
Biological and Genetic Mechanisms of COPD, Its Diagnosis, Treatment, and Relationship with Lung Cancer

 , ,
, ,  and
and 
Abstract
1. Introduction
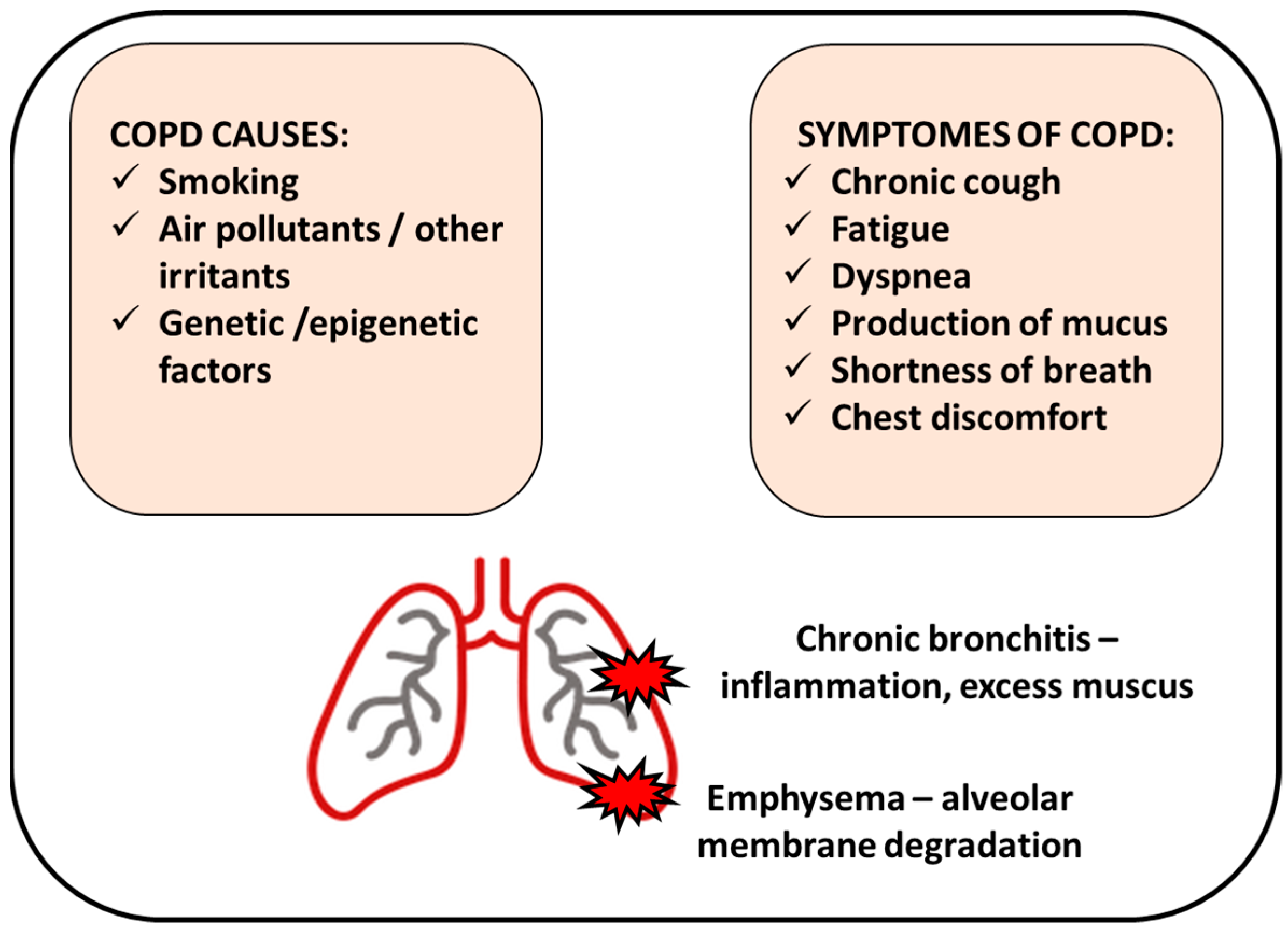
2. COPD Causes
3. Genetics of COPD
3.1. Gene Polymorphisms

{kind=link}
{kind=link}
{kind=link}
| Gene Identified | Location of Polymorphisms | Critical Effects | Reference |
|---|---|---|---|
| α-1-antitrypsin (AAT) | ATT*Z allele (Pi Z) homozygosity, single amino acid substitution causing base pair changes | low levels of AAT in serum, accumulation in hepatocytes leading to liver damage, neutrophil inactivity, emphysema | [26,27,28,29] |
| Alpha-nicotinic acetylcholine receptor | 2 SNPs (rs8034191 and rs1051730) at locus of CHRNA3/5 in chromosome 5 | lung dysfunction (deviations in FEV1 parameter) | [31,32] |
| HHIP (Hedgehog interacting protein) | chromosome 4q31 (HHIP mutations) | developmental problems in the lung and abnormality during morphogenesis | [31,32,33] |
| IREB2 (Iron responsive element binding protein 2) | chromosome 15q25.1 (SNP rs7937) | lung developmental changes and emphysema | [34,40] |
| ADPHD1 (Aspartate beta-hydroxylase domain containing 1) | chromosome 15q25.1 | airflow obstruction, AAT deficiency | [34] |
| HTR4 (5-hydroxytryptamine receptor 4) | chromosome 5q31-q33 | FEV1/FVC changes, airflow obstruction | [36] |
| CYP2A6 (Cytochrome P450 family 2 subfamily A member 6) | chromosome 19q13 | nicotine metabolism affected | [34,37] |
| EGLN2 (Egl-9 family hypoxia inducible factor 2) | chromosome 19q13.2 | hypoxia response destroyed | [37] |
3.2. Epigenetic Regulation (Methylation and Deacetylation)
| Epigenetic Mechanism | Altering Factors | Targets | Phenotype/Function in COPD Context | Reference |
|---|---|---|---|---|
| Methylation | cigarette smoking, air pollution | HSH2D, SNX10, CLIP4, TYKZ | regulation of lung macrophage activity, maintaining lung metabolic balance | [43] |
| mtTFA | hypermethylation of the promoter is associated with the initiation and progression of COPD | [44] | ||
| NEGR1, ARID5A, FOXl2, WDR46, AKNA, SYTL2 | air pollution-dependent regulation of gene expression in Asians | [46] | ||
| HLX, SPON2 | alteration of functional gene expression in parenchymal fibroblasts | [57] | ||
| IREB2, PSMA4 | smoke-independent association of COPD with genetic variants in chromosome 15q25.1 | [58] | ||
| Acetylation | cigarette smoking, regulators of HDACs activity (Trichostatin A) | Histones: H3K9, H3, H4 | increased levels are associated with inflammation, active gene transcription | [56] |
3.3. Transcriptional Regulation and Splicing
4. Pathogenesis of Lung Cancers
4.1. Lung Cancer Epidemiology and Subtypes
4.2. Genetics of Lung Cancer
4.2.1. Genetic Susceptibilities to Lung Cancers
4.2.2. Genetic Factors Involved in the Pathogenesis of NSCLC
4.3. Epigenetic Susceptibilities to Lung Cancers
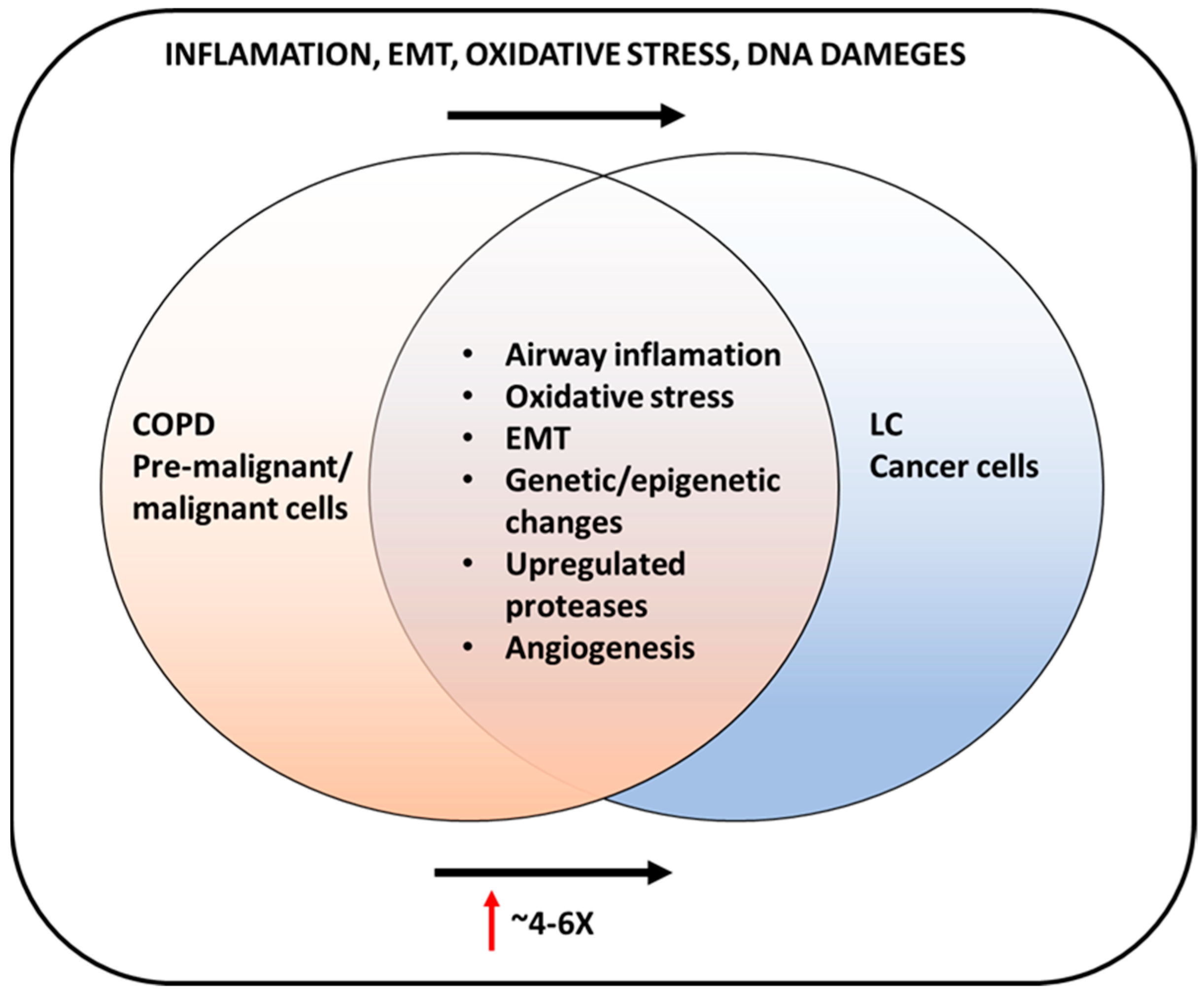
5. Role of Lung Inflammation in COPD Development and Lung Cancer
6. Biological Mechanisms of COPD and Lung Cancer Development
6.1. Smoking in COPD and Lung Cancers
6.2. Oxidative Stress
6.3. Protease Involvement and Matrix Remodeling
Role of Proteases in COPD and Lung Cancer
7. Diagnosis and Treatment of COPD
7.1. Detection of COPD
7.2. COPD Patient Classification and Treatment
8. Targeted Therapy for Lung Cancer
9. COPD and Lung Cancer Connection
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- May, S.M.; Li, J.T.C. Burden of Chronic Obstructive Pulmonary Disease: Healthcare Costs and Beyond. Allergy Asthma Proc. 2015, 36, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Turner, A.M. Chronic Obstructive Pulmonary Disease: The Present and Future. Biomedicines 2022, 10, 499. [Google Scholar] [CrossRef]
- Bhatt, S.P.; Kim, Y.; Harrington, K.F.; Hokanson, J.E.; Lutz, S.M.; Cho, M.H.; DeMeo, D.L.; Wells, J.M.; Make, B.J.; Rennard, S.I.; et al. Smoking Duration Alone Provides Stronger Risk Estimates of Chronic Obstructive Pulmonary Disease than Pack-Years. Thorax 2018, 73, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Diette, G.B.; Accinelli, R.A.; Balmes, J.R.; Buist, A.S.; Checkley, W.; Garbe, P.; Hansel, N.N.; Kapil, V.; Gordon, S.; Lagat, D.K.; et al. Obstructive lung disease and exposure to burning biomass fuel in the indoor environment. Glob. Heart 2012, 7, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Lopez, A.D. Alternative Projections of Mortality and Disability by Cause 1990–2020: Global Burden of Disease Study. Lancet 1997, 349, 1498–1504. [Google Scholar] [CrossRef] [PubMed]
- Young, R.P.; Hopkins, R.J.; Christmas, T.; Black, P.N.; Metcalf, P.; Gamble, G.D. COPD Prevalence Is Increased in Lung Cancer, Independent of Age, Sex and Smoking History. Eur. Respir. J. 2009, 34, 380–386. [Google Scholar] [CrossRef]
- Barreiro, E.; Bustamante, V.; Curull, V.; Gea, J.; López-Campos, J.L.; Muñoz, X. Relationships between Chronic Obstructive Pulmonary Disease and Lung Cancer: Biological Insights. J. Thorac. Dis. 2016, 8, E1122–E1135. [Google Scholar] [CrossRef]
- Singh, D.; Agusti, A.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Criner, G.J.; Frith, P.; Halpin, D.M.G.; Han, M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: The GOLD Science Committee Report 2019. Eur. Respir. J. 2019, 53, 1900164. [Google Scholar] [CrossRef]
- Lopez, A.D.; Murray, C.C.J.L. The Global Burden of Disease, 1990–2020. Nat. Med. 1998, 4, 1241–1243. [Google Scholar] [CrossRef]
- Qureshi, H.; Sharafkhaneh, A.; Hanania, N.A. Chronic Obstructive Pulmonary Disease Exacerbations: Latest Evidence and Clinical Implications. Ther. Adv. Chronic. Dis. 2014, 5, 212–227. [Google Scholar] [CrossRef]
- Bednarek, M.; Maciejewski, J.; Wozniak, M.; Kuca, P.; Zielinski, J. Prevalence, Severity and Underdiagnosis of COPD in the Primary Care Setting. Thorax 2008, 63, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Denden, S.; Khelil, A.H.; Knani, J.; Lakhdar, R.; Perrin, P.; Lefranc, G.; Chibani, J.B. Alpha-1 Antitrypsin Gene Polymorphism in Chronic Obstructive Pulmonary Disease (COPD). Genet. Mol. Biol. 2010, 33, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Molloy, K.; Hersh, C.P.; Morris, V.B.; Carroll, T.P.; O’Connor, C.A.; Lasky-Su, J.A.; Greene, C.M.; O’Neill, S.J.; Silverman, E.K.; McElvaney, N.G. Clarification of the Risk of Chronic Obstructive Pulmonary Disease in A1-Antitrypsin Deficiency PiMZ Heterozygotes. Am. J. Respir. Crit. Care Med. 2014, 189, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.M.; Parker, M.M.; Oster, R.A.; Bowler, R.P.; Dransfield, M.T.; Bhatt, S.P.; Cho, M.H.; Kim, V.; Curtis, J.L.; Martinez, F.J.; et al. Elevated Circulating MMP-9 Is Linked to Increased COPD Exacerbation Risk in SPIROMICS and COPDGene. JCI Insight 2018, 3, e123614. [Google Scholar] [CrossRef]
- Bentley, A.R.; Emrani, P.; Cassano, P.A. Genetic Variation and Gene Expression in Antioxidant Related Enzymes and Risk of COPD: A Systematic Review. Thorax 2008, 63, 956–961. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Ganbold, C.; Jamiyansuren, J.; Tumurbaatar, A.; Bayarmaa, A.; Enebish, T.; Dashtseren, I.; Jav, S. The Cumulative Effect of Gene–Gene Interactions Between GSTM1, CHRNA3, CHRNA5 and SOD3 Gene Polymorphisms Combined with Smoking on COPD Risk. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 2857–2868. [Google Scholar] [CrossRef]
- Brøgger, J.; Steen, V.M.; Eiken, H.G.; Gulsvik, A.; Bakke, P. Genetic Association between COPD and Polymorphisms in TNF, ADRB2 and EPHX1. Eur. Respir. J. 2006, 27, 682–688. [Google Scholar] [CrossRef]
- Sparrow, D.; Glynn, R.J.; Cohen, M.; Weiss, S.T. The Relationship of the Peripheral Leukocyte Count and Cigarette Smoking to Pulmonary Function among Adult Men. Chest 1984, 86, 383–386. [Google Scholar] [CrossRef]
- Saha, S.; Brightling, C.E. Eosinophilic Airway Inflammation in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2006, 1, 39–47. [Google Scholar] [CrossRef]
- Cornwell, W.D.; Kim, V.; Song, C.; Rogers, T.J. Pathogenesis of Inflammation and Repair in Advanced COPD. Semin. Respir. Crit. Care Med. 2010, 31, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Polosukhin, V.V.; Gutor, S.S.; Du, R.-H.; Richmond, B.W.; Massion, P.P.; Wu, P.; Cates, J.M.; Sandler, K.L.; Rennard, S.I.; Blackwell, T.S. Small Airway Determinants of Airflow Limitation in Chronic Obstructive Pulmonary Disease. Thorax 2021, 76, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-W.; Yau, T.; Fulgar, C.C.; Mack, S.M.; Revilla, A.M.; Kenyon, N.J.; Pinkerton, K.E. Long-Term Sequelae of Smoking and Cessation in Spontaneously Hypertensive Rats. Toxicol. Pathol. 2020, 48, 422–436. [Google Scholar] [CrossRef] [PubMed]
- Janssens, J.-P. Aging of the Respiratory System: Impact on Pulmonary Function Tests and Adaptation to Exertion. Clin. Chest Med. 2005, 26, 469–484. [Google Scholar] [CrossRef]
- Silverman, E.K. Applying Functional Genomics to Chronic Obstructive Pulmonary Disease. Ann. Am. Thorac. Soc. 2018, 15, S239–S242. [Google Scholar] [CrossRef] [PubMed]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992, 18, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Silverman, E.K. Genetics of COPD. Annu. Rev. Physiol. 2020, 82, 413–431. [Google Scholar] [CrossRef]
- Ekeowa, U.I.; Gooptu, B.; Belorgey, D.; Hägglöf, P.; Karlsson-Li, S.; Miranda, E.; Pérez, J.; MacLeod, I.; Kroger, H.; Marciniak, S.J.; et al. Alpha1-Antitrypsin Deficiency, Chronic Obstructive Pulmonary Disease and the Serpinopathies. Clin. Sci. (Lond.) 2009, 116, 837–850. [Google Scholar] [CrossRef]
- Stoller, J.K.; Aboussouan, L.S. A Review of A1-Antitrypsin Deficiency. Am. J. Respir. Crit. Care Med. 2012, 185, 246–259. [Google Scholar] [CrossRef]
- Sun, W.; Kechris, K.; Jacobson, S.; Drummond, M.B.; Hawkins, G.A.; Yang, J.; Chen, T.-H.; Quibrera, P.M.; Anderson, W.; Barr, R.G.; et al. Common Genetic Polymorphisms Influence Blood Biomarker Measurements in COPD. PLoS Genet. 2016, 12, e1006011. [Google Scholar] [CrossRef]
- Pillai, S.G.; Ge, D.; Zhu, G.; Kong, X.; Shianna, K.V.; Need, A.C.; Feng, S.; Hersh, C.P.; Bakke, P.; Gulsvik, A.; et al. A Genome-Wide Association Study in Chronic Obstructive Pulmonary Disease (COPD): Identification of Two Major Susceptibility Loci. PLoS Genet. 2009, 5, e1000421. [Google Scholar] [CrossRef]
- Cho, M.H.; McDonald, M.-L.N.; Zhou, X.; Mattheisen, M.; Castaldi, P.J.; Hersh, C.P.; Demeo, D.L.; Sylvia, J.S.; Ziniti, J.; Laird, N.M.; et al. Risk Loci for Chronic Obstructive Pulmonary Disease: A Genome-Wide Association Study and Meta-Analysis. Lancet Respir. Med. 2014, 2, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Wilk, J.B.; Shrine, N.R.G.; Loehr, L.R.; Zhao, J.H.; Manichaikul, A.; Lopez, L.M.; Smith, A.V.; Heckbert, S.R.; Smolonska, J.; Tang, W.; et al. Genome-Wide Association Studies Identify CHRNA5/3 and HTR4 in the Development of Airflow Obstruction. Am. J. Respir. Crit. Care Med. 2012, 186, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Minematsu, N.; Nakamura, H.; Iwata, M.; Tateno, H.; Nakajima, T.; Takahashi, S.; Fujishima, S.; Yamaguchi, K. Association of CYP2A6 Deletion Polymorphism with Smoking Habit and Development of Pulmonary Emphysema. Thorax 2003, 58, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Castaldi, P.J.; Wan, E.S.; Siedlinski, M.; Hersh, C.P.; Demeo, D.L.; Himes, B.E.; Sylvia, J.S.; Klanderman, B.J.; Ziniti, J.P.; et al. A Genome-Wide Association Study of COPD Identifies a Susceptibility Locus on Chromosome 19q13. Hum. Mol. Genet. 2012, 21, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Siedlinski, M.; Cho, M.H.; Bakke, P.; Gulsvik, A.; Lomas, D.A.; Anderson, W.; Kong, X.; Rennard, S.I.; Beaty, T.H.; Hokanson, J.E.; et al. Genome-Wide Association Study of Smoking Behaviours in Patients with COPD. Thorax 2011, 66, 894–902. [Google Scholar] [CrossRef]
- Bloom, A.J.; Baker, T.B.; Chen, L.-S.; Breslau, N.; Hatsukami, D.; Bierut, L.J.; Goate, A. Variants in Two Adjacent Genes, EGLN2 and CYP2A6, Influence Smoking Behavior Related to Disease Risk via Different Mechanisms. Hum. Mol. Genet. 2014, 23, 555–561. [Google Scholar] [CrossRef]
- Hobbs, B.D.; de Jong, K.; Lamontagne, M.; Bossé, Y.; Shrine, N.; Artigas, M.S.; Wain, L.V.; Hall, I.P.; Jackson, V.E.; Wyss, A.B.; et al. Genetic Loci Associated with Chronic Obstructive Pulmonary Disease Overlap with Loci for Lung Function and Pulmonary Fibrosis. Nat. Genet. 2017, 49, 426–432. [Google Scholar] [CrossRef]
- Sakornsakolpat, P.; Prokopenko, D.; Lamontagne, M.; Reeve, N.F.; Guyatt, A.L.; Jackson, V.E.; Shrine, N.; Qiao, D.; Bartz, T.M.; Kim, D.K.; et al. Genetic Landscape of Chronic Obstructive Pulmonary Disease Identifies Heterogeneous Cell Type and Phenotype Associations. Nat. Genet. 2019, 51, 494–505. [Google Scholar] [CrossRef]
- Schirmer, M.; Garner, A.; Vlamakis, H.; Xavier, R.J. Microbial Genes and Pathways in Inflammatory Bowel Disease. Nat. Rev. Microbiol. 2019, 17, 497–511. [Google Scholar] [CrossRef]
- Jin, B.; Li, Y.; Robertson, K.D. DNA Methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.; van der Plaat, D.A.; Nedeljkovic, I.; Verkaik-Schakel, R.N.; Kooistra, W.; Amin, N.; van Duijn, C.M.; Brandsma, C.-A.; van Diemen, C.C.; Vonk, J.M.; et al. From Blood to Lung Tissue: Effect of Cigarette Smoke on DNA Methylation and Lung Function. Respir. Res. 2018, 19, 212. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.A.; Chen, Y.; Dessaint, J.A.; Aridgides, D.S.; Channon, J.Y.; Mellinger, D.L.; Christensen, B.C.; Ashare, A. DNA Methylation Changes in Regional Lung Macrophages Are Associated with Metabolic Differences. Immunohorizons 2019, 3, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Guo, T.; Chen, Z.; Zhang, H.; Cai, S.; Yang, M.; Chen, P.; Guan, C.; Fang, X. Hypermethylation of Mitochondrial Transcription Factor A Induced by Cigarette Smoke Is Associated with Chronic Obstructive Pulmonary Disease. Exp. Lung. Res. 2019, 45, 101–111. [Google Scholar] [CrossRef]
- Rider, C.F.; Carlsten, C. Air Pollution and DNA Methylation: Effects of Exposure in Humans. Clin. Epigenetics 2019, 11, 131. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Xu, C.-J.; Carnes, M.U.; Nichols, C.E.; Ward, J.M.; BIOS consortium; Kwon, S.O.; Kim, S.-Y.; Kim, W.J.; London, S.J. Genome-Wide DNA Methylation and Long-Term Ambient Air Pollution Exposure in Korean Adults. Clin. Epigenet. 2019, 11, 37. [Google Scholar] [CrossRef]
- Clifford, R.L.; Fishbane, N.; Patel, J.; MacIsaac, J.L.; McEwen, L.M.; Fisher, A.J.; Brandsma, C.-A.; Nair, P.; Kobor, M.S.; Hackett, T.-L.; et al. Altered DNA Methylation Is Associated with Aberrant Gene Expression in Parenchymal but Not Airway Fibroblasts Isolated from Individuals with COPD. Clin. Epigenet. 2018, 10, 32. [Google Scholar] [CrossRef]
- Busch, R.; Hobbs, B.D.; Zhou, J.; Castaldi, P.J.; McGeachie, M.J.; Hardin, M.E.; Hawrylkiewicz, I.; Sliwinski, P.; Yim, J.-J.; Kim, W.J.; et al. Genetic Association and Risk Scores in a Chronic Obstructive Pulmonary Disease Meta-Analysis of 16,707 Subjects. Am. J. Respir. Cell Mol. Biol. 2017, 57, 35–46. [Google Scholar] [CrossRef]
- Hazari, Y.M.; Bashir, A.; Habib, M.; Bashir, S.; Habib, H.; Qasim, M.A.; Shah, N.N.; Haq, E.; Teckman, J.; Fazili, K.M. Alpha-1-Antitrypsin Deficiency: Genetic Variations, Clinical Manifestations and Therapeutic Interventions. Mutat. Res. Rev. Mutat. Res. 2017, 773, 14–25. [Google Scholar] [CrossRef]
- Tian, L.; Fong, M.P.; Wang, J.J.; Wei, N.E.; Jiang, H.; Doerge, R.W.; Chen, Z.J. Reversible Histone Acetylation and Deacetylation Mediate Genome-Wide, Promoter-Dependent and Locus-Specific Changes in Gene Expression during Plant Development. Genetics 2005, 169, 337–345. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.M.; Tsaprouni, L.; Bhavsar, P.; Ito, K. Epigenetic Regulation of Airway Inflammation. Curr. Opin. Immunol. 2007, 19, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Szulakowski, P.; Crowther, A.J.L.; Jiménez, L.A.; Donaldson, K.; Mayer, R.; Leonard, T.B.; MacNee, W.; Drost, E.M. The Effect of Smoking on the Transcriptional Regulation of Lung Inflammation in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2006, 174, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Tao, F.; Zhou, Y.; Wang, M.; Wang, C.; Zhu, W.; Han, Z.; Sun, N.; Wang, D. Metformin Alleviates Chronic Obstructive Pulmonary Disease and Cigarette Smoke Extract-Induced Glucocorticoid Resistance by Activating the Nuclear Factor E2-Related Factor 2/Heme Oxygenase-1 Signaling Pathway. Korean J. Physiol. Pharmacol. 2022, 26, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, F.; Cong, Y.; Miao, J.; Wu, D.; Liu, B.; Wang, L. Trichostatin A Inhibits Skeletal Muscle Atrophy Induced by Cigarette Smoke Exposure in Mice. Life Sci. 2019, 235, 116800. [Google Scholar] [CrossRef]
- Sundar, I.K.; Nevid, M.Z.; Friedman, A.E.; Rahman, I. Cigarette Smoke Induces Distinct Histone Modifications in Lung Cells: Implications for the Pathogenesis of COPD and Lung Cancer. J. Proteome Res. 2014, 13, 982–996. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, N.S.; Nazli, R.; Zafar, H.; Fatima, S. Effects of Lipid Based Multiple Micronutrients Supplement on the Birth Outcome of Underweight Pre-Eclamptic Women: A Randomized Clinical Trial. Pak. J. Med. Sci. 2022, 38, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Nedeljkovic, I.; Carnero-Montoro, E.; Lahousse, L.; van der Plaat, D.A.; de Jong, K.; Vonk, J.M.; van Diemen, C.C.; Faiz, A.; van den Berge, M.; Obeidat, M.; et al. Understanding the Role of the Chromosome 15q25.1 in COPD through Epigenetics and Transcriptomics. Eur. J. Hum. Genet. 2018, 26, 709–722. [Google Scholar] [CrossRef]
- Sohal, S.S.; Walters, E.H. Role of Epithelial Mesenchymal Transition (EMT) in Chronic Obstructive Pulmonary Disease (COPD). Respir. Res. 2013, 14, 120. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Mahmood, M.Q.; Walters, E.H.; Shukla, S.D.; Weston, S.; Muller, H.K.; Ward, C.; Sohal, S.S. β-Catenin, Twist and Snail: Transcriptional Regulation of EMT in Smokers and COPD, and Relation to Airflow Obstruction. Sci. Rep. 2017, 7, 10832. [Google Scholar] [CrossRef] [PubMed]
- Aloufi, N.; Alluli, A.; Eidelman, D.H.; Baglole, C.J. Aberrant Post-Transcriptional Regulation of Protein Expression in the Development of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 11963. [Google Scholar] [CrossRef] [PubMed]
- DeMeo, D.L.; Mariani, T.; Bhattacharya, S.; Srisuma, S.; Lange, C.; Litonjua, A.; Bueno, R.; Pillai, S.G.; Lomas, D.A.; Sparrow, D.; et al. Integration of Genomic and Genetic Approaches Implicates IREB2 as a COPD Susceptibility Gene. Am. J. Hum. Genet. 2009, 85, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.F. Non-Coding RNAs: Meet Thy Masters. Bioessays 2010, 32, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Chen, H.; Fu, B.; Huang, Z.; Mou, Y.; Liu, J.; Xu, Y.; Xiong, W.; Cao, Y. LncRNAs NR-026690 and ENST00000447867 Are Upregulated in CD4+ T Cells in Patients with Acute Exacerbation of COPD. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 699–711. [Google Scholar] [CrossRef]
- Donaldson, A.; Natanek, S.A.; Lewis, A.; Man, W.D.-C.; Hopkinson, N.S.; Polkey, M.I.; Kemp, P.R. Increased Skeletal Muscle-Specific MicroRNA in the Blood of Patients with COPD. Thorax 2013, 68, 1140–1149. [Google Scholar] [CrossRef]
- Sun, Y.; An, N.; Li, J.; Xia, J.; Tian, Y.; Zhao, P.; Liu, X.; Huang, H.; Gao, J.; Zhang, X. MiRNA-206 Regulates Human Pulmonary Microvascular Endothelial Cell Apoptosis via Targeting in Chronic Obstructive Pulmonary Disease. J. Cell. Biochem. 2019, 120, 6223–6236. [Google Scholar] [CrossRef]
- Chatila, W.M.; Criner, G.J.; Hancock, W.W.; Akimova, T.; Moldover, B.; Chang, J.-K.; Cornwell, W.; Santerre, M.; Rogers, T.J. Blunted Expression of MiR-199a-5p in Regulatory T Cells of Patients with Chronic Obstructive Pulmonary Disease Compared to Unaffected Smokers. Clin. Exp. Immunol. 2014, 177, 341–352. [Google Scholar] [CrossRef]
- Velasco-Torres, Y.; Ruiz-López, V.; Pérez-Bautista, O.; Buendía-Roldan, I.; Ramírez-Venegas, A.; Pérez-Ramos, J.; Falfán-Valencia, R.; Ramos, C.; Montaño, M. MiR-34a in Serum Is Involved in Mild-to-Moderate COPD in Women Exposed to Biomass Smoke. BMC Pulm. Med. 2019, 19, 227. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Glass, K.; Laucho-Contreras, M.E.; Bhashyam, A.R.; Cervo, M.; Pabón, M.A.; Konrad, C.; Polverino, F.; Siempos, I.I.; Perez, E.; et al. Mitochondrial Iron Chelation Ameliorates Cigarette Smoke-Induced Bronchitis and Emphysema in Mice. Nat. Med. 2016, 22, 163–174. [Google Scholar] [CrossRef]
- Sun, J.; Gu, X.; Wu, N.; Zhang, P.; Liu, Y.; Jiang, S. Human Antigen R Enhances the Epithelial-Mesenchymal Transition via Regulation of ZEB-1 in the Human Airway Epithelium. Respir. Res. 2018, 19, 109. [Google Scholar] [CrossRef] [PubMed]
- Wigington, C.P.; Jung, J.; Rye, E.A.; Belauret, S.L.; Philpot, A.M.; Feng, Y.; Santangelo, P.J.; Corbett, A.H. Post-Transcriptional Regulation of Programmed Cell Death 4 (PDCD4) MRNA by the RNA-Binding Proteins Human Antigen R (HuR) and T-Cell Intracellular Antigen 1 (TIA1). J. Biol. Chem. 2015, 290, 3468–3487. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.; Lecuona, E.; Sznajder, J.I. Role of MicroRNAs in Lung Disease. Arch. Bronconeumol. 2012, 48, 325–330. [Google Scholar] [CrossRef]
- Conickx, G.; Mestdagh, P.; Avila Cobos, F.; Verhamme, F.M.; Maes, T.; Vanaudenaerde, B.M.; Seys, L.J.M.; Lahousse, L.; Kim, R.Y.; Hsu, A.C.; et al. MicroRNA Profiling Reveals a Role for MicroRNA-218-5p in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Lackey, L.; McArthur, E.; Laederach, A. Increased Transcript Complexity in Genes Associated with Chronic Obstructive Pulmonary Disease. PLoS ONE 2015, 10, e0140885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kuo, C.C.J.; Chen, L. GC Content around Splice Sites Affects Splicing through Pre-MRNA Secondary Structures. BMC Genom. 2011, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Corley, M.; Solem, A.; Phillips, G.; Lackey, L.; Ziehr, B.; Vincent, H.A.; Mustoe, A.M.; Ramos, S.B.V.; Weeks, K.M.; Moorman, N.J.; et al. An RNA Structure-Mediated, Posttranscriptional Model of Human α-1-Antitrypsin Expression. Proc. Natl. Acad. Sci. USA 2017, 114, E10244–E10253. [Google Scholar] [CrossRef]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, Classification, and Expression of RAGE Gene Splice Variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global Cancer Statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, H.; Gesthalter, Y.; Spira, A.; Brody, J.S.; Steiling, K. Updates and Controversies in the Rapidly Evolving Field of Lung Cancer Screening, Early Detection, and Chemoprevention. Cancers 2014, 6, 1157–1179. [Google Scholar] [CrossRef] [PubMed]
- Blandin Knight, S.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and Prospects of Early Detection in Lung Cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef] [PubMed]
- Bade, B.C.; Dela Cruz, C.S. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef]
- Krzakowski, M.; Jassem, J. Cancer of the lung, pleura and mediastinum. Oncol. Clin. Pract. 2019, 15, 20–50. [Google Scholar] [CrossRef]
- Raso, M.G.; Bota-Rabassedas, N.; Wistuba, I.I. Pathology and Classification of SCLC. Cancers 2021, 13, 820. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The Biology and Management of Non-Small Cell Lung Cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Petersen, I. The Morphological and Molecular Diagnosis of Lung Cancer. Dtsch. Arztebl. Int. 2011, 108, 525–531. [Google Scholar] [CrossRef]
- Ha, S.Y.; Choi, S.-J.; Cho, J.H.; Choi, H.J.; Lee, J.; Jung, K.; Irwin, D.; Liu, X.; Lira, M.E.; Mao, M.; et al. Lung Cancer in Never-Smoker Asian Females Is Driven by Oncogenic Mutations, Most Often Involving EGFR. Oncotarget 2015, 6, 5465–5474. [Google Scholar] [CrossRef]
- Czarnecka, K.H.; Migdalska-Sęk, M.; Antczak, A.; Pastuszak-Lewandoska, D.; Kordiak, J.; Nawrot, E.; Domańska, D.; Kaleta, D.; Górski, P.; Brzeziańska, E.B. Allelic Imbalance in 1p, 7q, 9p, 11p, 12q and 16q Regions in Non-Small Cell Lung Carcinoma and Its Clinical Association: A Pilot Study. Mol. Biol. Rep. 2013, 40, 6671–6684. [Google Scholar] [CrossRef]
- Popanda, O.; Edler, L.; Waas, P.; Schattenberg, T.; Butkiewicz, D.; Muley, T.; Dienemann, H.; Risch, A.; Bartsch, H.; Schmezer, P. Elevated risk of squamous-cell carcinoma of the lung in heavy smokers carrying the variant alleles of the TP53 Arg72Pro and p21 Ser31Arg polymorphisms. Lung Cancer 2007, 55, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Kiyohara, C.; Horiuchi, T.; Miyake, Y.; Takayama, K.; Nakanishi, Y. Cigarette Smoking, TP53 Arg72Pro, TP53BP1 Asp353Glu and the Risk of Lung Cancer in a Japanese Population. Oncol. Rep. 2010, 23, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Sonoyama, T.; Sakai, A.; Mita, Y.; Yasuda, Y.; Kawamoto, H.; Yagi, T.; Yoshioka, M.; Mimura, T.; Nakachi, K.; Ouchida, M.; et al. TP53 Codon 72 Polymorphism Is Associated with Pancreatic Cancer Risk in Males, Smokers and Drinkers. Mol. Med. Rep. 2011, 4, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Kiyohara, C.; Ohno, Y. Sex Differences in Lung Cancer Susceptibility: A Review. Gend. Med. 2010, 7, 381–401. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Castelli, G.; Pelosi, E. Lung Cancers: Molecular Characterization, Clonal Heterogeneity and Evolution, and Cancer Stem Cells. Cancers 2018, 10, 248. [Google Scholar] [CrossRef]
- Gkountakos, A.; Sartori, G.; Falcone, I.; Piro, G.; Ciuffreda, L.; Carbone, C.; Tortora, G.; Scarpa, A.; Bria, E.; Milella, M.; et al. PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around. Cancers 2019, 11, 1141. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [CrossRef]
- Ferraro, B.; Bepler, G.; Sharma, S.; Cantor, A.; Haura, E.B. EGR1 Predicts PTEN and Survival in Patients with Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2005, 23, 1921–1926. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Ma, J.; Khanna, A.; Lyons, G.; Rinsurongkawong, W.; Bassett, R.; Guo, M.; Routbort, M.J.; Zhang, J.; Skoulidis, F.; et al. Simplified Molecular Classification of Lung Adenocarcinomas Based on EGFR, KRAS, and TP53 Mutations. BMC Cancer 2020, 20, 83. [Google Scholar] [CrossRef] [PubMed]
- El-Telbany, A.; Ma, P.C. Cancer Genes in Lung Cancer: Racial Disparities: Are There Any? Genes Cancer 2012, 3, 467–480. [Google Scholar] [CrossRef]
- Jin, R.; Peng, L.; Shou, J.; Wang, J.; Jin, Y.; Liang, F.; Zhao, J.; Wu, M.; Li, Q.; Zhang, B.; et al. EGFR-Mutated Squamous Cell Lung Cancer and Its Association with Outcomes. Front. Oncol. 2021, 11, 680804. [Google Scholar] [CrossRef] [PubMed]
- Fathi, Z.; Mousavi, S.A.J.; Roudi, R.; Ghazi, F. Distribution of KRAS, DDR2, and TP53 Gene Mutations in Lung Cancer: An Analysis of Iranian Patients. PLoS ONE 2018, 13, e0200633. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Maruoka, S.; Shimizu, T.; Gon, Y. Role of Nrf2 in the Pathogenesis of Respiratory Diseases. Respir. Investig. 2020, 58, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ortega, M.; Carrera, A.C.; Garrido, A. Role of NRF2 in Lung Cancer. Cells 2021, 10, 1879. [Google Scholar] [CrossRef]
- Pinto, J.A.; Vallejos, C.S.; Raez, L.E.; Mas, L.A.; Ruiz, R.; Torres-Roman, J.S.; Morante, Z.; Araujo, J.M.; Gómez, H.L.; Aguilar, A.; et al. Gender and Outcomes in Non-Small Cell Lung Cancer: An Old Prognostic Variable Comes Back for Targeted Therapy and Immunotherapy? ESMO Open 2018, 3, e000344. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Aisner, D.L.; Wood, D.E.; Akerley, W.; Bauman, J.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; Dilling, T.J.; Dobelbower, M.; et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 807–821. [Google Scholar] [CrossRef]
- Clavé, S.; Rodon, N.; Pijuan, L.; Díaz, O.; Lorenzo, M.; Rocha, P.; Taus, Á.; Blanco, R.; Bosch-Barrera, J.; Reguart, N.; et al. Next-Generation Sequencing for ALK and ROS1 Rearrangement Detection in Patients with Non-Small-Cell Lung Cancer: Implications of FISH-Positive Patterns. Clin. Lung Cancer 2019, 20, e421–e429. [Google Scholar] [CrossRef]
- Bergethon, K.; Shaw, A.T.; Ignatius Ou, S.-H.; Katayama, R.; Lovly, C.M.; McDonald, N.T.; Massion, P.P.; Siwak-Tapp, C.; Gonzalez, A.; Fang, R.; et al. ROS1 Rearrangements Define a Unique Molecular Class of Lung Cancers. J. Clin. Oncol. 2012, 30, 863–870. [Google Scholar] [CrossRef]
- Wistuba, I.I.; Behrens, C.; Virmani, A.K.; Mele, G.; Milchgrub, S.; Girard, L.; Fondon, J.W.; Garner, H.R.; McKay, B.; Latif, F.; et al. High Resolution Chromosome 3p Allelotyping of Human Lung Cancer and Preneoplastic/Preinvasive Bronchial Epithelium Reveals Multiple, Discontinuous Sites of 3p Allele Loss and Three Regions of Frequent Breakpoints. Cancer Res. 2000, 60, 1949–1960. [Google Scholar] [PubMed]
- Senchenko, V.N.; Liu, J.; Loginov, W.; Bazov, I.; Angeloni, D.; Seryogin, Y.; Ermilova, V.; Kazubskaya, T.; Garkavtseva, R.; Zabarovska, V.I.; et al. Discovery of Frequent Homozygous Deletions in Chromosome 3p21.3 LUCA and AP20 Regions in Renal, Lung and Breast Carcinomas. Oncogene 2004, 23, 5719–5728. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ortega, J.; Saez, M.C.; Sierra, E.; Torres, A.; Balibrea, J.L.; Hernando, F.; Sanz-Esponera, J.; Merino, M.J. 3p21, 5q21, and 9p21 Allelic Deletions Are Frequently Found in Normal Bronchial Cells Adjacent to Non-Small-Cell Lung Cancer, While They Are Unusual in Patients with No Evidence of Malignancy. J. Pathol. 2001, 195, 429–434. [Google Scholar] [CrossRef]
- Antczak, A.; Migdalska-Sęk, M.; Pastuszak-Lewandoska, D.; Czarnecka, K.; Nawrot, E.; Domańska, D.; Kordiak, J.; Górski, P.; Brzeziańska, E. Significant Frequency of Allelic Imbalance in 3p Region Covering RARβ and MLH1 Loci Seems to Be Essential in Molecular Non-Small Cell Lung Cancer Diagnosis. Med. Oncol. 2013, 30, 532. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. DNA Methylation Errors and Cancer. Cancer Res. 1996, 56, 2463–2467. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The Epigenetic Progenitor Origin of Human Cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Brzeziańska, E.; Dutkowska, A.; Antczak, A. The Significance of Epigenetic Alterations in Lung Carcinogenesis. Mol. Biol. Rep. 2013, 40, 309–325. [Google Scholar] [CrossRef]
- Saito, K.; Kawakami, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Minamoto, T. Long Interspersed Nuclear Element 1 Hypomethylation Is a Marker of Poor Prognosis in Stage IA Non-Small Cell Lung Cancer. Clin. Cancer Res. 2010, 16, 2418–2426. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA Methylation and Human Disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Balgkouranidou, I.; Liloglou, T.; Lianidou, E.S. Lung Cancer Epigenetics: Emerging Biomarkers. Biomark. Med. 2013, 7, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Dobersch, S.; Romero-Olmedo, A.J.; Barreto, G. Epigenetics in Lung Cancer Diagnosis and Therapy. Cancer Metastasis Rev. 2015, 34, 229–241. [Google Scholar] [CrossRef]
- Czarnecka, K.H.; Migdalska-Sęk, M.; Domańska, D.; Pastuszak-Lewandoska, D.; Dutkowska, A.; Kordiak, J.; Nawrot, E.; Kiszałkiewicz, J.; Antczak, A.; Brzeziańska-Lasota, E. FHIT Promoter Methylation Status, Low Protein and High MRNA Levels in Patients with Non-Small Cell Lung Cancer. Int. J. Oncol. 2016, 49, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.F.; Frühwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomäki, P.; Lang, J.C.; et al. Aberrant CpG-Island Methylation Has Non-Random and Tumour-Type-Specific Patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Heller, G.; Babinsky, V.N.; Ziegler, B.; Weinzierl, M.; Noll, C.; Altenberger, C.; Müllauer, L.; Dekan, G.; Grin, Y.; Lang, G.; et al. Genome-Wide CpG Island Methylation Analyses in Non-Small Cell Lung Cancer Patients. Carcinogenesis 2013, 34, 513–521. [Google Scholar] [CrossRef]
- Bjaanæs, M.M.; Fleischer, T.; Halvorsen, A.R.; Daunay, A.; Busato, F.; Solberg, S.; Jørgensen, L.; Kure, E.; Edvardsen, H.; Børresen-Dale, A.-L.; et al. Genome-wide DNA Methylation Analyses in Lung Adenocarcinomas: Association with EGFR, KRAS and TP53 Mutation Status, Gene Expression and Prognosis. Mol. Oncol. 2016, 10, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Colarusso, C.; Terlizzi, M.; Molino, A.; Pinto, A.; Sorrentino, R. Role of the Inflammasome in Chronic Obstructive Pulmonary Disease (COPD). Oncotarget 2017, 8, 81813–81824. [Google Scholar] [CrossRef]
- Rovina, N.; Koutsoukou, A.; Koulouris, N.G. Inflammation and Immune Response in COPD: Where Do We Stand? Mediat. Inflamm. 2013, 2013, 413735. [Google Scholar] [CrossRef]
- King, P.T. Inflammation in Chronic Obstructive Pulmonary Disease and Its Role in Cardiovascular Disease and Lung Cancer. Clin. Transl. Med. 2015, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Burge, S.; Wedzicha, J.A. COPD Exacerbations: Definitions and Classifications. Eur. Respir. J. Suppl. 2003, 41, 46s–53s. [Google Scholar] [CrossRef]
- Seemungal, T.A.; Hurst, J.R.; Wedzicha, J.A. Exacerbation Rate, Health Status and Mortality in COPD–a Review of Potential Interventions. Int. J. Chronic Obstr. Pulm. Dis. 2009, 4, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, W.; Guo, Y.; Liu, Y.; Li, Q.; Yang, K.; Wang, S.; Zeng, N.; Duan, W.; Chen, Z.; et al. Early COPD Risk Decision for Adults Aged from 40 to 79 Years Based on Lung Radiomics Features. Front. Med. 2022, 9, 845286. [Google Scholar] [CrossRef] [PubMed]
- Kukrety, S.P.; Parekh, J.D.; Bailey, K.L. Chronic Obstructive Pulmonary Disease and the Hallmarks of Aging. Lung India 2018, 35, 321–327. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Meng, Y.; Adcock, I.M.; Yao, X. Role of Inflammatory Cells in Airway Remodeling in COPD. Int. J. Chronic Obstr. Pulm. Dis. 2018, 13, 3341–3348. [Google Scholar] [CrossRef]
- Agustí, A.; MacNee, W.; Donaldson, K.; Cosio, M. Hypothesis: Does COPD Have an Autoimmune Component? Thorax 2003, 58, 832–834. [Google Scholar] [CrossRef]
- Sethi, S.; Evans, N.; Grant, B.J.B.; Murphy, T.F. New Strains of Bacteria and Exacerbations of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2002, 347, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.; Rogers, T.J.; Criner, G.J. New Concepts in the Pathobiology of Chronic Obstructive Pulmonary Disease. Proc. Am. Thorac. Soc. 2008, 5, 478–485. [Google Scholar] [CrossRef]
- O’Donnell, R.; Breen, D.; Wilson, S.; Djukanovic, R. Inflammatory Cells in the Airways in COPD. Thorax 2006, 61, 448–454. [Google Scholar] [CrossRef]
- Vlahos, R.; Bozinovski, S. Role of Alveolar Macrophages in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2014, 5, 435. [Google Scholar] [CrossRef]
- Trivedi, A.; Khan, M.A.; Bade, G.; Talwar, A. Orchestration of Neutrophil Extracellular Traps (Nets), a Unique Innate Immune Function during Chronic Obstructive Pulmonary Disease (COPD) Development. Biomedicines 2021, 9, 53. [Google Scholar] [CrossRef]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic Obstructive Pulmonary Disease: Molecular and Cellular Mechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef] [PubMed]
- de Boer, W.I.; Sont, J.K.; van Schadewijk, A.; Stolk, J.; van Krieken, J.H.; Hiemstra, P.S. Monocyte Chemoattractant Protein 1, Interleukin 8, and Chronic Airways Inflammation in COPD. J. Pathol. 2000, 190, 619–626. [Google Scholar] [CrossRef]
- Selvarajah, S.; Todd, I.; Tighe, P.J.; John, M.; Bolton, C.E.; Harrison, T.; Fairclough, L.C. Multiple Circulating Cytokines Are Coelevated in Chronic Obstructive Pulmonary Disease. Mediat. Inflamm. 2016, 2016, 3604842. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, P.; Ramlau, R.; Błach, J.; Kieszko, R.; Roszkowski-Ślisz, K.; Kucharczyk, T.; Kieszko, S.; Milanowski, J. Czynniki ryzyka i profilaktyka pierwotna raka płuca. Leczenie chorych uzależnionych od palenia tytoniu. Onkol. Prakt. Klin.-Eduk. 2021, 7, 160–173. [Google Scholar]
- Talhout, R.; Schulz, T.; Florek, E.; van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous Compounds in Tobacco Smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Besaratinia, A.; Pfeifer, G.P. Second-Hand Smoke and Human Lung Cancer. Lancet Oncol. 2008, 9, 657–666. [Google Scholar] [CrossRef]
- Pugmire, J.; Sweeting, H.; Moore, L. Environmental Tobacco Smoke Exposure among Infants, Children and Young People: Now Is No Time to Relax. Arch. Dis. Child. 2017, 102, 117–118. [Google Scholar] [CrossRef]
- Glantz, S.A.; Parmley, W.W. Passive Smoking and Heart Disease. Epidemiology, Physiology, and Biochemistry. Circulation 1991, 83, 1–12. [Google Scholar] [CrossRef]
- Kaleta, D.; Usidame, B.; Dziankowska-Zaborszczyk, E.; Makowiec-Dąbrowska, T. Correlates of Cessation Success among Romanian Adults. Biomed. Res. Int. 2014, 2014, 675496. [Google Scholar] [CrossRef]
- Taylor, J.D. COPD and the Response of the Lung to Tobacco Smoke Exposure. Pulm. Pharmacol. Ther. 2010, 23, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco Smoke: Involvement of Reactive Oxygen Species and Stable Free Radicals in Mechanisms of Oxidative Damage, Carcinogenesis and Synergistic Effects with Other Respirable Particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Bowler, R.P.; Barnes, P.J.; Crapo, J.D. The Role of Oxidative Stress in Chronic Obstructive Pulmonary Disease. COPD 2004, 1, 255–277. [Google Scholar] [CrossRef]
- Białas, A.J.; Sitarek, P.; Miłkowska-Dymanowska, J.; Piotrowski, W.J.; Górski, P. The Role of Mitochondria and Oxidative/Antioxidative Imbalance in Pathobiology of Chronic Obstructive Pulmonary Disease. Oxid. Med. Cell. Longev. 2016, 2016, 7808576. [Google Scholar] [CrossRef]
- Boukhenouna, S.; Wilson, M.A.; Bahmed, K.; Kosmider, B. Reactive Oxygen Species in Chronic Obstructive Pulmonary Disease. Oxid. Med. Cell. Longev 2018, 2018, 5730395. [Google Scholar] [CrossRef] [PubMed]
- Kryczka, J.; Kryczka, J.; Czarnecka-Chrebelska, K.H.; Brzeziańska-Lasota, E. Molecular Mechanisms of Chemoresistance Induced by Cisplatin in NSCLC Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 8885. [Google Scholar] [CrossRef]
- Cohen, B.H.; Ball, W.C.; Brashears, S.; Diamond, E.L.; Kreiss, P.; Levy, D.A.; Menkes, H.A.; Permutt, S.; Tockman, M.S. Risk Factors in Chronic Obstructive Pulmonary Disease (COPD). Am. J. Epidemiol. 1977, 105, 223–232. [Google Scholar] [CrossRef]
- Finaud, J.; Lac, G.; Filaire, E. Oxidative Stress. Sports Med. 2006, 36, 327–358. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative Stress, Aging, and Diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Cross, C.E.; van der Vliet, A.; O’Neill, C.A.; Louie, S.; Halliwell, B. Oxidants, Antioxidants, and Respiratory Tract Lining Fluids. Environ. Health Perspect. 1994, 102 (Suppl. S10), 185–191. [Google Scholar] [CrossRef]
- Davies, K.J. Oxidative Stress, Antioxidant Defenses, and Damage Removal, Repair, and Replacement Systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, A.J.A.; Sapey, E. Oxidative Stress in COPD: Sources, Markers, and Potential Mechanisms. J. Clin. Med. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Delsignore, M.E. Protein Damage and Degradation by Oxygen Radicals. III. Modification of Secondary and Tertiary Structure. J. Biol. Chem. 1987, 262, 9908–9913. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Koppal, T.; Howard, B.; Subramaniam, R.; Hall, N.; Hensley, K.; Yatin, S.; Allen, K.; Aksenov, M.; Aksenova, M.; et al. Structural and Functional Changes in Proteins Induced by Free Radical-Mediated Oxidative Stress and Protective Action of the Antioxidants N-Tert-Butyl-Alpha-Phenylnitrone and Vitamin E. Ann. N. Y. Acad. Sci. 1998, 854, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Kim, S.R.; Lee, Y.C. Impact of Oxidative Stress on Lung Diseases. Respirology 2009, 14, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A.; Young, L.R.; Wang, Y.; Horger, T.; Rose, M.C.; Fischer, B.M. Neutrophil Elastase Increases MUC5AC MRNA and Protein Expression in Respiratory Epithelial Cells. Am. J. Physiol. 1999, 276, L835–L843. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.C.; De, S.; Mishra, P.K. Role of Proteases in Chronic Obstructive Pulmonary Disease. Front. Pharmacol. 2017, 8, 512. [Google Scholar] [CrossRef]
- Gadek, J.E.; Pacht, E.R. The Protease-Antiprotease Balance within the Human Lung: Implications for the Pathogenesis of Emphysema. Lung 1990, 168, 552–564. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Belaaouaj, A.; McCarthy, R.; Baumann, M.; Gao, Z.; Ley, T.J.; Abraham, S.N.; Shapiro, S.D. Mice Lacking Neutrophil Elastase Reveal Impaired Host Defense against Gram Negative Bacterial Sepsis. Nat. Med. 1998, 4, 615–618. [Google Scholar] [CrossRef]
- Tsai, Y.-F.; Hwang, T.-L. Neutrophil Elastase Inhibitors: A Patent Review and Potential Applications for Inflammatory Lung Diseases (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, Y.; Kravis, T.C.; Moser, K.M.; Taylor, J.C.; Crawford, I.P. Relationship of Leukocyte Elastase Concentration to Severity of Emphysema in Homozygous Alpha1-Antitrypsin-Deficient Persons. Am. Rev. Respir. Dis. 1977, 115, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.M.; Voynow, J.A. Neutrophil Elastase Induces MUC5AC Gene Expression in Airway Epithelium via a Pathway Involving Reactive Oxygen Species. Am. J. Respir. Cell Mol. Biol. 2002, 26, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Kohri, K.; Ueki, I.F.; Nadel, J.A. Neutrophil Elastase Induces Mucin Production by Ligand-Dependent Epidermal Growth Factor Receptor Activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L531–L540. [Google Scholar] [CrossRef]
- Kim, K.C.; Wasano, K.; Niles, R.M.; Schuster, J.E.; Stone, P.J.; Brody, J.S. Human Neutrophil Elastase Releases Cell Surface Mucins from Primary Cultures of Hamster Tracheal Epithelial Cells. Proc. Natl. Acad. Sci. USA 1987, 84, 9304–9308. [Google Scholar] [CrossRef]
- Kao, S.S.-T.; Ramezanpour, M.; Bassiouni, A.; Wormald, P.-J.; Psaltis, A.J.; Vreugde, S. The Effect of Neutrophil Serine Proteases on Human Nasal Epithelial Cell Barrier Function. Int. Forum Allergy Rhinol. 2019, 9, 1220–1226. [Google Scholar] [CrossRef]
- Vermeer, P.D.; Einwalter, L.A.; Moninger, T.O.; Rokhlina, T.; Kern, J.A.; Zabner, J.; Welsh, M.J. Segregation of Receptor and Ligand Regulates Activation of Epithelial Growth Factor Receptor. Nature 2003, 422, 322–326. [Google Scholar] [CrossRef]
- Boxio, R.; Wartelle, J.; Nawrocki-Raby, B.; Lagrange, B.; Malleret, L.; Hirche, T.; Taggart, C.; Pacheco, Y.; Devouassoux, G.; Bentaher, A. Neutrophil Elastase Cleaves Epithelial Cadherin in Acutely Injured Lung Epithelium. Respir. Res. 2016, 17, 129. [Google Scholar] [CrossRef]
- Voynow, J.A.; Fischer, B.M.; Malarkey, D.E.; Burch, L.H.; Wong, T.; Longphre, M.; Ho, S.B.; Foster, W.M. Neutrophil Elastase Induces Mucus Cell Metaplasia in Mouse Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L1293–L1302. [Google Scholar] [CrossRef]
- Amitani, R.; Wilson, R.; Rutman, A.; Read, R.; Ward, C.; Burnett, D.; Stockley, R.A.; Cole, P.J. Effects of Human Neutrophil Elastase and Pseudomonas Aeruginosa Proteinases on Human Respiratory Epithelium. Am. J. Respir. Cell Mol. Biol. 1991, 4, 26–32. [Google Scholar] [CrossRef]
- Le Gars, M.; Descamps, D.; Roussel, D.; Saussereau, E.; Guillot, L.; Ruffin, M.; Tabary, O.; Hong, S.-S.; Boulanger, P.; Paulais, M.; et al. Neutrophil Elastase Degrades Cystic Fibrosis Transmembrane Conductance Regulator via Calpains and Disables Channel Function in Vitro and in Vivo. Am. J. Respir. Crit. Care Med. 2013, 187, 170–179. [Google Scholar] [CrossRef]
- Chua, F.; Laurent, G.J. Neutrophil Elastase: Mediator of Extracellular Matrix Destruction and Accumulation. Proc. Am. Thorac. Soc. 2006, 3, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Mecham, R.P. Elastin in Lung Development and Disease Pathogenesis. Matrix. Biol. 2018, 73, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-Y.; Ho, S.-C.; Lin, H.-C.; Lin, S.-M.; Liu, C.-Y.; Huang, C.-D.; Wang, C.-H.; Chung, K.F.; Kuo, H.-P. Neutrophil-Derived Elastase Induces TGF-Beta1 Secretion in Human Airway Smooth Muscle via NF-KappaB Pathway. Am. J. Respir. Cell Mol. Biol. 2006, 35, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Geraghty, P.; Campos, M.; Garcia-Arcos, I.; Dabo, A.J.; Gaffney, A.; Eden, E.; Jiang, X.-C.; D’Armiento, J.; Foronjy, R. Cathepsin G Degradation of Phospholipid Transfer Protein (PLTP) Augments Pulmonary Inflammation. FASEB J. 2014, 28, 2318–2331. [Google Scholar] [CrossRef]
- Gudmann, N.S.; Manon-Jensen, T.; Sand, J.M.B.; Diefenbach, C.; Sun, S.; Danielsen, A.; Karsdal, M.A.; Leeming, D.J. Lung Tissue Destruction by Proteinase 3 and Cathepsin G Mediated Elastin Degradation Is Elevated in Chronic Obstructive Pulmonary Disease. Biochem. Biophys. Res. Commun. 2018, 503, 1284–1290. [Google Scholar] [CrossRef]
- Finlay, G.A.; O’Driscoll, L.R.; Russell, K.J.; D’Arcy, E.M.; Masterson, J.B.; FitzGerald, M.X.; O’Connor, C.M. Matrix Metalloproteinase Expression and Production by Alveolar Macrophages in Emphysema. Am. J. Respir. Crit. Care Med. 1997, 156, 240–247. [Google Scholar] [CrossRef]
- Houghton, A.M. Matrix Metalloproteinases in Destructive Lung Disease. Matrix. Biol. 2015, 44–46, 167–174. [Google Scholar] [CrossRef]
- Hautamaki, R.D.; Kobayashi, D.K.; Senior, R.M.; Shapiro, S.D. Requirement for Macrophage Elastase for Cigarette Smoke-Induced Emphysema in Mice. Science 1997, 277, 2002–2004. [Google Scholar] [CrossRef]
- Chaudhuri, R.; McSharry, C.; Brady, J.; Donnelly, I.; Grierson, C.; McGuinness, S.; Jolly, L.; Weir, C.J.; Messow, C.M.; Spears, M.; et al. Sputum Matrix Metalloproteinase-12 in Patients with Chronic Obstructive Pulmonary Disease and Asthma: Relationship to Disease Severity. J. Allergy Clin. Immunol. 2012, 129, 655–663.e8. [Google Scholar] [CrossRef]
- Haq, I.; Chappell, S.; Johnson, S.R.; Lotya, J.; Daly, L.; Morgan, K.; Guetta-Baranes, T.; Roca, J.; Rabinovich, R.; Millar, A.B.; et al. Association of MMP-2 Polymorphisms with Severe and Very Severe COPD: A Case Control Study of MMPs-1, 9 and 12 in a European Population. BMC Med. Genet. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Friedland, J.S.; Ugarte-Gil, C.A.; Elkington, P.T. Series “Matrix Metalloproteinases in Lung Health and Disease”: Matrix Metalloproteinases in Tuberculosis. DSace Repository. 2011. Available online: https://hdl.handle.net/20.500.12866/11034 (accessed on 15 December 2022).
- O’Connor, C.M.; FitzGerald, M.X. Matrix Metalloproteases and Lung Disease. Thorax 1994, 49, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Zhong, N. Expression of Matrix Metalloproteinases MMP-9 within the Airways in Asthma. Respir. Med. 2003, 97, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, D.; Munaut, C.; Noël, A.; Frankenne, F.; Bartsch, P.; Foidart, J.M.; Louis, R. MMP-2- and MMP-9-Linked Gelatinolytic Activity in the Sputum from Patients with Asthma and Chronic Obstructive Pulmonary Disease. Int. Arch. Allergy Immunol. 2000, 123, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Sng, J.J.; Prazakova, S.; Thomas, P.S.; Herbert, C. MMP-8, MMP-9 and Neutrophil Elastase in Peripheral Blood and Exhaled Breath Condensate in COPD. COPD 2017, 14, 238–244. [Google Scholar] [CrossRef]
- Linder, R.; Rönmark, E.; Pourazar, J.; Behndig, A.; Blomberg, A.; Lindberg, A. Serum Metalloproteinase-9 Is Related to COPD Severity and Symptoms-Cross-Sectional Data from a Population Based Cohort-Study. Respir. Res. 2015, 16, 28. [Google Scholar] [CrossRef]
- Devereux, G.; Steele, S.; Jagelman, T.; Fielding, S.; Muirhead, R.; Brady, J.; Grierson, C.; Brooker, R.; Winter, J.; Fardon, T.; et al. An Observational Study of Matrix Metalloproteinase (MMP)-9 in Cystic Fibrosis. J. Cyst. Fibros. 2014, 13, 557–563. [Google Scholar] [CrossRef]
- Atkinson, J.J.; Senior, R.M. Matrix Metalloproteinase-9 in Lung Remodeling. Am. J. Respir. Cell Mol. Biol. 2003, 28, 12–24. [Google Scholar] [CrossRef]
- Lee, E.J.; In, K.H.; Kim, J.H.; Lee, S.Y.; Shin, C.; Shim, J.J.; Kang, K.H.; Yoo, S.H.; Kim, C.H.; Kim, H.-K.; et al. Proteomic Analysis in Lung Tissue of Smokers and COPD Patients. Chest 2009, 135, 344–352. [Google Scholar] [CrossRef]
- Gogebakan, B.; Bayraktar, R.; Ulaslı, M.; Oztuzcu, S.; Tasdemir, D.; Bayram, H. The Role of Bronchial Epithelial Cell Apoptosis in the Pathogenesis of COPD. Mol. Biol. Rep. 2014, 41, 5321–5327. [Google Scholar] [CrossRef]
- Nakajima, T.; Nakamura, H.; Owen, C.A.; Yoshida, S.; Tsuduki, K.; Chubachi, S.; Shirahata, T.; Mashimo, S.; Nakamura, M.; Takahashi, S.; et al. Plasma Cathepsin S and Cathepsin S/Cystatin C Ratios Are Potential Biomarkers for COPD. Dis. Markers 2016, 2016, e4093870. [Google Scholar] [CrossRef] [PubMed]
- Golovatch, P.; Mercer, B.A.; Lemaître, V.; Wallace, A.; Foronjy, R.F.; D’Armiento, J. Role for Cathepsin K in Emphysema in Smoke-Exposed Guinea Pigs. Exp. Lung Res. 2009, 35, 631–645. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Homer, R.; Zhang, Y.; Petrache, I.; Mannam, P.; Lee, P.J. Cathepsin E Promotes Pulmonary Emphysema via Mitochondrial Fission. Am. J. Pathol. 2014, 184, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.-J.; Li, M.-H.; Li, J.-X.; Xu, X.; Ren, S.-X.; Rajbanshi, B.; Xu, J.-F. High Expression of Cathepsin E Is Associated with the Severity of Airflow Limitation in Patients with COPD. COPD 2016, 13, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Bamlet, W.R.; Sun, Z.; Ebbert, J.O.; Aubry, M.-C.; Taylor, W.R.; Marks, R.S.; Deschamps, C.; Swensen, S.J.; Wieben, E.D.; et al. A1-Antitrypsin and Neutrophil Elastase Imbalance and Lung Cancer Risk. Chest 2005, 128, 445–452. [Google Scholar] [CrossRef]
- Perlmutter, D.H. Alpha-1-Antitrypsin Deficiency: Biochemistry and Clinical Manifestations. Ann. Med. 1996, 28, 385–394. [Google Scholar] [CrossRef]
- Petrache, I.; Fijalkowska, I.; Medler, T.R.; Skirball, J.; Cruz, P.; Zhen, L.; Petrache, H.I.; Flotte, T.R.; Tuder, R.M. α-1 Antitrypsin Inhibits Caspase-3 Activity, Preventing Lung Endothelial Cell Apoptosis. Am. J. Pathol. 2006, 169, 1155–1166. [Google Scholar] [CrossRef]
- Strange, C. Alpha-1 Antitrypsin Deficiency Associated COPD. Clin. Chest Med. 2020, 41, 339–345. [Google Scholar] [CrossRef]
- Gooptu, B.; Ekeowa, U.I.; Lomas, D.A. Mechanisms of Emphysema in A1-Antitrypsin Deficiency: Molecular and Cellular Insights. Eur. Respir. J. 2009, 34, 475–488. [Google Scholar] [CrossRef]
- Moreau, T.; Baranger, K.; Dadé, S.; Dallet-Choisy, S.; Guyot, N.; Zani, M.-L. Multifaceted Roles of Human Elafin and Secretory Leukocyte Proteinase Inhibitor (SLPI), Two Serine Protease Inhibitors of the Chelonianin Family. Biochimie 2008, 90, 284–295. [Google Scholar] [CrossRef]
- Yoshizaki, K.; de Vega, S.; Yamada, Y. Gene Evolution and Functions of Extracellular Matrix Proteins in Teeth. Orthod. Waves 2013, 72, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Trojanek, J. [Matrix metalloproteinases and their tissue inhibitors]. Postep. Biochem. 2012, 58, 353–362. [Google Scholar]
- Kumaki, F.; Matsui, K.; Kawai, T.; Ozeki, Y.; Yu, Z.-X.; Ferrans, V.J.; Travis, W.D. Expression of Matrix Metalloproteinases in Invasive Pulmonary Adenocarcinoma with Bronchioloalveolar Component and Atypical Adenomatous Hyperplasia. Am. J. Pathol. 2001, 159, 2125–2135. [Google Scholar] [CrossRef]
- Ishikawa, S.; Takenaka, K.; Yanagihara, K.; Miyahara, R.; Kawano, Y.; Otake, Y.; Hasegawa, S.; Wada, H.; Tanaka, F. Matrix Metalloproteinase-2 Status in Stromal Fibroblasts, Not in Tumor Cells, Is a Significant Prognostic Factor in Non–Small-Cell Lung Cancer. Clin. Cancer Res. 2004, 10, 6579–6585. [Google Scholar] [CrossRef]
- Galateau-Salle, F.B.; Luna, R.E.; Horiba, K.; Sheppard, M.N.; Hayashi, T.; Fleming, M.V.; Colby, T.V.; Bennett, W.; Harris, C.C.; Stetler-Stevenson, W.G.; et al. Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases in Bronchial Squamous Preinvasive Lesions. Hum. Pathol. 2000, 31, 296–305. [Google Scholar] [CrossRef]
- Pastuszak-Lewandoska, D.; Kordiak, J.; Czarnecka, K.H.; Migdalska-Sęk, M.; Nawrot, E.; Domańska-Senderowska, D.; Kiszałkiewicz, J.M.; Antczak, A.; Górski, P.; Brzeziańska-Lasota, E. Expression Analysis of Three MiRNAs, MiR-26a, MiR-29b and MiR-519d, in Relation to MMP-2 Expression Level in Non-Small Cell Lung Cancer Patients: A Pilot Study. Med. Oncol. 2016, 33, 96. [Google Scholar] [CrossRef]
- Kallio, J.P.; Hopkins-Donaldson, S.; Baker, A.H.; Kähäri, V.-M. TIMP-3 Promotes Apoptosis in Nonadherent Small Cell Lung Carcinoma Cells Lacking Functional Death Receptor Pathway. Int. J. Cancer 2011, 128, 991–996. [Google Scholar] [CrossRef]
- Hadler-Olsen, E.; Winberg, J.-O.; Uhlin-Hansen, L. Matrix Metalloproteinases in Cancer: Their Value as Diagnostic and Prognostic Markers and Therapeutic Targets. Tumor. Biol. 2013, 34, 2041–2051. [Google Scholar] [CrossRef]
- Guan, Z.; Zhang, J.; Song, S.; Dai, D. Promoter Methylation and Expression of TIMP3 Gene in Gastric Cancer. Diagn. Pathol. 2013, 8, 110. [Google Scholar] [CrossRef]
- Lei, Y.; Liu, Z.; Yang, W. Negative Correlation of Cytoplasm TIMP3 with MiR-222 Indicates a Good Prognosis for NSCLC. Onco Targets Ther. 2018, 11, 5551–5557. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, K.H.; Szmyd, B.; Barańska, M.; Kaszkowiak, M.; Kordiak, J.; Antczak, A.; Pastuszak-Lewandoska, D.; Brzeziańska-Lasota, E. A Strong Decrease in TIMP3 Expression Mediated by the Presence of MiR-17 and 20a Enables Extracellular Matrix Remodeling in the NSCLC Lesion Surroundings. Front. Oncol. 2019, 9, 1372. [Google Scholar] [CrossRef] [PubMed]
- Rennard, S.I.; Drummond, M.B. Early Chronic Obstructive Pulmonary Disease: Definition, Assessment, and Prevention. Lancet 2015, 385, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Soriano, J.B.; Polverino, F.; Cosio, B.G. What Is Early COPD and Why Is It Important? Eur. Respir. J. 2018, 52, 1801448. [Google Scholar] [CrossRef] [PubMed]
- Fazleen, A.; Wilkinson, T. Early COPD: Current Evidence for Diagnosis and Management. Ther. Adv. Respir. Dis. 2020, 14, 1753466620942128. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Barr, R.G.; Bleecker, E.; Christenson, S.A.; Couper, D.; Curtis, J.L.; Gouskova, N.A.; Hansel, N.N.; Hoffman, E.A.; Kanner, R.E.; et al. Clinical Significance of Symptoms in Smokers with Preserved Pulmonary Function. N. Engl. J. Med. 2016, 374, 1811–1821. [Google Scholar] [CrossRef]
- Agusti, A.; Calverley, P.M.; Celli, B.; Coxson, H.O.; Edwards, L.D.; Lomas, D.A.; MacNee, W.; Miller, B.E.; Rennard, S.; Silverman, E.K.; et al. Characterisation of COPD Heterogeneity in the ECLIPSE Cohort. Respir. Res. 2010, 11, 122. [Google Scholar] [CrossRef]
- Deng, Z.; Wu, S.; Wang, Y.; Shi, D. Circulating Tumor Cell Isolation for Cancer Diagnosis and Prognosis. EBioMedicine 2022, 83, 104237. [Google Scholar] [CrossRef]
- Ilie, M.; Hofman, V.; Long-Mira, E.; Selva, E.; Vignaud, J.-M.; Padovani, B.; Mouroux, J.; Marquette, C.-H.; Hofman, P. “Sentinel” Circulating Tumor Cells Allow Early Diagnosis of Lung Cancer in Patients with Chronic Obstructive Pulmonary Disease. PLoS ONE 2014, 9, e111597. [Google Scholar] [CrossRef]
- Romero-Palacios, P.J.; Alcázar-Navarrete, B.; Díaz Mochón, J.J.; de Miguel-Pérez, D.; López Hidalgo, J.L.; Garrido-Navas, M.D.C.; Quero Valenzuela, F.; Lorente, J.A.; Serrano, M.J. Liquid Biopsy beyond of Cancer: Circulating Pulmonary Cells as Biomarkers of COPD Aggressivity. Crit. Rev. Oncol. Hematol. 2019, 136, 31–36. [Google Scholar] [CrossRef]
- Bai, J.-W.; Chen, X.; Liu, S.; Yu, L.; Xu, J.-F. Smoking Cessation Affects the Natural History of COPD. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Sanei, F.; Wilkinson, T. Influenza Vaccination for Patients with Chronic Obstructive Pulmonary Disease: Understanding Immunogenicity, Efficacy and Effectiveness. Ther. Adv. Respir. Dis. 2016, 10, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Aitio, M.-L. N-Acetylcysteine--Passe-Partout or Much Ado about Nothing? Br. J. Clin. Pharmacol. 2006, 61, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-H.; Chen, Y.-C.; Wang, J.-J.; Liao, K.-M. Incidence and Relative Risk for Developing Cancer among Patients with COPD: A Nationwide Cohort Study in Taiwan. BMJ Open 2017, 7, e013195. [Google Scholar] [CrossRef] [PubMed]
- Uliński, R.; Kwiecień, I.; Domagała-Kulawik, J. Lung Cancer in the Course of COPD-Emerging Problems Today. Cancers 2022, 14, 3819. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Z.; Fang, Z.; Zhao, J.; Zhou, Y.; Tang, C. Multi-Omics Study on Biomarker and Pathway Discovery of Chronic Obstructive Pulmonary Disease. J. Breath Res. 2021, 15, 044001. [Google Scholar] [CrossRef]
- Wiegman, C.H.; Li, F.; Ryffel, B.; Togbe, D.; Chung, K.F. Oxidative Stress in Ozone-Induced Chronic Lung Inflammation and Emphysema: A Facet of Chronic Obstructive Pulmonary Disease. Front. Immunol. 2020, 11, 1957. [Google Scholar] [CrossRef]
- Wang, Z.; Locantore, N.; Haldar, K.; Ramsheh, M.Y.; Beech, A.S.; Ma, W.; Brown, J.R.; Tal-Singer, R.; Barer, M.R.; Bafadhel, M.; et al. Inflammatory Endotype-Associated Airway Microbiome in Chronic Obstructive Pulmonary Disease Clinical Stability and Exacerbations: A Multicohort Longitudinal Analysis. Am. J. Respir. Crit. Care Med. 2021, 203, 1488–1502. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Qian, J.; Massion, P.P. Next-Generation Molecular Therapy in Lung Cancer. Transl. Lung Cancer Res. 2018, 7, S31–S34. [Google Scholar] [CrossRef]
- Domagała-Kulawik, J. Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer-towards Daily Practice. Adv. Respir. Med. 2018, 86, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Yuan, J.-Q.; Wang, K.-F.; Fu, X.-H.; Han, X.-R.; Threapleton, D.; Yang, Z.-Y.; Mao, C.; Tang, J.-L. The Prevalence of EGFR Mutation in Patients with Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Oncotarget 2016, 7, 78985–78993. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus Standard Chemotherapy as First-Line Treatment for European Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (EURTAC): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Khaddour, K.; Jonna, S.; Deneka, A.; Patel, J.D.; Abazeed, M.E.; Golemis, E.; Borghaei, H.; Boumber, Y. Targeting the Epidermal Growth Factor Receptor in EGFR-Mutated Lung Cancer: Current and Emerging Therapies. Cancers 2021, 13, 3164. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-Q.; Zeng, L.-S.; Wang, L.-F.; Wang, Y.-Y.; Cheng, J.-T.; Zhang, Y.; Han, Z.-W.; Zhou, Y.; Huang, S.-L.; Wang, X.-W.; et al. The Latest Battles Between EGFR Monoclonal Antibodies and Resistant Tumor Cells. Front. Oncol. 2020, 10, 1249. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, T. Downregulation of MicroRNA-135 Promotes Sensitivity of Non-Small Cell Lung Cancer to Gefitinib by Targeting TRIM16. Oncol. Res. 2018, 26, 1005–1014. [Google Scholar] [CrossRef]
- Yonesaka, K.; Takegawa, N.; Watanabe, S.; Haratani, K.; Kawakami, H.; Sakai, K.; Chiba, Y.; Maeda, N.; Kagari, T.; Hirotani, K.; et al. An HER3-Targeting Antibody-Drug Conjugate Incorporating a DNA Topoisomerase I Inhibitor U3-1402 Conquers EGFR Tyrosine Kinase Inhibitor-Resistant NSCLC. Oncogene 2019, 38, 1398–1409. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The Emerging Treatment Landscape of Targeted Therapy in Non-Small-Cell Lung Cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef]
- Gu, L.; Deng, Z.J.; Roy, S.; Hammond, P.T. A Combination RNAi-Chemotherapy Layer-by-Layer Nanoparticle for Systemic Targeting of KRAS/P53 with Cisplatin to Treat Non-Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 7312–7323. [Google Scholar] [CrossRef]
- Hu, Y.; Hong, Y.; Xu, Y.; Liu, P.; Guo, D.-H.; Chen, Y. Inhibition of the JAK/STAT Pathway with Ruxolitinib Overcomes Cisplatin Resistance in Non-Small-Cell Lung Cancer NSCLC. Apoptosis 2014, 19, 1627–1636. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus Crizotinib in Patients with ALK-Positive Non-Small-Cell Lung Cancer (J-ALEX): An Open-Label, Randomised Phase 3 Trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Ou, S.-H.I.; Bang, Y.-J.; Camidge, D.R.; Solomon, B.J.; Salgia, R.; Riely, G.J.; Varella-Garcia, M.; Shapiro, G.I.; Costa, D.B.; et al. Crizotinib in ROS1-Rearranged Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2014, 371, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Stencel, K.; Świniuch, D.; Ramlau, R. Molecular Targeted Therapy of Patients with Non-Small-Cell Lung Cancer. Oncol. Clin. Pract. 2019, 15, 289–296. [Google Scholar] [CrossRef]
- Gonzalez, J.; Marín, M.; Sánchez-Salcedo, P.; Zulueta, J.J. Lung Cancer Screening in Patients with Chronic Obstructive Pulmonary Disease. Ann. Transl. Med. 2016, 4, 160. [Google Scholar] [CrossRef]
- Brenner, D.R.; McLaughlin, J.R.; Hung, R.J. Previous Lung Diseases and Lung Cancer Risk: A Systematic Review and Meta-Analysis. PLoS ONE 2011, 6, e17479. [Google Scholar] [CrossRef]
- Wang, P.; Zhu, M.; Zhang, D.; Guo, X.-G.; Zhao, S.; Zhang, X.-L.; Wang, D.-L.; Liu, C.-T. The Relationship between Chronic Obstructive Pulmonary Disease and Non-Small Cell Lung Cancer in the Elderly. Cancer Med. 2019, 8, 4124–4134. [Google Scholar] [CrossRef]
- Günay, E.; Sarınç Ulaşlı, S.; Akar, O.; Ahsen, A.; Günay, S.; Koyuncu, T.; Unlü, M. Neutrophil-to-Lymphocyte Ratio in Chronic Obstructive Pulmonary Disease: A Retrospective Study. Inflammation 2014, 37, 374–380. [Google Scholar] [CrossRef]
- Bagley, S.J.; Kothari, S.; Aggarwal, C.; Bauml, J.M.; Alley, E.W.; Evans, T.L.; Kosteva, J.A.; Ciunci, C.A.; Gabriel, P.E.; Thompson, J.C.; et al. Pretreatment Neutrophil-to-Lymphocyte Ratio as a Marker of Outcomes in Nivolumab-Treated Patients with Advanced Non-Small-Cell Lung Cancer. Lung Cancer 2017, 106, 1–7. [Google Scholar] [CrossRef]
- Silverman, E.K.; Mosley, J.D.; Palmer, L.J.; Barth, M.; Senter, J.M.; Brown, A.; Drazen, J.M.; Kwiatkowski, D.J.; Chapman, H.A.; Campbell, E.J.; et al. Genome-Wide Linkage Analysis of Severe, Early-Onset Chronic Obstructive Pulmonary Disease: Airflow Obstruction and Chronic Bronchitis Phenotypes. Hum. Mol. Genet. 2002, 11, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Tessema, M.; Yingling, C.M.; Picchi, M.A.; Wu, G.; Liu, Y.; Weissfeld, J.L.; Siegfried, J.M.; Tesfaigzi, Y.; Belinsky, S.A. Epigenetic Repression of CCDC37 and MAP1B Links Chronic Obstructive Pulmonary Disease to Lung Cancer. J. Thorac. Oncol. 2015, 10, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Sunny, J.S.; Rakshit, S.; George, M.; Leela, K.V.; Sarkar, K. Involvement of Inflammatory Cytokines and Epigenetic Modification of the MtTFA Complex in T-Helper Cells of Patients’ Suffering from Non-Small Cell Lung Cancer and Chronic Obstructive Pulmonary Disease. Mol. Immunol. 2022, 151, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, H.; Zhang, L.; Luo, H.; Chen, Q.; Zuo, X. HSP90AA1, ADRB2, TBL1XR1 and HSPB1 Are Chronic Obstructive Pulmonary Disease-Related Genes That Facilitate Squamous Cell Lung Cancer Progression. Oncol. Lett. 2020, 19, 2115–2122. [Google Scholar] [CrossRef]
- Williams, A.E.; Perry, M.M.; Moschos, S.A.; Larner-Svensson, H.M.; Lindsay, M.A. Role of MiRNA-146a in the Regulation of the Innate Immune Response and Cancer. Biochem. Soc. Trans. 2008, 36, 1211–1215. [Google Scholar] [CrossRef]
- Fathinavid, A.; Ghobadi, M.Z.; Najafi, A.; Masoudi-Nejad, A. Identification of Common MicroRNA between COPD and Non-Small Cell Lung Cancer through Pathway Enrichment Analysis. BMC Genom. Data 2021, 22, 41. [Google Scholar] [CrossRef]
- Suzuki, M.; Wada, H.; Yoshino, M.; Tian, L.; Shigematsu, H.; Suzuki, H.; Alaa, M.; Tamura, H.; Fujiwara, T.; Nagato, K.; et al. Molecular Characterization of Chronic Obstructive Pulmonary Disease-Related Non-Small Cell Lung Cancer through Aberrant Methylation and Alterations of EGFR Signaling. Ann. Surg. Oncol. 2010, 17, 878–888. [Google Scholar] [CrossRef]
- Ellis, P.M.; Vandermeer, R. Delays in the Diagnosis of Lung Cancer. J. Thorac. Dis. 2011, 3, 183–188. [Google Scholar] [CrossRef]
- Jeganathan, V.; Knight, S.; Bricknell, M.; Ridgers, A.; Wong, R.; Brazzale, D.J.; Ruehland, W.R.; Rahman, M.A.; Leong, T.L.; McDonald, C.F. Impact of Smoking Status and Chronic Obstructive Pulmonary Disease on Pulmonary Complications Post Lung Cancer Surgery. PLoS ONE 2022, 17, e0266052. [Google Scholar] [CrossRef]



| A | B | E | |||
|---|---|---|---|---|---|
| Pharmacological therapy | Initial therapy | SABA or SAMA or LABA or LAMA | LABA or LAMA or LABA + LAMA | LAMA or LABA + LAMA or LABA + LAMA + ICS | |
| Follow-up therapy |
Persistent dyspnea: (1) LAMA or LABA monotherapy → LAMA + LABA (2) LABA + LAMA → LAMA + LABA + ICS (3) Investigation and treatment of comorbid conditions | ||||
|
Exacerbations: (1) LABA or LAMA → LABA + LAMA or LABA + LAMA + ICS (2) LABA + LAMA → LABA + LAMA + ICS or roflumilast or azithromycin (3) LABA + LAMA + ICS → roflumilast or macrolide or LABA + LAMA | |||||
| Non-pharmacological therapy | Essential | Smoking cessation |
Smoking cessation Pulmonary rehabilitation | ||
| Recommended | Physical activity | ||||
| Optional | Flu vaccination Pneumococcal vaccination Pertussis vaccination COVID-19 vaccination | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarnecka-Chrebelska, K.H.; Mukherjee, D.; Maryanchik, S.V.; Rudzinska-Radecka, M. Biological and Genetic Mechanisms of COPD, Its Diagnosis, Treatment, and Relationship with Lung Cancer. Biomedicines 2023, 11, 448. https://doi.org/10.3390/biomedicines11020448
Czarnecka-Chrebelska KH, Mukherjee D, Maryanchik SV, Rudzinska-Radecka M. Biological and Genetic Mechanisms of COPD, Its Diagnosis, Treatment, and Relationship with Lung Cancer. Biomedicines. 2023; 11(2):448. https://doi.org/10.3390/biomedicines11020448
Chicago/Turabian StyleCzarnecka-Chrebelska, Karolina H., Debjita Mukherjee, Sofya V. Maryanchik, and Magdalena Rudzinska-Radecka. 2023. "Biological and Genetic Mechanisms of COPD, Its Diagnosis, Treatment, and Relationship with Lung Cancer" Biomedicines 11, no. 2: 448. https://doi.org/10.3390/biomedicines11020448
APA StyleCzarnecka-Chrebelska, K. H., Mukherjee, D., Maryanchik, S. V., & Rudzinska-Radecka, M. (2023). Biological and Genetic Mechanisms of COPD, Its Diagnosis, Treatment, and Relationship with Lung Cancer. Biomedicines, 11(2), 448. https://doi.org/10.3390/biomedicines11020448