
The New Era of Therapeutic Strategies for the Treatment of Retinitis Pigmentosa: A Narrative Review of Pathomolecular Mechanisms for the Development of Cell-Based Therapies

 , ,
, ,
Abstract
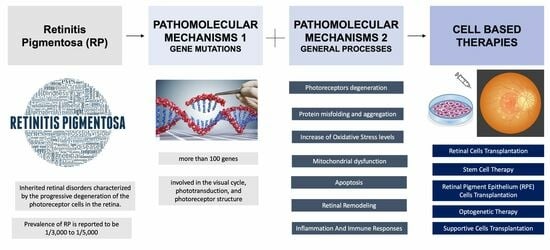
:
1. Introduction
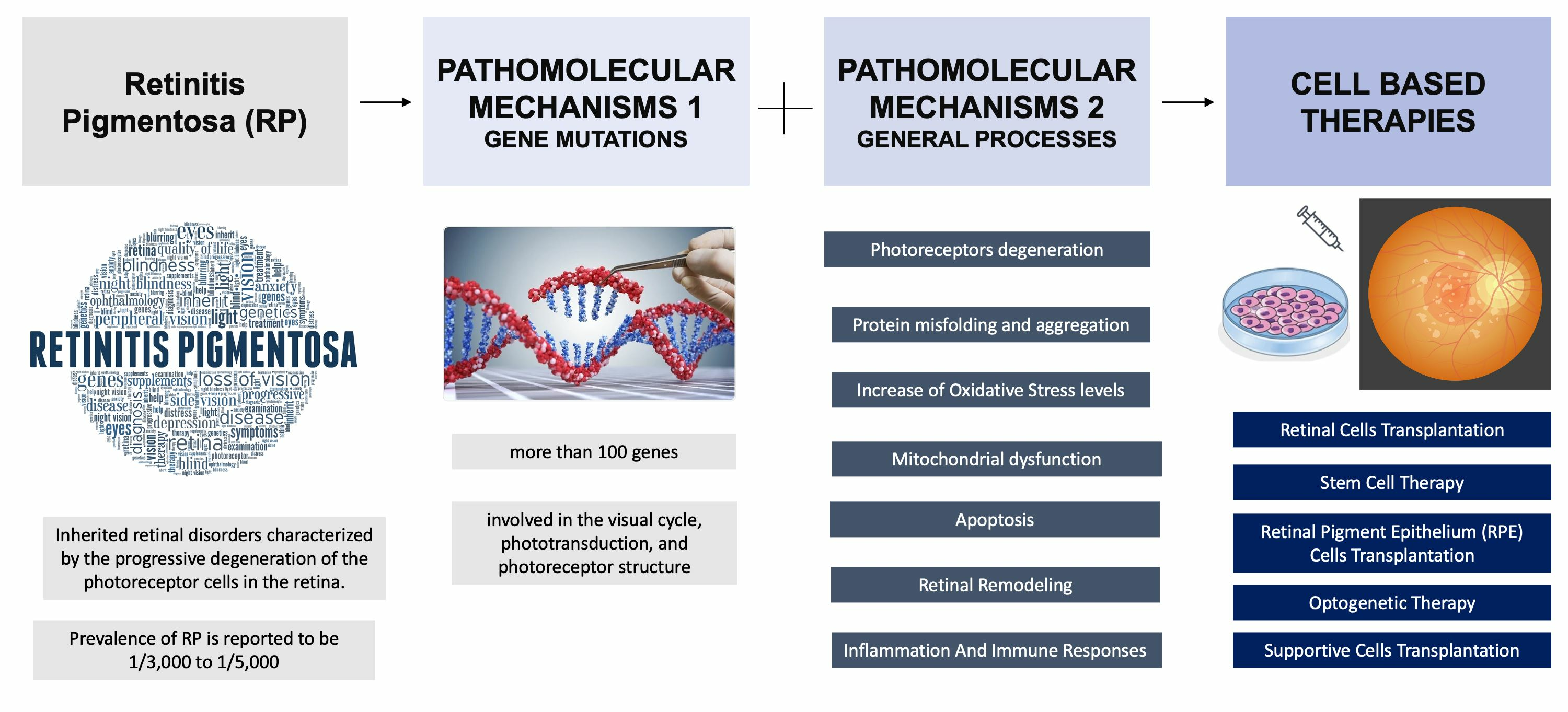
2. Pathomolecular Mechanisms of Retinitis Pigmentosa
2.1. Major Genes Involved in RP
2.1.1. Rhodopsin (RHO)
2.1.2. Peripherin/RDS (PRPH2)
2.1.3. Cyclic Nucleotide-Gated (CNG) Channels
2.1.4. Retinal Pigment Epithelium-Specific 65 kDa Protein (RPE65)
2.1.5. Retinitis Pigmentosa GTPase Regulator (RPGR)
2.1.6. Cone–Rod Homeobox Protein (CRX)
2.1.7. Usher Syndrome Genes
2.2. Mechanisms Involved in RP
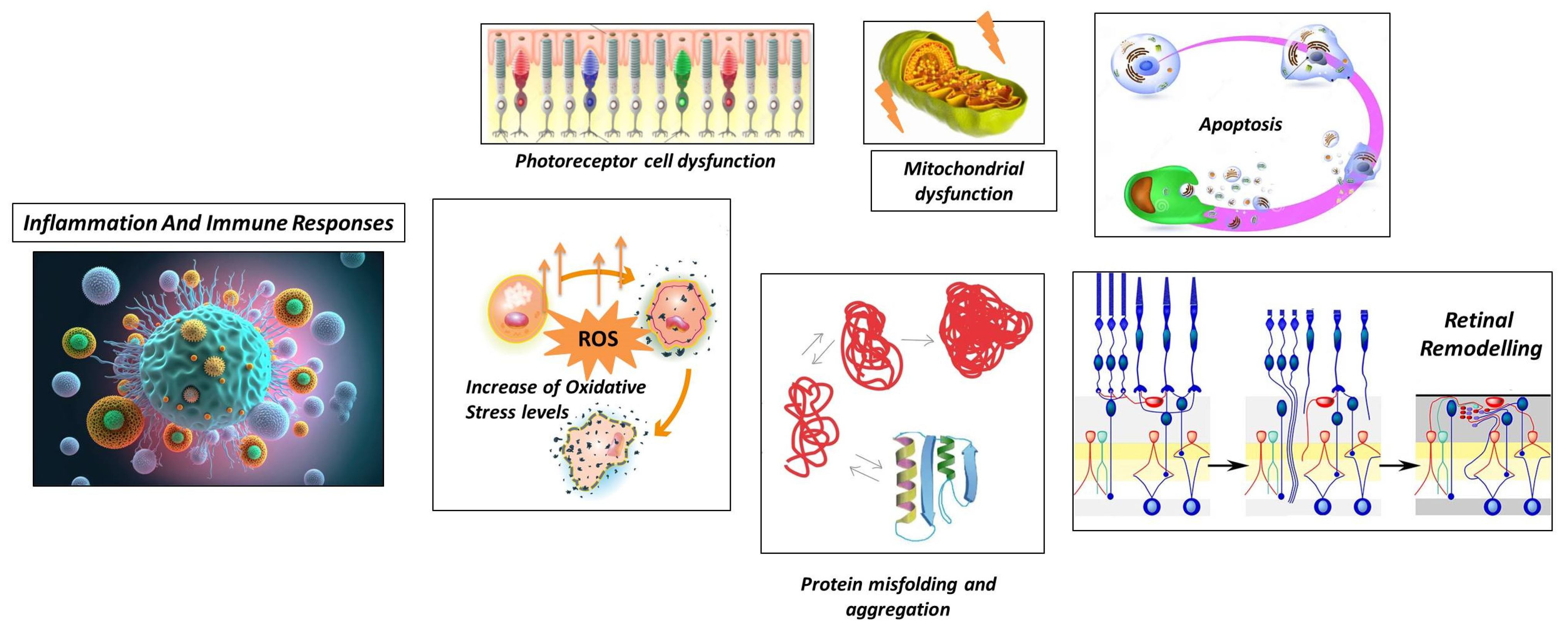
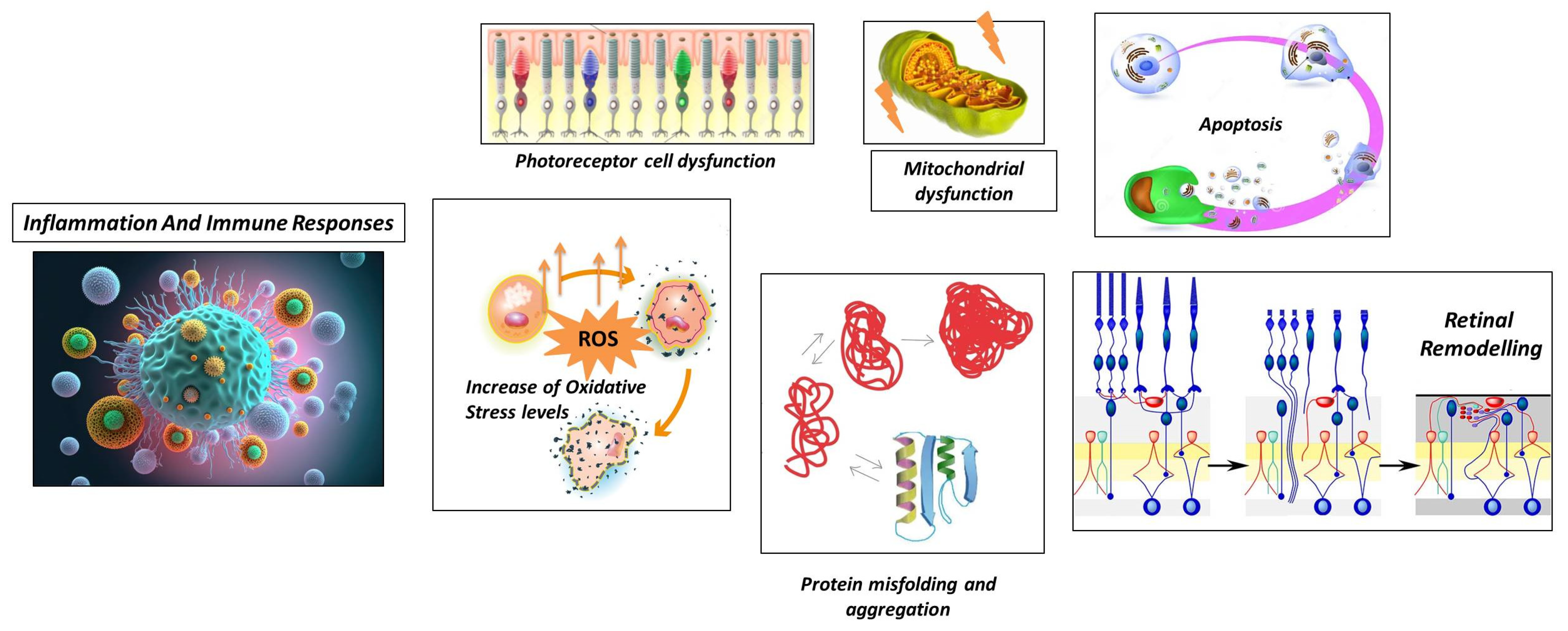
2.2.1. Photoreceptor Cell Dysfunction
2.2.2. Protein Misfolding and Aggregation
2.2.3. Increase in Oxidative Stress Levels
2.2.4. Mitochondrial Dysfunction
2.2.5. Apoptosis
2.2.6. Retinal Remodeling
- Neuronal Rearrangement: As photoreceptor cells degenerate, the remaining retinal neurons, including bipolar cells, horizontal cells, and amacrine cells, undergo reorganization. These neurons undergo structural changes and establish new connections with each other to compensate for the loss of photoreceptor input [79].
- Bipolar Cell Dystrophy: The second-order neurons in the visual pathway, bipolar cells, also undergo structural and functional changes in RP. Abnormal dendritic sprouting or retraction may occur, leading to the formation of ectopic synapses [80]. These changes can result in altered signal processing and contribute to visual abnormalities in RP [81].
- Müller Cell Gliosis: Müller cells are the major glial cells in the retina and play a crucial role in maintaining retinal homeostasis. In response to photoreceptor cell degeneration, Müller cells undergo gliotic changes, becoming activated and hypertrophic [82]. This gliosis involves changes in gene expression, increased production of glial fibrillary acidic protein (GFAP), and alterations to their structural morphology. Müller cell gliosis can have both protective and detrimental effects on retinal function and can influence the survival and function of the remaining retinal neurons [83].
- Synaptic Remodeling: Synaptic connections in the retina are reorganized in RP. As photoreceptor cells degenerate, the synaptic connections between photoreceptor cells and downstream neurons, such as bipolar cells and horizontal cells, alter. As a consequence, bipolar cells and surviving cones or bipolar cells and other retinal neurons may form new synaptic connections [84]. This synaptic remodeling can lead to altered signal processing and contribute to the rewiring of the retinal circuitry [85].
2.2.7. Inflammation and Immune Responses
- Microglial Activation: In response to photoreceptor cell death and degeneration, microglia, the resident immune cells of the retina, become activated. Activated microglia release pro-inflammatory cytokines, chemokines, and reactive oxygen species. While microglial activation initially aims to clear debris and promote tissue repair, chronic or excessive activation can lead to neuroinflammation and further damage to the retina [89,90].
- Infiltration of Immune Cells: Immune cells from the bloodstream can infiltrate the retina in some cases of RP, which further contributes to the inflammatory response. The release of inflammatory mediators by immune cells, including macrophages and T cells, can worsen retinal damage [91].
- Cytokine Imbalance: In RP, there is evidence of an imbalance in cytokine signaling in the retina. Pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), are upregulated, while anti-inflammatory cytokines, such as interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), are downregulated. This imbalance can perpetuate the inflammatory response and contribute to the degeneration of photoreceptor cells [92,93].
- Complement System Activation: Activation of the complement system can lead to the deposition of complement proteins on photoreceptor cells and subsequent immune-mediated damage [91].
- Oxidative Stress and Inflammation: The imbalance between reactive oxygen species production and antioxidant defense mechanisms can lead to oxidative stress, which can further cause inflammation in RP. The inflammatory cascade and retinal damage can be exacerbated by ROS activating various intracellular signaling pathways involved in inflammatory responses [94].
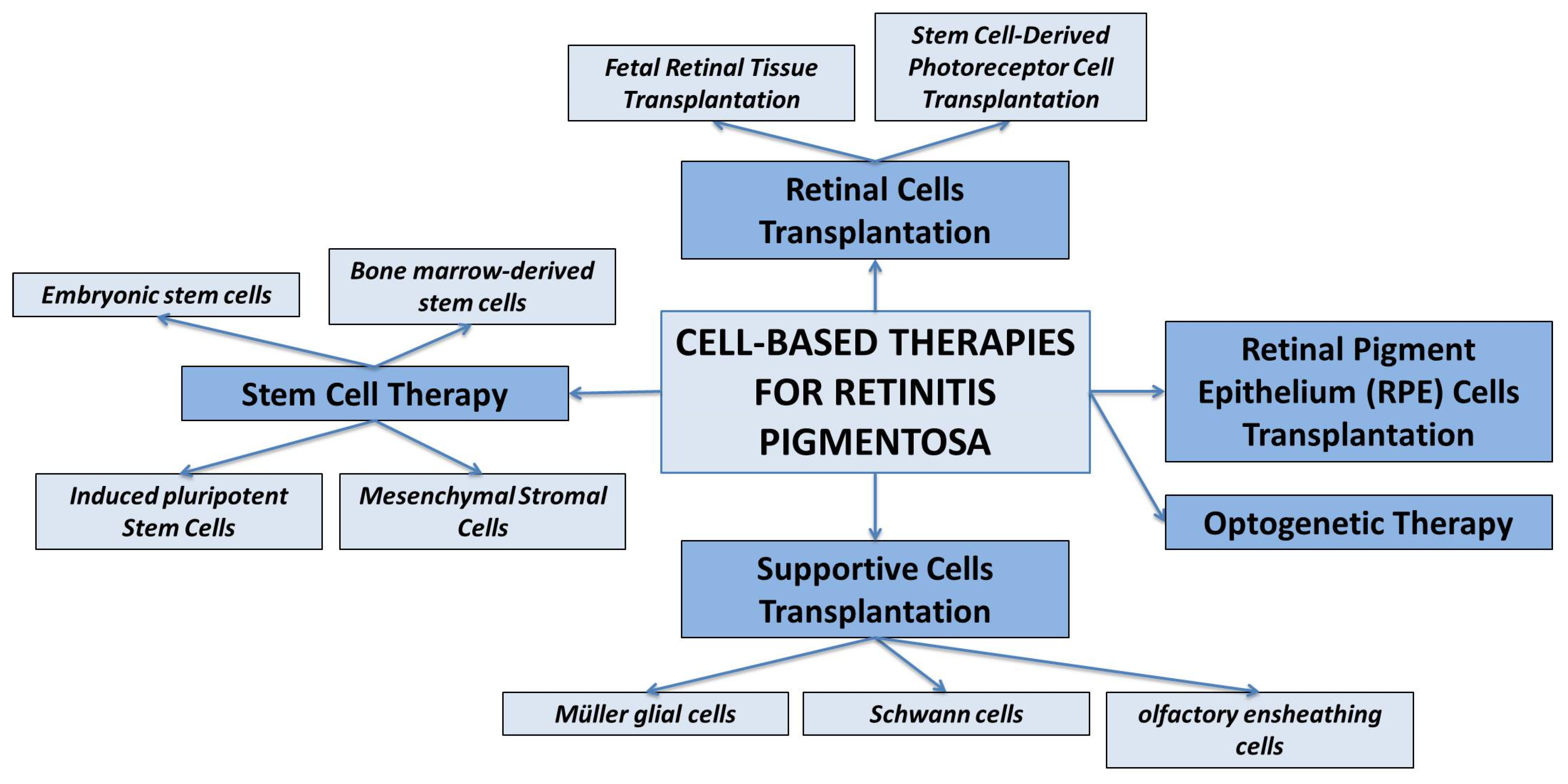
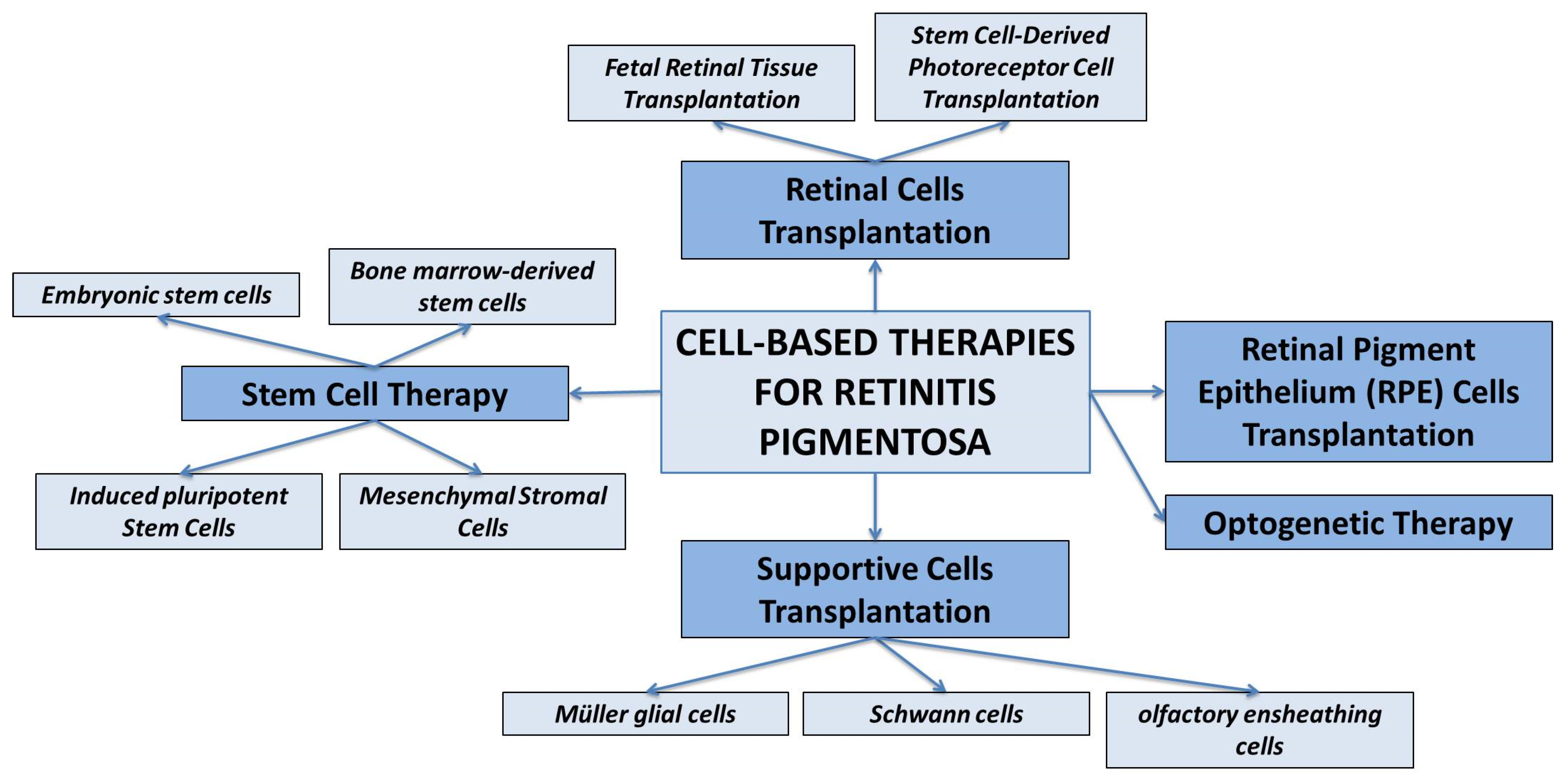
3. Cell-Based Therapies for Retinitis Pigmentosa
3.1. Retinal Cells Transplantation
- Fetal Retinal Tissue Transplantation. Fetal retinal tissue, obtained from donor fetuses, can be transplanted into the subretinal space of RP patients. The transplanted cells can integrate into the host retina and potentially improve visual function. However, the availability of fetal tissue is limited, and immunological compatibility needs to be considered [98].
- Stem Cell-Derived Photoreceptor Cell Transplantation. Pluripotent stem cells, such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), can be differentiated into photoreceptor-like cells in vitro [99,100]. These cells can then be transplanted into the retina to replace the degenerated photoreceptor cells.
3.2. RPE Cell Transplantation
3.3. Supportive Cell Transplantation
3.4. Stem Cell Therapy
3.5. Optogenetic Therapy
- Channelrhodopsin-based therapy: Channelrhodopsin-2 (ChR2) is a light-sensitive protein derived from algae. In optogenetic therapy for RP, ChR2 is introduced into RGCs or bipolar cells. When activated by light of specific wavelengths, ChR2 can depolarize the cells and initiate electrical signals, mimicking the function of photoreceptor cells. This approach aims to restore light sensitivity and enable visual information to be transmitted to the brain [138].
- Halorhodopsin-based therapy: Halorhodopsin (NpHR) is a light-sensitive protein that responds to yellow or amber light. In optogenetic therapy, NpHR can be introduced into bipolar cells or RGCs to allow the cells to be inhibited in response to light stimulation. By selectively inhibiting specific cell types, such as ON or OFF bipolar cells, the retinal circuitry can be modulated to enhance visual processing and restore functional vision [139,140].
- Red-shifted opsin-based therapy: In addition to ChR2 and NpHR, other light-sensitive proteins with red-shifted absorption spectra are being explored for optogenetic therapy in RP. These proteins, such as ReaChR or ChrimsonR, can be activated by longer wavelengths of light, including red or near-infrared light. By utilizing these red-shifted opsins, optogenetic therapy can potentially penetrate deeper into the retina and improve light sensitivity in RP patients [141].
3.6. Strategies for Promoting the Survival and Function of Existing Retinal Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| RP | Retinitis pigmentosa |
| RPE | Retinal pigment epithelium |
| CME | Cystoid macular edema |
| adRP | Autosomal dominant retinitis pigmentosa |
| NpHR | Halorhodopsin |
| BDNF | Brain-derived neurotrophic factor |
| ChR2 | Channelrhodopsin-2 |
| GDNF | Glial cell line-derived neurotrophic factor |
| GFAP | Glial fibrillary acidic protein |
| ESCs | Embryonic stem cells |
| iPSCs | Induced pluripotent stem cells |
| ROS | Reactive oxygen species |
| OECs | Olfactory ensheathing cells |
| MSCs | Mesenchymal stromal cells |
| RGCs | Retinal ganglion cells |
| CNTF | Ciliary neurotrophic factor |
References
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Anderson, R.J.; Lourenco, P. Prevalence of posterior subcapsular lens opacities in patients with retinitis pigmentosa. Br. J. Ophthalmol. 1985, 69, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Yamamoto, S.; Ogata, K.; Sugawara, T.; Hiramatsu, A.; Shibata, M.; Mitamura, Y. Macular abnormalities in patients with retinitis pigmentosa: Prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmol. 2011, 89, e122–e125. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef]
- Wu, K.Y.; Kulbay, M.; Toameh, D.; Xu, A.Q.; Kalevar, A.; Tran, S.D. Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development. Pharmaceutics 2023, 15, 685. [Google Scholar] [CrossRef]
- Maguire, A.M.; High, K.A.; Auricchio, A.; Wright, J.F.; Pierce, E.A.; Testa, F.; Mingozzi, F.; Bennicelli, J.L.; Ying, G.-S.; Rossi, S.; et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: A phase 1 dose-escalation trial. Lancet 2009, 374, 1597–1605. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Smith, A.J.; Barker, S.S.; Robbie, S.; Henderson, R.; Balaggan, K.; Viswanathan, A.; Holder, G.E.; Stockman, A.; Tyler, N.; et al. Effect of Gene Therapy on Visual Function in Leber’s Congenital Amaurosis. N. Engl. J. Med. 2008, 358, 2231–2239. [Google Scholar] [CrossRef]
- Sodi, A.; Banfi, S.; Testa, F.; Della Corte, M.; Passerini, I.; Pelo, E.; Rossi, S.; Simonelli, F.; Italian IRD Working Group. RPE65-associated inherited retinal diseases: Consensus recommendations for eligibility to gene therapy. Orphanet J. Rare Dis. 2021, 16, 257. [Google Scholar] [CrossRef]
- Acland, G.M.; Aguirre, G.D.; Ray, J.; Zhang, Q.; Aleman, T.S.; Cideciyan, A.V.; Pearce-Kelling, S.E.; Anand, V.; Zeng, Y.; Maguire, A.M.; et al. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001, 28, 92–95. [Google Scholar] [CrossRef]
- Pierce, E.A.; Bennett, J. The Status of RPE65 Gene Therapy Trials: Safety and Efficacy. Cold Spring Harb. Perspect. Med. 2015, 5, a017285. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Pang, J.-J.; Zhang, H.; Mansfield, D. Retinitis Pigmentosa: Disease Mechanisms, Diagnosis, and Therapies. J. Ophthalmol. 2015, 2015, 819452. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Sullivan, L.S.; Bowne, S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013, 84, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef]
- Sakmar, T.P.; Huber, T. Rhodopsin. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Cambridge, MA, USA, 2009; pp. 365–372. ISBN 9780080450469. [Google Scholar] [CrossRef]
- Xiao, T.; Xu, K.; Zhang, X.; Xie, Y.; Li, Y. Sector Retinitis Pigmentosa caused by mutations of the RHO gene. Eye 2019, 33, 592–599. [Google Scholar] [CrossRef]
- Lewin, A.S.; Rossmiller, B.; Mao, H. Gene Augmentation for adRP Mutations in RHO. Cold Spring Harb. Perspect. Med. 2014, 4, a017400. [Google Scholar] [CrossRef]
- Chuang, J.-Z.; Vega, C.; Jun, W.; Sung, C.-H. Structural and functional impairment of endocytic pathways by retinitis pigmentosa mutant rhodopsin-arrestin complexes. J. Clin. Investig. 2004, 114, 131–140. [Google Scholar] [CrossRef]
- Trofimova, S. Molecular Mechanisms of Retina Pathology and Ways of Its Correction; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar] [CrossRef]
- Liu, X.; Feng, B.; Vats, A.; Tang, H.; Seibel, W.; Swaroop, M.; Tawa, G.; Zheng, W.; Byrne, L.; Schurdak, M.; et al. Pharmacological clearance of misfolded rhodopsin for the treatment of RHO-associated retinitis pigmentosa. FASEB J. 2020, 34, 10146–10167. [Google Scholar] [CrossRef]
- Tebbe, L.; Sakthivel, H.; Makia, M.S.; Kakakhel, M.; Conley, S.M.; Al-Ubaidi, M.R.; Naash, M.I. Prph2 disease mutations lead to structural and functional defects in the RPE. FASEB J. 2022, 36, e22284. [Google Scholar] [CrossRef]
- Peeters, M.H.; Khan, M.; Rooijakkers, A.A.; Mulders, T.; Haer-Wigman, L.; Boon, C.J.; Klaver, C.C.; van den Born, L.I.; Hoyng, C.B.; Cremers, F.P.; et al. PRPH2 mutation update: In silico assessment of 245 reported and 7 novel variants in patients with retinal disease. Hum. Mutat. 2021, 42, 1521–1547. [Google Scholar] [CrossRef]
- Boon, C.J.; den Hollander, A.I.; Hoyng, C.B.; Cremers, F.P.; Klevering, B.J.; Keunen, J.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 2008, 27, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Coco-Martin, R.M.; Sanchez-Tocino, H.T.; Desco, C.; Usategui-Martín, R.; Tellería, J.J. PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation. Genes 2020, 11, 773. [Google Scholar] [CrossRef] [PubMed]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog. Retin. Eye Res. 2016, 52, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, M.J.; Priglinger, S.G.; Biel, M.; Michalakis, S. Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies. Biomedicines 2023, 11, 269. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.M.R.; Torre, V.; Marchesi, A. CNG channel structure, function, and gating: A tale of conformational flexibility. Pflügers Arch. -Eur. J. Physiol. 2021, 473, 1423–1435. [Google Scholar] [CrossRef]
- Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749. [Google Scholar] [CrossRef]
- Gerhardt, M.J.; Petersen-Jones, S.M.; Michalakis, S. CNG channel-related retinitis pigmentosa. Vis. Res. 2023, 208, 108232. [Google Scholar] [CrossRef]
- Dryja, T.P.; Finn, J.T.; Peng, Y.W.; McGee, T.L.; Berson, E.L.; Yau, K.W. Mutations in the gene encoding the alpha subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 10177–10181. [Google Scholar] [CrossRef]
- Duricka, D.L.; Brown, R.L.; Varnum, M.D. Defective trafficking of cone photoreceptor CNG channels induces the unfolded protein response and ER-stress-associated cell death. Biochem. J. 2011, 441, 685–696. [Google Scholar] [CrossRef]
- Morimura, H.; Fishman, G.A.; Grover, S.A.; Fulton, A.B.; Berson, E.L.; Dryja, T.P. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc. Natl. Acad. Sci. USA 1998, 95, 3088–3093. [Google Scholar] [CrossRef]
- Wolf, G. Function of the Protein RPE65 in the Visual Cycle. Nutr. Rev. 2005, 63, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Sallum, J.M.F.; Kaur, V.P.; Shaikh, J.; Banhazi, J.; Spera, C.; Aouadj, C.; Viriato, D.; Fischer, M.D. Epidemiology of Mutations in the 65-kDa Retinal Pigment Epithelium (RPE65) Gene-Mediated Inherited Retinal Dystrophies: A Systematic Literature Review. Adv. Ther. 2022, 39, 1179–1198. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.A.; Gyürüs, P.; Fleischer, L.L.; Bingham, E.L.; McHenry, C.L.; Apfelstedt-Sylla, E.; Zrenner, E.; Lorenz, B.; Richards, J.E.; Jacobson, S.G.; et al. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4293–4299. [Google Scholar]
- He, S.; Parapuram, S.K.; Hurd, T.W.; Behnam, B.; Margolis, B.; Swaroop, A.; Khanna, H. Retinitis Pigmentosa GTPase Regulator (RPGR) protein isoforms in mammalian retina: Insights into X-linked Retinitis Pigmentosa and associated ciliopathies. Vis. Res. 2008, 48, 366–376. [Google Scholar] [CrossRef]
- Vinikoor-Imler, L.C.; Simpson, C.; Narayanan, D.; Abbasi, S.; Lally, C. Prevalence of RPGR-mutated X-linked retinitis pigmentosa among males. Ophthalmic Genet. 2022, 43, 581–588. [Google Scholar] [CrossRef]
- Khanna, H. Photoreceptor Sensory Cilium: Traversing the Ciliary Gate. Cells 2015, 4, 674–686. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.A.; Atkins, S.J.; Peranen, J.; Swaroop, A.; Khanna, H. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: Implications for cilia dysfunction and photoreceptor degeneration. Hum. Mol. Genet. 2010, 19, 3591–3598. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.; Swaroop, A.; Khanna, H. Multiprotein Complexes of Retinitis Pigmentosa GTPase Regulator (RPGR), a Ciliary Protein Mutated in X-Linked Retinitis Pigmentosa (XLRP). Adv. Exp. Med. Biol. 2010, 664, 105–114. [Google Scholar] [CrossRef]
- Sun, C.; Chen, S. Gene Augmentation for Autosomal Dominant CRX-Associated Retinopathies. Adv. Exp. Med. Biol. 2023, 1415, 135–141. [Google Scholar] [CrossRef]
- Clanor, P.B.; Buchholz, C.N.; Hayes, J.E.; Friedman, M.A.; White, A.M.; Enke, R.A.; Berndsen, C.E. Structural and functional analysis of the human cone-rod homeobox transcription factor. Proteins Struct. Funct. Bioinform. 2022, 90, 1584–1593. [Google Scholar] [CrossRef]
- Swain, P.K.; Chen, S.; Wang, Q.-L.; Affatigato, L.M.; Coats, C.L.; Brady, K.D.; Fishman, G.A.; Jacobson, S.G.; Swaroop, A.; Stone, E.; et al. Mutations in the Cone-Rod Homeobox Gene Are Associated with the Cone-Rod Dystrophy Photoreceptor Degeneration. Neuron 1997, 19, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Freund, C.L.; Gregory-Evans, C.Y.; Furukawa, T.; Papaioannou, M.; Looser, J.; Ploder, L.; Bellingham, J.; Ng, D.; Herbrick, J.-A.S.; Duncan, A.; et al. Loutradis-Anagnostou A, Jacobson SG, Cepko CL, Bhattacharya SS, McInnes RR. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997, 91, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, Q.-L.; Xu, S.; Liu, I.; Li, L.Y.; Wang, Y.; Zack, D.J. Functional analysis of cone-rod homeobox (CRX) mutations associated with retinal dystrophy. Hum. Mol. Genet. 2002, 11, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Fuster-García, C.; García-Bohórquez, B.; Rodríguez-Muñoz, A.; Aller, E.; Jaijo, T.; Millán, J.M.; García-García, G. Usher Syndrome: Genetics of a Human Ciliopathy. Int. J. Mol. Sci. 2021, 22, 6723. [Google Scholar] [CrossRef] [PubMed]
- Lenassi, E.; Saihan, Z.; Cipriani, V.; Stabej, P.L.Q.; Moore, A.T.; Luxon, L.M.; Bitner-Glindzicz, M.; Webster, A.R. Natural History and Retinal Structure in Patients with Usher Syndrome Type 1 Owing to MYO7A Mutation. Ophthalmology 2013, 121, 580–587. [Google Scholar] [CrossRef]
- Well, D.; Blanchard, S.; Kaplan, J.; Guilford, P.; Gibson, F.; Walsh, J.; Mburu, P.; Varela, A.; Levilliers, J.; Weston, M.D.; et al. Defective myosin VIIA gene responsible for Usher syndrome type IB. Nature 1995, 374, 60–61. [Google Scholar] [CrossRef]
- Nagel-Wolfrum, K.; Fadl, B.R.; Becker, M.M.; Wunderlich, K.A.; Schäfer, J.; Sturm, D.; Fritze, J.; Gür, B.; Kaplan, L.; Andreani, T.; et al. Expression and subcellular localization of USH1C/harmonin in human retina provides insights into pathomechanisms and therapy. Hum. Mol. Genet. 2022, 32, 431–449. [Google Scholar] [CrossRef]
- Sun, J.-P.; Li, R.; Ren, H.-Z.; Xu, A.-T.; Yu, X.; Xu, Z.-G. The Very Large G Protein Coupled Receptor (Vlgr1) in Hair Cells. J. Mol. Neurosci. 2012, 50, 204–214. [Google Scholar] [CrossRef]
- Ratnam, K.; Västinsalo, H.; Roorda, A.; Sankila, E.-M.K.; Duncan, J.L. Cone Structure in Patients with Usher Syndrome Type III and Mutations in the Clarin 1 Gene. JAMA Ophthalmol. 2013, 131, 67–74. [Google Scholar] [CrossRef]
- Liu, W.; Liu, S.; Li, P.; Yao, K. Retinitis Pigmentosa: Progress in Molecular Pathology and Biotherapeutical Strategies. Int. J. Mol. Sci. 2022, 23, 4883. [Google Scholar] [CrossRef]
- Manley, A.; Meshkat, B.I.; Jablonski, M.M.; Hollingsworth, T. Cellular and Molecular Mechanisms of Pathogenesis Underlying Inherited Retinal Dystrophies. Biomolecules 2023, 13, 271. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.M.; Nassisi, M.; Hernandez, C.S.; Méjécase, C.; El Shamieh, S.; Condroyer, C.; Antonio, A.; Meunier, I.; Andrieu, C.; Defoort-Dhellemmes, S.; et al. Retinal Phenotype of Patients with Isolated Retinal Degeneration Due to CLN3 Pathogenic Variants in a French Retinitis Pigmentosa Cohort. JAMA Ophthalmol. 2021, 139, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Lin, J.H.; LaVail, M.M. Misfolded Proteins and Retinal Dystrophies. Adv. Exp. Med. Biol. 2010, 664, 115–121. [Google Scholar] [CrossRef]
- Tzekov, R.; Stein, L.; Kaushal, S. Protein Misfolding and Retinal Degeneration. Cold Spring Harb. Perspect. Biol. 2011, 3, a007492. [Google Scholar] [CrossRef]
- Vingolo, E.M.; Casillo, L.; Contento, L.; Toja, F.; Florido, A. Retinitis Pigmentosa (RP): The Role of Oxidative Stress in the Degenerative Process Progression. Biomedicines 2022, 10, 582. [Google Scholar] [CrossRef]
- Wang, J.; Li, M.; Geng, Z.; Khattak, S.; Ji, X.; Wu, D.; Dang, Y. Role of Oxidative Stress in Retinal Disease and the Early Intervention Strategies: A Review. Oxidative Med. Cell. Longev. 2022, 2022, 7836828. [Google Scholar] [CrossRef]
- Gallenga, C.E.; Lonardi, M.; Pacetti, S.; Violanti, S.S.; Tassinari, P.; Di Virgilio, F.; Tognon, M.; Perri, P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants 2021, 10, 848. [Google Scholar] [CrossRef]
- Domènech, E.B.; Marfany, G. The Relevance of Oxidative Stress in the Pathogenesis and Therapy of Retinal Dystrophies. Antioxidants 2020, 9, 347. [Google Scholar] [CrossRef]
- Plafker, S.M.; O’Mealey, G.B.; Szweda, L.I. Mechanisms for countering oxidative stress and damage in retinal pigment epithelium. Int. Rev. Cell Mol. Biol. 2012, 298, 135–177. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305. [Google Scholar] [CrossRef]
- Ren, X.; Léveillard, T. Modulating antioxidant systems as a therapeutic approach to retinal degeneration. Redox Biol. 2022, 57, 102510. [Google Scholar] [CrossRef]
- Lefevere, E.; Toft-Kehler, A.K.; Vohra, R.; Kolko, M.; Moons, L.; Van Hove, I. Mitochondrial dysfunction underlying outer retinal diseases. Mitochondrion 2017, 36, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Barot, M.; Gokulgandhi, M.R.; Mitra, A.K. Mitochondrial Dysfunction in Retinal Diseases. Curr. Eye Res. 2011, 36, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; Haelterman, N.A.; Sandoval, H.; Xiong, B.; Donti, T.; Kalsotra, A.; Yamamoto, S.; Cooper, T.A.; Graham, B.H.; Bellen, H.J. Correction: Impaired Mitochondrial Energy Production Causes Light-Induced Photoreceptor Degeneration Independent of Oxidative Stress. PLoS Biol. 2018, 16, e1002622. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, X.; Gao, A.L.H.; Zhao, M.; Ge, L.; Li, M.; Yang, C.; Gong, Y.; Gu, Z.; Xu, H. Alleviation of Photoreceptor Degeneration Based on Fullerenols in rd1 Mice by Reversing Mitochondrial Dysfunction via Modulation of Mitochondrial DNA Transcription and Leakage. Small 2023, 5, e2205998. [Google Scholar] [CrossRef] [PubMed]
- Vlachantoni, D.; Bramall, A.N.; Murphy, M.P.; Taylor, R.W.; Shu, X.; Tulloch, B.; Van Veen, T.; Turnbull, D.M.; McInnes, R.R.; Wright, A.F. Evidence of severe mitochondrial oxidative stress and a protective effect of low oxygen in mouse models of inherited photoreceptor degeneration. Hum. Mol. Genet. 2011, 20, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Marigo, V.; Kutluer, M.; Huang, L.; Comitato, A.; Schiroli, D.; Schwede, F.; Rentsch, A.; Ekstrom, P.A.R.; Paquet-Durand, F. Decrease of intracellular calcium to restrain rod cell death in retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4866. [Google Scholar]
- Das, S.; Chen, Y.; Yan, J.; Christensen, G.; Belhadj, S.; Tolone, A.; Paquet-Durand, F. The role of cGMP-signalling and calcium-signalling in photoreceptor cell death: Perspectives for therapy development. Pflugers Arch. Eur. J. Physiol. 2021, 473, 1411–1421. [Google Scholar] [CrossRef]
- Beeson, C.; Peterson, Y.K.; Perron, N.; Bandyopadhyay, M.; Nasarre, C.; Beeson, G.; Comer, R.F.; Lindsey, C.C.; Schnellmann, R.G.; Rohrer, B. Newly Identified Chemicals Preserve Mitochondrial Capacity and Decelerate Loss of Photoreceptor Cells in Murine Retinal Degeneration Models. J. Ocul. Pharmacol. Ther. 2021, 37, 367–378. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Milam, A.H. Apoptosis in Retinitis Pigmentosa. In Degenerative Diseases of the Retina; Anderson, R.E., LaVail, M.M., Hollyfield, J.G., Eds.; Springer: Boston, MA, USA, 1995. [Google Scholar] [CrossRef]
- Wong, P. Apoptosis, retinitis pigmentosa, and degeneration. Biochem. Cell Biol. 1994, 72, 489–498. [Google Scholar] [CrossRef]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Remé, C.E.; Grimm, C.; Hafezi, F.; Wenzel, A.; Williams, T.P. Apoptosis in the Retina: The Silent Death of Vision. News Physiol. Sci. 2000, 15, 120–124. [Google Scholar] [CrossRef]
- Jones, B.W.; Kondo, M.; Terasaki, H.; Lin, Y.; McCall, M.; Marc, R.E. Retinal remodeling. Jpn. J. Ophthalmol. 2012, 56, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.W.; Pfeiffer, R.L.; Ferrell, W.D.; Watt, C.B.; Marmor, M.; Marc, R.E. Retinal remodeling in human retinitis pigmentosa. Exp. Eye Res. 2016, 150, 149–165. [Google Scholar] [CrossRef]
- Marc, R.E.; Jones, B.W.; Watt, C.B.; Strettoi, E. Neural remodeling in retinal degeneration. Prog. Retin. Eye Res. 2003, 22, 607–655. [Google Scholar] [CrossRef]
- Gilhooley, M.J.; Hickey, D.G.; Lindner, M.; Palumaa, T.; Hughes, S.; Peirson, S.N.; MacLaren, R.E.; Hankins, M.W. ON-bipolar cell gene expression during retinal degeneration: Implications for optogenetic visual restoration. Exp. Eye Res. 2021, 207, 108553. [Google Scholar] [CrossRef]
- Martínez-Gil, N.; Maneu, V.; Kutsyr, O.; Fernández-Sánchez, L.; Sánchez-Sáez, X.; Sánchez-Castillo, C.; Campello, L.; Lax, P.; Pinilla, I.; Cuenca, N. Cellular and molecular alterations in neurons and glial cells in inherited retinal degeneration. Front. Neuroanat. 2022, 16, 984052. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Huang, X.; He, J.; Zou, T.; Chen, X.; Xu, H. The roles of microglia in neural remodeling during retinal degeneration. Histol. Histopathol. 2021, 37, 1–10. [Google Scholar] [CrossRef]
- Gordon, W.C.; Knott, E.J.; Sheets, K.G.; Regan, C.E., Jr.; Bazan, N.G. Müller Cell Reactive Gliosis Contributes to Retinal Degeneration in Ccl2-/-/Cx3cr1-/- Mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1375. [Google Scholar]
- Phillips, M.J.; Otteson, D.C.; Sherry, D.M. Progression of neuronal and synaptic remodeling in the rd10 mouse model of retinitis pigmentosa. J. Comp. Neurol. 2010, 518, 2071–2089. [Google Scholar] [CrossRef] [PubMed]
- Soto, F.; Kerschensteiner, D. Synaptic remodeling of neuronal circuits in early retinal degeneration. Front. Cell. Neurosci. 2015, 9, 395. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog. Retin Eye Res. 2020, 74, 100771. [Google Scholar] [CrossRef] [PubMed]
- Telias, M.; Nawy, S.; Kramer, R.H. Degeneration-Dependent Retinal Remodeling: Looking for the Molecular Trigger. Front. Neurosci. 2020, 14, 618019. [Google Scholar] [CrossRef]
- Pfeiffer, R.L.; Jones, B.W. Current perspective on retinal remodeling: Implications for therapeutics. Front. Neuroanat. 2022, 16, 1099348. [Google Scholar] [CrossRef]
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef]
- Peng, B.; Xiao, J.; Wang, K.; So, K.-F.; Tipoe, G.L.; Lin, B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J. Neurosci. 2014, 34, 8139–8150. [Google Scholar] [CrossRef]
- Mohan, K.V.; Mishra, A.; Muniyasamy, A.; Sinha, P.; Sahu, P.; Kesarwani, A.; Jain, K.; Nagarajan, P.; Scaria, V.; Agarwal, M.; et al. Immunological consequences of compromised ocular immune privilege accelerate retinal degeneration in retinitis pigmentosa. Orphanet J. Rare Dis. 2022, 17, 378. [Google Scholar] [CrossRef]
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2021, 22, 2096. [Google Scholar] [CrossRef]
- Okita, A.; Murakami, Y.; Shimokawa, S.; Funatsu, J.; Fujiwara, K.; Nakatake, S.; Koyanagi, Y.; Akiyama, M.; Takeda, A.; Hisatomi, T.; et al. Changes of Serum Inflammatory Molecules and Their Relationships with Visual Function in Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2020, 61, 30. [Google Scholar] [CrossRef]
- Murakami, Y.; Nakabeppu, Y.; Sonoda, K.-H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 7170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Hou, C.; Yan, N. Neuroinflammation in retinitis pigmentosa: Therapies targeting the innate immune system. Front. Immunol. 2022, 13, 1059947. [Google Scholar] [CrossRef] [PubMed]
- Bull, N.D.; Martin, K.R. Concise review: Toward stem cell-based therapies for retinal neurodegenerative diseases. Stem Cells. 2011, 29, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Tezel, T.; Ruff, A. Retinal cell transplantation in retinitis pigmentosa. Taiwan J. Ophthalmol. 2021, 11, 336–347. [Google Scholar] [CrossRef] [PubMed]
- MacLaren, R.; Pearson, R.; MacNeil, A.; Douglas, R.H.; Salt, T.E.; Akimoto, M.; Swaroop, A.; Sowden, J.; Ali, R. Retinal repair by transplantation of photoreceptor precursors. Nature 2006, 444, 203–207. [Google Scholar] [CrossRef]
- Yanai, A.; Laver, C.; Joe, A.W.; Gregory-Evans, K. Efficient Production of Photoreceptor Precursor Cells from Human Embryonic Stem Cells. Methods Mol Biol. 2013, 1307, 357–369. [Google Scholar] [CrossRef]
- Li, Y.; Tsai, Y.-T.; Hsu, C.-W.; Erol, D.; Yang, J.; Wu, W.-H.; Davis, R.J.; Egli, D.; Tsang, S.H. Long-term safety and efficacy of human-induced Pluripotent Stem cell (iPS) grafts in a preclinical model of retinitis pigmentosa. Mol. Med. 2012, 18, 1312–1319. [Google Scholar] [CrossRef]
- Khan, A.Z.; Utheim, T.P.; Eidet, J.R. Retinal Pigment Epithelium Transplantation: Past, Present, and Future. J. Ophthalmic Vis. Res. 2022, 17, 574–580. [Google Scholar] [CrossRef]
- Alexander, P.; Thomson, H.A.J.; Luff, A.J.; Lotery, A.J. Retinal pigment epithelium transplantation: Concepts, challenges, and future prospects. Eye 2015, 29, 992–1002. [Google Scholar] [CrossRef]
- Heravi, M.; Rasoulinejad, S.A. Potential of Müller Glial Cells in Regeneration of Retina; Clinical and Molecular Approach. Int. J. Organ. Transplant. Med. 2022, 13, 50–59. [Google Scholar]
- Eastlake, K.; Lamb, W.; Luis, J.; Khaw, P.; Jayaram, H.; Limb, G. Prospects for the application of Müller glia and their derivatives in retinal regenerative therapies. Prog. Retin. Eye Res. 2021, 85, 100970. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, Z.; Gu, P. Stem/progenitor cell-based transplantation for retinal degeneration: A review of clinical trials. Cell Death Dis. 2020, 11, 793. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, R.C. Stem cell therapy for retinal diseases: Update. Stem Cell Res. Ther. 2011, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Wong, I.Y.-H.; Poon, M.-W.; Pang, R.T.-W.; Lian, Q.; Wong, D. Promises of stem cell therapy for retinal degenerative diseases. Graefe’s Arch. Clin. Exp. Ophthalmol. 2011, 249, 1439–1448. [Google Scholar] [CrossRef]
- He, Y.; Zhang, Y.; Liu, X.; Ghazaryan, E.; Li, Y.; Xie, J.; Su, G. Recent advances of stem cell therapy for retinitis pigmentosa. Int. J. Mol. Sci. 2014, 15, 14456–14474. [Google Scholar] [CrossRef]
- Jin, Z.-B.; Okamoto, S.; Xiang, P.; Takahashi, M. Integration-free induced pluripotent stem cells derived from retinitis pigmentosa patient for disease modeling. Stem Cells Transl. Med. 2012, 1, 503–509. [Google Scholar] [CrossRef]
- Idelson, M.; Alper, R.; Obolensky, A.; Ben-Shushan, E.; Hemo, I.; Yachimovich-Cohen, N.; Khaner, H.; Smith, Y.; Wiser, O.; Gropp, M.; et al. Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell 2009, 5, 396–408. [Google Scholar] [CrossRef]
- Vugler, A.; Lawrence, J.; Walsh, J.; Carr, A.; Gias, C.; Semo, M.; Ahmado, A.; da Cruz, L.; Andrews, P.; Coffey, P. Embryonic stem cells and retinal repair. Mech. Dev. 2007, 124, 807–829. [Google Scholar] [CrossRef]
- Wang, N.-K.; Tosi, J.; Kasanuki, J.M.; Chou, C.L.; Kong, J.; Parmalee, N.; Wert, K.J.; Allikmets, R.; Lai, C.-C.; Chien, C.-L.; et al. Transplantation of reprogrammed embryonic stem cells improves visual function in a mouse model for retinitis pigmentosa. Transplantation 2010, 89, 911–919. [Google Scholar] [CrossRef]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef]
- Mannino, G.; Russo, C.; Longo, A.; Anfuso, C.D.; Lupo, G.; Furno, D.L.; Giuffrida, R.; Giurdanella, G. Potential therapeutic applications of mesenchymal stem cells for the treatment of eye diseases. World J. Stem Cells 2021, 13, 632–644. [Google Scholar] [CrossRef]
- Adak, S.; Magdalene, D.; Deshmukh, S.; Das, D.; Jaganathan, B.G. A Review on Mesenchymal Stem Cells for Treatment of Retinal Diseases. Stem Cell Rev. Rep. 2021, 17, 1154–1173. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Jiang, Y.; Duan, Y.; Zhang, X.; Li, X. Mesenchymal-Stem-Cell-Based Strategies for Retinal Diseases. Genes 2022, 13, 1901. [Google Scholar] [CrossRef] [PubMed]
- Holan, V.; Palacka, K.; Hermankova, B. Mesenchymal Stem Cell-Based Therapy for Retinal Degenerative Diseases: Experimental Models and Clinical Trials. Cells 2021, 10, 588. [Google Scholar] [CrossRef]
- Reboussin, É.; Buffault, J.; Brignole-Baudouin, F.; Goazigo, A.R.-L.; Riancho, L.; Olmiere, C.; Sahel, J.-A.; Parsadaniantz, S.M.; Baudouin, C. Evaluation of neuroprotective and immunomodulatory properties of mesenchymal stem cells in an ex vivo retinal explant model. J. Neuroinflamm. 2022, 19, 63. [Google Scholar] [CrossRef]
- Stavely, R.; Nurgali, K. The emerging antioxidant paradigm of mesenchymal stem cell therapy. Stem Cells Transl. Med. 2020, 9, 985–1006. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Gatti, M.; Prata, C.; Hrelia, S.; Maraldi, T. Role of Mesenchymal Stem Cells in Counteracting Oxidative Stress—Related Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3299. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.L.S.; Kumar, S.; Mok, P.L. Cellular Reparative Mechanisms of Mesenchymal Stem Cells for Retinal Diseases. Int. J. Mol. Sci. 2017, 18, 1406. [Google Scholar] [CrossRef]
- Ruitenberg, M.J.; Vukovic, J.; Sarich, J.; Busfield, S.J.; Plant, G.W.; Yang, H.; He, B.-R.; Hao, D.-J.; Su, Z.; He, C.; et al. Olfactory ensheathing cells: Characteristics, genetic engineering, and therapeutic potential. J. Neurotrauma 2006, 23, 468–478. [Google Scholar] [CrossRef]
- Huo, S.J.; Li, Y.C.; Xie, J.; Li, Y.; Raisman, G.; Zeng, Y.X.; He, J.R.; Weng, C.H.; Yin, Z.Q. Transplanted olfactory ensheathing cells reduce retinal degeneration in royal college of surgeons rats. Curr. Eye Res. 2012, 37, 749–758. [Google Scholar] [CrossRef]
- Ajgaonkar, B.S.; Kumaran, A.; Kumar, S.; Jain, R.D.; Dandekar, P.P. Cell-based Therapies for Corneal and Retinal Disorders. Stem Cell Rev. Rep. 2023; Epub ahead of print. [Google Scholar] [CrossRef]
- Klassen, H.J.; Ng, T.F.; Kurimoto, Y.; Kirov, I.; Shatos, M.; Coffey, P.; Young, M.J. Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-mediated behavior. Investig. Opthalmol. Vis. Sci. 2004, 45, 4167–4173. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Klassen, H.; Zhang, X.; Young, M. Laser injury promotes migration and integration of retinal progenitor cells into host retina. Mol. Vis. 2010, 16, 983–990. [Google Scholar] [PubMed]
- West, E.; Pearson, R.; Tschernutter, M.; Sowden, J.; MacLaren, R.; Ali, R. Pharmacological disruption of the outer limiting membrane leads to increased retinal integration of transplanted photoreceptor precursors. Exp. Eye Res. 2008, 86, 601–611. [Google Scholar] [CrossRef]
- Chang, A.Y. Challenges of Treatment Methodologies and the Future of Gene Therapy and Stem Cell Therapy to Treat Retinitis Pigmentosa; Springer: New York, NY, USA, 2022; Volume 2560, pp. 363–374. [Google Scholar] [CrossRef]
- Sahel, J.-A.; Boulanger-Scemama, E.; Pagot, C.; Arleo, A.; Galluppi, F.; Martel, J.N.; Degli Esposti, S.; Delaux, A.; de Saint Aubert, J.-B.; de Montleau, C.; et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat. Med. 2021, 27, 1223–1229. [Google Scholar] [CrossRef]
- Busskamp, V.; Picaud, S.; Sahel, J.A.; Roska, B. Optogenetic therapy for retinitis pigmentosa. Gene Ther. 2012, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.R.; Moore, A.T. Optogenetic approaches to therapy for inherited retinal degenerations. J. Physiol. 2022, 600, 4623–4632. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Duebel, J.; Balya, D.; Fradot, M.; Viney, T.J.; Siegert, S.; Groner, A.C.; Cabuy, E.; Forster, V.; Seeliger, M.; et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 2010, 329, 413–417. [Google Scholar] [CrossRef]
- Sakai, D.; Tomita, H.; Maeda, A. Optogenetic Therapy for Visual Restoration. Int. J. Mol. Sci. 2022, 23, 15041. [Google Scholar] [CrossRef]
- Parnami, K.; Bhattacharyya, A. Current approaches to vision restoration using optogenetic therapy. Front. Cell. Neurosci. 2023, 17, 1236826. [Google Scholar] [CrossRef]
- Prosseda, P.P.; Tran, M.; Kowal, T.; Wang, B.; Sun, Y. Advances in Ophthalmic Optogenetics: Approaches and Applications. Biomolecules 2022, 12, 269. [Google Scholar] [CrossRef]
- Yan, B.; Viswanathan, S.; Brodie, S.E.; Deng, W.-T.; Coleman, K.E.; Hauswirth, W.W.; Nirenberg, S. A clinically viable approach to restoring visual function using optogenetic gene therapy. Mol. Ther. Methods Clin. Dev. 2023, 29, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.H.; Tang, P.H.; De la Huerta, I.; Korot, E.; Muscat, S.B.; Palanker, D.A.; Williams, G.A. Stem cell therapies, gene-based therapies, optogenetics, and retinal prosthetics: Current State and Implications for the Future. Retina 2019, 39, 820–835. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, A.; Gordeliy, V.; Bamberg, E. Rhodopsin-Based Optogenetics: Basics and Applications. Methods Mol. Biol. 2022, 2501, 71–100. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.Y.; Han, X.; Dobry, A.S.; Qian, X.; Chuong, A.S.; Li, M.; Henninger, M.A.; Belfort, G.M.; Lin, Y.; Monahan, P.E.; et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 2010, 463, 98–102. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, S.; Flossmann, T.; Gao, S.; Witte, O.W.; Nagel, G.; Holthoff, K.; Kirmse, K. Optimized photo-stimulation of halorhodopsin for long-term neuronal inhibition. BMC Biol. 2019, 17, 95. [Google Scholar] [CrossRef]
- Shen, Y.; Campbell, R.E.; Côté, D.C.; Paquet, M.-E. Challenges for Therapeutic Applications of Opsin-Based Optogenetic Tools in Humans. Front. Neural Circuits 2020, 14, 41. [Google Scholar] [CrossRef]
- Yin, X.; He, T.; Chen, R.; Cui, H.; Li, G. Impact of neurotrophic factors combination therapy on retinitis pigmentosa. J. Int. Med. Res. 2020, 48, 0300060520967833. [Google Scholar] [CrossRef]
- Falsini, B.; Iarossi, G.; Chiaretti, A.; Ruggiero, A.; Manni, L.; Galli-Resta, L.; Corbo, G.; Abed, E. NGF eye-drops topical administration in patients with retinitis pigmentosa, a pilot study. J. Transl. Med. 2016, 14, 8. [Google Scholar] [CrossRef]
- Sieving, P.A.; Caruso, R.C.; Tao, W.; Coleman, H.R.; Thompson, D.J.S.; Fullmer, K.R.; Bush, R.A. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: Phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc. Natl. Acad. Sci. USA 2006, 103, 3896–3901. [Google Scholar] [CrossRef]
- Boia, R.; Ruzafa, N.; Aires, I.D.; Pereiro, X.; Ambrósio, A.F.; Vecino, E.; Santiago, A.R. Neuroprotective Strategies for Retinal Ganglion Cell Degeneration: Current Status and Challenges Ahead. Int. J. Mol. Sci. 2020, 21, 2262. [Google Scholar] [CrossRef]
- Ortega, J.T.; Jastrzebska, B. Neuroinflammation as a Therapeutic Target in Retinitis Pigmentosa and Quercetin as Its Potential Modulator. Pharmaceutics 2021, 13, 1935. [Google Scholar] [CrossRef] [PubMed]
- Komeima, K.; Rogers, B.S.; Campochiaro, P.A. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J. Cell. Physiol. 2007, 213, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Usui, S.; Zafar, A.-B.; Oveson, B.C.; Jo, Y.-J.; Lu, L.; Masoudi, S.; Campochiaro, P.A. N-acetylcysteine promotes long-term survival of cones in a model of retinitis pigmentosa. J. Cell. Physiol. 2010, 226, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Jaganathan, B.G. Stem Cell Therapy for Retinal Degeneration: The Evidence to Date. Biol. Targets Ther. 2021, 15, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, J.W.; Mahmoudzadeh, R.; Kuriyan, A.E. Cell-based therapies for retinal diseases: A review of clinical trials and direct to consumer “cell therapy” clinics. Stem Cell Res. Ther. 2021, 12, 538. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE/PROTEIN | FUNCTION | EFFECTS OF MUTATIONS | EFFECTS ON RETINA’S STRUCTURE |
|---|---|---|---|
| Rhodopsin (RHO) | Found in rod cells, plays a central role in phototransduction and rod photoreceptor cell health | Alteration of the protein’s structure/function, abnormalities in the protein’s folding or trafficking | Disruption and degeneration of rod photoreceptor cells, defects in phototransduction, reduced sensitivity to light |
| Peripherin/RDS (PRPH2) | Found in rod and cone cells, plays a crucial role in the structural integrity and organization of the photoreceptor outer segments (essential for disk morphogenesis) | Alteration of protein folding, stability, and interactions with other proteins | Alteration of integrity and function of the outer segments, progressive degeneration of the photoreceptor associated with peripheral vision loss, and central vision impairment |
| Cyclic Nucleotide-Gated (CNG) Channels | Nonselective cation channels located in the outer segment of rod and cone photoreceptor cells, involved in the regulation of ion influx in response to light stimulation | Impairs the normal function of these channels | Abnormalities in the phototransduction process, reduced sensitivity to light, decreased visual acuity, and progressive vision loss; dysfunctional CNG channels can lead to cellular stress, oxidative damage |
| Retinal Pigment Epithelium-Specific 65 kDa Protein (RPE65) | Component of the vitamin A visual cycle of the retina, which supplies the 11-cis retinal chromophore of the photoreceptors opsin visual pigments | Loss or dysfunction of the RPE65 protein, disrupting the visual cycle and impairing the regeneration of 11-cis-retinal | Progressive loss of photoreceptors reduced sensitivity to light, decreased visual acuity |
| Retinitis Pigmentosa GTPase Regulator (RPGR) | Predominantly localized in the connecting cilium and outer segment of photoreceptor cells in the retina, plays critical roles in the phototransduction cascade | Impairs the normal function | Impaired ciliary transport, altered protein–protein interactions, or disrupted signaling pathways, leading to photoreceptor cell death and vision loss |
| Cone-Rod Homeobox Protein (CRX) | Photoreceptor-specific transcription factor, which plays a role in the differentiation of photoreceptor cells; this homeodomain protein is necessary for the maintenance of normal cone and rod function | Disrupts the normal function | Impaired development and function of photoreceptor cells associated with degeneration of cones and rods |
| Usher Syndrome Genes (MYO7A, USH1C, USH2A, GPR98, CLRN1) | MYO7A encodes the protein myosin VIIA, involved in the development and maintenance of photoreceptor cells | Impairs the normal function | Impaired development and function of photoreceptor cells associated with degeneration |
| USH1C encodes a scaffold protein involved in the organization of hair cell stereocilia and synaptic connections in the retina | |||
| USH2A encodes a protein that contains laminin EGF motifs involved in the maintenance of the structure and function of photoreceptor cells (maintenance of periciliary membrane complex) | |||
| GPR98 encodes the protein ADGRV1 involved in the development of photoreceptors (maintenance of periciliary membrane complex) | |||
| CLRN1 encodes the protein Clarin 1, which plays an important role in the development and homeostasis of photoreceptor cells (regulatory element for the synapses within the retina) |
| ID | NAME | PHASE | AIM | METHODS |
|---|---|---|---|---|
| NCT02320812 | A Prospective, Multicenter, Open-Label, Single-Arm Study of the Safety and Tolerability of a Single, Intravitreal Injection of Human Retinal Progenitor Cells (jCell) in Adult Subjects With Retinitis Pigmentosa (RP) | 1/2 | Test the safety, tolerability, and efficacy (impact on visual status) of the administration of a single dose of jCell | Single intravitreal injection of 0.5–3.0 × 106 human retinal progenitor cells (hRPC-jCell) |
| NCT04925687 | Phase 1 Study of Intravitreal Autologous CD34+ Stem Cell Therapy for Retinitis Pigmentosa (BMSCRP1) | 1 | Determine the safety and feasibility of injection of autologous CD34+ stem cells harvested from bone marrow | Intravitreal injection of autologous CD34+ cells harvested from bone marrow under GMP conditions |
| NCT04763369 | Investigation of Therapeutic Efficacy and Safety of UMSCs for the Management of Retinitis Pigmentosa (RP) | 1/2 | Investigate the safety and therapeutic efficacy of umbilical cord-derived mesenchymal stem cell (UC-MSC) injection, employing two different routes (sub-tenon injection versus suprachoroidal injection) | Sub-tenon and suprachoroidal injection of UC-MSCs |
| NCT05909488 | Role of UC-MSC and CM to Inhibit Vision Loss in Retinitis Pigmentosa Phase I/II | 2/3 | Investigate the safety and therapeutic efficacy of peribulbar injection of umbilical cord-derived mesenchymal stem cell (UC-MSC) with conditioned medium (CM) | Peribulbar injection of 1.5–5 × 106 UC-MSC + CM |
| NCT03944239 | Safety and Efficacy of Subretinal Transplantation of Clinical Human Embryonic Stem Cell-Derived Retinal Pigment Epitheliums in Treatment of Retinitis Pigmentosa | 1/2 | Test the safety and therapeutic efficacy of clinical-level human embryonic stem cell-derived retinal pigment epithelium transplantation | Subretinal transplantation of clinical human embryonic stem cell-derived retinal pigment epitheliums |
| NCT01531348 | Intravitreal Injection of MSCs in Retinitis Pigmentosa | 1 | Determine feasibility and safety of adult human bone marrow-derived mesenchymal stem cells (BM-MSC) by intravitreal injection | Intravitreal injection of 1 × 106 BM-MSC in a balanced salt solution |
| NCT03073733 | Safety and Efficacy of Intravitreal Injection of Human Retinal Progenitor Cells in Adults With Retinitis Pigmentosa | 2 | Evaluation of safety and efficacy of intravitreal injection of human retinal progenitor (hRPC) | Intravitreal injection of 3.0–6.0 × 106 of human retinal progenitor cells (hRPC) suspended in clinical-grade medium |
| NCT04284293 | CNS10-NPC for the Treatment of RP | 1 | Assess the safety and tolerability of two escalating doses of clinical-grade human fetal cortical-derived neural progenitor cells (CNS10-NPC); determine if CNS10-NPC can engraft and survive long-term in the retina of transiently immunosuppressed subjects; obtain evidence that subretinal injection of CNS10-NPC can favorably impact the progression of vision loss in subjects with moderate RP | Human neural progenitor cell (CNS10-NPC) sub-retinal space implantation |
| NCT02709876 | Autologous Bone Marrow-Derived CD34+, CD133+, and CD271+ Stem Cell Transplantation for Retinitis Pigmentosa | 1/2 | Assess the safety and efficacy of purified adult autologous bone marrow-derived CD34+, CD133+, and CD271+ stem cells through a 48-month follow-up period. The combination of these three cell types was based on their diverse potentialities to differentiate into specific functional cell types to regenerate damaged retinal tissue | Intravitreal injection of bone marrow-derived CD34+, CD133+, CD271+ stem cells in 1.0 mL normal saline |
| NCT03963154 | Interventional Study of Implantation of hESC-derived RPE in Patients With RP Due to Monogenic Mutation | 1/2 | Study the safety, tolerability, and preliminary efficacy of implantation into one eye of human embryonic stem cell-derived retinal pigment epithelium (hESC-derived RPE)) | Implantation into one eye of human embryonic stem cell-derived retinal pigment epithelium (hESC-derived RPE) |
| NCT03566147 | Treatment of RP and LCA by Primary RPE Transplantation | Early 1 | Study the safety and preliminary efficacy of human primary retinal pigment epithelial (HuRPE) cells subretinal transplantation | Subretinal space transplantation of 0.3–1 × 106 HuRPE cells through a standard surgical approach |
| NCT03772938 | Stem Cells Therapy in Degenerative Diseases of the Retina | 1 | Investigation of the safety and efficacy of intravitreal injection of autologous bone marrow-isolated stem/progenitor cells with different selected phenotypes; this clinical trial was specially designed to test the therapeutic (pro-regenerative and neuro-protective) functions of different stem/progenitor cell populations able to secrete bioactive neurotrophic factors | Intravitreal injection of human autologous bone marrow-derived stem/progenitor cell |
| NCT05147701 | Safety of Cultured Allogeneic Adult Umbilical Cord-Derived Mesenchymal Stem Cells for Eye Diseases | 1 | Study the safety and efficacy of intravenous and sub-tenon delivery of cultured allogeneic adult umbilical cord-derived mesenchymal stem cells (UC-MSCs) | Intravenous and sub-tenon injection of 1 × 106 allogeneic adult umbilical cord-derived mesenchymal stem cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becherucci, V.; Bacci, G.M.; Marziali, E.; Sodi, A.; Bambi, F.; Caputo, R. The New Era of Therapeutic Strategies for the Treatment of Retinitis Pigmentosa: A Narrative Review of Pathomolecular Mechanisms for the Development of Cell-Based Therapies. Biomedicines 2023, 11, 2656. https://doi.org/10.3390/biomedicines11102656
Becherucci V, Bacci GM, Marziali E, Sodi A, Bambi F, Caputo R. The New Era of Therapeutic Strategies for the Treatment of Retinitis Pigmentosa: A Narrative Review of Pathomolecular Mechanisms for the Development of Cell-Based Therapies. Biomedicines. 2023; 11(10):2656. https://doi.org/10.3390/biomedicines11102656
Chicago/Turabian StyleBecherucci, Valentina, Giacomo Maria Bacci, Elisa Marziali, Andrea Sodi, Franco Bambi, and Roberto Caputo. 2023. "The New Era of Therapeutic Strategies for the Treatment of Retinitis Pigmentosa: A Narrative Review of Pathomolecular Mechanisms for the Development of Cell-Based Therapies" Biomedicines 11, no. 10: 2656. https://doi.org/10.3390/biomedicines11102656
APA StyleBecherucci, V., Bacci, G. M., Marziali, E., Sodi, A., Bambi, F., & Caputo, R. (2023). The New Era of Therapeutic Strategies for the Treatment of Retinitis Pigmentosa: A Narrative Review of Pathomolecular Mechanisms for the Development of Cell-Based Therapies. Biomedicines, 11(10), 2656. https://doi.org/10.3390/biomedicines11102656