

Inborn Errors of Immunity in Children with Autoimmune and Allergic Complaints: A Single Center Experience from Diagnosis to Treatment
 , , , , , ,
, , , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Recruitment Criteria
- One or more autoimmune disorders associated with at least one of the following: hypergammaglobulinemia or hypogammaglobulinemia, hypereosinophilia (>1500/mcL) or chronic lymphoproliferation (lymphadenopathy and/or splenomegaly);
- Severe dermatitis plus hyper-IgE (>2000 UI/mL) and at least one of the following: recurrent infections, skeletal and joint disorders, recurrent pneumonia or pyogenic cutaneous abscesses;
- Idiopathic hypereosinophilia (>5000/mcL) plus hypergammaglobulinemia (values above the 97.5% for age and sex for at least one among IgG, IgA and IgM) and at least one of dermatitis, enteropathy and chronic lymphoproliferation;
- Severe dermatitis refractory to topical glucocorticoid treatment; dermatitis is considered severe if it affects at least 10 percent of the body surface and it affects quality of life and disrupts sleep, despite continuous topical treatment with glucocorticoid-based ointments;
- Severe food anaphylaxis refractory to oral desensitizing treatments. At our hospital, oral desensitization is proposed to all patients who experienced episodes of anaphylaxis to foods for which it is not easy to avoid the risk of allergic reactions for accidental exposures, such as for cow’s milk and egg proteins and that contain high levels of food-specific IgE antibodies.
2.2. Flow Cytometry Analysis
2.2.1. Immunophenotype
2.2.2. CTLA4 Expression
2.2.3. LRBA Expression
2.2.4. STAT1 Expression
2.2.5. S6 Phosphorylation
2.3. Genetic Analysis
2.4. RNA Sequencing (RNAseq) Analysis
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCusker, C.; Upton, J.; Warrington, R. Primary immunodeficiency. Allergy Asthma Clin. Immunol. 2018, 14 (Suppl. S2), 61. [Google Scholar] [CrossRef] [PubMed]
- Arkwright, P.D.; Gennery, A.R. Ten warning signs of primary immunodeficiency: A new paradigm is needed for the 21st century. Ann. N. Y. Acad. Sci. 2011, 1238, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Delmonte, O.M.; Castagnoli, R.; Calzoni, E.; Notarangelo, L.D. Inborn Errors of Immunity with Immune Dysregulation: From Bench to Bedside. Front. Pediatr. 2019, 7, 353. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.E.; Ayala, I.A.; Milojevic, D. Autoimmunity as a continuum in primary immunodeficiency. Curr. Opin. Pediatr. 2019, 31, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Boz, V.; Zanchi, C.; Levantino, L.; Riccio, G.; Tommasini, A. Druggable monogenic immune defects hidden in diverse medical specialties: Focus on overlap syndromes. World J. Clin. Pediatr. 2022, 11, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- van Gent, R.; van Tilburg, C.M.; Nibbelke, E.E.; Otto, S.A.; Gaiser, J.F.; Janssens-Korpela, P.L.; Sanders, E.A.M.; Borghans, J.A.M.; Wulffraat, N.M.; Bierings, M.B.; et al. Refined characterization and reference values of the pediatric T- and B-cell compartments. Clin. Immunol. 2009, 133, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.B.; Bleesing, J.J.; Dianzani, U.; Fleisher, T.A.; Jaffe, E.S.; Lenardo, M.J.; Rieux-Laucat, F.; Siegel, R.M.; Su, H.C.; Teachey, D.T.; et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood 2010, 116, e35–e40. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Lo, B.; Zhang, K.; Lu, W.; Zheng, L.; Zhang, Q.; Kanellopoulou, C.; Zhang, Y.; Liu, Z.; Fritz, J.M.; Marsh, R.; et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015, 349, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Boz, V.; Valencic, E.; Girardelli, M.; Pin, A.; Gàmez-Diaz, L.; Tommasini, A.; Lega, S.; Bramuzzo, M. Case Report: Refractory Autoimmune Gastritis Responsive to Abatacept in LRBA Deficiency. Front. Immunol. 2021, 12, 619246. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, M.; Dell’Orso, G.; Grossi, A.; Ceccherini, I.; Viola, S.; Terranova, P.; Micalizzi, C.; Guardo, D.; Massaccesi, E.; Palmisani, E.; et al. Underlying CTLA4 Deficiency in a Patient with Juvenile Idiopathic Arthritis and Autoimmune Lymphoproliferative Syndrome Features Successfully Treated With Abatacept-A Case Report. J. Pediatr. Hematol. Oncol. 2021, 43, e1168–e1172. [Google Scholar] [CrossRef] [PubMed]
- Angelino, G.; Cifaldi, C.; Zangari, P.; Di Cesare, S.; Di Matteo, G.; Chiriaco, M.; Francalanci, P.; Faraci, S.; Rea, F.; Romeo, E.F.; et al. Gastric cancer, inflammatory bowel disease and polyautoimmunity in a 17-year-old boy: CTLA-4 deficiency successfully treated with Abatacept. Eur. J. Gastroenterol. Hepatol. 2021, 33 (Suppl. S1), e1051–e1056. [Google Scholar] [CrossRef] [PubMed]
- Lanz, A.L.; Riester, M.; Peters, P.; Schwerd, T.; Lurz, E.; Hajji, M.S.; Rohlfs, M.; Ley-Zaporozhan, J.; Walz, C.; Kotlarz, D.; et al. Abatacept for treatment-refractory pediatric CTLA4-haploinsufficiency. Clin. Immunol. 2021, 229, 108779. [Google Scholar] [CrossRef] [PubMed]
- Meesilpavikkai, K.; Dik, W.A.; Schrijver, B.; Nagtzaam, N.M.A.; Posthumus-van Sluijs, S.J.; van Hagen, P.M.; Dalm, V.A.S.H. Baricitinib treatment in a patient with a gain-of-function mutation in signal transducer and activator of transcription 1 (STAT1). J. Allergy Clin. Immunol. 2018, 142, 328–330.e2. [Google Scholar] [CrossRef] [PubMed]
- Girardelli, M.; Valencic, E.; Moressa, V.; Margagliotta, R.; Tesser, A.; Pastore, S.; Spadola, O.; Athanasakis, E.; Severini, G.M.; Taddio, A.; et al. Genetic and immunologic findings in children with recurrent aphthous stomatitis with systemic inflammation. Pediatr. Rheumatol. 2021, 19, 70. [Google Scholar] [CrossRef] [PubMed]
- Zama, D.; Cocchi, I.; Masetti, R.; Specchia, F.; Alvisi, P.; Gambineri, E.; Lima, M.; Pession, A. Late-onset of immunodysregulation, polyendocrinopathy, enteropathy, x-linked syndrome (IPEX) with intractable diarrhea. Ital. J. Pediatr. 2014, 40, 68. [Google Scholar] [CrossRef] [PubMed]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Al-Herz, W.; Ailal, F.; Chatila, T.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; Holland, S.M.; et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J. Clin. Immunol. 2020, 40, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Rispoli, F.; Valencic, E.; Girardelli, M.; Pin, A.; Tesser, A.; Piscianz, E.; Boz, V.; Faletra, F.; Severini, G.M.; Taddio, A.; et al. Immunity and Genetics at the Revolving Doors of Diagnostics in Primary Immunodeficiencies. Diagnostics 2021, 11, 532. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
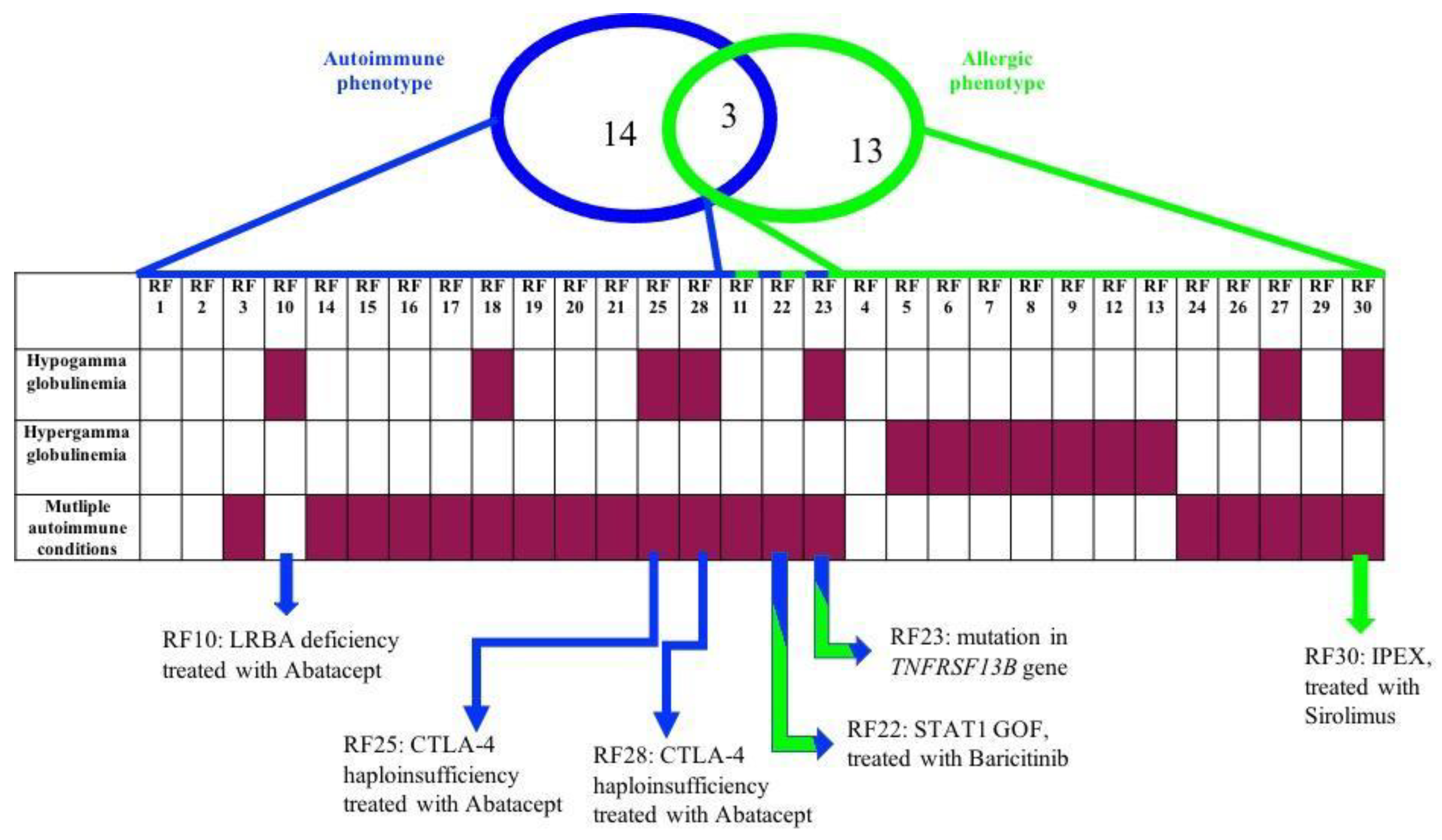
| Pt | Age at Onset (years) | Clinical Features | Mutations | Inheritance | Segregation Analysis | Immune Phenotype and Immunoglobulins | Functional Assays | Transcriptome Analysis | Targeted Treatment |
|---|---|---|---|---|---|---|---|---|---|
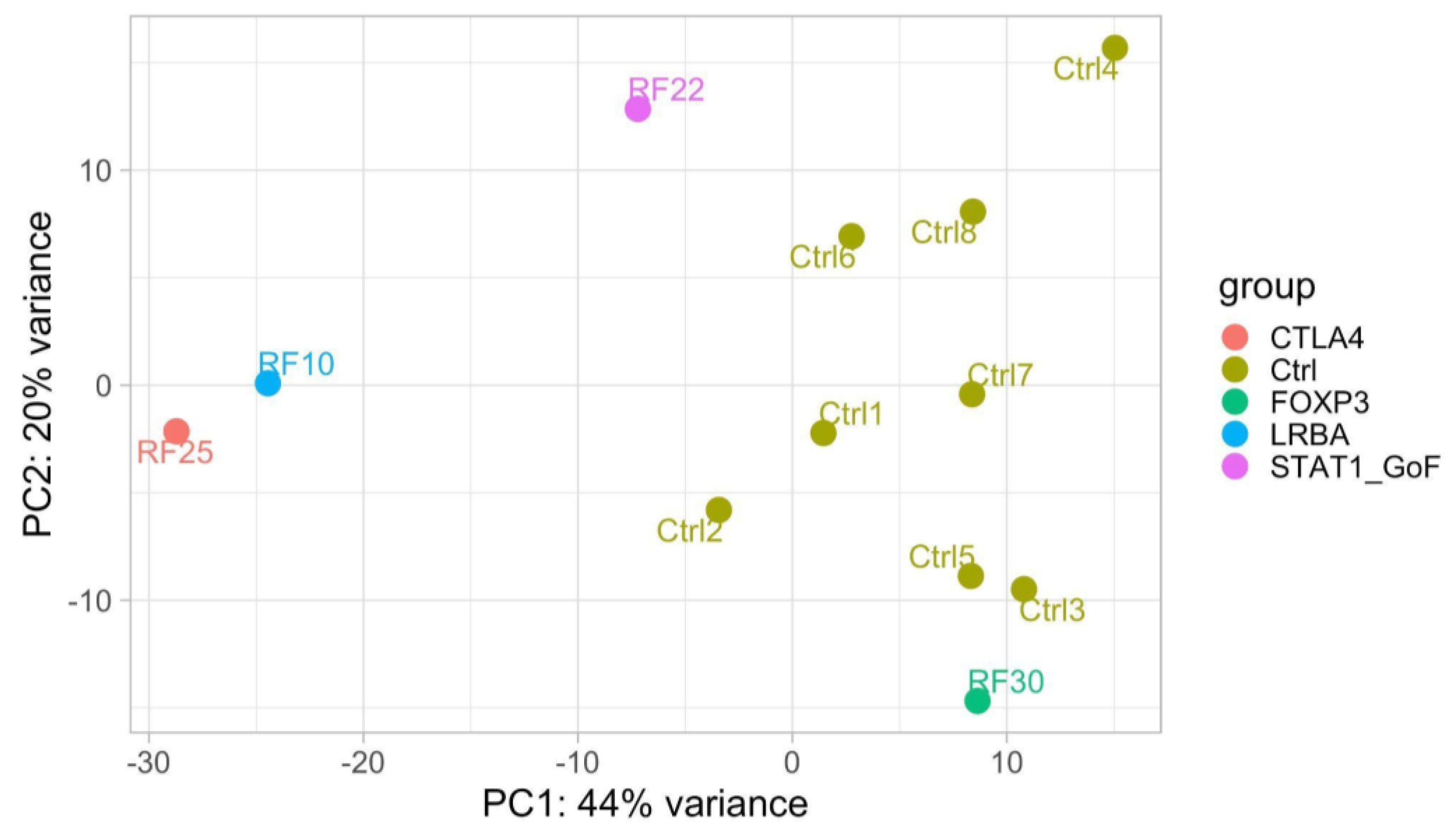
| RF10 | 2 | autoimmune gastritis; splenomegaly; multiple lymphadenopathy; hypogammaglobulinemia; anemia | LRBA c.C6415T p.R2139X; c.C7315T p.R2439X | AR | LRBA c.C6415T p.R2139X (father) c.C7315T p.R2439X (mother) | DNT αβ↑; IgA↓ | LRBA, CTLA4 and pS6 expression | LRBA ↓ | Abatacept |
| RF25 | 15 | recurrent fevers; autoimmune thrombocytopenia; autoimmune hemolytic anemia; leukopenia; recurrent infections; severe hypogammaglobulinemia | CTLA4 c.G160A p.A54T | AD | CTLA4 c.G160A p.A54T (mother and brother); father, daughter and maternal aunt wild-type | B lymphocytes ↓; RTE↓; IgG↓; IgM↓; IgA↓ | CTLA4 and pS6 expression | signaling related to B cells population | Abatacept |
| RF28 | 13 | autoimmune thrombocytopenia; severe hypogammaglobulinemia; chronic enteropathy; insulin-dependent type 1 diabetes mellitus; autoimmune thyroiditis; frequent infections | CTLA4 c.C223T p.R75W | AD | nd | B lymphocytes ↓; RTE↓; IgG↓; IgM↓; IgA↓ | CTLA4 and pS6 expression | nd | Abatacept |
| RF22 | 2 | recurrent oral candidiasis; aphthous stomatitis (RAS); autoimmune thyroiditis, autoimmune gastritis; hemolytic anemia | STAT1 c.A1721C p.N574T | AD | nd | normal | STAT1 expression | √ | Baricitinib |
| RF30 | 0.5 | diarrhea; atopic eczema; leukocytosis; hypereosinophilia; coeliac disease; hypogammaglobulinemia (IgG) | FOXP3 c.748_750ofAAG p.K250del | X-linked | nd | T lymphocytes CD8+↑; IgG↓ | nd | √ | Sirolimus |
| RF23 | 2 | autoimmune thyroiditis; autoimmune gastritis; alopecia; polyarthritis; hypogammaglobulinemia; anemia | TNFRSF13B c.T310C p.C104R | AD | nd | switched memory B cells↓; IgG↓; IgA↓ | nd | nd | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boz, V.; Tesser, A.; Girardelli, M.; Burlo, F.; Pin, A.; Severini, G.M.; De Marchi, G.; Verzegnassi, F.; Naviglio, S.; Tommasini, A.; et al. Inborn Errors of Immunity in Children with Autoimmune and Allergic Complaints: A Single Center Experience from Diagnosis to Treatment. Biomedicines 2023, 11, 1299. https://doi.org/10.3390/biomedicines11051299
Boz V, Tesser A, Girardelli M, Burlo F, Pin A, Severini GM, De Marchi G, Verzegnassi F, Naviglio S, Tommasini A, et al. Inborn Errors of Immunity in Children with Autoimmune and Allergic Complaints: A Single Center Experience from Diagnosis to Treatment. Biomedicines. 2023; 11(5):1299. https://doi.org/10.3390/biomedicines11051299
Chicago/Turabian StyleBoz, Valentina, Alessandra Tesser, Martina Girardelli, Francesca Burlo, Alessia Pin, Giovanni Maria Severini, Ginevra De Marchi, Federico Verzegnassi, Samuele Naviglio, Alberto Tommasini, and et al. 2023. "Inborn Errors of Immunity in Children with Autoimmune and Allergic Complaints: A Single Center Experience from Diagnosis to Treatment" Biomedicines 11, no. 5: 1299. https://doi.org/10.3390/biomedicines11051299
APA StyleBoz, V., Tesser, A., Girardelli, M., Burlo, F., Pin, A., Severini, G. M., De Marchi, G., Verzegnassi, F., Naviglio, S., Tommasini, A., & Valencic, E. (2023). Inborn Errors of Immunity in Children with Autoimmune and Allergic Complaints: A Single Center Experience from Diagnosis to Treatment. Biomedicines, 11(5), 1299. https://doi.org/10.3390/biomedicines11051299