heart rate (Table 1 and Table 2) and associated in vitro data on (changes in) HCN4 current characteristics (Table 3) for a total of 12 mutations (Figure 1). This rather small number of mutations highlights the unfortunate situation that clinical studies identifying mutations in HCN4 with potentially bradycardic effects are not always accompanied by in vitro data on their functional effects. Perhaps the best example is the extensive clinical study by Hategan et al. [95], which identified the novel c.1737 + 1 G > T splice-site mutation in HCN4 in a large family with familial bradycardia. Although it is highly likely that the c.1737 + 1 G > T mutation is disease-causing, it would be very interesting to know its functional effects. This is all the more important for less extensive clinical studies, where only one or a few mutation carriers have been identified. The importance of in vitro data in such studies is, for example, underscored in the study of Erlenhardt et al. [96], who identified the V759I mutation in HCN4, which had previously been identified in cases of sudden infant death syndrome [97] and sudden unexpected death in epilepsy in a patient with severe sinus bradycardia [98]. Erlenhardt et al. [96] were the first to perform functional studies on the mutation they identified. Patch clamp experiments on Xenopus oocytes showed that voltage dependence, activation kinetics, sensitivity to cAMP, and cell surface expression of mutant channels were all indistinguishable from wild type channels.
After obtaining our clinical and in vitro data, we used the in vitro data to compute Qf—i.e., the charge carried by If during the diastolic depolarization phase of a prerecorded human sinus node action potential waveform (Figure 3)—in simulated AP clamp experiments for each of the mutations as a potential measure of their severity. Also, we used these in vitro data to test the functional effects of each of the mutations on the beating rate of the comprehensive Fabbri–Severi model of a human sinus node cell (Figure 4). These in silico experiments revealed a very strong correlation between the beating rate and Qf (Figure 5), suggesting that this (relatively) readily obtained Qf may prove a useful tool for risk stratification of sinus bradycardia due to loss-of-function mutations in HCN4. A high correlation could be anticipated from the use of identical If equations [63] in our reconstructions of If under action potential clamp conditions and in the Fabbri–Severi model [62].
With R2 values of 0.73 and 0.71, respectively, the clinically observed minimum or resting heart rates (Table 1 and Table 2) show a strong correlation with Qf (Figure 6 and Figure 7). However, the maximum heart rate during 24 h Holter recordings (Figure 6) and during exercise testing (Figure 7) do not show a clear correlation with Qf. This may reflect a less pronounced role of If under β-adrenergic tone, when other ion currents contributing to diastolic depolarization are substantially upregulated, in particular the L-type and T-type calcium currents [99,100]. Alternatively, or simultaneously, the loss of a clear correlation between the clinical data and Qf at high rates may point to shortcomings in the ‘High Iso’ settings that we applied to the Fabbri–Severi model to simulate β-adrenergic tone [65]. One such shortcoming is that the T-type calcium current is not considered a β-adrenergic target. Fabbri et al. [62] had to develop their model in the absence of an electrophysiological characterization of the β-adrenergic targets in the human sinus node, thus potentially requiring updates when quantitative data from human tissue become available. Furthermore, it should be kept in mind that the Fabbri–Severi model is a single-cell model, whereas the interaction of the human sinus node with its atrial surroundings is also dependent on β-adrenergic tone, as individual ion currents of the atrial myocytes are also up- or downregulated under β-adrenergic tone [101,102].
In the present study, we based our computations of Qf on the in vitro data that are summarized in Table 3. Thus, we implicitly assumed that all of the experimentally observed mutation-induced effects on HCN4 currents in expression systems also apply to If in human SAN pacemaker cells from mutation carriers under physiological conditions. Although all mutation-induced effects listed in Table 3 are relative changes (i.e., reductions in fully activated conductance and/or shifts in the voltage dependence of steady-state activation and time constants of (de)activation), it may well be that these mutation-induced effects are quantitatively or even qualitatively dependent on the specific expression system and recording temperature or other recording conditions, which show essential differences between studies from different laboratories (Table 3). In a direct comparison of the effects of the p.T1620M mutation in the SCN5A gene, encoding the pore-forming α-subunit of the cardiac NaV1.5 fast sodium channel, Baroudi et al. [103] even obtained opposite phenotypes depending on the expression system (Xenopus oocytes vs. mammalian tsA201 cells; both at room temperature). Studying the same p.T1620M mutation in SCN5A in tsA201 cells at 22 °C and 32 °C, Dumaine et al. [104] observed a mutation-induced acceleration of current decay at 32 °C but not 22 °C, directly demonstrating a temperature dependence of the nature of the mutation-induced effects. Differences in the expression system (CHO vs. HEK-293 cells) and recording conditions (37 ± 0.2 °C vs. room temperature; amphotericin-perforated vs. whole cell patch clamp technique) may explain, at least in part, why Milano et al. [71] and Schweizer et al. [74] obtained essentially different data on WT + G482R channels (−39 mV shift in the steady-state activation curve vs. 65% reduction in fully activated conductance). In contrast, Campostrini et al. [78] obtained very similar data on WT + R550C channels in CHO cells at room temperature and in NRVCs at 36 ± 1 °C (both whole cell patch clamp recordings).
5. Conclusions
While a translational perspective remains to be seen, we conclude that AP clamp on transfected cells, without the need for further voltage clamp experiments and data analysis to determine individual biophysical parameters of If, is a promising tool for risk stratification of sinus bradycardia due to loss-of-function mutations in HCN4. In combination with an If blocker, this tool may also prove useful when applied to human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) obtained from mutation carriers and non-carriers.
Author Contributions
Conceptualization, A.O.V. and R.W.; all other aspects, including methodology, investigation, and writing, A.O.V. and R.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are available from the academic researchers upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef]
- Reddy, A.; Sessums, L.; Gupta, R.; Jin, J.; Day, T.; Finke, B.; Bitton, A. Risk stratification methods and provision of care management services in comprehensive primary care initiative practices. Ann. Fam. Med. 2017, 15, 451–454. [Google Scholar] [CrossRef]
- Dom Dera, J. Risk stratification: A two-step process for identifying your sickest patients. Fam. Pract. Manag. 2019, 26, 21–26. [Google Scholar]
- Girwar, S.-A.M.; Jabroer, R.; Fiocco, M.; Sutch, S.P.; Numans, M.E.; Bruijnzeels, M.A. A systematic review of risk stratification tools internationally used in primary care settings. Health Sci. Rep. 2021, 4, e329. [Google Scholar] [CrossRef]
- Taylor, L.C.; Law, K.; Hutchinson, A.; Dennison, R.A.; Usher-Smith, J.A. Acceptability of risk stratification within population-based cancer screening from the perspective of healthcare professionals: A mixed methods systematic review and recommendations to support implementation. PLoS ONE 2023, 18, e0279201. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Schwartz, P.J.; Napolitano, C.; Bloise, R.; Ronchetti, E.; Grillo, M.; Vicentini, A.; Spazzolini, C.; Nastoli, J.; Bottelli, G.; et al. Risk stratification in the long-QT syndrome. N. Engl. J. Med. 2003, 348, 1866–1874. [Google Scholar] [CrossRef]
- Daliento, L.; Mazzotti, E.; Pomiato, E.; Spadotto, V.; Bauce, B. Arrhythmic risk stratification in adults with congenital heart disease. J. Cardiovasc. Disord. 2015, 2, 1013. [Google Scholar]
- Arevalo, H.J.; Vadakkumpadan, F.; Guallar, E.; Jebb, A.; Malamas, P.; Wu, K.C.; Trayanova, N.A. Arrhythmia risk stratification of patients after myocardial infarction using personalized heart models. Nat. Commun. 2016, 7, 11437. [Google Scholar] [CrossRef] [PubMed]
- Gatzoulis, K.A.; Tsiachris, D.; Arsenos, P.; Antoniou, C.-K.; Dilaveris, P.; Sideris, S.; Kanoupakis, E.; Simantirakis, E.; Korantzopoulos, P.; Goudevenos, I.; et al. Arrhythmic risk stratification in post-myocardial infarction patients with preserved ejection fraction: The PRESERVE EF study. Eur. Heart J. 2019, 40, 2940–2949. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Ackerman, M.J.; Antzelevitch, C.; Bezzina, C.R.; Borggrefe, M.; Cuneo, B.F.; Wilde, A.A.M. Inherited cardiac arrhythmias. Nat. Rev. Dis. Primers 2020, 6, 58. [Google Scholar] [CrossRef]
- Du, D.; Li, C.O.Y.; Ong, K.; Parsa, A.; Weissler-Snir, A.; Geske, J.B.; Laksman, Z. Arrhythmia monitoring for risk stratification in hypertrophic cardiomyopathy. CJC Open 2022, 4, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Tse, G.; Zhang, N.; Song, W.; Letsas, K.P.; Ngarmukos, T.; Jeevaratnam, K.; Liu, T. Editorial: Risk stratification strategies for cardiac rhythm abnormalities. Front. Cardiovasc. Med. 2022, 9, 887461. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, C.; Nederend, I.; Hofman, N.; Van Geloven, N.; Ebink, C.; Frohn-Mulder, I.M.E.; Alings, A.M.W.; Bosker, H.A.; Bracke, F.A.; Van den Heuvel, F.; et al. Familial evaluation in catecholaminergic polymorphic ventricular tachycardia: Disease penetrance and expression in cardiac ryanodine receptor mutation-carrying relatives. Circ. Arrhythm. Electrophysiol. 2012, 5, 748–756. [Google Scholar] [CrossRef] [PubMed]
- El-Battrawy, I.; Besler, J.; Liebe, V.; Schimpf, R.; Tülümen, E.; Rudic, B.; Lang, S.; Wolpert, C.; Zhou, X.; Akin, I.; et al. Long-term follow-up of patients with short QT syndrome: Clinical profile and outcome. J. Am. Heart Assoc. 2018, 7, e010073. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Amin, A.S.; Postema, P.G. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 2022, 108, 332–338. [Google Scholar] [CrossRef]
- Arsenos, P.; Gatzoulis, K.A.; Tsiachris, D.; Dilaveris, P.; Sideris, S.; Sotiropoulos, I.; Archontakis, S.; Antoniou, C.K.; Kordalis, A.; Skiadas, I.; et al. Arrhythmic risk stratification in ischemic, non-ischemic and hypertrophic cardiomyopathy: A two-step multifactorial, electrophysiology study inclusive approach. World J. Cardiol. 2022, 14, 139–151. [Google Scholar] [CrossRef]
- Darar, C.; Mohammed, E.-A.; Mohammed, B.; Noha, E.O.; Zakaria, B. Risk stratification of sudden cardiac death in Brugada syndrome: An updated review of literature. Egypt. Heart J. 2022, 74, 25. [Google Scholar] [CrossRef]
- Rieder, M.; Kreifels, P.; Stuplich, J.; Ziupa, D.; Servatius, H.; Nicolai, L.; Castiglione, A.; Zweier, C.; Asatryan, B.; Odening, K.E. Genotype-specific ECG-based risk stratification approaches in patients with long-QT syndrome. Front. Cardiovasc. Med. 2022, 9, 916036. [Google Scholar] [CrossRef]
- Leung, J.; Lee, S.; Zhou, J.; Jeevaratnam, K.; Lakhani, I.; Radford, D.; Coakley-Youngs, E.; Pay, L.; Çinier, G.; Altinsoy, M.; et al. Clinical characteristics, genetic findings and arrhythmic outcomes of patients with catecholaminergic polymorphic ventricular tachycardia from China: A systematic review. Life 2022, 12, 1104. [Google Scholar] [CrossRef]
- Aizawa, T.; Wada, Y.; Hasegawa, K.; Huang, H.; Imamura, T.; Gao, J.; Kashiwa, A.; Kohjitani, H.; Fukuyama, M.; Kato, K.; et al. Non-missense variants of KCNH2 show better outcomes in type 2 long QT syndrome. Europace 2023, 25, 1491–1499. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Wells, Q.S.; Stallings, S.; Bush, W.S.; Gordon, A.; Nickerson, D.A.; Kim, J.H.; Crosslin, D.R.; Jarvik, G.P.; Carrell, D.S.; et al. Association of arrhythmia-related genetic variants with phenotypes documented in electronic medical records. JAMA 2016, 315, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, A.; Brown, P.F.; Bradley, J.; Nucifora, G.; Claver, E.; De Frutos, F.; Dallaglio, P.D.; Comin-Colet, J.; Anguera, I.; Miller, C.A.; et al. Improved risk stratification for ventricular arrhythmias and sudden death in patients with nonischemic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2021, 77, 2890–2905. [Google Scholar] [CrossRef]
- Chung, C.T.; Bazoukis, G.; Lee, S.; Liu, Y.; Liu, T.; Letsas, K.P.; Armoundas, A.A.; Tse, G. Machine learning techniques for arrhythmic risk stratification: A review of the literature. Int. J. Arrhythmia 2022, 23, 10. [Google Scholar] [CrossRef]
- Napolitano, C.; Mazzanti, A.; Priori, S.G. Genetic risk stratification in cardiac arrhythmias. Curr. Opin. Cardiol. 2018, 33, 298–303. [Google Scholar] [CrossRef]
- Glazer, A.M.; Davogustto, G.; Shaffer, C.M.; Vanoye, C.G.; Desai, R.R.; Farber-Eger, E.H.; Dikilitas, O.; Shang, N.; Pacheco, J.A.; Yang, T.; et al. Arrhythmia variant associations and reclassifications in the eMERGE-III sequencing study. Circulation 2022, 145, 877–891. [Google Scholar] [CrossRef] [PubMed]
- De Brouwer, R.; Bosman, L.P.; Gripenstedt, S.; Wilde, A.A.M.; Van den Berg, M.P.; Van Tintelen, J.P.; De Boer, R.A.; Te Riele, A.S.J.M.; on behalf of the Netherlands ACM Registry. Value of genetic testing in the diagnosis and risk stratification of arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 2022, 19, 1659–1665. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hamill, O.P.; Marty, A.; Neher, E.; Sakmann, B.; Sigworth, F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981, 391, 85–100. [Google Scholar] [CrossRef]
- Kornreich, B.G. The patch clamp technique: Principles and technical considerations. J. Vet. Cardiol. 2007, 9, 25–37. [Google Scholar] [CrossRef]
- Marbán, E. Cardiac channelopathies. Nature 2002, 415, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.O.; Remme, C.A. Zebrafish: A novel research tool for cardiac (patho)electrophysiology and ion channel disorders. Front. Physiol. 2012, 3, 255. [Google Scholar] [CrossRef]
- Clauss, S.; Bleyer, C.; Schüttler, D.; Tomsits, P.; Renner, S.; Klymiuk, N.; Wakili, R.; Massberg, S.; Wolf, E.; Kääb, S. Animal models of arrhythmia: Classic electrophysiology to genetically modified large animals. Nat. Rev. Cardiol. 2019, 16, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Odening, K.E.; Gomez, A.-M.; Dobrev, D.; Fabritz, L.; Heinzel, F.R.; Mangoni, M.E.; Molina, C.E.; Sacconi, L.; Smith, G.; Stengl, M.; et al. ESC Working Group on Cardiac Cellular Electrophysiology position paper: Relevance, opportunities, and limitations of experimental models for cardiac electrophysiology research. Europace 2021, 23, 1795–1814. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, M.; Mummery, C.L.; Wilde, A.A.M.; Bezzina, C.R.; Verkerk, A.O. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front. Physiol. 2012, 3, 346. [Google Scholar] [CrossRef]
- Van den Brink, L.; Grandela, C.; Mummery, C.L.; Davis, R.P. Inherited cardiac diseases, pluripotent stem cells, and genome editing combined—The past, present, and future. Stem Cells 2020, 38, 174–186. [Google Scholar] [CrossRef]
- Melgari, D.; Calamaio, S.; Frosio, A.; Prevostini, R.; Anastasia, L.; Pappone, C.; Rivolta, I. Automated patch-clamp and induced pluripotent stem cell-derived cardiomyocytes: A synergistic approach in the study of Brugada syndrome. Int. J. Mol. Sci. 2023, 24, 6687. [Google Scholar] [CrossRef]
- Gamper, N.; Stockand, J.D.; Shapiro, M.S. The use of Chinese hamster ovary (CHO) cells in the study of ion channels. J. Pharmacol. Toxicol. Methods. 2005, 51, 177–185. [Google Scholar] [CrossRef]
- Mathie, A.; Veale, E.L.; Holden, R.G. Heterologous expression of ion channels in mammalian cell lines. Methods Mol. Biol. 2021, 2188, 51–65. [Google Scholar] [CrossRef]
- Vanoye, C.G.; Desai, R.R.; Fabre, K.L.; Gallagher, S.L.; Potet, F.; DeKeyser, J.-M.; Macaya, D.; Meiler, J.; Sanders, C.R.; George, A.L., Jr. High-throughput functional evaluation of KCNQ1 decrypts variants of unknown significance. Circ. Genom. Precis. Med. 2018, 11, e002345. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.-A.; Perry, M.D.; Liang, W.; Smith, N.J.; Foo, B.; Shrier, A.; Lukacs, G.L.; Hill, A.P.; Vandenberg, J.I. High-throughput phenotyping of heteromeric human ether-à-go-go-related gene potassium channel variants can discriminate pathogenic from rare benign variants. Heart Rhythm 2020, 17, 492–500. [Google Scholar] [CrossRef]
- Glazer, A.M.; Wada, Y.; Li, B.; Muhammad, A.; Kalash, O.R.; O’Neill, M.J.; Shields, T.; Hall, L.; Short, L.; Blair, M.A.; et al. High-throughput reclassification of SCN5A variants. Am. J. Hum. Genet. 2020, 107, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Kozek, K.A.; Glazer, A.M.; Ng, C.-A.; Blackwell, D.; Egly, C.L.; Vanags, L.R.; Blair, M.; Mitchell, D.; Matreyek, K.A.; Fowler, D.M.; et al. High-throughput discovery of trafficking-deficient variants in the cardiac potassium channel KV11.1. Heart Rhythm 2020, 17, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.-A.; Ullah, R.; Farr, J.; Hill, A.P.; Kozek, K.A.; Vanags, L.R.; Mitchell, D.W.; Kroncke, B.M.; Vandenberg, J.I. A massively parallel assay accurately discriminates between functionally normal and abnormal variants in a hotspot domain of KCNH2. Am. J. Hum. Genet. 2022, 109, 1208–1216. [Google Scholar] [CrossRef]
- Copier, J.S.; Bootsma, M.; Ng, C.A.; Wilde, A.A.M.; Bertels, R.A.; Bikker, H.; Christiaans, I.; Van der Crabben, S.N.; Hol, J.A.; Koopmann, T.T.; et al. Reclassification of a likely pathogenic Dutch founder variant in KCNH2; implications of reduced penetrance. Hum. Mol. Genet. 2023, 32, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Kroncke, B.M.; Glazer, A.M.; Smith, D.K.; Blume, J.D.; Roden, D.M. SCN5A (NaV1.5) variant functional perturbation and clinical presentation: Variants of a certain significance. Circ. Genom. Precis. Med. 2018, 11, e002095. [Google Scholar] [CrossRef]
- Ono, M.; Burgess, D.E.; Schroder, E.A.; Elayi, C.S.; Anderson, C.L.; January, C.T.; Sun, B.; Immadisetty, K.; Kekenes-Huskey, P.M.; Delisle, B.P. Long QT syndrome type 2: Emerging strategies for correcting class 2 KCNH2 (hERG) mutations and identifying new patients. Biomolecules 2020, 10, 1144. [Google Scholar] [CrossRef]
- Adán, V.; Crown, L.A. Diagnosis and treatment of sick sinus syndrome. Am. Fam. Physician. 2003, 67, 1725–1732. [Google Scholar]
- Dobrzynski, H.; Boyett, M.R.; Anderson, R.H. New insights into pacemaker activity: Promoting understanding of sick sinus syndrome. Circulation 2007, 115, 1921–1932. [Google Scholar] [CrossRef] [PubMed]
- Monfredi, O.; Boyett, M.R. Sick sinus syndrome and atrial fibrillation in older persons—A view from the sinoatrial nodal myocyte. J. Mol. Cell. Cardiol. 2015, 83, 88–100. [Google Scholar] [CrossRef]
- Roberts-Thomson, K.C.; Sanders, P.; Kalman, J.M. Sinus node disease: An idiopathic right atrial myopathy. Trends Cardiovasc. Med. 2007, 17, 211–214. [Google Scholar] [CrossRef]
- Chang, H.-Y.; Lin, Y.-J.; Lo, L.-W.; Chang, S.-L.; Hu, Y.-F.; Li, C.-H.; Chao, T.-F.; Yin, W.-H.; Chen, S.-A. Sinus node dysfunction in atrial fibrillation patients: The evidence of regional atrial substrate remodelling. Europace 2013, 15, 205–211. [Google Scholar] [CrossRef]
- Ishikawa, T.; Tsuji, Y.; Makita, N. Inherited bradyarrhythmia: A diverse genetic background. J. Arrhythm. 2016, 32, 352–358. [Google Scholar] [CrossRef]
- Chandler, N.J.; Greener, I.D.; Tellez, J.O.; Inada, S.; Musa, H.; Molenaar, P.; DiFrancesco, D.; Baruscotti, M.; Longhi, R.; Anderson, R.H.; et al. Molecular architecture of the human sinus node: Insights into the function of the cardiac pacemaker. Circulation 2009, 119, 1562–1575. [Google Scholar] [CrossRef]
- Aminu, A.J.; Petkova, M.; Atkinson, A.J.; Yanni, J.; Morris, A.D.; Simms, R.T.; Chen, W.; Yin, Z.; Kuniewicz, M.; Holda, M.K.; et al. Further insights into the molecular complexity of the human sinus node—The role of ‘novel’ transcription factors and microRNAs. Prog. Biophys. Mol. Biol. 2021, 166, 86–104. [Google Scholar] [CrossRef]
- Hoekstra, M.; Van Ginneken, A.C.G.; Wilders, R.; Verkerk, A.O. HCN4 current during human sinoatrial node-like action potentials. Prog. Biophys. Mol. Biol. 2021, 166, 105–118. [Google Scholar] [CrossRef]
- Baruscotti, M.; Bottelli, G.; Milanesi, R.; DiFrancesco, J.C.; DiFrancesco, D. HCN-related channelopathies. Pflügers Arch. 2010, 460, 405–415. [Google Scholar] [CrossRef]
- DiFrancesco, D. Funny channel gene mutations associated with arrhythmias. J. Physiol. 2013, 591, 4117–4124. [Google Scholar] [CrossRef]
- DiFrancesco, D. HCN4, sinus bradycardia and atrial fibrillation. Arrhythm. Electrophysiol. Rev. 2015, 4, 9–13. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R. Pacemaker activity of the human sinoatrial node: Effects of HCN4 mutations on the hyperpolarization-activated current. Europace 2014, 16, 384–395. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R. Pacemaker activity of the human sinoatrial node: An update on the effects of mutations in HCN4 on the hyperpolarization-activated current. Int. J. Mol. Sci. 2015, 16, 3071–3094. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R.; Van Borren, M.M.G.J.; Peters, R.J.G.; Broekhuis, E.; Lam, K.; Coronel, R.; De Bakker, J.M.T.; Tan, H.L. Pacemaker current (If) in the human sinoatrial node. Eur. Heart J. 2007, 28, 2472–2478. [Google Scholar] [CrossRef]
- Fabbri, A.; Fantini, M.; Wilders, R.; Severi, S. Computational analysis of the human sinus node action potential: Model development and effects of mutations. J. Physiol. 2017, 595, 2365–2396. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Van Borren, M.M.G.J.; Peters, R.J.G.; Broekhuis, E.; Lam, K.; Coronel, R.; De Bakker, J.M.T.; Tan, H.L.; Wilders, R. Single cells isolated from human sinoatrial node: Action potentials and numerical reconstruction of pacemaker current. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2007, 2007, 904–907. [Google Scholar] [CrossRef]
- Rush, S.; Larsen, H. A practical algorithm for solving dynamic membrane equations. IEEE Trans. Biomed. Eng. 1978, 25, 389–392. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Wilders, R. Human sinoatrial node pacemaker activity: Role of the slow component of the delayed rectifier K.+ current, IKs. Int. J. Mol. Sci. 2023, 24, 7264. [Google Scholar] [CrossRef]
- Cuellar, A.A.; Lloyd, C.M.; Nielsen, P.F.; Bullivant, D.P.; Nickerson, D.P.; Hunter, P.J. An overview of CellML 1.1, a biological model description language. Simulation 2003, 79, 740–747. [Google Scholar] [CrossRef]
- Lloyd, C.M.; Lawson, J.R.; Hunter, P.J.; Nielsen, P.F. The CellML Model Repository. Bioinformatics 2008, 24, 2122–2123. [Google Scholar] [CrossRef]
- Garny, A.; Kohl, P.; Noble, D. Cellular open resource (COR): A public CellML based environment for modelling biological function. Int. J. Bifurcat. Chaos 2003, 13, 3579–3590. [Google Scholar] [CrossRef]
- Alonso-Fernández-Gatta, M.; Gallego-Delgado, M.; Caballero, R.; Villacorta, E.; Díaz-Peláez, E.; García-BerrocaL, B.; Crespo-García, T.; Plata-Izquierdo, B.; Marcos-Vadillo, E.; García-Cuenllas, L.; et al. A rare HCN4 variant with combined sinus bradycardia, left atrial dilatation, and hypertrabeculation/left ventricular noncompaction phenotype. Rev. Esp. Cardiol. 2021, 74, 781–789. [Google Scholar] [CrossRef]
- Möller, M.; Silbernagel, N.; Wrobel, E.; Stallmayer, B.; Amedonu, E.; Rinné, S.; Peischard, S.; Meuth, S.G.; Wünsch, B.; Strutz-Seebohm, N.; et al. In vitro analyses of novel HCN4 gene mutations. Cell Physiol. Biochem. 2018, 49, 1197–1207. [Google Scholar] [CrossRef]
- Milano, A.; Vermeer, A.M.C.; Lodder, E.M.; Barc, J.; Verkerk, A.O.; Postma, A.V.; Van der Bilt, I.A.C.; Baars, M.J.H.; Van Haelst, P.L.; Caliskan, K.; et al. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J. Am. Coll. Cardiol. 2014, 64, 745–756. [Google Scholar] [CrossRef]
- Nof, E.; Luria, D.; Brass, D.; Marek, D.; Lahat, H.; Reznik-Wolf, H.; Pras, E.; Dascal, N.; Eldar, M.; Glikson, M. Point mutation in the HCN4 cardiac ion channel pore affecting synthesis, trafficking, and functional expression is associated with familial asymptomatic sinus bradycardia. Circulation 2007, 116, 463–470. [Google Scholar] [CrossRef]
- Vermeer, A.M.C.; Lodder, E.M.; Thomas, D.; Duijkers, F.A.M.; Marcelis, C.; Van Gorselen, E.O.F.; Fortner, P.; Buss, S.J.; Mereles, D.; Katus, H.A.; et al. Dilation of the aorta ascendens forms part of the clinical spectrum of HCN4 mutations. J. Am. Coll. Cardiol. 2016, 67, 2313–2315. [Google Scholar] [CrossRef]
- Schweizer, P.A.; Schröter, J.; Greiner, S.; Haas, J.; Yampolsky, P.; Mereles, D.; Buss, S.J.; Seyler, C.; Bruehl, C.; Draguhn, A.; et al. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J. Am. Coll. Cardiol. 2014, 64, 757–767. [Google Scholar] [CrossRef]
- Brunet-Garcia, L.; Odori, A.; Fell, H.; Field, E.; Roberts, A.M.; Starling, L.; Kaski, J.P.; Cervi, E. Noncompaction cardiomyopathy, sick sinus disease, and aortic dilatation: Too much for a single diagnosis? JACC Case Rep. 2022, 4, 287–293. [Google Scholar] [CrossRef]
- Laish-Farkash, A.; Glikson, M.; Brass, D.; Marek-Yagel, D.; Pras, E.; Dascal, N.; Antzelevitch, C.; Nof, E.; Reznik, H.; Eldar, M.; et al. A novel mutation in the HCN4 gene causes symptomatic sinus bradycardia in Moroccan Jews. J. Cardiovasc. Electrophysiol. 2010, 21, 1365–1372. [Google Scholar] [CrossRef]
- Duhme, N.; Schweizer, P.A.; Thomas, D.; Becker, R.; Schröter, J.; Barends, T.R.; Schlichting, I.; Draguhn, A.; Bruehl, C.; Katus, H.A.; et al. Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur. Heart J. 2013, 34, 2768–2775. [Google Scholar] [CrossRef]
- Campostrini, G.; DiFrancesco, J.C.; Castellotti, B.; Milanesi, R.; Gnecchi-Ruscone, T.; Bonzanni, M.; Bucchi, A.; Baruscotti, M.; Ferrarese, C.; Franceschetti, S.; et al. A loss-of-function HCN4 mutation associated with familial benign myoclonic epilepsy in infancy causes increased neuronal excitability. Front. Mol. Neurosci. 2018, 11, 269. [Google Scholar] [CrossRef]
- Wang, H.; Wu, T.; Huang, Z.; Huang, J.; Geng, Z.; Cui, B.; Yan, Y.; Zhang, Y.; Wang, Y. Channel HCN4 mutation R666Q associated with sporadic arrhythmia decreases channel electrophysiological function and increases protein degradation. J. Biol. Chem. 2022, 298, 102599. [Google Scholar] [CrossRef]
- Milanesi, R.; Baruscotti, M.; Gnecchi-Ruscone, T.; DiFrancesco, D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N. Engl. J. Med. 2006, 354, 151–157. [Google Scholar] [CrossRef]
- Schweizer, P.A.; Duhme, N.; Thomas, D.; Becker, R.; Zehelein, J.; Draguhn, A.; Bruehl, C.; Katus, H.A.; Koenen, M. cAMP sensitivity of HCN pacemaker channels determines basal heart rate but is not critical for autonomic rate control. Circ. Arrhythm. Electrophysiol. 2010, 3, 542–552. [Google Scholar] [CrossRef]
- Schulze-Bahr, E.; Neu, A.; Friederich, P.; Kaupp, U.B.; Breithardt, G.; Pongs, O.; Isbrandt, D. Pacemaker channel dysfunction in a patient with sinus node disease. J. Clin. Investig. 2003, 111, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Nakamura, K.; Hayashi, T.; Inagaki, N.; Takahashi, M.; Arimura, T.; Morita, H.; Higashiuesato, Y.; Hirano, Y.; Yasunami, M.; et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J. Biol. Chem. 2004, 279, 27194–27198. [Google Scholar] [CrossRef] [PubMed]
- Chanavat, V.; Janin, A.; Millat, G. A fast and cost-effective molecular diagnostic tool for genetic diseases involved in sudden cardiac death. Clin. Chim. Acta. 2016, 30, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Ader, F.; Roux, M.; Donal, E.; Eicher, J.-C.; Aoutil, N.; Huttin, O.; Selton-Suty, C.; Coisne, D.; Jondeau, G.; et al. Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin. Genet. 2019, 95, 356–367. [Google Scholar] [CrossRef]
- Cambon-Viala, M.; Gerard, H.; Nguyen, K.; Richard, P.; Ader, F.; Pruny, J.-F.; Donal, E.; Eicher, J.-C.; Huttin, O.; Selton-Suty, C.; et al. Phenotype/genotype relationship in left ventricular noncompaction: Ion channel gene mutations are associated with preserved left ventricular systolic function and biventricular noncompaction: Phenotype/genotype of noncompaction. J. Card. Fail. 2021, 27, 677–681. [Google Scholar] [CrossRef]
- Nicole, S.; Lory, P. New challenges resulting from the loss of function of Nav1.4 in neuromuscular diseases. Front. Pharmacol. 2021, 12, 751095. [Google Scholar] [CrossRef]
- Zheng, H.; Yan, X.; Li, G.; Lin, H.; Deng, S.; Zhuang, W.; Yao, F.; Lu, Y.; Xia, X.; Yuan, H.; et al. Proactive functional classification of all possible missense single-nucleotide variants in KCNQ4. Genome Res. 2022, 32, 1573–1584. [Google Scholar] [CrossRef]
- Baez-Nieto, D.; Allen, A.; Akers-Campbell, S.; Yang, L.; Budnik, N.; Pupo, A.; Shin, Y.-C.; Genovese, G.; Liao, M.; Pérez-Palma, E.; et al. Analysing an allelic series of rare missense variants of CACNA1I in a Swedish schizophrenia cohort. Brain 2022, 145, 1839–1853. [Google Scholar] [CrossRef]
- Parihar, R.; Ganesh, S. The SCN1A gene variants and epileptic encephalopathies. J. Hum. Genet. 2013, 58, 573–580. [Google Scholar] [CrossRef]
- Kang, S.K.; Vanoye, C.G.; Misra, S.N.; Echevarria, D.M.; Calhoun, J.D.; O’Connor, J.B.; Fabre, K.L.; McKnight, D.; Demmer, L.; Goldenberg, P.; et al. Spectrum of KV2.1 dysfunction in KCNB1-associated neurodevelopmental disorders. Ann. Neurol. 2019, 86, 899–912. [Google Scholar] [CrossRef] [PubMed]
- Porro, A.; Abbandonato, G.; Veronesi, V.; Russo, A.; Binda, A.; Antolini, L.; Granata, T.; Castellotti, B.; Marini, C.; Moroni, A.; et al. Do the functional properties of HCN1 mutants correlate with the clinical features in epileptic patients? Prog. Biophys. Mol. Biol. 2021, 166, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Vanoye, C.G.; Desai, R.R.; Ji, Z.; Adusumilli, S.; Jairam, N.; Ghabra, N.; Joshi, N.; Fitch, E.; Helbig, K.L.; McKnight, D.; et al. High-throughput evaluation of epilepsy-associated KCNQ2 variants reveals functional and pharmacological heterogeneity. JCI Insight 2022, 7, e156314. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.H.; Potet, F.; Abramova, T.V.; DeKeyser, J.-M.; Ghabra, N.F.; Vanoye, C.G.; Millichap, J.J.; George, A.L., Jr. Epilepsy-associated SCN2A (NaV1.2) variants exhibit diverse and complex functional properties. J. Gen. Physiol. 2023, 155, e202313375. [Google Scholar] [CrossRef]
- Hategan, L.; Csányi, B.; Ördög, B.; Kákonyi, K.; Tringer, A.; Kiss, O.; Orosz, A.; Sághy, L.; Nagy, I.; Hegedűs, Z.; et al. A novel ‘splice site’ HCN4 gene mutation, c.1737+1 G > T, causes familial bradycardia, reduced heart rate response, impaired chronotropic competence and increased short-term heart rate variability. Int. J. Cardiol. 2017, 241, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Erlenhardt, N.; Kletke, O.; Wohlfarth, F.; Komadowski, M.A.; Clasen, L.; Makimoto, H.; Rinné, S.; Kelm, M.; Jungen, C.; Decher, N.; et al. Disease-associated HCN4 V759I variant is not sufficient to impair cardiac pacemaking. Pflügers Arch. 2020, 472, 1733–1742. [Google Scholar] [CrossRef]
- Evans, A.; Bagnall, R.D.; Duflou, J.; Semsarian, C. Postmortem review and genetic analysis in sudden infant death syndrome: An 11-year review. Hum. Pathol. 2013, 44, 1730–1736. [Google Scholar] [CrossRef]
- Tu, E.; Waterhouse, L.; Duflou, J.; Bagnall, R.D.; Semsarian, C. Genetic analysis of hyperpolarization-activated cyclic nucleotide-gated cation channels in sudden unexpected death in epilepsy cases. Brain Pathol. 2011, 21, 692–698. [Google Scholar] [CrossRef]
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-type CaV1.2 calcium channels: From in vitro findings to in vivo function. Physiol. Rev. 2014, 94, 303–326. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Zhang, C.; Zhang, X.; Li, Y.; Qi, Z.; Szeto, C.; Tang, M.; Peng, Y.; Molkentin, J.D. Increasing T-type calcium channel activity by β-adrenergic stimulation contributes to β-adrenergic regulation of heart rates. J. Physiol. 2018, 596, 1137–1151. [Google Scholar] [CrossRef]
- Workman, A.J. Cardiac adrenergic control and atrial fibrillation. Naunyn-Schmied. Arch. Pharmacol. 2010, 381, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Varró, A.; Tomek, J.; Nagy, N.; Virág, L.; Passini, E.; Rodriguez, B.; Baczkó, I. Cardiac transmembrane ion channels and action potentials: Cellular physiology and arrhythmogenic behavior. Physiol. Rev. 2021, 101, 1083–1176. [Google Scholar] [CrossRef] [PubMed]
- Baroudi, G.; Carbonneau, E.; Pouliot, V.; Chahine, M. SCN5A mutation (T1620M) causing Brugada syndrome exhibits different phenotypes when expressed in Xenopus oocytes and mammalian cells. FEBS Lett. 2000, 467, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Dumaine, R.; Towbin, J.A.; Brugada, P.; Vatta, M.; Nesterenko, D.V.; Nesterenko, V.V.; Brugada, J.; Brugada, R.; Antzelevitch, C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ. Res. 1999, 85, 803–809. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).