
Ion Channel Genes in Painful Neuropathies
 , , and
on behalf of the PainNet Study Group
, , and
on behalf of the PainNet Study Group
Abstract
:1. Introduction
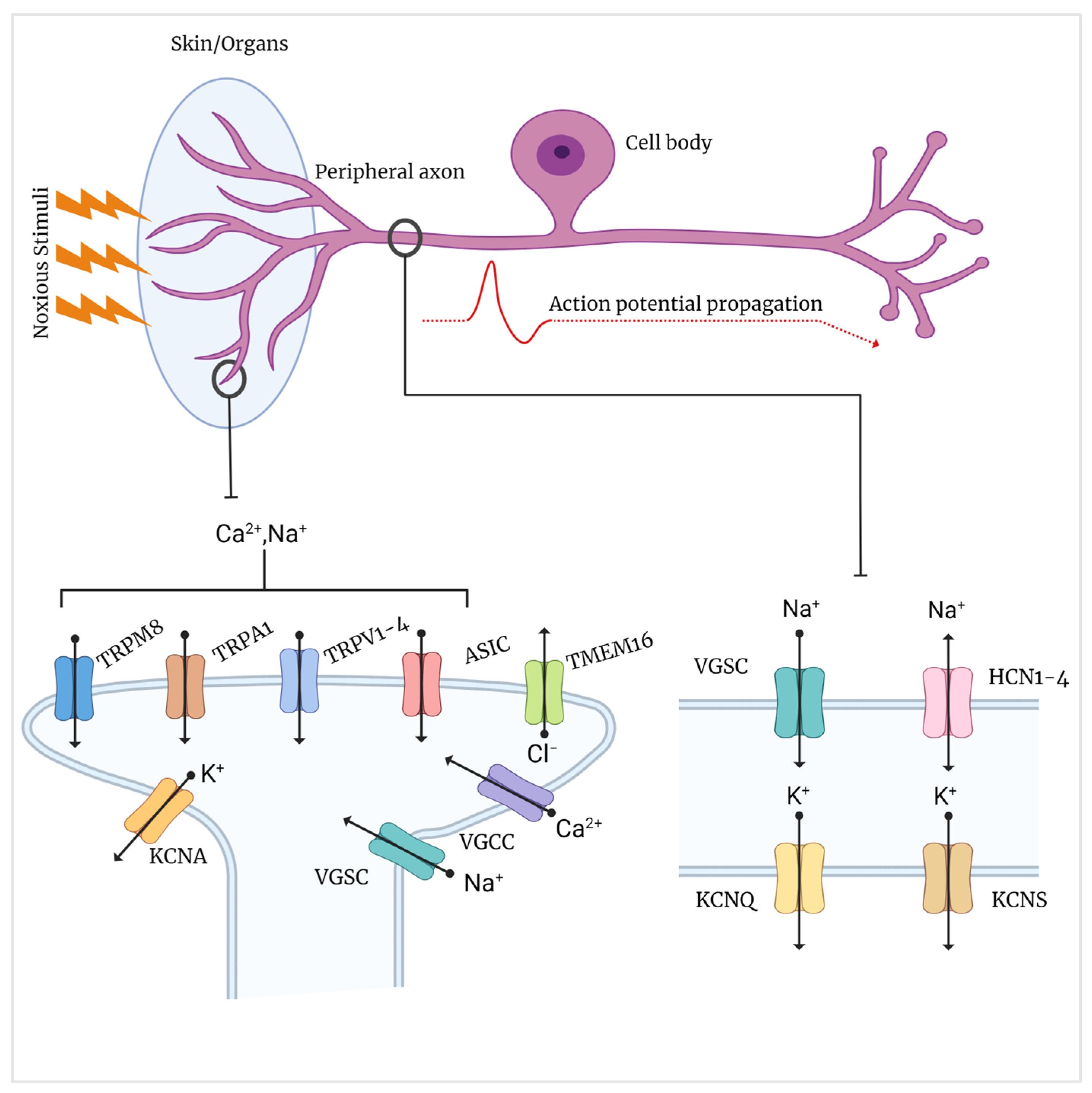
2. Role of Ion Channels in Painful Peripheral Neuropathy
2.1. Voltage-Gated Ca2+ Channels and Calcium Signaling
2.1.1. CACNA1A and CACNA1B
2.1.2. CACNA1G/1H/1I
2.1.3. CACNA2D1/D2/D3/D4
2.2. Potassium (K+) Channels
2.2.1. KCNA1 and KCNA2
2.2.2. KCNQ2/3/5
2.2.3. KCNS1 (Kv9.1)
2.3. Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Channels
2.3.1. HCN1
2.3.2. HCN2
2.3.3. HCN3 and HCN4
2.4. Anoctamin Gene Family
2.4.1. ANO1
2.4.2. ANO3
2.5. Transient Receptor Potential (TRP) Cation Channels
2.5.1. TRPA1
2.5.2. TRPM2
2.5.3. TRPM3
2.5.4. TRPM8
2.5.5. TRPV1

{kind=link}
| Disease/Condition | Therapeutic Approach | References |
|---|---|---|
| CIPN | TRPV1 siRNA into a druggable approach | [152] |
| PDPN | Potential target blocking GPR177-WNT5a-TRPV1 axis | [153] |
| PDPN | Alpha-lipoic acid and capsazepine together inhibits TRPV1 channel | [154] |
| PDPN | Ajugarin-I treatment reduces TRPV1/TRPM8 expression | [145] |
| NP | Pregablin reduces pain perception via PKCε/TRPV1 pathway | [155] |
| Side-effects of opioids post-NP | Potential target such as β-arrestin 2 that regulates bi-directionally TRPV1 and μ-opioid receptors | [156] |
| NP | Cannabidiol for acute NP partially inhibits 5-HT1A and TRPV1 | [157] |
2.5.6. TRPV3
2.5.7. TRPV4
2.6. Acid-Sensing Ion Channels (ASICs)
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watson, J.C.; Dyck, P.J.B. Peripheral Neuropathy: A Practical Approach to Diagnosis and Symptom Management. Mayo Clin. Proc. 2015, 90, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Mammana, S.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The Neuropathic Pain: An Overview of the Current Treatment and Future Therapeutic Approaches. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419838383. [Google Scholar] [CrossRef]
- Yan, Y.Y.; Li, C.Y.; Zhou, L.; Ao, L.Y.; Fang, W.R.; Li, Y.M. man Research Progress of Mechanisms and Drug Therapy for Neuropathic Pain. Life Sci. 2017, 190, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Zorina-Lichtenwalter, K.; Parisien, M.; Diatchenko, L. Genetic Studies of Human Neuropathic Pain Conditions: A Review. Pain 2018, 159, 583. [Google Scholar] [CrossRef] [PubMed]
- Shinu, P.; Morsy, M.A.; Nair, A.B.; Al Mouslem, A.K.; Venugopala, K.N.; Goyal, M.; Bansal, M.; Jacob, S.; Deb, P.K. Novel Therapies for the Treatment of Neuropathic Pain: Potential and Pitfalls. J. Clin. Med. 2022, 11, 3002. [Google Scholar] [CrossRef] [PubMed]
- Sopacua, M.; Hoeijmakers, J.G.J.; Merkies, I.S.J.; Lauria, G.; Waxman, S.G.; Faber, C.G. Small-Fiber Neuropathy: Expanding the Clinical Pain Universe. J. Peripher. Nerv. Syst. 2019, 24, 19–33. [Google Scholar] [CrossRef]
- Bennett, D.L.; Clark, X.A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 2019, 99, 1079–1151. [Google Scholar] [CrossRef]
- Ślęczkowska, M.; Almomani, R.; Marchi, M.; de Greef, B.T.A.; Sopacua, M.; Hoeijmakers, J.G.J.; Lindsey, P.; Salvi, E.; Bönhof, G.J.; Ziegler, D.; et al. Peripheral Ion Channel Gene Screening in Painful-and Painless-Diabetic Neuropathy. Int. J. Mol. Sci. 2022, 23, 7190. [Google Scholar] [CrossRef]
- Ślęczkowska, M.; Almomani, R.; Marchi, M.; Salvi, E.; de Greef, B.T.A.; Sopacua, M.; Hoeijmakers, J.G.J.; Lindsey, P.; Waxman, S.G.; Lauria, G.; et al. Peripheral Ion Channel Genes Screening in Painful Small Fiber Neuropathy. Int. J. Mol. Sci. 2022, 23, 14095. [Google Scholar] [CrossRef]
- Goodwin, G.; McMahon, S.B. The Physiological Function of Different Voltage-Gated Sodium Channels in Pain. Nat. Rev. Neurosci. 2021, 22, 263–274. [Google Scholar] [CrossRef]
- Bigsby, S.; Neapetung, J.; Campanucci, V.A. Voltage-Gated Sodium Channels in Diabetic Sensory Neuropathy: Function, Modulation, and Therapeutic Potential. Front. Cell. Neurosci. 2022, 16, 994585. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G.; Zamponi, G.W. Regulating Excitability of Peripheral Afferents: Emerging Ion Channel Targets. Nat. Neurosci. 2014, 17, 153–163. [Google Scholar] [CrossRef]
- González-Ramírez, R.; Chen, Y.; Liedtke, W.B.; Morales-Lázaro, S.L. TRP Channels and Pain. In Neurobiology of TRP Channels; CRC Press: Boca Raton, FL, USA, 2017; pp. 125–148. [Google Scholar] [CrossRef]
- Dolphin, A.C.; Lee, A. Presynaptic Calcium Channels: Specialized Control of Synaptic Neurotransmitter Release. Nat. Rev. Neurosci. 2020, 21, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Ponnusamy, M. Voltage-Dependent Calcium Channels: From Physiology to Diseases. In Calcium Signaling: From Physiology to Diseases; Springer: Singapore, 2017; pp. 61–72. [Google Scholar] [CrossRef]
- Haworth, A.S.; Brackenbury, W.J. Emerging Roles for Multifunctional Ion Channel Auxiliary Subunits in Cancer. Cell Calcium 2019, 80, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Gambeta, E.; Gandini, M.A.; Souza, I.A.; Ferron, L.; Zamponi, G.W. A CACNA1A Variant Associated with Trigeminal Neuralgia Alters the Gating of Cav2.1 Channels. Mol. Brain 2021, 14, 4. [Google Scholar] [CrossRef]
- Huang, M.; Nibbeling, E.A.R.; Lagrand, T.J.; Souza, I.A.; Groen, J.L.; Gandini, M.A.; Zhang, F.X.; Koelman, J.H.T.M.; Adir, N.; Sinke, R.J.; et al. Rare Functional Missense Variants in CACNA1H: What Can We Learn from Writer’s Cramp? Mol. Brain 2021, 14, 18. [Google Scholar] [CrossRef]
- Costigan, M.; Belfer, I.; Griffin, R.S.; Dai, F.; Barrett, L.B.; Coppola, G.; Wu, T.; Kiselycznyk, C.; Poddar, M.; Lu, Y.; et al. Multiple Chronic Pain States Are Associated with a Common Amino Acid–Changing Allele in KCNS1. Brain 2010, 133, 2519–2527. [Google Scholar] [CrossRef]
- Meents, J.E.; Fischer, M.J.M.; McNaughton, P.A. Sensitization of TRPA1 by Protein Kinase A. PLoS ONE 2017, 12, e0170097. [Google Scholar] [CrossRef]
- Zhang, Z.; Schmelz, M.; Segerdahl, M.; Quiding, H.; Centerholt, C.; Juréus, A.; Carr, T.H.; Whiteley, J.; Salter, H.; Kvernebo, M.S.; et al. Exonic Mutations in SCN9A (NaV1.7) Are Found in a Minority of Patients with Erythromelalgia. Scand. J. Pain 2014, 5, 217–225. [Google Scholar] [CrossRef]
- Di Stefano, G.; Yuan, J.H.; Cruccu, G.; Waxman, S.G.; Dib-Hajj, S.D.; Truini, A. Familial Trigeminal Neuralgia—A Systematic Clinical Study with a Genomic Screen of the Neuronal Electrogenisome. Cephalalgia 2020, 40, 767–777. [Google Scholar] [CrossRef]
- Fan, L.; Guan, X.; Wang, W.; Zhao, J.Y.; Zhang, H.; Tiwari, V.; Hoffman, P.N.; Li, M.; Tao, Y.X. Impaired Neuropathic Pain and Preserved Acute Pain in Rats Overexpressing Voltage-Gated Potassium Channel Subunit Kv1.2 in Primary Afferent Neurons. Mol. Pain 2014, 10, 1744–8069. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.H.; Schulman, B.R.; Effraim, P.R.; Sulayman, D.H.; Jacobs, D.S.; Waxman, S.G. Genomic Analysis of 21 Patients with Corneal Neuralgia after Refractive Surgery. Pain Rep. 2020, 5, e826. [Google Scholar] [CrossRef] [PubMed]
- Gualdani, R.; Yuan, J.-H.; Effraim, P.R.; Di Stefano, G.; Truini, A.; Cruccu, G.; Dib-Hajj, S.D.; Gailly, P.; Waxman, S.G. Trigeminal Neuralgia TRPM8 Mutation: Enhanced Activation, Basal [Ca2+]i and Menthol Response. Neurol. Genet. 2021, 7, e550. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Hu, L.; Cao, X.; Zhao, J.; Zhang, X.; Lee, M.; Wang, H.; Zhang, J.; Chen, Q.; Feng, C.; et al. Genotype–Phenotype Correlation of TRPV3-Related Olmsted Syndrome. J. Investig. Dermatol. 2021, 141, 545–554. [Google Scholar] [CrossRef]
- Yoshioka, T.; Imura, K.; Asakawa, M.; Suzuki, M.; Oshima, I.; Hirasawa, T.; Sakata, T.; Horikawa, T.; Arimura, A. Impact of the Gly573Ser Substitution in TRPV3 on the Development of Allergic and Pruritic Dermatitis in Mice. J. Investig. Dermatol. 2009, 129, 714–722. [Google Scholar] [CrossRef]
- Landouré, G.; Zdebik, A.A.; Martinez, T.L.; Burnett, B.G.; Stanescu, H.C.; Inada, H.; Shi, Y.; Taye, A.A.; Kong, L.; Munns, C.H.; et al. Mutations in TRPV4 Cause Charcot-Marie-Tooth Disease Type 2C. Nat. Genet. 2010, 42, 170. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Dubourg, O.; Carlier, P.; Carlier, R.Y.; Sabouraud, P.; Péréon, Y.; Chapon, F.; Thauvin-Robinet, C.; Laforêt, P.; Eymard, B.; et al. Phenotypic Spectrum and Incidence of TRPV4 Mutations in Patients with Inherited Axonal Neuropathy. Neurology 2014, 82, 1919–1926. [Google Scholar] [CrossRef]
- Klein, C.J.; Shi, Y.; Fecto, F.; Donaghy, M.; Nicholson, G.; McEntagart, M.E.; Crosby, A.H.; Wu, Y.; Lou, H.; McEvoy, K.M.; et al. TRPV4 Mutations and Cytotoxic Hypercalcemia in Axonal Charcot-Marie-Tooth Neuropathies. Neurology 2011, 76, 887–894. [Google Scholar] [CrossRef]
- Auer-Grumbach, M.; Olschewski, A.; Papi, L.; Kremer, H.; McEntagart, M.E.; Uhrig, S.; Fischer, C.; Fröhlich, E.; Bálint, Z.; Tang, B.; et al. Alterations in the Ankyrin Domain of TRPV4 Cause Congenital Distal SMA, Scapuloperoneal SMA and HMSN2C. Nat. Genet. 2010, 42, 160. [Google Scholar] [CrossRef]
- Deng, H.X.; Klein, C.J.; Yan, J.; Shi, Y.; Wu, Y.; Fecto, F.; Yau, H.J.; Yang, Y.; Zhai, H.; Siddique, N.; et al. Scapuloperoneal Spinal Muscular Atrophy and CMT2C Are Allelic Disorders Caused by Alterations in TRPV4. Nat. Genet. 2010, 42, 165–169. [Google Scholar] [CrossRef]
- Hommersom, M.P.; van Prooije, T.H.; Pennings, M.; Schouten, M.I.; van Bokhoven, H.; Kamsteeg, E.J.; van de Warrenburg, B.P.C. The Complexities of CACNA1A in Clinical Neurogenetics. J. Neurol. 2022, 269, 3094–3108. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.T.T.; Campos, M.M.; Carvalho, V.D.P.R.; da Silva Junior, C.A.; Magno, L.A.V.; de Souza, A.H.; Gomez, M.V. Current Drug Development Overview: Targeting Voltage-Gated Calcium Channels for the Treatment of Pain. Int. J. Mol. Sci. 2023, 24, 9223. [Google Scholar] [CrossRef] [PubMed]
- Hoppanova, L.; Lacinova, L. Voltage-Dependent CaV3.2 and CaV2.2 Channels in Nociceptive Pathways. Pflügers Arch.—Eur. J. Physiol. 2022, 474, 421–434. [Google Scholar] [CrossRef] [PubMed]
- DuBreuil, D.M.; Soto, E.J.L.; Daste, S.; Meir, R.; Li, D.; Wainger, B.; Fleischmann, A.; Lipscombe, D. Heat But Not Mechanical Hypersensitivity Depends on Voltage-Gated CaV2.2 Calcium Channel Activity in Peripheral Axon Terminals Innervating Skin. J. Neurosci. 2021, 41, 7546–7560. [Google Scholar] [CrossRef]
- Nieto-Rostro, M.; Patel, R.; Dickenson, A.H.; Dolphin, A.C. Nerve Injury Increases Native CaV2.2 Trafficking in Dorsal Root Ganglion Mechanoreceptors. Pain 2023, 164, 1264. [Google Scholar] [CrossRef]
- Tonello, R.; Trevisan, G.; Luckemeyer, D.; Castro-Junior, C.J.; Gomez, M.V.; Ferreira, J. Phα1β, a Dual Blocker of TRPA1 and Cav2.2, as an Adjuvant Drug in Opioid Therapy for Postoperative Pain. Toxicon 2020, 188, 80–88. [Google Scholar] [CrossRef]
- Gorman, K.M.; Meyer, E.; Grozeva, D.; Spinelli, E.; McTague, A.; Sanchis-Juan, A.; Carss, K.J.; Bryant, E.; Reich, A.; Schneider, A.L.; et al. Bi-Allelic Loss-of-Function CACNA1B Mutations in Progressive Epilepsy-Dyskinesia. Am. J. Hum. Genet. 2019, 104, 948–956. [Google Scholar] [CrossRef]
- Groen, J.L.; Andrade, A.; Ritz, K.; Jalalzadeh, H.; Haagmans, M.; Bradley, T.E.J.; Jongejan, A.; Verbeek, D.S.; Nürnberg, P.; Denome, S.; et al. CACNA1B Mutation Is Linked to Unique Myoclonus-Dystonia Syndrome. Hum. Mol. Genet. 2015, 24, 987–993. [Google Scholar] [CrossRef]
- Cherian, A.; Chandarana, M.; Susvirkar, A.A.; Divya, K.P.; Saraf, U.U.; Krishnan, S. Abnormal Saccades Differentiate Adolescent Onset Variant Ataxia Telangiectasia from Other Myoclonus Dystonia. Ann. Indian Acad. Neurol. 2021, 24, 630. [Google Scholar] [CrossRef]
- Loke, M.F.; Wei, H.; Yeo, J.; Sng, B.L.; Sia, A.T.; Tan, E.C. Deep Sequencing Analysis to Identify Novel and Rare Variants in Pain-Related Genes in Patients with Acute Postoperative Pain and High Morphine Use. J. Pain Res. 2019, 12, 2755. [Google Scholar] [CrossRef]
- Weiss, N.; Zamponi, G.W. Genetic T-Type Calcium Channelopathies. J. Med. Genet. 2020, 57, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Montera, M.; Goins, A.; Cmarko, L.; Weiss, N.; Westlund, K.N.; Alles, S.R.A. Trigeminal Neuropathic Pain Is Alleviated by Inhibition of Cav3.3 T-Type Calcium Channels in Mice. Channels 2020, 15, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tatsui, C.E.; Rhines, L.D.; North, R.Y.; Harrison, D.S.; Cassidy, R.M.; Johansson, C.A.; Kosturakis, A.K.; Edwards, D.D.; Zhang, H.; et al. Dorsal Root Ganglion Neurons Become Hyperexcitable and Increase Expression of Voltage-Gated T-Type Calcium Channels (Cav3.2) in Paclitaxel-Induced Peripheral Neuropathy. Pain 2017, 158, 417. [Google Scholar] [CrossRef] [PubMed]
- Ovsepian, S.V.; Waxman, S.G. Gene Therapy for Chronic Pain: Emerging Opportunities in Target-Rich Peripheral Nociceptors. Nat. Rev. Neurosci. 2023, 24, 252–265. [Google Scholar] [CrossRef]
- Snutch, T.P.; Zamponi, G.W. Recent Advances in the Development of T-type Calcium Channel Blockers for Pain Intervention. Br. J. Pharmacol. 2018, 175, 2375. [Google Scholar] [CrossRef]
- Cai, S.; Gomez, K.; Moutal, A.; Khanna, R. Targeting T-Type/CaV3.2 Channels for Chronic Pain. Transl. Res. 2021, 234, 20. [Google Scholar] [CrossRef]
- Souza, I.A.; Gandini, M.A.; Zamponi, G.W. Splice-Variant Specific Effects of a CACNA1H Mutation Associated with Writer’s Cramp. Mol. Brain 2021, 14, 145. [Google Scholar] [CrossRef]
- Nieto-Rostro, M.; Ramgoolam, K.; Pratt, W.S.; Kulik, A.; Dolphin, A.C. Ablation of α 2 δ-1 Inhibits Cell-Surface Trafficking of Endogenous N-Type Calcium Channels in the Pain Pathway in Vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E12043–E12052. [Google Scholar] [CrossRef]
- Dolphin, A.C. Voltage-Gated Calcium Channel α2δ Subunits: An Assessment of Proposed Novel Roles. F1000Research 2018, 7, 1830. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic Pain: From Mechanisms to Treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.R.; Zhou, M.H.; Jin, D.; Chen, H.; Wang, L.; DePinho, R.A.; Pan, H.L. HDAC2 in Primary Sensory Neurons Constitutively Restrains Chronic Pain by Repressing A2δ-1 Expression and Associated NMDA Receptor Activity. J. Neurosci. 2022, 42, 8918–8935. [Google Scholar] [CrossRef]
- Landmann, J.; Richter, F.; Oros-Peusquens, A.M.; Shah, N.J.; Classen, J.; Neely, G.G.; Richter, A.; Penninger, J.M.; Bechmann, I. Neuroanatomy of Pain-Deficiency and Cross-Modal Activation in Calcium Channel Subunit (CACN) A2δ3 Knockout Mice. Brain Struct. Funct. 2018, 223, 111–130. [Google Scholar] [CrossRef]
- Andres-Bilbe, A.; Castellanos, A.; Pujol-Coma, A.; Callejo, G.; Comes, N.; Gasull, X. The Background K+ Channel TRESK in Sensory Physiology and Pain. Int. J. Mol. Sci. 2020, 21, 5206. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.T.; Jayanthi, S.; Cadet, J.L. Potassium Channels and Their Potential Roles in Substance Use Disorders. Int. J. Mol. Sci. 2021, 22, 1249. [Google Scholar] [CrossRef] [PubMed]
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; García, J.A.L. Potassium Channels in Neuropathic Pain: Advances, Challenges, and Emerging Ideas. Pain 2016, 157 (Suppl. 1), S7–S14. [Google Scholar] [CrossRef]
- Takeda, M.; Tsuboi, Y.; Kitagawa, J.; Nakagawa, K.; Iwata, K.; Matsumoto, S. Potassium Channels as a Potential Therapeutic Target for Trigeminal Neuropathic and Inflammatory Pain. Mol. Pain 2011, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.D.; Tempel, B.L. Hyperalgesia in Mice Lacking the Kv1.1 Potassium Channel Gene. Neurosci. Lett. 1998, 251, 121–124. [Google Scholar] [CrossRef]
- Zhao, Q.; Fan, L.; Wang, J.; Deng, X.; Zou, J. Relationship between Pain Behavior and Changes in KCNA2 Expression in the Dorsal Root Ganglia of Rats with Osteoarthritis. Pain Res. Manag. 2020, 2020, 4636838. [Google Scholar] [CrossRef]
- Zhao, X.; Tang, Z.; Zhang, H.; Atianjoh, F.E.; Zhao, J.Y.; Liang, L.; Wang, W.; Guan, X.; Kao, S.C.; Tiwari, V.; et al. A Long Noncoding RNA Contributes to Neuropathic Pain by Silencing Kcna2 in Primary Afferent Neurons. Nat. Neurosci. 2013, 16, 1024–1031. [Google Scholar] [CrossRef]
- Liang, L.; Gu, X.; Zhao, J.Y.; Wu, S.; Miao, X.; Xiao, J.; Mo, K.; Zhang, J.; Lutz, B.M.; Bekker, A.; et al. G9a Participates in Nerve Injury-Induced Kcna2 Downregulation in Primary Sensory Neurons. Sci. Rep. 2016, 6, 37704. [Google Scholar] [CrossRef]
- Mo, K.; Wu, S.; Gu, X.; Xiong, M.; Cai, W.; Atianjoh, F.E.; Jobe, E.E.; Zhao, X.; Tu, W.F.; Tao, Y.X. MBD1 Contributes to the Genesis of Acute Pain and Neuropathic Pain by Epigenetic Silencing of Oprm1 and Kcna2 Genes in Primary Sensory Neurons. J. Neurosci. 2018, 38, 9883–9899. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.Y.; Liang, L.; Gu, X.; Li, Z.; Wu, S.; Sun, L.; Atianjoh, F.E.; Feng, J.; Mo, K.; Jia, S.; et al. DNA Methyltransferase DNMT3a Contributes to Neuropathic Pain by Repressing Kcna2 in Primary Afferent Neurons. Nat. Commun. 2017, 8, 14712. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Gu, X.; Pan, Z.; Guo, X.; Liu, J.; Atianjoh, F.E.; Wu, S.; Mo, K.; Xu, B.; Liang, L.; et al. Contribution of DNMT1 to Neuropathic Pain Genesis Partially through Epigenetically Repressing Kcna2 in Primary Afferent Neurons. J. Neurosci. 2019, 39, 6595–6607. [Google Scholar] [CrossRef]
- Zhang, J.; Rong, L.; Shao, J.; Zhang, Y.; Liu, Y.; Zhao, S.; Li, L.; Yu, W.; Zhang, M.; Ren, X.; et al. Epigenetic Restoration of Voltage-Gated Potassium Channel Kv1.2 Alleviates Nerve Injury-Induced Neuropathic Pain. J. Neurochem. 2021, 156, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Wang, Y.; Wang, J.; Feng, S.; Wang, X. The Etiological Roles of MiRNAs, LncRNAs, and CircRNAs in Neuropathic Pain: A Narrative Review. J. Clin. Lab. Anal. 2022, 36, e24592. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Arconada, I.; Roza, C.; Lopez-Garcia, J.A. Enhancing M Currents: A Way out for Neuropathic Pain? Front. Mol. Neurosci. 2009, 2, 10. [Google Scholar] [CrossRef]
- Yu, T.; Li, L.; Liu, H.; Li, H.; Liu, Z.; Li, Z. KCNQ2/3/5 Channels in Dorsal Root Ganglion Neurons Can Be Therapeutic Targets of Neuropathic Pain in Diabetic Rats. Mol. Pain 2018, 14, 1744806918793229. [Google Scholar] [CrossRef]
- Ling, J.; Erol, F.; Viatchenko-Karpinski, V.; Kanda, H.; Gu, J.G. Orofacial Neuropathic Pain Induced by Oxaliplatin: Downregulation of KCNQ2 Channels in V2 Trigeminal Ganglion Neurons and Treatment by the KCNQ2 Channel Potentiator Retigabine. Mol. Pain 2017, 13, 1744806917724715. [Google Scholar] [CrossRef]
- Mis, M.A.; Yang, Y.; Tanaka, B.S.; Gomis-Perez, C.; Liu, S.; Dib-Hajj, F.; Adi, T.; Garcia-Milian, R.; Schulman, B.R.; Dib-Hajj, S.D.; et al. Resilience to Pain: A Peripheral Component Identified Using Induced Pluripotent Stem Cells and Dynamic Clamp. J. Neurosci. 2019, 39, 382–392. [Google Scholar] [CrossRef]
- Yuan, J.-H.; Estacion, M.; Mis, M.A.; Tanaka, B.S.; Schulman, B.R.; Chen, L.; Liu, S.; Dib-Hajj, F.B.; Dib-Hajj, S.D.; Waxman, S.G. KCNQ Variants and Pain Modulation: A Missense Variant in Kv7.3 Contributes to Pain Resilience. Brain Commun. 2021, 3, fcab212. [Google Scholar] [CrossRef]
- Richardson, F.C.; Kaczmarek, L.K. Modification of Delayed Rectifier Potassium Currents by the Kv9.1 Potassium Channel Subunit. Hear. Res. 2000, 147, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulas, C.; Zhu, L.; Shaifta, Y.; Grist, J.; Ward, J.P.T.; Raouf, R.; Michael, G.J.; McMahon, S.B. Sensory Neuron Downregulation of the Kv9.1 Potassium Channel Subunit Mediates Neuropathic Pain Following Nerve Injury. J. Neurosci. 2012, 32, 17502–17513. [Google Scholar] [CrossRef] [PubMed]
- Tsantoulas, C.; Denk, F.; Signore, M.; Nassar, M.A.; Futai, K.; McMahon, S.B. Mice Lacking Kcns1 in Peripheral Neurons Show Increased Basal and Neuropathic Pain Sensitivity. Pain 2018, 159, 1641–1651. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, J.; Siegelbaum, S.A. Properties of Hyperpolarization-Activated Pacemaker Current Defined by Coassembly of HCN1 and HCN2 Subunits and Basal Modulation by Cyclic Nucleotide. J. Gen. Physiol. 2001, 117, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Sartiani, L.; Mannaioni, G.; Masi, A.; Romanelli, M.N.; Cerbai, E. The Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels: From Biophysics to Pharmacology of a Unique Family of Ion Channels. Pharmacol. Rev. 2017, 69, 354–395. [Google Scholar] [CrossRef]
- Santoro, B.; Chen, S.; Lüthi, A.; Pavlidis, P.; Shumyatsky, G.P.; Tibbs, G.R.; Siegelbaum, S.A. Molecular and Functional Heterogeneity of Hyperpolarization-Activated Pacemaker Channels in the Mouse CNS. J. Neurosci. 2000, 20, 5264–5275. [Google Scholar] [CrossRef]
- Jiang, Y.Q.; Sun, Q.; Tu, H.Y.; Wan, Y. Characteristics of HCN Channels and Their Participation in Neuropathic Pain. Neurochem. Res. 2008, 33, 1979–1989. [Google Scholar] [CrossRef]
- Rivolta, I.; Binda, A.; Masi, A.; DiFrancesco, J.C. Cardiac and Neuronal HCN Channelopathies. Pflugers Arch. Eur. J. Physiol. 2020, 472, 931–951. [Google Scholar] [CrossRef]
- Tu, H.; Deng, L.; Sun, Q.; Yao, L.; Han, J.S.; Wan, Y. Hyperpolarization-Activated, Cyclic Nucleotide-Gated Cation Channels: Roles in the Differential Electrophysiological Properties of Rat Primary Afferent Neurons. J. Neurosci. Res. 2004, 76, 713–722. [Google Scholar] [CrossRef]
- Chaplan, S.R.; Guo, H.Q.; Lee, D.H.; Luo, L.; Liu, C.; Kuei, C.; Velumian, A.A.; Butler, M.P.; Brown, S.M.; Dubin, A.E. Neuronal Hyperpolarization-Activated Pacemaker Channels Drive Neuropathic Pain. J. Neurosci. 2003, 23, 1169–1178. [Google Scholar] [CrossRef]
- Schnorr, S.; Eberhardt, M.; Kistner, K.; Rajab, H.; Käßer, J.; Hess, A.; Reeh, P.; Ludwig, A.; Herrmann, S. HCN2 Channels Account for Mechanical (but Not Heat) Hyperalgesia during Long-Standing Inflammation. Pain 2014, 155, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.; McMullan, S.; Djouhri, L.; Gao, L.; Watkins, R.; Berry, C.; Dempsey, K.; Lawson, S.N.; Zhang, Z. HCN1 and HCN2 in Rat DRG Neurons: Levels in Nociceptors and Non-Nociceptors, NT3-Dependence and Influence of CFA-Induced Skin Inflammation on HCN2 and NT3 Expression. PLoS ONE 2012, 7, e50442. [Google Scholar] [CrossRef] [PubMed]
- Momin, A.; Cadiou, H.; Mason, A.; McNaughton, P.A. Role of the Hyperpolarization-Activated Current I h in Somatosensory Neurons. J. Physiol. 2008, 586, 5911–5929. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, J.; Gu, L.; Zuo, Y. The Change of HCN1/HCN2 MRNA Expression in Peripheral Nerve after Chronic Constriction Injury Induced Neuropathy Followed by Pulsed Electromagnetic Field Therapy. Oncotarget 2017, 8, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Donnelly, D.F.; Ma, C.; LaMotte, R.H. Upregulation of the Hyperpolarization-Activated Cation Current after Chronic Compression of the Dorsal Root Ganglion. J. Neurosci. 2003, 23, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, J.; Takeda, M.; Suzuki, I.; Kadoi, J.; Tsuboi, Y.; Honda, K.; Matsumoto, S.; Nakagawa, H.; Tanabe, A.; Iwata, K. Mechanisms Involved in Modulation of Trigeminal Primary Afferent Activity in Rats with Peripheral Mononeuropathy. Eur. J. Neurosci. 2006, 24, 1976–1986. [Google Scholar] [CrossRef]
- Ramírez, D.; Zúñiga, R.; Concha, G.; Zúñiga, L. HCN Channels: New Therapeutic Targets for Pain Treatment. Molecules 2018, 23, 2094. [Google Scholar] [CrossRef]
- Mayar, S.; Memarpoor-Yazdi, M.; Makky, A.; Eslami Sarokhalil, R.; D’Avanzo, N. Direct Regulation of Hyperpolarization-Activated Cyclic-Nucleotide Gated (HCN1) Channels by Cannabinoids. Front. Mol. Neurosci. 2022, 15, 848540. [Google Scholar] [CrossRef]
- Resta, F.; Micheli, L.; Laurino, A.; Spinelli, V.; Mello, T.; Sartiani, L.; Di Cesare Mannelli, L.; Cerbai, E.; Ghelardini, C.; Romanelli, M.N.; et al. Selective HCN1 Block as a Strategy to Control Oxaliplatin-Induced Neuropathy. Neuropharmacology 2018, 131, 403–413. [Google Scholar] [CrossRef]
- Ludwig, A.; Budde, T.; Stieber, J.; Moosmang, S.; Wahl, C.; Holthoff, K.; Langebartels, A.; Wotjak, C.; Munsch, T.; Zong, X.; et al. Absence Epilepsy and Sinus Dysrhythmia in Mice Lacking the Pacemaker Channel HCN2. EMBO J. 2003, 22, 216–224. [Google Scholar] [CrossRef]
- Emery, E.C.; Young, G.T.; Berrocoso, E.M.; Chen, L.; McNaughton, P.A. HCN2 Ion Channels Play a Central Role in Inflammatory and Neuropathic Pain. Science 2011, 333, 1462–1466. [Google Scholar] [CrossRef]
- Tsantoulas, C.; Lainez, S.; Wong, S.; Mehta, I.; Vilar, B.; McNaughton, P.A. Hyperpolarization-Activated Cyclic Nucleotide-Gated 2 (Hcn2) Ion Channels Drive Pain in Mouse Models of Diabetic Neuropathy. Sci. Transl. Med. 2017, 9, eaam6072. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, L.; Jin, L.; Tan, Y.; Li, W.; Tang, J. HCN2 Contributes to Oxaliplatin-Induced Neuropathic Pain through Activation of the CaMKII/CREB Cascade in Spinal Neurons. Mol. Pain 2018, 14, 1744806918778490. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, Z.; Huang, D. Decreased HCN2 Channel Expression Attenuates Neuropathic Pain by Inhibiting Pro-Inflammatory Reactions and NF-ΚB Activation in Mice. Int. J. Clin. Exp. Pathol. 2019, 12, 154–163. [Google Scholar]
- Lainez, S.; Tsantoulas, C.; Biel, M.; McNaughton, P.A. HCN3 Ion Channels: Roles in Sensory Neuronal Excitability and Pain. J. Physiol. 2019, 597, 4661–4675. [Google Scholar] [CrossRef] [PubMed]
- Becker, F.; Reid, C.A.; Hallmann, K.; Tae, H.S.; Phillips, A.M.; Teodorescu, G.; Weber, Y.G.; Kleefuss-Lie, A.; Elger, C.; Perez-Reyes, E.; et al. Functional Variants in HCN4 and CACNA1H May Contribute to Genetic Generalized Epilepsy. Epilepsia Open 2017, 2, 334–342. [Google Scholar] [CrossRef]
- Campostrini, G.; Difrancesco, J.C.; Castellotti, B.; Milanesi, R.; Gnecchi-Ruscone, T.; Bonzanni, M.; Bucchi, A.; Baruscotti, M.; Ferrarese, C.; Franceschetti, S.; et al. A Loss-of-Function HCN4 Mutation Associated with Familial Benign Myoclonic Epilepsy in Infancy Causes Increased Neuronal Excitability. Front. Mol. Neurosci. 2018, 11, 269. [Google Scholar] [CrossRef] [PubMed]
- Pedemonte, N.; Galietta, L.J. Structure and Function of TMEM16 Proteins (Anoctamins). Physiol. Rev. 2014, 94, 419–459. [Google Scholar] [CrossRef]
- Huang, F.; Wang, X.; Ostertag, E.M.; Nuwal, T.; Huang, B.; Jan, Y.N.; Basbaum, A.I.; Jan, L.Y. TMEM16C Facilitates Na(+)-Activated K+ Currents in Rat Sensory Neurons and Regulates Pain Processing. Nat. Neurosci. 2013, 16, 1284–1290. [Google Scholar] [CrossRef]
- Takayama, Y.; Uta, D.; Furue, H.; Tominaga, M. Pain-Enhancing Mechanism through Interaction between TRPV1 and Anoctamin 1 in Sensory Neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 5213–5218. [Google Scholar] [CrossRef]
- Cho, H.; Oh, U. Anoctamin 1 Mediates Thermal Pain as a Heat Sensor. Curr. Neuropharmacol. 2013, 11, 641. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Cho, H.; Jung, J.; Yang, Y.D.; Yang, D.J.; Oh, U. Anoctamin 1 Contributes to Inflammatory and Nerve-Injury Induced Hypersensitivity. Mol. Pain 2014, 10, 1744–8069. [Google Scholar] [CrossRef]
- Cho, H.; Yang, Y.D.; Lee, J.; Lee, B.; Kim, T.; Jang, Y.; Back, S.K.; Na, H.S.; Harfe, B.D.; Wang, F.; et al. The Calcium-Activated Chloride Channel Anoctamin 1 Acts as a Heat Sensor in Nociceptive Neurons. Nat. Neurosci. 2012, 15, 1015–1021. [Google Scholar] [CrossRef]
- Liu, B.; Linley, J.E.; Du, X.; Zhang, X.; Ooi, L.; Zhang, H.; Gamper, N. The Acute Nociceptive Signals Induced by Bradykinin in Rat Sensory Neurons Are Mediated by Inhibition of M-Type K+ Channels and Activation of Ca2+-Activated Cl- Channels. J. Clin. Investig. 2010, 120, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Flockerzi, V. (Eds.) Handbook of Experimental Pharmacology 222; Springer: Berlin/Heidelberg, Germany, 2014; Volume 222, ISBN 9783642542145. [Google Scholar]
- Meents, J.E.; Ciotu, C.I.; Fischer, M.J.M. Trpa1: A Molecular View. J. Neurophysiol. 2019, 121, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Kremeyer, B.; Lopera, F.; Cox, J.J.; Momin, A.; Rugiero, F.; Marsh, S.; Woods, C.G.; Jones, N.G.; Paterson, K.J.; Fricker, F.R.; et al. A Gain-of-Function Mutation in TRPA1 Causes Familial Episodic Pain Syndrome. Neuron 2010, 66, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Gouin, O.; L’Herondelle, K.; Lebonvallet, N.; Le Gall-Ianotto, C.; Sakka, M.; Buhé, V.; Plée-Gautier, E.; Carré, J.L.; Lefeuvre, L.; Misery, L.; et al. TRPV1 and TRPA1 in Cutaneous Neurogenic and Chronic Inflammation: Pro-Inflammatory Response Induced by Their Activation and Their Sensitization. Protein Cell 2017, 8, 644–661. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.; Udit, S.; Chiu, I.M. Pain and Immunity: Implications for Host Defence. Nat. Rev. Immunol. 2019, 19, 433–447. [Google Scholar] [CrossRef]
- Marcotti, A.; Fernández-Trillo, J.; González, A.; Vizcaíno-Escoto, M.; Ros-Arlanzón, P.; Romero, L.; Vela, J.M.; Gomis, A.; Viana, F.; De La Peña, E. TRPA1 Modulation by Sigma-1 Receptor Prevents Oxaliplatin-Induced Painful Peripheral Neuropathy. Brain 2023, 146, 475–491. [Google Scholar] [CrossRef]
- De Logu, F.; De Siena, G.; Landini, L.; Marini, M.; Souza Monteiro de Araujo, D.; Albanese, V.; Preti, D.; Romitelli, A.; Chieca, M.; Titiz, M.; et al. Non-Neuronal TRPA1 Encodes Mechanical Allodynia Associated with Neurogenic Inflammation and Partial Nerve Injury in Rats. Br. J. Pharmacol. 2023, 180, 1232–1246. [Google Scholar] [CrossRef]
- Zhou, F.; Metzner, K.; Engel, P.; Balzulat, A.; Sisignano, M.; Ruth, P.; Lukowski, R.; Schmidtko, A.; Lu, R. Slack Potassium Channels Modulate TRPA1-Mediated Nociception in Sensory Neurons. Cells 2022, 11, 1693. [Google Scholar] [CrossRef] [PubMed]
- Vandewauw, I.; De Clercq, K.; Mulier, M.; Held, K.; Pinto, S.; Van Ranst, N.; Segal, A.; Voet, T.; Vennekens, R.; Zimmermann, K.; et al. A TRP Channel Trio Mediates Acute Noxious Heat Sensing. Nature 2018, 555, 662–666. [Google Scholar] [CrossRef]
- Marchi, M.; Salvi, E.; Andelic, M.; Mehmeti, E.; D’Amato, I.; Cazzato, D.; Chiappori, F.; Lombardi, R.; Cartelli, D.; Devigili, G.; et al. TRPA1 Rare Variants in Chronic Neuropathic and Nociplastic Pain Patients. Pain 2022, 164, 2048–2059. [Google Scholar] [CrossRef] [PubMed]
- Nirenberg, M.J.; Chaouni, R.; Biller, T.M.; Gilbert, R.M.; Paisán-Ruiz, C. A Novel TRPA1 Variant Is Associated with Carbamazepine-Responsive Cramp-Fasciculation Syndrome. Clin. Genet. 2018, 93, 164–168. [Google Scholar] [CrossRef]
- Wang, H.; Song, T.; Wang, W.; Zhang, Z. TRPM2 Participates the Transformation of Acute Pain to Chronic Pain during Injury-Induced Neuropathic Pain. Synapse 2019, 73, e22117. [Google Scholar] [CrossRef]
- Haraguchi, K.; Kawamoto, A.; Isami, K.; Maeda, S.; Kusano, A.; Asakura, K.; Shirakawa, H.; Mori, Y.; Nakagawa, T.; Kaneko, S. TRPM2 Contributes to Inflammatory and Neuropathic Pain through the Aggravation of Pronociceptive Inflammatory Responses in Mice. J. Neurosci. 2012, 32, 3931. [Google Scholar] [CrossRef]
- Jang, Y.; Cho, P.S.; Yang, Y.D.; Hwang, S.W. Nociceptive Roles of TRPM2 Ion Channel in Pathologic Pain. Mol. Neurobiol. 2018, 55, 6589–6600. [Google Scholar] [CrossRef] [PubMed]
- Malko, P.; Syed Mortadza, S.A.; McWilliam, J.; Jiang, L.H. TRPM2 Channel in Microglia as a New Player in Neuroinflammation Associated With a Spectrum of Central Nervous System Pathologies. Front. Pharmacol. 2019, 10, 239. [Google Scholar] [CrossRef]
- Nazıroğlu, M. Activation of TRPM2 and TRPV1 Channels in Dorsal Root Ganglion by NADPH Oxidase and Protein Kinase C Molecular Pathways: A Patch Clamp Study. J. Mol. Neurosci. 2017, 61, 425–435. [Google Scholar] [CrossRef]
- Bayir, M.H.; Yıldızhan, K.; Altındağ, F. Effect of Hesperidin on Sciatic Nerve Damage in STZ-Induced Diabetic Neuropathy: Modulation of TRPM2 Channel. Neurotox. Res. 2023, 1–10. [Google Scholar] [CrossRef]
- Sita, G.; Hrelia, P.; Graziosi, A.; Ravegnini, G.; Morroni, F. TRPM2 in the Brain: Role in Health and Disease. Cells 2018, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rubil, S.; Lesch, A.; Guethlein, L.A.; Rössler, O.G. Transient Receptor Potential TRPM3 Channels: Pharmacology, Signaling, and Biological Functions. Pharmacol. Res. 2017, 124, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, A.A.; Javeed, M.K.; Javed, Z.; Riaz, A.M.; Mukhtar, S.; Minhaj, S.; Abbas, S.; Bhatti, S. TRPM Channels: Same Ballpark, Different Players, and Different Rules in Immunogenetics. Immunogenetics 2011, 63, 773–787. [Google Scholar] [CrossRef]
- Vriens, J.; Voets, T.; Vriens JorisVriens, J. Sensing the Heat with TRPM3. Pflügers Arch.—Eur. J. Physiol. 2018, 470, 799–807. [Google Scholar] [CrossRef]
- Wagner, T.F.J.; Loch, S.; Lambert, S.; Straub, I.; Mannebach, S.; Mathar, I.; Düfer, M.; Lis, A.; Flockerzi, V.; Philipp, S.E.; et al. Transient Receptor Potential M3 Channels Are Ionotropic Steroid Receptors in Pancreatic Beta Cells. Nat. Cell Biol. 2008, 10, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Krügel, U.; Straub, I.; Beckmann, H.; Schaefer, M. Primidone Inhibits TRPM3 and Attenuates Thermal Nociception in Vivo. Pain 2017, 158, 856. [Google Scholar] [CrossRef] [PubMed]
- Silverman, H.A.; Chen, A.; Kravatz, N.L.; Chavan, S.S.; Chang, E.H. Involvement of Neural Transient Receptor Potential Channels in Peripheral Inflammation. Front. Immunol. 2020, 11, 2742. [Google Scholar] [CrossRef]
- Behrendt, M.; Gruss, F.; Enzeroth, R.; Dembla, S.; Zhao, S.; Crassous, P.A.; Mohr, F.; Nys, M.; Louros, N.; Gallardo, R.; et al. The Structural Basis for an On-off Switch Controlling Gβγ-Mediated Inhibition of TRPM3 Channels. Proc. Natl. Acad. Sci. USA 2020, 117, 29090–29100. [Google Scholar] [CrossRef]
- Dyment, D.A.; Terhal, P.A.; Rustad, C.F.; Tveten, K.; Griffith, C.; Jayakar, P.; Shinawi, M.; Ellingwood, S.; Smith, R.; van Gassen, K.; et al. De Novo Substitutions of TRPM3 Cause Intellectual Disability and Epilepsy. Eur. J. Hum. Genet. 2019, 27, 1611–1618. [Google Scholar] [CrossRef]
- Aloi, V.D.; Coutinho Pinto, S.J.P.; Van Bree, R.; Luyten, K.; Voets, T.; Vriens, J. TRPM3 as a Novel Target to Alleviate Acute Oxaliplatin-Induced Peripheral Neuropathic Pain. Pain 2023, 164, 2060–2069. [Google Scholar] [CrossRef]
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. Identification of a Cold Receptor Reveals a General Role for TRP Channels in Thermosensation. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lü, W.; Du, J. A Structural Overview of the Ion Channels of the TRPM Family. Cell Calcium 2020, 85, 102111. [Google Scholar] [CrossRef] [PubMed]
- Weyer, A.D.; Lehto, S.G. Development of TRPM8 Antagonists to Treat Chronic Pain and Migraine. Pharmaceuticals 2017, 10, 37. [Google Scholar] [CrossRef]
- De Caro, C.; Cristiano, C.; Avagliano, C.; Bertamino, A.; Ostacolo, C.; Campiglia, P.; Gomez-Monterrey, I.; La Rana, G.; Gualillo, O.; Calignano, A.; et al. Characterization of New TRPM8 Modulators in Pain Perception. Int. J. Mol. Sci. 2019, 20, 5544. [Google Scholar] [CrossRef] [PubMed]
- González-Muñiz, R.; Bonache, M.A.; Martín-Escura, C.; Gómez-Monterrey, I. Recent Progress in TRPM8 Modulation: An Update. Int. J. Mol. Sci. 2019, 20, 2618. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.A.; Chase, H.W.N.; Bevan, S. TRPM8 Activation by Menthol, Icilin, and Cold Is Differentially Modulated by Intracellular PH. J. Neurosci. 2004, 24, 5364–5369. [Google Scholar] [CrossRef]
- Wu, B.; Su, X.; Zhang, W.; Zhang, Y.-H.; Feng, X.; Ji, Y.-H.; Tan, Z.-Y. Oxaliplatin Depolarizes the IB4– Dorsal Root Ganglion Neurons to Drive the Development of Neuropathic Pain Through TRPM8 in Mice. Front. Mol. Neurosci. 2021, 14, 690858. [Google Scholar] [CrossRef]
- Khan, A.; Shal, B.; Khan, A.U.; Ullah, R.; Baig, M.W.; ul Haq, I.; Seo, E.K.; Khan, S. Suppression of TRPV1/TRPM8/P2Y Nociceptors by Withametelin via Downregulating MAPK Signaling in Mouse Model of Vincristine-Induced Neuropathic Pain. Int. J. Mol. Sci. 2021, 22, 6084. [Google Scholar] [CrossRef]
- Caudle, R.M.; Neubert, J.K. Effects of Oxaliplatin on Facial Sensitivity to Cool Temperatures and TRPM8 Expressing Trigeminal Ganglion Neurons in Mice. Front. Pain Res. 2022, 3, 868547. [Google Scholar] [CrossRef]
- Iftinca, M.; Basso, L.; Flynn, R.; Kwok, C.; Roland, C.; Hassan, A.; Defaye, M.; Ramachandran, R.; Trang, T.; Altier, C. Chronic Morphine Regulates TRPM8 Channels via MOR-PKCβ Signaling. Mol. Brain 2020, 13, 61. [Google Scholar] [CrossRef]
- Martín-Escura, C.; Medina-Peris, A.; Spear, L.A.; de la Torre Martínez, R.; Olivos-Oré, L.A.; Barahona, M.V.; González-Rodríguez, S.; Fernández-Ballester, G.; Fernández-Carvajal, A.; Artalejo, A.R.; et al. β-Lactam TRPM8 Antagonist RGM8-51 Displays Antinociceptive Activity in Different Animal Models. Int. J. Mol. Sci. 2022, 23, 2692. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Wang, F.; Shal, B.; Khan, A.U.; Zahra, S.S.; ul Haq, I.; Khan, S.; Rengasamy, K.R. Anti-Neuropathic Pain Activity of Ajugarin-I via Activation of Nrf2 Signaling and Inhibition of TRPV1/TRPM8 Nociceptors in STZ-Induced Diabetic Neuropathy. Pharmacol. Res. 2022, 183, 106392. [Google Scholar] [CrossRef] [PubMed]
- Messeguer, A.; Planells-Cases, R.; Ferrer-Montiel, A. Physiology and Pharmacology of the Vanilloid Receptor. Curr. Neuropharmacol. 2005, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.R.; Beyer, C.E.; Stahl, S.M. TRPV1 Antagonists and Chronic Pain: Beyond Thermal Perception. Pharmaceuticals 2012, 5, 114–132. [Google Scholar] [CrossRef]
- Kumar, R. Endogenous and Exogenous Vanilloids Evoke Disparate TRPV1 Activation to Produce Distinct Neuronal Responses. Front. Pharmacol. 2020, 11, 903. [Google Scholar] [CrossRef]
- Iftinca, M.; Defaye, M.; Altier, C. TRPV1-Targeted Drugs in Development for Human Pain Conditions. Drugs 2021, 81, 7–27. [Google Scholar] [CrossRef]
- Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P.; Edu, A.D.M.S.; Szallasi, A.; Huang, S.M. Nociceptive TRP Channels: Sensory Detectors and Transducers in Multiple Pain Pathologies. Pharmaceuticals 2016, 9, 72. [Google Scholar] [CrossRef]
- Katz, B.; Zaguri, R.; Edvardson, S.; Maayan, C.; Elpeleg, O.; Lev, S.; Davidson, E.; Peters, M.; Kfir-Erenfeld, S.; Berger, E.; et al. Nociception and Pain in Humans Lacking a Functional TRPV1 Channel. J. Clin. Investig. 2023, 133, e153558. [Google Scholar] [CrossRef]
- Akhilesh; Uniyal, A.; Gadepalli, A.; Tiwari, V.; Allani, M.; Chouhan, D.; Ummadisetty, O.; Verma, N.; Tiwari, V. Unlocking the Potential of TRPV1 Based SiRNA Therapeutics for the Treatment of Chemotherapy-Induced Neuropathic Pain. Life Sci. 2022, 288, 120187. [Google Scholar] [CrossRef]
- Xie, Y.K.; Luo, H.; Zhang, S.X.; Chen, X.Y.; Guo, R.; Qiu, X.Y.; Liu, S.; Wu, H.; Chen, W.B.; Zhen, X.H.; et al. GPR177 in A-Fiber Sensory Neurons Drives Diabetic Neuropathic Pain via WNT-Mediated TRPV1 Activation. Sci. Transl. Med. 2022, 14, eabh2557. [Google Scholar] [CrossRef]
- Yazğan, B.; Yazğan, Y.; Nazıroğlu, M. Alpha-Lipoic Acid Modulates the Diabetes Mellitus-Mediated Neuropathic Pain via Inhibition of the TRPV1 Channel, Apoptosis, and Oxidative Stress in Rats. J. Bioenerg. Biomembr. 2023, 55, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Peng, L.; Liu, D. Pregabalin Alleviates Neuropathic Pain via Inhibition of the PKCε/TRPV1 Pathway. Neurosci. Lett. 2022, 766, 136348. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bao, C.; Li, Z.; Yue, L.; Hu, L. Side Effects of Opioids Are Ameliorated by Regulating TRPV1 Receptors. Int. J. Environ. Res. Public Health 2022, 19, 2387. [Google Scholar] [CrossRef]
- Aguiar, D.D.; da Costa Oliveira, C.; Fonseca, F.C.S.; de Almeida, D.L.; Campos Pereira, W.V.; Guimarães, F.S.; Perez, A.C.; Duarte, I.D.G.; Romero, T.R.L. Peripherally Injected Canabidiol Reduces Neuropathic Pain in Mice: Role of the 5-HT1A and TRPV1 Receptors. Biochem. Biophys. Res. Commun. 2023, 660, 58–64. [Google Scholar] [CrossRef]
- Xiao, R.; Tang, J.; Wang, C.; Colton, C.K.; Tian, J.; Zhu, M.X. Calcium Plays a Central Role in the Sensitization of TRPV3 Channel to Repetitive Stimulations. J. Biol. Chem. 2008, 283, 6162–6174. [Google Scholar] [CrossRef] [PubMed]
- Zubcevic, L.; Herzik, M.A.; Wu, M.; Borschel, W.F.; Hirschi, M.; Song, A.S.; Lander, G.C.; Lee, S.-Y. Conformational Ensemble of the Human TRPV3 Ion Channel. Nat. Commun. 2018, 9, 4773. [Google Scholar] [CrossRef]
- Singh, A.K.; McGoldrick, L.L.; Sobolevsky, A.I. Structure and Gating Mechanism of the Transient Receptor Potential Channel TRPV3. Nat. Struct. Mol. Biol. 2018, 25, 805–813. [Google Scholar] [CrossRef]
- Deng, Z.; Maksaev, G.; Rau, M.; Xie, Z.; Hu, H.; Fitzpatrick, J.A.J.; Yuan, P. Gating of Human TRPV3 in a Lipid Bilayer. Nat. Struct. Mol. Biol. 2020, 27, 635–644. [Google Scholar] [CrossRef]
- Larkin, C.; Chen, W.; Szabó, I.L.; Shan, C.; Dajnoki, Z.; Szegedi, A.; Buhl, T.; Fan, Y.; O’Neill, S.; Walls, D.; et al. Novel Insights into the TRPV3-Mediated Itch in Atopic Dermatitis. J. Allergy Clin. Immunol. 2021, 147, 1110–1114.e5. [Google Scholar] [CrossRef]
- Peters, F.; Kopp, J.; Fischer, J.; Tantcheva-Poór, I. Mutation in TRPV3 Causes Painful Focal Plantar Keratoderma. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e620–e622. [Google Scholar] [CrossRef]
- Han, Y.; Luo, A.; Kamau, P.M.; Takomthong, P.; Hu, J.; Boonyarat, C.; Luo, L.; Lai, R. A Plant-Derived TRPV3 Inhibitor Suppresses Pain and Itch. Br. J. Pharmacol. 2021, 178, 1669–1683. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Paknejad, N.; Maksaev, G.; Sala-Rabanal, M.; Nichols, C.G.; Hite, R.K.; Yuan, P. Cryo-EM and X-Ray Structures of TRPV4 Reveal Insight into Ion Permeation and Gating Mechanisms. Nat. Struct. Mol. Biol. 2018, 25, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Todaka, H.; Taniguchi, J.; Satoh, J.I.; Mizuno, A.; Suzuki, M. Warm Temperature-Sensitive Transient Receptor Potential Vanilloid 4 (TRPV4) Plays an Essential Role in Thermal Hyperalgesia. J. Biol. Chem. 2004, 279, 35133–35138. [Google Scholar] [CrossRef] [PubMed]
- Aroke, E.N.; Powell-Roach, K.L.; Jaime-Lara, R.B.; Tesfaye, M.; Roy, A.; Jackson, P.; Joseph, P.V. Taste the Pain: The Role of TRP Channels in Pain and Taste Perception. Int. J. Mol. Sci. 2020, 21, 5929. [Google Scholar] [CrossRef]
- Bagnell, A.M.; Sumner, C.J.; McCray, B.A. TRPV4: A Trigger of Pathological RhoA Activation in Neurological Disease. Bioessays 2022, 44, e2100288. [Google Scholar] [CrossRef]
- Hu, X.; Du, L.; Liu, S.; Lan, Z.; Zang, K.; Feng, J.; Zhao, Y.; Yang, X.; Xie, Z.; Wang, P.L.; et al. A TRPV4-Dependent Neuroimmune Axis in the Spinal Cord Promotes Neuropathic Pain. J. Clin. Investig. 2023, 133, e161507. [Google Scholar] [CrossRef]
- Deng, S.; Feely, S.M.E.; Shi, Y.; Zhai, H.; Zhan, L.; Siddique, T.; Deng, H.X.; Shy, M.E. Incidence and Clinical Features of TRPV4-Linked Axonal Neuropathies in a USA Cohort of Charcot–Marie–Tooth Disease Type 2. NeuroMolecular Med. 2020, 22, 68–72. [Google Scholar] [CrossRef]
- De Logu, F.; Geppetti, P. Ion Channel Pharmacology for Pain Modulation. Handb. Exp. Pharmacol. 2019, 260, 161–186. [Google Scholar] [CrossRef]
- Staniland, A.A.; McMahon, S.B. Mice Lacking Acid-Sensing Ion Channels (ASIC) 1 or 2, but Not ASIC3, Show Increased Pain Behaviour in the Formalin Test. Eur. J. Pain 2009, 13, 554–563. [Google Scholar] [CrossRef]
- Duan, B.; Wu, L.J.; Yu, Y.Q.; Ding, Y.; Jing, L.; Xu, L.; Chen, J.; Xu, T. Le Upregulation of Acid-Sensing Ion Channel ASIC1a in Spinal Dorsal Horn Neurons Contributes to Inflammatory Pain Hypersensitivity. J. Neurosci. 2007, 27, 11139–11148. [Google Scholar] [CrossRef]
- Yang, Y.L.; Lai, T.W. Citric Acid in Drug Formulations Causes Pain by Potentiating Acid-Sensing Ion Channel 1. J. Neurosci. 2021, 41, 4596–4606. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Zimmer, A.; Sun, W.H.; Hall, J.; Brownstein, M.J.; Zimmer, A. A Role for ASIC3 in the Modulation of High-Intensity Pain Stimuli. Proc. Natl. Acad. Sci. USA 2002, 99, 8992–8997. [Google Scholar] [CrossRef] [PubMed]
- Hachisuka, J.; Chiang, M.C.; Ross, S.E. Itch and Neuropathic Itch. Pain 2018, 159, 603–609. [Google Scholar] [CrossRef] [PubMed]

| Gene | Mutation | Type of Evidence | Functional Effect | Phenotype | Reference |
|---|---|---|---|---|---|
| ANO3 | Ser213Phe | Targeted sequencing | N/A | PDPN | [8] |
| Ile453Val | Targeted sequencing | N/A | PDPN | [8] | |
| Leu984Phe | Targeted sequencing | N/A | PDPN | [8] | |
| Gly1034Arg | Targeted sequencing | N/A | SFN | [9] | |
| Met370Cysfs*? | Targeted sequencing | N/A | SFN | [9] | |
| CACNA1A | Pro2455His | WCV patch clamp | GOF | TN | [17] |
| CACNA1H | Arg481Cys | WES | N/A | Writer’s cramp and TN | [18] |
| HCN1 | Arg405Gln | Targeted sequencing | N/A | PDPN | [8] |
| KCNS1 | Ile48Val | SNPs association with greater pain outcome | N/A | Pain after nerve injury | [19] |
| KCNQ3 | Val629Ile | Targeted sequencing | N/A | SFN | [9] |
| Asp569Gly | Targeted sequencing | N/A | SFN | [9] | |
| TRPA1 | Ser86Ala | WCV patch clamp and calcium imaging | GOF | N/A | [20] |
| Leu118Val | Targeted sequencing | N/A | PDPN | [8] | |
| Thr311Asn | Targeted sequencing | N/A | SFN | [9] | |
| Ser317Ala | WCV patch clamp and calcium imaging | GOF | N/A | [20] | |
| Tyr327Cys | Targeted sequencing | N/A | SFN | [9] | |
| Arg343Cys | Targeted sequencing and Sanger sequencing | N/A | EM | [21] | |
| Arg393Gln | WES | N/A | TN | [22] | |
| Arg393* | Targeted sequencing | N/A | SFN | [9] | |
| Ser428Ala | WCV patch clamp and calcium imaging | GOF | N/A | [20] | |
| Asn460Ser | Targeted sequencing and Sanger sequencing | N/A | EM | [21] | |
| Cys608* | Targeted sequencing and Sanger sequencing | N/A | EM | [21] | |
| Arg652* | Targeted sequencing | N/A | PDPN, SFN | [8,9] | |
| Met689Val | Targeted sequencing | N/A | SFN | [9] | |
| Ala828Leufs*17 | Targeted sequencing | N/A | PDPN | [8] | |
| Asn855Ser | WCV patch clamp | GOF | Episodic pain syndrome | [23] | |
| Ser972Ala | WCV patch clamp and calcium imaging | GOF | N/A | [20] | |
| Lys1046Glu | Targeted sequencing | N/A | SFN | [9] | |
| Ser443Gly | WES | N/A | TN | [22] | |
| TRPM2 | Asp624Trp | NGS study | N/A | CN | [24] |
| Ala890Val | NGS study | N/A | CN | [24] | |
| Val934Ile | NGS study | N/A | CN | [24] | |
| Ala1645Val | WES | N/A | TN | [22] | |
| TRPM3 | Arg30Gln | WCV patch clamp and calcium ion imaging | GOF | TN | [22,25] |
| TRPM8 | Asn222Ser | Targeted sequencing | N/A | SFN | [9] |
| Arg368Trp | Targeted sequencing | N/A | SFN | [9] | |
| Glu479Asp | Targeted sequencing | N/A | PDPN | [8] | |
| Asp665Asn | NGS study | N/A | CN | [24] | |
| Val705Glyfs*79 | Targeted sequencing | N/A | PDPN | [8] | |
| Thr732Ile | Targeted sequencing | N/A | PDPN | [8] | |
| Val915Met | NGS study | N/A | CN | [24] | |
| Thr982Met | Targeted sequencing | N/A | SFN | [9] | |
| Gln85Arg | Whole-cell voltage clamp | GOF | CN after refractive surgery | [24] | |
| TRPV1 | Phe305Ser | Targeted sequencing and Sanger sequencing | N/A | EM | [21] |
| Phe305Cys | Targeted sequencing | N/A | SFN | [9] | |
| Thr450Ala | Targeted sequencing | N/A | SFN | [9] | |
| Gln498* | Targeted sequencing and Sanger sequencing | N/A | EM | [21] | |
| Arg579Cys | Targeted sequencing | N/A | SFN | [9] | |
| Gly568Asp | Case report | N/A | Painful focal plantar keratoderma | [26] | |
| TRPV3 | Gly568Cys | NGS study and WCV patch clamp | GOF | OSLP and EM | [26] |
| Gly568Val | Sanger sequencing | N/A | OSLP | [26] | |
| Gly573Ser | Animal model study | GOF | Severe itching | [27] | |
| Arg416Trp | Sanger sequencing | N/A | OSLP | [26] | |
| Arg416Gln | NGS study and WCV patch clamp | indirectly involved in channel gating | OSLP | [26] | |
| Leu673Phe | Sanger sequencing | moderately affects channel function | Atypical OSLP | [26] | |
| Trp692Ser | Sanger sequencing | severely affects channel function | OSLP | [26] | |
| Leu669Pro | Targeted sequencing | N/A | SFN | [9] | |
| p.? # | Targeted sequencing | N/A | SFN | [9] | |
| Arg186Gln | NGS study and qRT-PCR | N/A | HMSN2C and HMN8 | [28,29] | |
| TRPV4 | Arg232Cys | NGS study and qRT-PCR | N/A | HMSN2C and HMN8 | [30] |
| Arg269Cys | NGS study and qRT-PCR | N/A | CMT2C | [28] | |
| Arg269His | NGS study and qRT-PCR | GOF | CMT2C | [28,30] | |
| Tyr283Asn | WES | N/A | TN | [22] | |
| Arg316Trp | NGS study and WCV patch clamp | GOF | HMSN2C | [31,32] | |
| Arg316His | NGS study and WCV patch clamp | GOF | HMSN2C | [30] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ślęczkowska, M.; Misra, K.; Santoro, S.; Gerrits, M.M.; Hoeijmakers, J.G.J., on behalf of the PainNet Study Group. Ion Channel Genes in Painful Neuropathies. Biomedicines 2023, 11, 2680. https://doi.org/10.3390/biomedicines11102680
Ślęczkowska M, Misra K, Santoro S, Gerrits MM, Hoeijmakers JGJ on behalf of the PainNet Study Group. Ion Channel Genes in Painful Neuropathies. Biomedicines. 2023; 11(10):2680. https://doi.org/10.3390/biomedicines11102680
Chicago/Turabian StyleŚlęczkowska, Milena, Kaalindi Misra, Silvia Santoro, Monique M. Gerrits, and Janneke G. J. Hoeijmakers on behalf of the PainNet Study Group. 2023. "Ion Channel Genes in Painful Neuropathies" Biomedicines 11, no. 10: 2680. https://doi.org/10.3390/biomedicines11102680
APA StyleŚlęczkowska, M., Misra, K., Santoro, S., Gerrits, M. M., & Hoeijmakers, J. G. J., on behalf of the PainNet Study Group. (2023). Ion Channel Genes in Painful Neuropathies. Biomedicines, 11(10), 2680. https://doi.org/10.3390/biomedicines11102680