
Combined Presence in Heterozygosis of Two Variant Usher Syndrome Genes in Two Siblings Affected by Isolated Profound Age-Related Hearing Loss

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Patient and Materials and Methods
2.1. Subject
2.2. Genetic Analyses
2.3. Structural Predictions
3. Results
3.1. Genetic Analysis
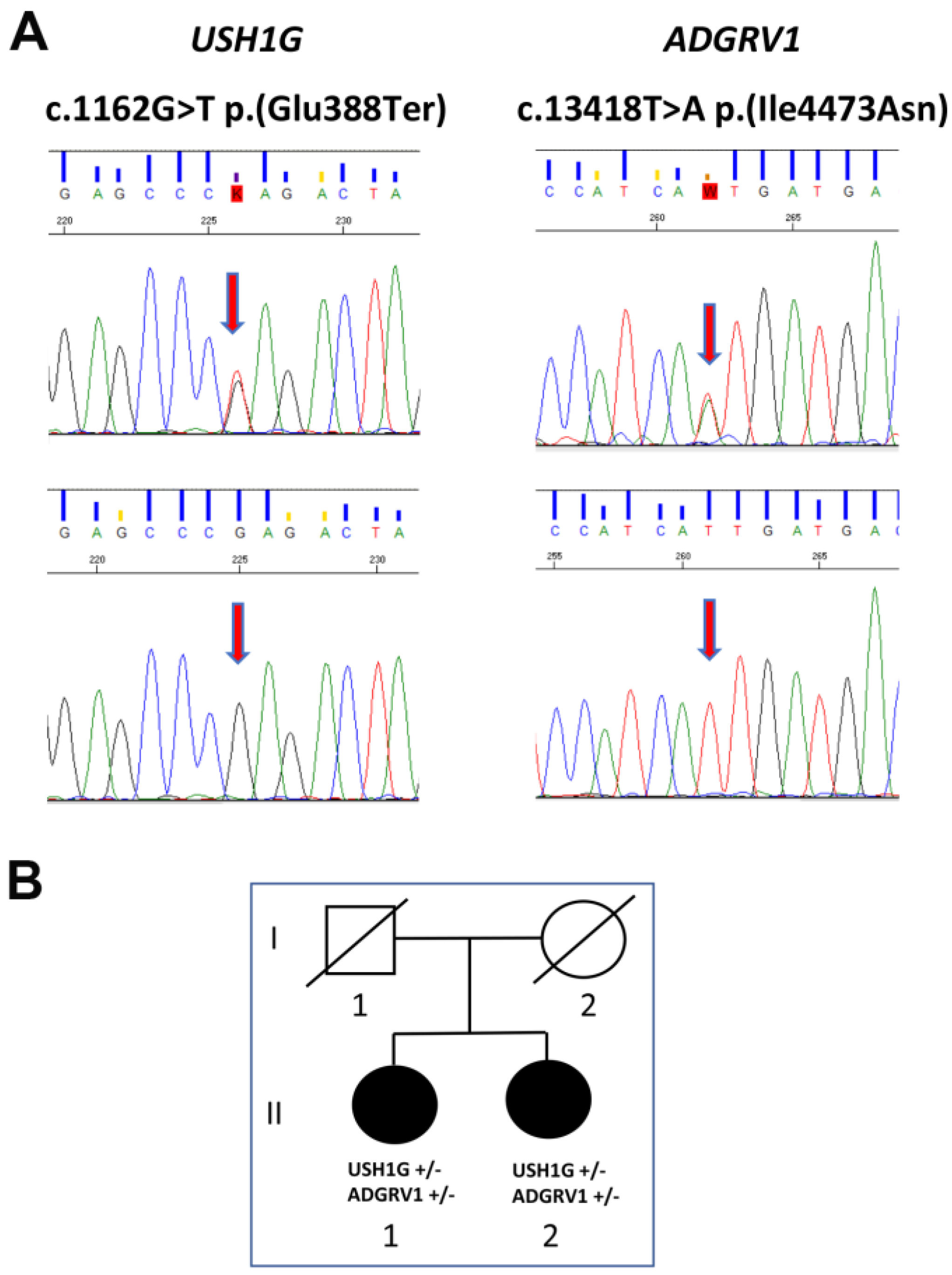
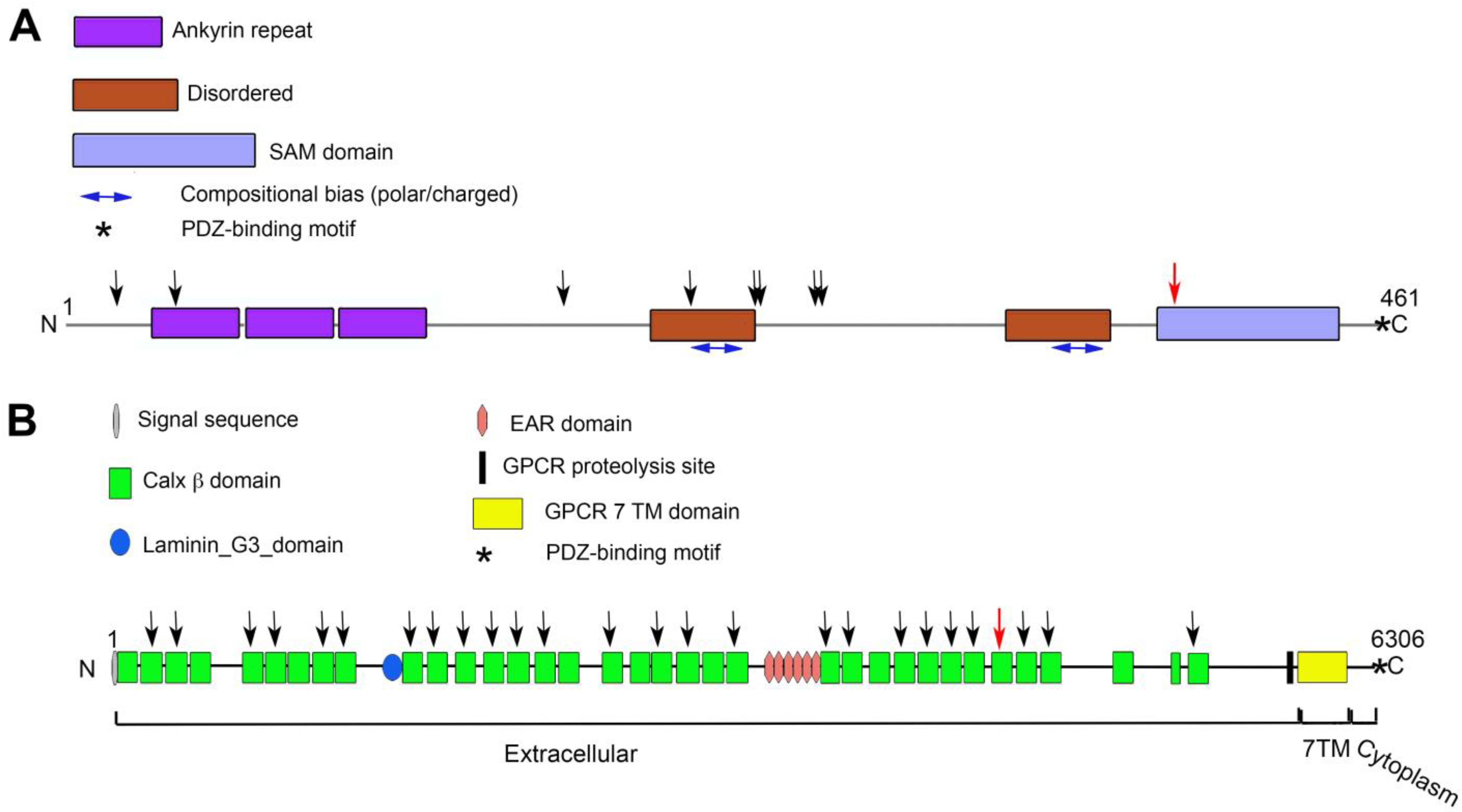
3.2. USH1G
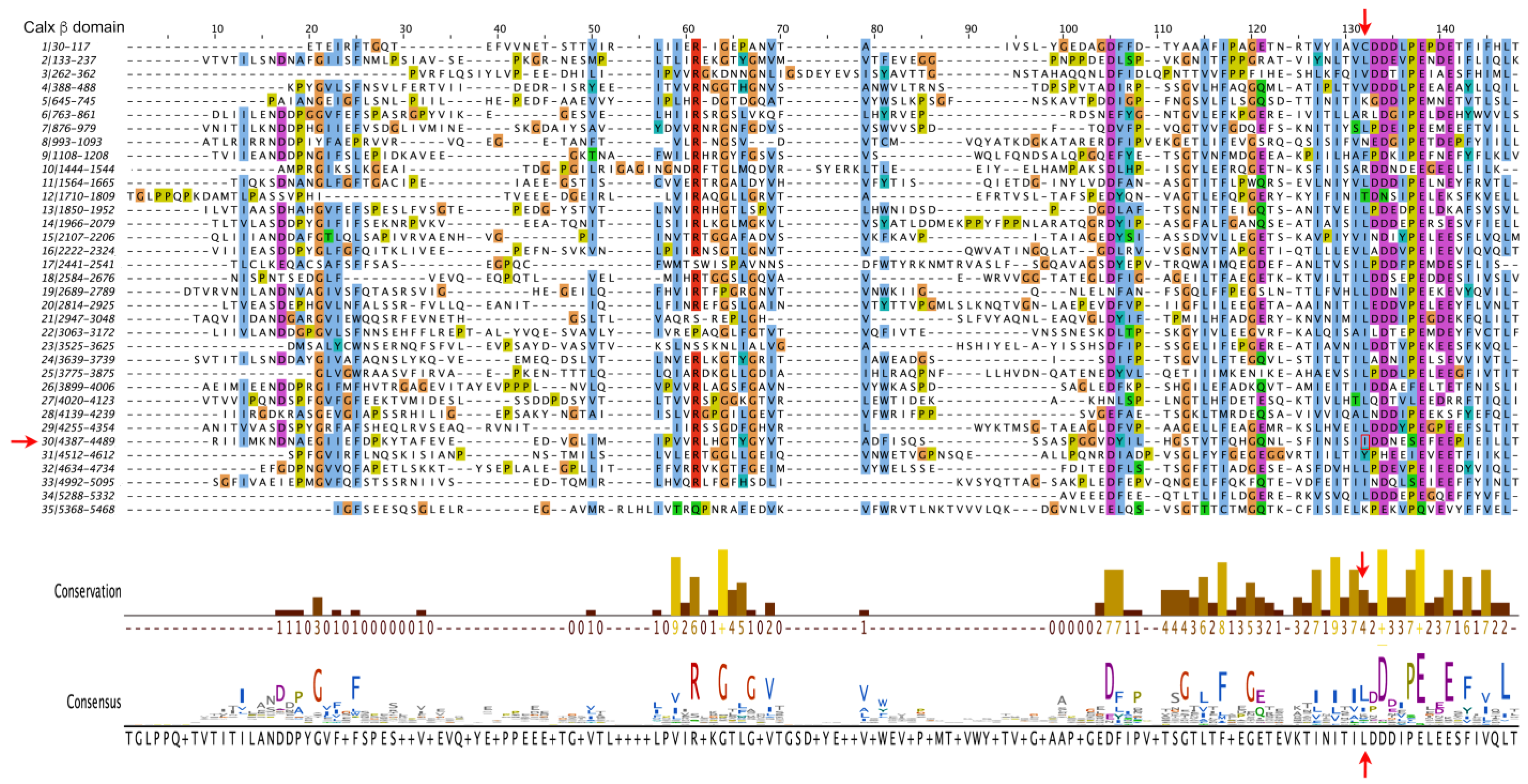
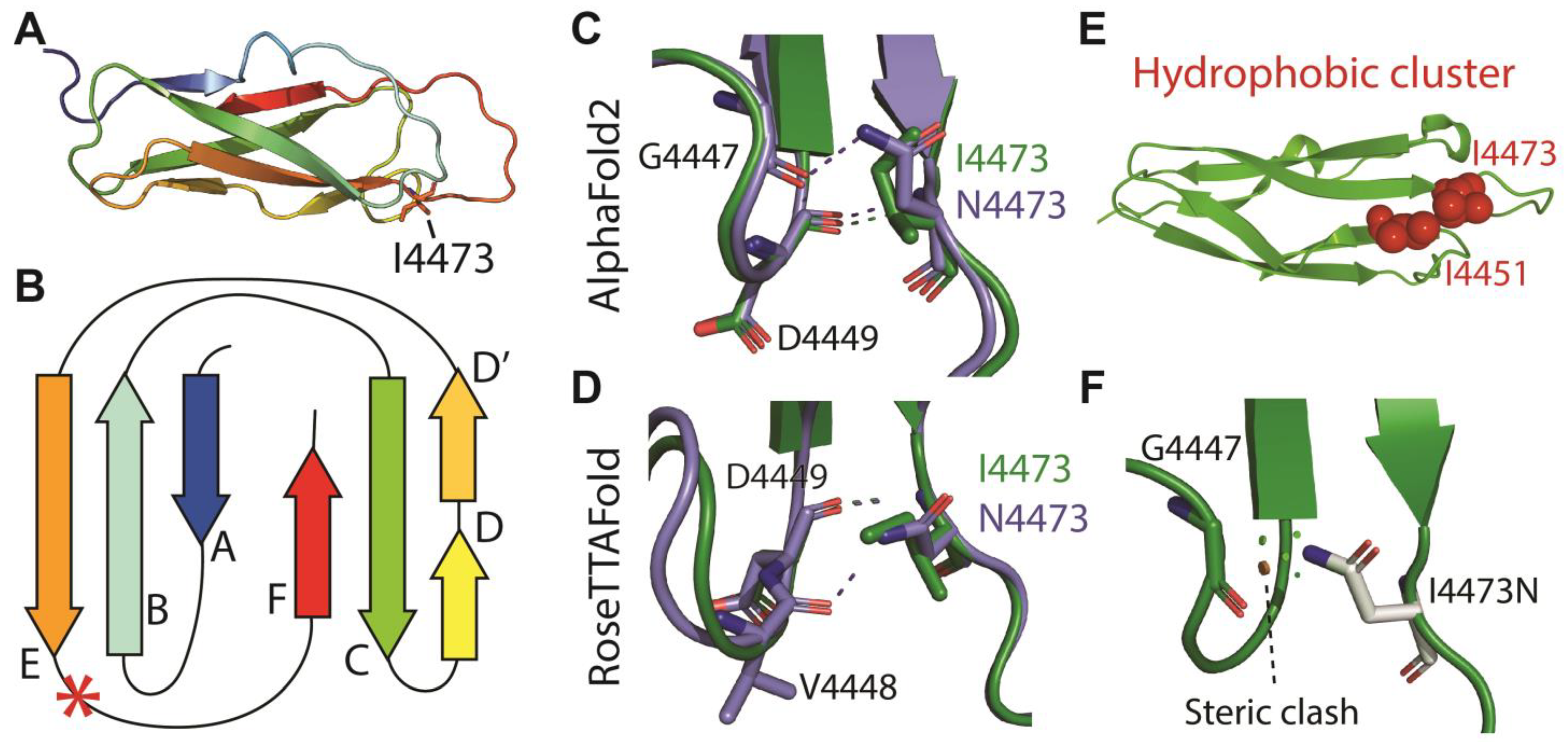
3.3. ADGRV1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Homans, N.C.; Metselaar, R.M.; Dingemanse, J.G.; van der Schroeff, M.P.; Brocaar, M.P.; Wieringa, M.H.; Baatenburg de Jong, R.J.; Hofman, A.; Goedegebure, A. Prevalence of age-related hearing loss, including sex differences, in older adults in a large cohort study. Laryngoscope 2017, 127, 725–730. [Google Scholar] [CrossRef]
- Roth, T.N.; Hanebuth, D.; Probst, R. Prevalence of age-related hearing loss in Europe: A review. Eur. Arch. Otorhinolaryngol. 2011, 268, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Amieva, H.; Ouvrard, C.; Meillon, C.; Rullier, L.; Dartigues, J.F. Death, Depression, Disability, and Dementia Associated With Self-reported Hearing Problems: A 25-Year Study. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Wells, H.R.R.; Newman, T.A.; Williams, F.M.K. Genetics of age-related hearing loss. J. Neurosci. Res. 2020, 98, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Praveen, K.; Dobbyn, L.; Gurski, L.; Ayer, A.H.; Staples, J.; Mishra, S.; Bai, Y.; Kaufman, A.; Moscati, A.; Benner, C.; et al. Population-scale analysis of common and rare genetic variation associated with hearing loss in adults. Commun. Biol. 2022, 5, 540. [Google Scholar] [CrossRef] [PubMed]
- Bowl, M.R.; Dawson, S.J. Age-Related Hearing Loss. Cold Spring Harb. Perspect. Med. 2019, 9, a033258. [Google Scholar] [CrossRef]
- Ahmadmehrabi, S.; Brant, J.; Epstein, D.J.; Ruckenstein, M.J.; Rader, D.J. Genetics of Postlingual Sensorineural Hearing Loss. Laryngoscope 2021, 131, 401–409. [Google Scholar] [CrossRef]
- Fransen, E.; Bonneux, S.; Corneveaux, J.J.; Schrauwen, I.; Di Berardino, F.; White, C.H.; Ohmen, J.D.; Van de Heyning, P.; Ambrosetti, U.; Huentelman, M.J.; et al. Genome-wide association analysis demonstrates the highly polygenic character of age-related hearing impairment. Eur. J. Hum. Genet. 2015, 23, 110–115. [Google Scholar] [CrossRef]
- Lewis, M.A.; Nolan, L.S.; Cadge, B.A.; Matthews, L.J.; Schulte, B.A.; Dubno, J.R.; Steel, K.P.; Dawson, S.J. Whole exome sequencing in adult-onset hearing loss reveals a high load of predicted pathogenic variants in known deafness-associated genes and identifies new candidate genes. BMC Med. Genom. 2018, 11, 77. [Google Scholar] [CrossRef]
- Ivarsdottir, E.V.; Holm, H.; Benonisdottir, S.; Olafsdottir, T.; Sveinbjornsson, G.; Thorleifsson, G.; Eggertsson, H.P.; Halldorsson, G.H.; Hjorleifsson, K.E.; Melsted, P.; et al. The genetic architecture of age-related hearing impairment revealed by genome-wide association analysis. Commun. Biol. 2021, 4, 706. [Google Scholar] [CrossRef]
- National Institute on Deafness and Other Communication Disorders. Fact Sheet; Hearing and Balance: “Age-Related Hearing Loss”; NIH Pub. No. 97-4235; NIDCD: Bethesda, MD, USA, 2016.
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, A.; Moller, C. Usher Syndrome. Audiol. Res. 2022, 12, 42–65. [Google Scholar] [CrossRef] [PubMed]
- Delmaghani, S.; El-Amraoui, A. The genetic and phenotypic landscapes of Usher syndrome: From disease mechanisms to a new classification. Hum. Genet. 2022, 141, 709–735. [Google Scholar] [CrossRef]
- Richardson, G.P.; Petit, C. Hair-Bundle Links: Genetics as the Gateway to Function. Cold Spring Harb. Perspect. Med. 2019, 9, a033142. [Google Scholar] [CrossRef] [PubMed]
- Dubno, J.R.; Eckert, M.A.; Lee, F.S.; Matthews, L.J.; Schmiedt, R.A. Classifying human audiometric phenotypes of age-related hearing loss from animal models. J. Assoc. Res. Otolaryngol. 2013, 14, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Pires, D.E.V.; Ascher, D.B. DynaMut2: Assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Kremer, H.; van Wijk, E.; Marker, T.; Wolfrum, U.; Roepman, R. Usher syndrome: Molecular links of pathogenesis, proteins and pathways. Hum. Mol. Genet. 2006, 15, R262–R270. [Google Scholar] [CrossRef]
- Caberlotto, E.; Michel, V.; Foucher, I.; Bahloul, A.; Goodyear, R.J.; Pepermans, E.; Michalski, N.; Perfettini, I.; Alegria-Prevot, O.; Chardenoux, S.; et al. Usher type 1G protein sans is a critical component of the tip-link complex, a structure controlling actin polymerization in stereocilia. Proc. Natl. Acad. Sci. USA 2011, 108, 5825–5830. [Google Scholar] [CrossRef]
- Grati, M.; Kachar, B. Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc. Natl. Acad. Sci. USA 2011, 108, 11476–11481. [Google Scholar] [CrossRef]
- Kikkawa, Y.; Shitara, H.; Wakana, S.; Kohara, Y.; Takada, T.; Okamoto, M.; Taya, C.; Kamiya, K.; Yoshikawa, Y.; Tokano, H.; et al. Mutations in a new scaffold protein Sans cause deafness in Jackson shaker mice. Hum. Mol. Genet. 2003, 12, 453–461. [Google Scholar] [CrossRef]
- Weil, D.; El-Amraoui, A.; Masmoudi, S.; Mustapha, M.; Kikkawa, Y.; Laine, S.; Delmaghani, S.; Adato, A.; Nadifi, S.; Zina, Z.B.; et al. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Hum. Mol. Genet. 2003, 12, 463–471. [Google Scholar] [CrossRef]
- Bjarnadottir, T.K.; Fredriksson, R.; Hoglund, P.J.; Gloriam, D.E.; Lagerstrom, M.C.; Schioth, H.B. The human and mouse repertoire of the adhesion family of G-protein-coupled receptors. Genomics 2004, 84, 23–33. [Google Scholar] [CrossRef]
- Knapp, B.; Wolfrum, U. Adhesion GPCR-Related Protein Networks. Handb. Exp. Pharmacol. 2016, 234, 147–178. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.R.; Kayes-Wandover, K.M.; Richardson, J.A.; White, P.C. Very large G protein-coupled receptor-1, the largest known cell surface protein, is highly expressed in the developing central nervous system. J. Biol. Chem. 2002, 277, 785–792. [Google Scholar] [CrossRef]
- McMillan, D.R.; White, P.C. Studies on the very large G protein-coupled receptor: From initial discovery to determining its role in sensorineural deafness in higher animals. Adv. Exp. Med. Biol. 2010, 706, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Hilge, M.; Aelen, J.; Vuister, G.W. Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol. Cell 2006, 22, 15–25. [Google Scholar] [CrossRef]
- McGee, J.; Goodyear, R.J.; McMillan, D.R.; Stauffer, E.A.; Holt, J.R.; Locke, K.G.; Birch, D.G.; Legan, P.K.; White, P.C.; Walsh, E.J.; et al. The very large G-protein-coupled receptor VLGR1: A component of the ankle link complex required for the normal development of auditory hair bundles. J. Neurosci. 2006, 26, 6543–6553. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Tokano, H.; Maeda, M.; Takabayashi, T.; Nagano, T.; Kiyama, H.; Fujieda, S.; Kitamura, K.; Sato, M. Vlgr1 is required for proper stereocilia maturation of cochlear hair cells. Genes Cells 2007, 12, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Weston, M.D.; Luijendijk, M.W.; Humphrey, K.D.; Moller, C.; Kimberling, W.J. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am. J. Hum. Genet. 2004, 74, 357–366. [Google Scholar] [CrossRef]
- Livingstone, C.D.; Barton, G.J. Protein sequence alignments: A strategy for the hierarchical analysis of residue conservation. Comput. Appl. Biosci. 1993, 9, 745–756. [Google Scholar] [CrossRef]
- Ferruz, N.; Schmidt, S.; Hocker, B. ProteinTools: A toolkit to analyze protein structures. Nucleic Acids Res. 2021, 49, W559–W566. [Google Scholar] [CrossRef]
- Bousfiha, A.; Bakhchane, A.; Charoute, H.; Detsouli, M.; Rouba, H.; Charif, M.; Lenaers, G.; Barakat, A. Novel compound heterozygous mutations in the GPR98 (USH2C) gene identified by whole exome sequencing in a Moroccan deaf family. Mol. Biol. Rep. 2017, 44, 429–434. [Google Scholar] [CrossRef]
- Richard, E.M.; Santos-Cortez, R.L.P.; Faridi, R.; Rehman, A.U.; Lee, K.; Shahzad, M.; Acharya, A.; Khan, A.A.; Imtiaz, A.; Chakchouk, I.; et al. Global genetic insight contributed by consanguineous Pakistani families segregating hearing loss. Hum. Mutat. 2019, 40, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, C.; Grati, M.; Marlin, S.; Levilliers, J.; Hardelin, J.P.; Parodi, M.; Niasme-Grare, M.; Zelenika, D.; Delepine, M.; Feldmann, D.; et al. Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J. Rare Dis. 2011, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Moteki, H.; Yoshimura, H.; Azaiez, H.; Booth, K.T.; Shearer, A.E.; Sloan, C.M.; Kolbe, D.L.; Murata, T.; Smith, R.J.; Usami, S. USH2 caused by GPR98 mutation diagnosed by massively parallel sequencing in advance of the occurrence of visual symptoms. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. S1), 123S–128S. [Google Scholar] [CrossRef] [PubMed]
- Fuster-Garcia, C.; Garcia-Garcia, G.; Jaijo, T.; Fornes, N.; Ayuso, C.; Fernandez-Burriel, M.; Sanchez-De la Morena, A.; Aller, E.; Millan, J.M. High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative. Sci. Rep. 2018, 8, 17113. [Google Scholar] [CrossRef] [PubMed]
- Maerker, T.; van Wijk, E.; Overlack, N.; Kersten, F.F.; McGee, J.; Goldmann, T.; Sehn, E.; Roepman, R.; Walsh, E.J.; Kremer, H.; et al. A novel Usher protein network at the periciliary reloading point between molecular transport machineries in vertebrate photoreceptor cells. Hum. Mol. Genet. 2008, 17, 71–86. [Google Scholar] [CrossRef]
- Cosgrove, D.; Zallocchi, M. Usher protein functions in hair cells and photoreceptors. Int. J. Biochem. Cell Biol. 2014, 46, 80–89. [Google Scholar] [CrossRef]
- Michalski, N.; Michel, V.; Bahloul, A.; Lefevre, G.; Barral, J.; Yagi, H.; Chardenoux, S.; Weil, D.; Martin, P.; Hardelin, J.P.; et al. Molecular characterization of the ankle-link complex in cochlear hair cells and its role in the hair bundle functioning. J. Neurosci. 2007, 27, 6478–6488. [Google Scholar] [CrossRef]
- Reiners, J.; Nagel-Wolfrum, K.; Jurgens, K.; Marker, T.; Wolfrum, U. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006, 83, 97–119. [Google Scholar] [CrossRef]
- Reiners, J.; van Wijk, E.; Marker, T.; Zimmermann, U.; Jurgens, K.; te Brinke, H.; Overlack, N.; Roepman, R.; Knipper, M.; Kremer, H.; et al. Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum. Mol. Genet. 2005, 14, 3933–3943. [Google Scholar] [CrossRef]
- Johnson, A.F.; Nguyen, H.T.; Veitia, R.A. Causes and effects of haploinsufficiency. Biol. Rev. 2019, 94, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Ebermann, I.; Phillips, J.B.; Liebau, M.C.; Koenekoop, R.K.; Schermer, B.; Lopez, I.; Schafer, E.; Roux, A.F.; Dafinger, C.; Bernd, A.; et al. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Investig. 2010, 120, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Eandi, C.M.; Dallorto, L.; Spinetta, R.; Micieli, M.P.; Vanzetti, M.; Mariottini, A.; Passerini, I.; Torricelli, F.; Alovisi, C.; Marchese, C. Targeted next generation sequencing in Italian patients with Usher syndrome: Phenotype-genotype correlations. Sci. Rep. 2017, 7, 15681. [Google Scholar] [CrossRef] [PubMed]
- Oonk, A.M.M.; van Huet, R.A.; Leijendeckers, J.M.; Oostrik, J.; Venselaar, H.; van Wijk, E.; Beynon, A.; Kunst, H.P.; Hoyng, C.B.; Kremer, H.; et al. Nonsyndromic hearing loss caused by USH1G mutations: Widening the USH1G disease spectrum. Ear Hear. 2015, 36, 205–211. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein HGVS | cDNA HGVS | Calxβ Domain N. | Grantham’s Distance | GnomAD Maximum MAF | ΔΔGStability (kcal/mol)-DynaMut2 Prediction § | ACMG Class | ACMG Evidence | Reported Clinical Phenotype |
|---|---|---|---|---|---|---|---|---|
| p.P352T | c.1054C>A | 3 | 38 | 0.005% NFE | −0.25 | 4 | PM1; PM5; PP1; PP3; PP5 | USH2C [41] |
| p.P352L | c.1055C>T | 3 | 98 | 0.0046% NFE | −0.26 | 4 | PM1; PM5; PP1; PP3; PP5 | Non-syndromic hearing loss [42] |
| p.D1944N | c.5830G>A | 13 | 23 | 0.5% NFE 1.5%Ashkenazi | 0.16 | 2 | PM1; PP3; BS1; BP6 | USH3/hearing loss [43,44] |
| p.A3155D | c.9464C>A | 22 | 126 | Not present | −1.59 | 4 | PM1; PM2; PM3; PP3 | USH2 [45] |
| p.I4473N | c.13418T>A | 30 | 149 | Not present | −0.22 | 4 | PM1; PM2; PP3; PP1 | Non-syndromic hearing loss (present study) |
| p.D4707Y | c.14119G>T | 32 | 160 | Not present | −0.55 | 4 | PM1; PM2; PP3; PP5 | Usher [43] |
| p.P4720L | c.14159C>T | 32 | 98 | Not present | −0.55 | 4 | PM1; PM2; PP3; PP5 | Usher [46] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borgese, N.; Guillén-Samander, A.; Colombo, S.F.; Mancassola, G.; Di Berardino, F.; Zanetti, D.; Carrera, P. Combined Presence in Heterozygosis of Two Variant Usher Syndrome Genes in Two Siblings Affected by Isolated Profound Age-Related Hearing Loss. Biomedicines 2023, 11, 2657. https://doi.org/10.3390/biomedicines11102657
Borgese N, Guillén-Samander A, Colombo SF, Mancassola G, Di Berardino F, Zanetti D, Carrera P. Combined Presence in Heterozygosis of Two Variant Usher Syndrome Genes in Two Siblings Affected by Isolated Profound Age-Related Hearing Loss. Biomedicines. 2023; 11(10):2657. https://doi.org/10.3390/biomedicines11102657
Chicago/Turabian StyleBorgese, Nica, Andrés Guillén-Samander, Sara Francesca Colombo, Giulia Mancassola, Federica Di Berardino, Diego Zanetti, and Paola Carrera. 2023. "Combined Presence in Heterozygosis of Two Variant Usher Syndrome Genes in Two Siblings Affected by Isolated Profound Age-Related Hearing Loss" Biomedicines 11, no. 10: 2657. https://doi.org/10.3390/biomedicines11102657
APA StyleBorgese, N., Guillén-Samander, A., Colombo, S. F., Mancassola, G., Di Berardino, F., Zanetti, D., & Carrera, P. (2023). Combined Presence in Heterozygosis of Two Variant Usher Syndrome Genes in Two Siblings Affected by Isolated Profound Age-Related Hearing Loss. Biomedicines, 11(10), 2657. https://doi.org/10.3390/biomedicines11102657