
Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment



Abstract
1. Introduction
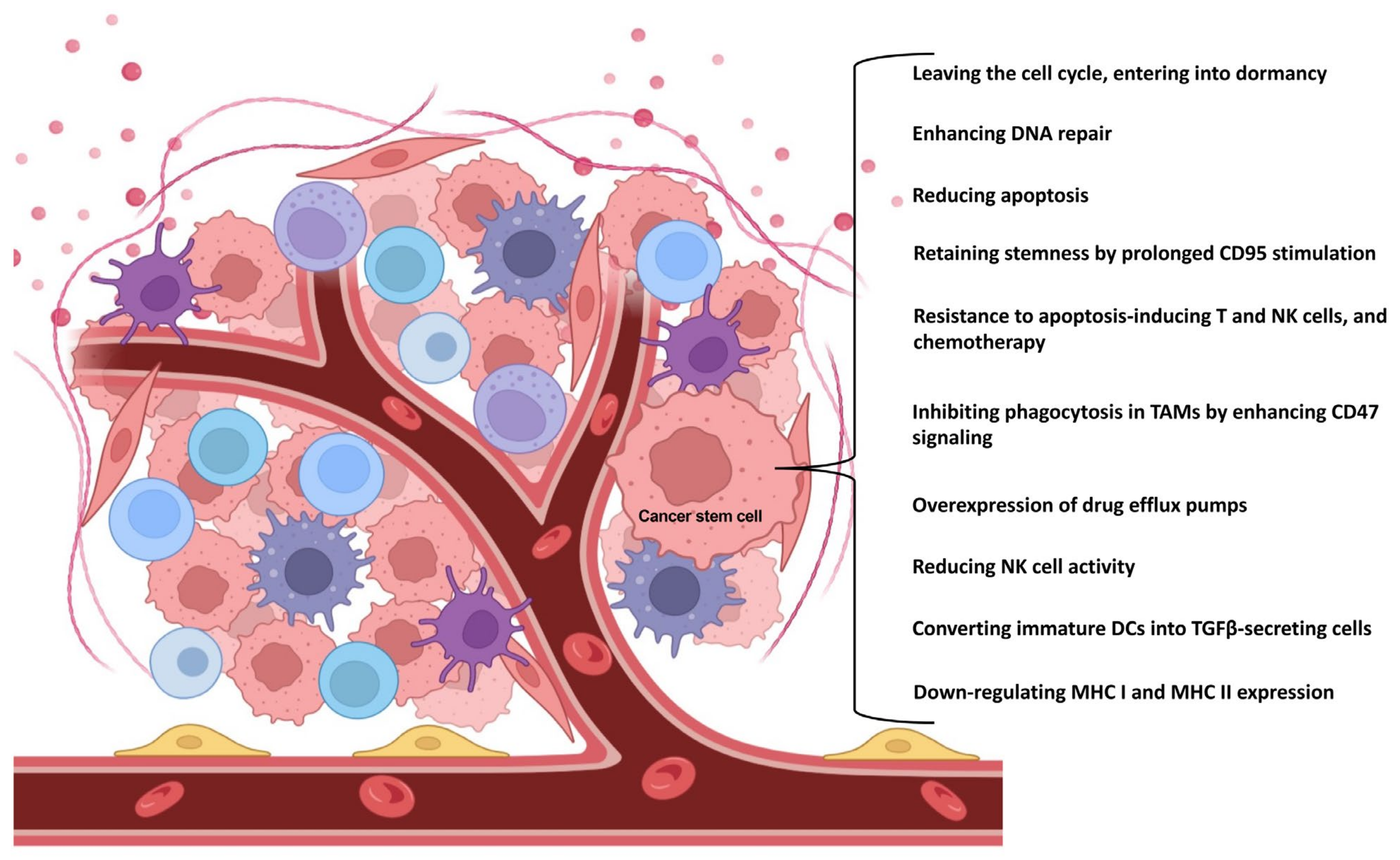
2. CSCs Influence Their Own Capabilities by Different Mechanisms
3. The Mutual Role of TME and CSCs in Immunomodulation and Stem Cell Niche Maintenance
4. The CSC-TME Crosstalk in Highly Inflammatory Cancers
5. Emergence of CSC Phenotype without TME
6. Utilization of the Inflammatory Process in Cancer Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, S.; Yang, X.; Wang, L.; Zhang, C. Interplay between inflammatory tumor microenvironment and cancer stem cells. Oncol. Lett. 2018, 16, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.; Noonan, D.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect. Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Lee-Theilen, M.; Fadini, D.D.; Hadhoud, J.R.; van Dongen, F.; Kroll, G.; Rolle, U.; Fiegel, H.C. Hepatoblastoma Cancer Stem Cells Express PD-L1, Reveal Plasticity and Can Emerge upon Chemotherapy. Cancers 2022, 14, 5825. [Google Scholar] [CrossRef] [PubMed]
- Gzil, A.; Zarębska, I.; Bursiewicz, W.; Antosik, P.; Grzanka, D.; Szylberg, Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol. Biol. Rep. 2019, 46, 6629–6645. [Google Scholar] [CrossRef]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Kise, K.; Kinugasa-Katayama, Y.; Takakura, N. Tumor microenvironment for cancer stem cells. Adv. Drug Deliv. Rev. 2016, 99 Pt B, 197–205. [Google Scholar] [CrossRef]
- Gasch, C.; Ffrench, B.; O’Leary, J.J.; Gallagher, M.F. Catching moving targets: Cancer stem cell hierarchies, therapy-resistance & considerations for clinical intervention. Mol. Cancer 2017, 16, 43. [Google Scholar]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95 (Suppl. 1), S2–S7. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [PubMed]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Blanpain, C.; Mohrin, M.; Sotiropoulou, P.A.; Passegué, E. DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell 2011, 8, 16–29. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef]
- Pastò, A.; Consonni, F.M.; Sica, A. Influence of Innate Immunity on Cancer Cell Stemness. Int. J. Mol. Sci. 2020, 21, 3352. [Google Scholar] [CrossRef]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef]
- Schatton, T.; Schütte, U.; Frank, N.Y.; Zhan, Q.; Hoerning, A.; Robles, S.C.; Zhou, J.; Hodi, F.S.; Spagnoli, G.C.; Murphy, G.F.; et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010, 70, 697–708. [Google Scholar] [CrossRef]
- Volonté, A.; Di Tomaso, T.; Spinelli, M.; Todaro, M.; Sanvito, F.; Albarello, L.; Bissolati, M.; Ghirardelli, L.; Orsenigo, E.; Ferrone, S.; et al. Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J. Immunol. 2014, 192, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Liu, Y.; Jiang, S.J.; Liu, Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, Q.; Wang, Z.; Jiang, J.; Yu, S.C.; Ping, Y.F.; Yang, J.; Xu, S.L.; Ye, X.Z.; Xu, C.; et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014, 74, 5746–5757. [Google Scholar] [CrossRef] [PubMed]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [PubMed]
- Drukker, M.; Katz, G.; Urbach, A.; Schuldiner, M.; Markel, G.; Itskovitz-Eldor, J.; Reubinoff, B.; Mandelboim, O.; Benvenisty, N. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9864–9869. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, L.; Yang, L.; Li, H.; Li, R.; Yu, J.; Yang, L.; Wei, F.; Yan, C.; Sun, Q.; et al. Anti-CD47 Antibody As a Targeted Therapeutic Agent for Human Lung Cancer and Cancer Stem Cells. Front. Immunol. 2017, 8, 404. [Google Scholar] [CrossRef]
- Cioffi, M.; Trabulo, S.; Hidalgo, M.; Costello, E.; Greenhalf, W.; Erkan, M.; Kleeff, J.; Sainz, B.; Heeschen, C., Jr. Inhibition of CD47 Effectively Targets Pancreatic Cancer Stem Cells via Dual Mechanisms. Clin. Cancer Res. 2015, 21, 2325–2337. [Google Scholar] [CrossRef]
- Lee, T.K.; Cheung, V.C.; Lu, P.; Lau, E.Y.; Ma, S.; Tang, K.H.; Tong, M.; Lo, J.; Ng, I.O. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology 2014, 60, 179–191. [Google Scholar] [CrossRef]
- Uger, R.; Johnson, L. Blockade of the CD47-SIRPα axis: A promising approach for cancer immunotherapy. Expert Opin. Biol. Ther. 2020, 20, 5–8. [Google Scholar] [CrossRef]
- Abdullah, L.N.; Chow, E.K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med. 2013, 2, 3. [Google Scholar] [CrossRef]
- Ni, J.; Cozzi, P.; Hao, J.; Duan, W.; Graham, P.; Kearsley, J.; Li, Y. Cancer stem cells in prostate cancer chemoresistance. Curr. Cancer Drug Targets 2014, 14, 225–240. [Google Scholar] [CrossRef]
- Qadir, A.S.; Ceppi, P.; Brockway, S.; Law, C.; Mu, L.; Khodarev, N.N.; Kim, J.; Zhao, J.C.; Putzbach, W.; Murmann, A.E.; et al. CD95/Fas Increases Stemness in Cancer Cells by Inducing a STAT1-Dependent Type I Interferon Response. Cell Rep. 2017, 18, 2373–2386. [Google Scholar] [CrossRef]
- Ceppi, P.; Hadji, A.; Kohlhapp, F.J.; Pattanayak, A.; Hau, A.; Liu, X.; Liu, H.; Murmann, A.E.; Peter, M.E. CD95 and CD95L promote and protect cancer stem cells. Nat. Commun. 2014, 5, 5238. [Google Scholar] [CrossRef]
- Relation, T.; Dominici, M.; Horwitz, E.M. Concise Review: An (Im)Penetrable Shield: How the Tumor Microenvironment Protects Cancer Stem Cells. Stem Cells 2017, 35, 1123–1130. [Google Scholar] [CrossRef]
- Ju, F.; Atyah, M.M.; Horstmann, N.; Gul, S.; Vago, R.; Bruns, C.J.; Zhao, Y.; Dong, Q.Z.; Ren, N. Characteristics of the cancer stem cell niche and therapeutic strategies. Stem Cell Res. Ther. 2022, 13, 233. [Google Scholar] [CrossRef]
- Maccalli, C.; Rasul, K.I.; Elawad, M.; Ferrone, S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin. Cancer Biol. 2018, 53, 189–200. [Google Scholar] [CrossRef]
- Shlush, L.I.; Chapal-Ilani, N.; Adar, R.; Pery, N.; Maruvka, Y.; Spiro, A.; Shouval, R.; Rowe, J.M.; Tzukerman, M.; Bercovich, D.; et al. Cell lineage analysis of acute leukemia relapse uncovers the role of replication-rate heterogeneity and microsatellite instability. Blood 2012, 120, 603–612. [Google Scholar] [CrossRef]
- Vahidian, F.; Duijf, P.H.G.; Safarzadeh, E.; Derakhshani, A.; Baghbanzadeh, A.; Baradaran, B. Interactions between cancer stem cells, immune system and some environmental components: Friends or foes? Immunol. Lett. 2019, 208, 19–29. [Google Scholar] [CrossRef]
- De Vlaeminck, Y.; González-Rascón, A.; Goyvaerts, C.; Breckpot, K. Cancer-Associated Myeloid Regulatory Cells. Front. Immunol. 2016, 7, 113. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Guarneri, V.; Gennari, A. Myelopoiesis, metabolism and therapy: A crucial crossroads in cancer progression. Cell Stress 2019, 3, 284–294. [Google Scholar] [CrossRef]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef]
- Ueha, S.; Shand, F.H.; Matsushima, K. Myeloid cell population dynamics in healthy and tumor-bearing mice. Int. Immunopharmacol. 2011, 11, 783–788. [Google Scholar] [CrossRef]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef]
- Ravindran, S.; Rasool, S.; Maccalli, C. The Cross Talk between Cancer Stem Cells/Cancer Initiating Cells and Tumor Microenvironment: The Missing Piece of the Puzzle for the Efficient Targeting of these Cells with Immunotherapy. Cancer Microenviron. 2019, 12, 133–148. [Google Scholar] [CrossRef]
- Lau, E.Y.; Ho, N.P.; Lee, T.K. Cancer Stem Cells and Their Microenvironment: Biology and Therapeutic Implications. Stem Cells Int. 2017, 2017, 3714190. [Google Scholar] [CrossRef]
- Inamoto, S.; Itatani, Y.; Yamamoto, T.; Minamiguchi, S.; Hirai, H.; Iwamoto, M.; Hasegawa, S.; Taketo, M.M.; Sakai, Y.; Kawada, K. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin. Cancer Res. 2016, 22, 492–501. [Google Scholar] [CrossRef]
- Panni, R.Z.; Sanford, D.E.; Belt, B.A.; Mitchem, J.B.; Worley, L.A.; Goetz, B.D.; Mukherjee, P.; Wang-Gillam, A.; Link, D.C.; Denardo, D.G.; et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic Cancer. Cancer Immunol. Immunother. 2014, 63, 513–528. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, K.; Tian, J.; Xia, X.; Ma, J.; Tang, X.; Xu, H.; Wang, S. Granulocytic Myeloid-Derived Suppressor Cells Promote the Stemness of Colorectal Cancer Cells through Exosomal S100A9. Adv. Sci. 2019, 6, 1901278. [Google Scholar] [CrossRef]
- Kuroda, H.; Mabuchi, S.; Yokoi, E.; Komura, N.; Kozasa, K.; Matsumoto, Y.; Kawano, M.; Takahashi, R.; Sasano, T.; Shimura, K.; et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical Cancer. Oncotarget 2018, 9, 36317–36330. [Google Scholar] [CrossRef]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef]
- Sainz BJr Martín, B.; Tatari, M.; Heeschen, C.; Guerra, S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014, 74, 7309–7320. [Google Scholar] [CrossRef]
- Chen, R.H.; Du, Y.; Han, P.; Wang, H.B.; Liang, F.Y.; Feng, G.K.; Zhou, A.J.; Cai, M.Y.; Zhong, Q.; Zeng, M.S.; et al. ISG15 predicts poor prognosis and promotes cancer stem cell phenotype in nasopharyngeal carcinoma. Oncotarget 2016, 7, 16910–16922. [Google Scholar] [CrossRef]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Elaimy, A.L.; Guru, S.; Chang, C.; Ou, J.; Amante, J.J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M. VEGF-neuropilin-2 signaling promotes stem-like traits in breast cancer cells by TAZ-mediated repression of the Rac GAP β2-chimaerin. Sci. Signal. 2018, 11, eaao6897. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, A.M. VEGF/Neuropilin Signaling in Cancer Stem Cells. Int. J. Mol. Sci. 2019, 20, 490. [Google Scholar] [CrossRef] [PubMed]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef]
- Whiteside, T.L. Regulatory T cell subsets in human cancer: Are they regulating for or against tumor progression? Cancer Immunol. Immunother. 2014, 63, 67–72. [Google Scholar] [CrossRef]
- Arneth, B. Tumor Microenvironment. Medicina (Kaunas) 2019, 56, 15. [Google Scholar] [CrossRef]
- Hirahara, K.; Poholek, A.; Vahedi, G.; Laurence, A.; Kanno, Y.; Milner, J.D.; O’Shea, J.J. Mechanisms underlying helper T-cell plasticity: Implications for immune-mediated disease. J. Allergy Clin. Immunol. 2013, 131, 1276–1287. [Google Scholar] [CrossRef]
- Liang, J.; Bi, G.; Shan, G.; Jin, X.; Bian, Y.; Wang, Q. Tumor-Associated Regulatory T Cells in Non-Small-Cell Lung Cancer: Current Advances and Future Perspectives. J. Immunol. Res. 2022, 2022, 4355386. [Google Scholar] [CrossRef]
- Peck, A.; Mellins, E.D. Plasticity of T-cell phenotype and function: The T helper type 17 example. Immunology 2010, 129, 147–153. [Google Scholar] [CrossRef]
- Cuiffo, B.G.; Campagne, A.; Bell, G.W.; Lembo, A.; Orso, F.; Lien, E.C.; Bhasin, M.K.; Raimo, M.; Hanson, S.E.; Marusyk, A.; et al. MSC-regulated microRNAs converge on the transcription factor FOXP2 and promote breast cancer metastasis. Cell Stem Cell 2014, 15, 762–774. [Google Scholar] [CrossRef]
- Gwak, J.M.; Kim, H.J.; Kim, E.J.; Chung, Y.R.; Yun, S.; Seo, A.N.; Lee, H.J.; Park, S.Y. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast Cancer. Breast Cancer Res. Treat. 2014, 147, 39–49. [Google Scholar] [CrossRef]
- Frisbie, L.; Buckanovich, R.J.; Coffman, L. Carcinoma-Associated Mesenchymal Stem/Stromal Cells: Architects of the Pro-tumorigenic Tumor Microenvironment. Stem Cells 2022, 40, 705–715. [Google Scholar] [CrossRef]
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; van Amersfoort, M.; et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 2011, 20, 370–383. [Google Scholar] [CrossRef]
- van der Velden, D.L.; Cirkel, G.A.; Houthuijzen, J.M.; van Werkhoven, E.; Roodhart, J.M.L.; Daenen, L.G.M.; Kaing, S.; Gerrits, J.; Verhoeven-Duif, N.M.; Grootscholten, C.; et al. Phase I study of combined indomethacin and platinum-based chemotherapy to reduce platinum-induced fatty acids. Cancer Chemother. Pharmacol. 2018, 81, 911–921. [Google Scholar] [CrossRef]
- Mallampati, S.; Leng, X.; Ma, H.; Zeng, J.; Li, J.; Wang, H.; Lin, K.; Lu, Y.; Yang, Y.; Sun, B.; et al. Tyrosine kinase inhibitors induce mesenchymal stem cell-mediated resistance in BCR-ABL+ acute lymphoblastic leukemia. Blood 2015, 125, 2968–2973. [Google Scholar] [CrossRef]
- Xue, B.Z.; Xiang, W.; Zhang, Q.; Wang, H.F.; Zhou, Y.J.; Tian, H.; Abdelmaksou, A.; Xue, J.; Sun, M.X.; Yi, D.Y.; et al. CD90low glioma-associated mesenchymal stromal/stem cells promote temozolomide resistance by activating FOXS1-mediated epithelial-mesenchymal transition in glioma cells. Stem Cell Res. Ther. 2021, 12, 394. [Google Scholar]
- Eliason, S.; Hong, L.; Sweat, Y.; Chalkley, C.; Cao, H.; Liu, Q.; Qi, H.; Xu, H.; Zhan, F.; Amendt, B.A. Extracellular vesicle expansion of PMIS-miR-210 expression inhibits colorectal tumour growth via apoptosis and an XIST/NME1 regulatory mechanism. Clin. Transl. Med. 2022, 12, e1037. [Google Scholar] [CrossRef]
- Ji, R.; Zhang, B.; Zhang, X.; Xue, J.; Yuan, X.; Yan, Y.; Wang, M.; Zhu, W.; Qian, H.; Xu, W. Exosomes derived from human mesenchymal stem cells confer drug resistance in gastric Cancer. Cell Cycle 2015, 14, 2473–2483. [Google Scholar] [CrossRef]
- Naito, Y.; Yoshioka, Y.; Ochiya, T. Intercellular crosstalk between cancer cells and cancer-associated fibroblasts via extracellular vesicles. Cancer Cell Int. 2022, 22, 367. [Google Scholar] [CrossRef]
- Kondo, R.; Sakamoto, N.; Harada, K.; Hashimoto, H.; Morisue, R.; Yanagihara, K.; Kinoshita, T.; Kojima, M.; Ishii, G. Cancer-associated fibroblast-dependent and -independent invasion of gastric cancer cells. J. Cancer Res. Clin. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Chen, Y.J.; Lin, C.Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.; Lo, J.; Cheng, B.Y.; Ma, M.K.; Lee, J.M.; Ng, J.K.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.S.; Hsu, C.C.; Pai, V.C.; Liao, W.Y.; Huang, S.S.; Tan, K.T.; Yen, C.J.; Hsu, S.C.; Chen, W.Y.; Shan, Y.S.; et al. Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J. Exp. Med. 2016, 213, 2967–2988. [Google Scholar] [CrossRef] [PubMed]
- Wilczyński, J.R.; Wilczyński, M.; Paradowska, E. Cancer Stem Cells in Ovarian Cancer-A Source of Tumor Success and a Challenging Target for Novel Therapies. Int. J. Mol. Sci. 2022, 23, 2496. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Zhang, Q.; Cai, D.J.; Li, B. Ovarian cancer stem-like cells elicit the polarization of M2 macrophages. Mol. Med. Rep. 2015, 11, 4685–4693. [Google Scholar] [CrossRef]
- Nusblat, L.M.; Carroll, M.J.; Roth, C.M. Crosstalk between M2 macrophages and glioma stem cells. Cell. Oncol. 2017, 40, 471–482. [Google Scholar] [CrossRef]
- Kokubu, Y.; Tabu, K.; Muramatsu, N.; Wang, W.; Murota, Y.; Nobuhisa, I.; Jinushi, M.; Taga, T. Induction of protumoral CD11c(high) macrophages by glioma cancer stem cells through GM-CSF. Genes Cells 2016, 21, 241–251. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Xie, Y.; Lei, Y.L.; Mehta, G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother. Cancer 2019, 7, 190. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef]
- Domenis, R.; Cesselli, D.; Toffoletto, B.; Bourkoula, E.; Caponnetto, F.; Manini, I.; Beltrami, A.P.; Ius, T.; Skrap, M.; Di Loreto, C.; et al. Systemic T Cells Immunosuppression of Glioma Stem Cell-Derived Exosomes Is Mediated by Monocytic Myeloid-Derived Suppressor Cells. PLoS ONE 2017, 12, e0169932. [Google Scholar] [CrossRef]
- Biswas, S.; Mandal, G.; Roy Chowdhury, S.; Purohit, S.; Payne, K.K.; Anadon, C.; Gupta, A.; Swanson, P.; Yu, X.; Conejo-Garcia, J.R.; et al. Exosomes Produced by Mesenchymal Stem Cells Drive Differentiation of Myeloid Cells into Immunosuppressive M2-Polarized Macrophages in Breast Cancer. J. Immunol. 2019, 203, 3447–3460. [Google Scholar] [CrossRef]
- Grange, C.; Tapparo, M.; Tritta, S.; Deregibus, M.C.; Battaglia, A.; Gontero, P.; Frea, B.; Camussi, G. Role of HLA-G and extracellular vesicles in renal cancer stem cell-induced inhibition of dendritic cell differentiation. BMC Cancer 2015, 15, 1009. [Google Scholar] [CrossRef]
- Hwang, W.L.; Lan, H.Y.; Cheng, W.C.; Huang, S.C.; Yang, M.H. Tumor stem-like cell-derived exosomal RNAs prime neutrophils for facilitating tumorigenesis of colon Cancer. J. Hematol. Oncol. 2019, 12, 10. [Google Scholar] [CrossRef]
- Anselmi, M.; Fontana, F.; Marzagalli, M.; Gagliano, N.; Sommariva, M.; Limonta, P. Melanoma Stem Cells Educate Neutrophils to Support Cancer Progression. Cancers 2022, 14, 3391. [Google Scholar] [CrossRef]
- Műzes, G.; Sipos, F. Mesenchymal Stem Cell-Derived Secretome: A Potential Therapeutic Option for Autoimmune and Immune-Mediated Inflammatory Diseases. Cells 2022, 11, 2300. [Google Scholar] [CrossRef]
- Alzahrani, F.A.; El-Magd, M.A.; Abdelfattah-Hassan, A.; Saleh, A.A.; Saadeldin, I.M.; El-Shetry, E.S.; Badawy, A.A.; Alkarim, S. Potential Effect of Exosomes Derived from Cancer Stem Cells and MSCs on Progression of DEN-Induced HCC in Rats. Stem Cells Int. 2018, 2018, 8058979. [Google Scholar] [CrossRef]
- Sun, Z.P.; Li, A.Q.; Jia, W.H.; Ye, S.; Van Eps, G.; Yu, J.M.; Yang, W.J. MicroRNA expression profiling in exosomes derived from gastric cancer stem-like cells. Oncotarget 2017, 8, 93839–93855. [Google Scholar] [CrossRef]
- Grange, C.; Tapparo, M.; Collino, F.; Vitillo, L.; Damasco, C.; Deregibus, M.C.; Tetta, C.; Bussolati, B.; Camussi, G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011, 7, 5346–5356. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, G.; Zhao, D.; Wang, J.; Bai, Y.; Peng, Q.; Wang, H.; Fang, R.; Chen, G.; Wang, Z.; et al. CD103-positive CSC exosome promotes EMT of clear cell renal cell carcinoma: Role of remote MiR-19b-3p. Mol. Cancer 2019, 18, 86. [Google Scholar] [CrossRef] [PubMed]
- Hardin, H.; Helein, H.; Meyer, K.; Robertson, S.; Zhang, R.; Zhong, W.; Lloyd, R.V. Thyroid cancer stem-like cell exosomes: Regulation of EMT via transfer of lncRNAs. Lab. Investig. 2018, 98, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Fröse, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Benezra, R.; Iavarone, A. The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Heidarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control. Re-lease 2018, 288, 62–83. [Google Scholar] [CrossRef]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell. Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef]
- Ji, C.; Yang, L.; Yi, W.; Xiang, D.; Wang, Y.; Zhou, Z.; Qian, F.; Ren, Y.; Cui, W.; Zhang, X.; et al. Capillary morphogenesis gene 2 maintains gastric cancer stem-like cell phenotype by activating a Wnt/β-catenin pathway. Oncogene 2018, 37, 3953–3966. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Tiwari, A.; Trivedi, R.; Lin, S.Y. Tumor microenvironment: Barrier or opportunity towards effective cancer therapy. J. Biomed. Sci. 2022, 29, 83. [Google Scholar] [CrossRef]
- Kordes, C.; Häussinger, D. Hepatic stem cell niches. J Clin Invest 2013, 123, 1874–1880. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, L.; Ge, D.; Tan, L.; Cao, B.; Fan, H.; Xue, L. Exosomal O-GlcNAc transferase from esophageal carcinoma stem cell promotes cancer immunosuppression through up-regulation of PD-1 in CD8+ T cells. Cancer Lett. 2021, 500, 98–106. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Ariyama, H.; Stancikova, J.; Sakitani, K.; Asfaha, S.; Renz, B.W.; Dubeykovskaya, Z.A.; Shibata, W.; Wang, H.; Westphalen, C.B.; et al. Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 2015, 28, 800–814. [Google Scholar] [CrossRef]
- Sphyris, N.; Hodder, M.C.; Sansom, O.J. Subversion of Niche-Signalling Pathways in Colorectal Cancer: What Makes and Breaks the Intestinal Stem Cell. Cancers 2021, 13, 1000. [Google Scholar] [CrossRef]
- Li, Y.; Tang, T.; Lee, H.J.; Song, K. Selective Anti-Cancer Effects of Plasma-Activated Medium and Its High Efficacy with Cisplatin on Hepatocellular Carcinoma with Cancer Stem Cell Characteristics. Int. J. Mol. Sci. 2021, 22, 3956. [Google Scholar] [CrossRef]
- Nimmakayala, R.K.; Leon, F.; Rachagani, S.; Rauth, S.; Nallasamy, P.; Marimuthu, S.; Shailendra, G.K.; Chhonker, Y.S.; Chugh, S.; Chirravuri, R.; et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene 2021, 40, 215–231. [Google Scholar] [CrossRef]
- Lin, X.; Chen, W.; Wei, F.; Xie, X. TV-circRGPD6 Nanoparticle Suppresses Breast Cancer Stem Cell-Mediated Metastasis via the miR-26b/YAF2 Axis. Mol. Ther. 2021, 29, 244–262. [Google Scholar] [CrossRef]
- Jain, S.; Annett, S.L.; Morgan, M.P.; Robson, T. The Cancer Stem Cell Niche in Ovarian Cancer and Its Impact on Immune Surveillance. Int. J. Mol. Sci. 2021, 22, 4091. [Google Scholar] [CrossRef]
- Hagiwara, M.; Yasumizu, Y.; Yamashita, N.; Rajabi, H.; Fushimi, A.; Long, M.D.; Li, W.; Bhattacharya, A.; Ahmad, R.; Oya, M.; et al. MUC1-C Activates the BAF (mSWI/SNF) Complex in Prostate Cancer Stem Cells. Cancer Res. 2021, 81, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Fendler, A.; Bauer, D.; Busch, J.; Jung, K.; Wulf-Goldenberg, A.; Kunz, S.; Song, K.; Myszczyszyn, A.; Elezkurtaj, S.; Erguen, B.; et al. Inhibiting WNT and NOTCH in renal cancer stem cells and the implications for human patients. Nat. Commun. 2020, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal Cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Billet, S.; Choudhury, D.; Cheng, R.; Haldar, S.; Fernandez, A.; Biondi, S.; Liu, Z.; Zhou, H.; Bhowmick, N.A. Bone marrow mesenchymal stem cells interact with head and neck squamous cell carcinoma cells to promote cancer progression and drug resistance. Neoplasia 2021, 23, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Raniszewska, A.; Vroman, H.; Dumoulin, D.; Cornelissen, R.; Aerts, J.G.J.V.; Domagała-Kulawik, J. PD-L1+ lung cancer stem cells modify the metastatic lymph-node immunomicroenvironment in nsclc patients. Cancer Immunol. Immunother. 2021, 70, 453–461. [Google Scholar] [CrossRef]
- Hsu, M.Y.; Yang, M.H.; Schnegg, C.I.; Hwang, S.; Ryu, B.; Alani, R.M. Notch3 signaling-mediated melanoma-endothelial crosstalk regulates melanoma stem-like cell homeostasis and niche morphogenesis. Lab. Investig. 2017, 97, 725–736. [Google Scholar] [CrossRef]
- Lacina, L.; Plzak, J.; Kodet, O.; Szabo, P.; Chovanec, M.; Dvorankova, B.; Smetana, K., Jr. Cancer Microenvironment: What Can We Learn from the Stem Cell Niche. Int. J. Mol. Sci. 2015, 16, 24094–24110. [Google Scholar] [CrossRef]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Tabu, K.; Taga, T. Cancer ego-system in glioma: An iron-replenishing niche network systemically self-organized by cancer stem cells. Inflamm. Regen. 2022, 42, 54. [Google Scholar] [CrossRef]
- Kim, M.; Jo, K.W.; Kim, H.; Han, M.E.; Oh, S.O. Genetic heterogeneity of liver cancer stem cells. Anat. Cell Biol. 2022. [Google Scholar] [CrossRef]
- Pedersen, R.K.; Andersen, M.; Skov, V.; Kjær, L.; Hasselbalch, H.C.; Ottesen, J.T.; Stiehl, T. HSC niche dynamics in regeneration, pre-malignancy and cancer: Insights from mathematical modeling. Stem Cells 2022, sxac079. [Google Scholar] [CrossRef]
- Akindona, F.A.; Frederico, S.C.; Hancock, J.C.; Gilbert, M.R. Exploring the origin of the cancer stem cell niche and its role in anti-angiogenic treatment for glioblastoma. Front. Oncol. 2022, 12, 947634. [Google Scholar] [CrossRef]
- Luo, S.; Yang, G.; Ye, P.; Cao, N.; Chi, X.; Yang, W.H.; Yan, X. Macrophages Are a Double-Edged Sword: Molecular Crosstalk between Tumor-Associated Macrophages and Cancer Stem Cells. Biomolecules 2022, 12, 850. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Nakamura, T.; Sakai, K.; Nakamura, T.; Matsumoto, K. Hepatocyte growth factor twenty years on: Much more than a growth factor. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. 1), 188–202. [Google Scholar] [CrossRef]
- Lau, C.K.; Yang, Z.F.; Ho, D.W.; Ng, M.N.; Yeoh, G.C.; Poon, R.T.; Fan, S.T. An Akt/hypoxia-inducible factor-1alpha/platelet-derived growth factor-BB autocrine loop mediates hypoxia-induced chemoresistance in liver cancer cells and tumorigenic hepatic progenitor cells. Clin. Cancer Res. 2009, 15, 3462–3471. [Google Scholar] [CrossRef]
- Wang, X.; Sun, W.; Shen, W.; Xia, M.; Chen, C.; Xiang, D.; Ning, B.; Cui, X.; Li, H.; Li, X.; et al. Long non-coding RNA DILC regulates liver cancer stem cells via IL-6/STAT3 axis. J. Hepatol. 2016, 64, 1283–1294. [Google Scholar] [CrossRef]
- Liu, G.; Yang, Z.F.; Sun, J.; Sun, B.Y.; Zhou, P.Y.; Zhou, C.; Guan, R.Y.; Wang, Z.T.; Yi, Y.; Qiu, S.J. The LINC00152/miR-205-5p/CXCL11 axis in hepatocellular carcinoma cancer-associated fibroblasts affects cancer cell phenotypes and tumor growth. Cell Oncol. 2022, 45, 1435–1449. [Google Scholar] [CrossRef]
- Huang, H.; Hu, M.; Li, P.; Lu, C.; Li, M. Mir-152 inhibits cell proliferation and colony formation of CD133(+) liver cancer stem cells by targeting KIT. Tumor Biol. 2015, 36, 921–928. [Google Scholar] [CrossRef]
- Sukowati, C.H.; Tiribelli, C. The biological implication of cancer stem cells in hepatocellular carcinoma: A possible target for future therapy. Expert Rev. Gastroenterol. Hepatol. 2013, 7, 749–757. [Google Scholar] [CrossRef]
- Singh, S.R. Gastric cancer stem cells: A novel therapeutic target. Cancer Lett. 2013, 338, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Xu, X.Y. Epithelial-mesenchymal transition and gastric cancer stem cell. Tumor Biol. 2017, 39, 1010428317698373. [Google Scholar] [CrossRef] [PubMed]
- Becerril-Rico, J.; Alvarado-Ortiz, E.; Toledo-Guzmán, M.E.; Pelayo, R.; Ortiz-Sánchez, E. The cross talk between gastric cancer stem cells and the immune microenvironment: A tumor-promoting factor. Stem Cell Res. Ther. 2021, 12, 498. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal Cancer. Oncotarget 2017, 9, 33403–33415. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, E.G.; Nasreddin, N.; Valbuena, G.N.; Mulholland, E.J.; Belnoue-Davis, H.L.; Eggington, H.R.; Schenck, R.O.; Wouters, V.M.; Wirapati, P.; Gilroy, K.; et al. Dynamic and adaptive cancer stem cell population admixture in colorectal neoplasia. Cell Stem Cell 2022, 29, 1213–1228.e8. [Google Scholar] [CrossRef]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef]
- Martin, M.L.; Zeng, Z.; Adileh, M.; Jacobo, A.; Li, C.; Vakiani, E.; Hua, G.; Zhang, L.; Haimovitz-Friedman, A.; Fuks, Z.; et al. Logarithmic expansion of LGR5+ cells in human colorectal Cancer. Cell Signal. 2018, 42, 97–105. [Google Scholar] [CrossRef]
- Iwaya, M.; Ota, H.; Nakajima, T.; Uehara, T.; Riddell, R.; Conner, J. Most colitis associated carcinomas lack expression of LGR5: A preliminary study with implications for unique pathways of carcinogenesis compared to sporadic colorectal carcinoma. BMC Cancer 2021, 21, 119. [Google Scholar] [CrossRef]
- Yaeger, R.; Shah, M.A.; Miller, V.A.; Kelsen, J.R.; Wang, K.; Heins, Z.J.; Ross, J.S.; He, Y.; Sanford, E.; Yantiss, R.K.; et al. Genomic Alterations Observed in Colitis-Associated Cancers Are Distinct From Those Found in Sporadic Colorectal Cancers and Vary by Type of Inflammatory Bowel Disease. Gastroenterology 2016, 151, 278–287.e6. [Google Scholar] [CrossRef]
- Klingler, S.; Hsu, K.S.; Hua, G.; Martin, M.L.; Adileh, M.; Baslan, T.; Zhang, Z.; Paty, P.B.; Fuks, Z.; Brown, A.M.C.; et al. Disruption of the crypt niche promotes outgrowth of mutated colorectal tumor stem cells. J. Clin. Investig. Insight. 2022, 7, e153793. [Google Scholar] [CrossRef]
- Han, H.; Davidson, L.A.; Fan, Y.Y.; Goldsby, J.S.; Yoon, G.; Jin, U.H.; Wright, G.A.; Landrock, K.K.; Weeks, B.R.; Wright, R.C.; et al. Loss of aryl hydrocarbon receptor potentiates FoxM1 signaling to enhance self-renewal of colonic stem and progenitor cells. EMBO J. 2020, 39, e104319. [Google Scholar] [CrossRef]
- Jeong, Y.J.; Oh, H.K.; Park, S.H.; Bong, J.G. Association between inflammation and cancer stem cell phenotype in breast Cancer. Oncol. Lett. 2018, 15, 2380–2386. [Google Scholar] [CrossRef]
- Guo, W. Concise review: Breast cancer stem cells: Regulatory networks, stem cell niches, and disease relevance. Stem Cells Transl. Med. 2014, 3, 942–948. [Google Scholar] [CrossRef]
- Proia, T.A.; Keller, P.J.; Gupta, P.B.; Klebba, I.; Jones, A.D.; Sedic, M.; Gilmore, H.; Tung, N.; Naber, S.P.; Schnitt, S.; et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 2011, 8, 149–163. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Li, X.Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA 2012, 109, 16654–16659. [Google Scholar] [CrossRef]
- Tiwari, N.; Tiwari, V.K.; Waldmeier, L.; Balwierz, P.J.; Arnold, P.; Pachkov, M.; Meyer-Schaller, N.; Schübeler, D.; van Nimwegen, E.; Christofori, G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 2013, 23, 768–783. [Google Scholar] [CrossRef]
- Chang, C.J.; Yang, J.Y.; Xia, W.; Chen, C.T.; Xie, X.; Chao, C.H.; Woodward, W.A.; Hsu, J.M.; Hortobagyi, G.N.; Hung, M.C. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-β-catenin signaling. Cancer Cell 2011, 19, 86–100. [Google Scholar] [CrossRef]
- Műzes, G.; Bohusné Barta, B.; Szabó, O.; Horgas, V.; Sipos, F. Cell-Free DNA in the Pathogenesis and Therapy of Non-Infectious Inflammations and Tumors. Biomedicines 2022, 10, 2853. [Google Scholar] [CrossRef]
- Sipos, F.; Kiss, A.L.; Constantinovits, M.; Tulassay, Z.; Műzes, G. Modified Genomic Self-DNA Influences In Vitro Survival of HT29 Tumor Cells via TLR9- and Autophagy Signaling. Pathol. Oncol. Res. 2019, 25, 1505–1517. [Google Scholar] [CrossRef]
- Sipos, F.; Bohusné Barta, B.; Simon, Á.; Nagy, L.; Dankó, T.; Raffay, R.E.; Petővári, G.; Zsiros, V.; Wichmann, B.; Sebestyén, A.; et al. Survival of HT29 Cancer Cells Is Affected by IGF1R Inhibition via Modulation of Self-DNA-Triggered TLR9 Signaling and the Autophagy Response. Pathol. Oncol. Res. 2022, 28, 1610322. [Google Scholar] [CrossRef]
- Qi, Y.; Zou, H.; Zhao, X.; Kapeleris, J.; Monteiro, M.; Li, F.; Xu, Z.P.; Deng, Y.; Wu, Y.; Tang, Y.; et al. Inhibition of colon cancer K-RasG13D mutation reduces cancer cell proliferation but promotes stemness and inflammation via RAS/ERK pathway. Front. Pharmacol. 2022, 13, 996053. [Google Scholar] [CrossRef] [PubMed]
- Muraro, M.G.; Mele, V.; Däster, S.; Han, J.; Heberer, M.; Cesare Spagnoli, G.; Iezzi, G. CD133+, CD166+CD44+, and CD24+CD44+ phenotypes fail to reliably identify cell populations with cancer stem cell functional features in established human colorectal cancer cell lines. Stem Cells Transl. Med. 2012, 1, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Karami Fath, M.; Ebrahimi, M.; Nourbakhsh, E.; Zia Hazara, A.; Mirzaei, A.; Shafieyari, S.; Salehi, A.; Hoseinzadeh, M.; Payandeh, Z.; Barati, G. PI3K/Akt/mTOR signaling pathway in cancer stem cells. Pathol. Res. Pract. 2022, 237, 154010. [Google Scholar] [CrossRef] [PubMed]
- Nengroo, M.A.; Verma, A.; Datta, D. Cytokine chemokine network in tumor microenvironment: Impact on CSC properties and therapeutic applications. Cytokine 2022, 156, 155916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, D.G.; Rycaj, K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin Cancer Biol. 2018, 52, 94–106. [Google Scholar] [CrossRef]
- Mortezaee, K. Human hepatocellular carcinoma: Protection by melatonin. J. Cell. Physiol. 2018, 233, 6486–6508. [Google Scholar] [CrossRef]
- Zappasodi, R.; Budhu, S.; Hellmann, M.D.; Postow, M.A.; Senbabaoglu, Y.; Manne, S.; Gasmi, B.; Liu, C.; Zhong, H.; Li, Y.; et al. Non-conventional Inhibitory CD4+Foxp3-PD-1hi T Cells as a Biomarker of Immune Checkpoint Blockade Activity. Cancer Cell 2018, 33, 1017–1032.e7. [Google Scholar] [CrossRef]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416–432. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Y.; Meng, L.; Li, L.; Beatson, R.; Deng, J.; Zhang, T.; Liu, J.; Han, X. Epigenetic Signaling of Cancer Stem Cells During Inflammation. Front. Cell Dev. Biol. 2021, 9, 772211. [Google Scholar] [CrossRef]
- Pang, X.; Tang, Y.L.; Liang, X.H. Transforming growth factor-β signaling in head and neck squamous cell carcinoma: Insights into cellular responses. Oncol. Lett. 2018, 16, 4799–4806. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Alraouji, N.N.; Al-Mohanna, F.H.; Ghebeh, H.; Arafah, M.; Almeer, R.; Al-Tweigeri, T.; Aboussekhra, A. Tocilizumab potentiates cisplatin cytotoxicity and targets cancer stem cells in triple-negative breast Cancer. Mol. Carcinog. 2020, 59, 1041–1051. [Google Scholar] [CrossRef]
- Sun, L.; Mathews, L.A.; Cabarcas, S.M.; Zhang, X.; Yang, A.; Zhang, Y.; Young, M.R.; Klarmann, K.D.; Keller, J.R.; Farrar, W.L. Epigenetic regulation of SOX9 by the NF-κB signaling pathway in pancreatic cancer stem cells. Stem Cells 2013, 31, 1454–1466. [Google Scholar] [CrossRef]
- Loh, J.J.; Ma, S. The Role of Cancer-Associated Fibroblast as a Dynamic Player in Mediating Cancer Stemness in the Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 727640. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate Cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal Cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar]
- Ferrer-Mayorga, G.; Gómez-López, G.; Barbáchano, A.; Fernández-Barral, A.; Peña, C.; Pisano, D.G.; Cantero, R.; Rojo, F.; Muñoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal Cancer. Gut 2017, 66, 1449–1462. [Google Scholar] [CrossRef]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; deSouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic Cancer. Nat. Commun. 2020, 11, 4841. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Galbo, P.M., Jr.; Zang, X.; Zheng, D. Molecular Features of Cancer-associated Fibroblast Subtypes and their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clin. Cancer Res. 2021, 27, 2636–2647. [Google Scholar] [CrossRef]
- Ribatti, D.; Solimando, A.G.; Pezzella, F. The Anti-VEGF(R) Drug Discovery Legacy: Improving Attrition Rates by Breaking the Vicious Cycle of Angiogenesis in Cancer. Cancers 2021, 13, 3433. [Google Scholar] [CrossRef] [PubMed]
- Saygin, C.; Matei, D.; Majeti, R.; Reizes, O.; Lathia, J.D. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell 2019, 24, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, S.J.; Sargent, D.J. Genomic advances and their impact on clinical trial design. Genome Med. 2009, 1, 69. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Deng, Y.; Li, Z.; Chen, Y.; Zhu, X.; Tan, X.; Cao, G. Cancer Evo-Dev: A Theory of Inflammation-Induced Oncogenesis. Front. Immunol. 2021, 12, 768098. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cancer Stem Cells | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lung | Breast | Gastric | Colorectal | Liver | Pancreas | Melanoma | Glioblastoma | Prostate | Acute Myeloid Leukemia | Chronic Myeloid Leukemia | ||
| Surface CD markers | CD15 | + | ||||||||||
| CD24 | + | + | + | + | + | |||||||
| CD25 | + | + | ||||||||||
| CD26 | + | |||||||||||
| CD29 | + | |||||||||||
| CD33 | + | + | ||||||||||
| CD36 | + | |||||||||||
| CD44 (and variants) | + | + | + | + | + | + | + | + | ||||
| CD49f | + | |||||||||||
| CD61 | + | |||||||||||
| CD70 | + | |||||||||||
| CD87 | + | |||||||||||
| CD90 | + | + | + | + | + | |||||||
| CD117 | + | + | ||||||||||
| CD123 | + | + | ||||||||||
| CD133 | + | + | + | + | + | + | + | + | + | |||
| CD166 | + | |||||||||||
| CD271 | + | |||||||||||
| CD274 | + | |||||||||||
| Other surface markers | EpCAM | + | + | + | + | + | + | |||||
| CXCR4 | + | + | + | |||||||||
| Lgr5 | + | + | + | |||||||||
| ProC-R | + | |||||||||||
| LINGO2 | + | |||||||||||
| CLL-1 | + | |||||||||||
| TIM3 | + | |||||||||||
| IL1RAP | + | |||||||||||
| cell surface vimentin | + | |||||||||||
| OV-6 | + | |||||||||||
| ESA | + | |||||||||||
| DCLK1 | + | |||||||||||
| ABCB1 | + | |||||||||||
| CHL1 | + | |||||||||||
| TACSTD2 | + | |||||||||||
| Intracellular markers | ALDH | + | + | + | + | + | + | + | + | |||
| Nanog | + | + | + | + | + | + | + | + | ||||
| Oct-3/4 | + | + | + | + | + | + | + | + | ||||
| BMI-1 | + | |||||||||||
| SOX2 | + | + | + | + | + | + | + | + | ||||
| Letm1 | + | + | ||||||||||
| Musashi2 | + | |||||||||||
| FOXO | + | |||||||||||
| Sall4 | + | + | ||||||||||
| AFP | + | |||||||||||
| KLF4 | + | |||||||||||
| NES | + | |||||||||||
| TGM2 | + | |||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sipos, F.; Műzes, G. Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment. Biomedicines 2023, 11, 189. https://doi.org/10.3390/biomedicines11010189
Sipos F, Műzes G. Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment. Biomedicines. 2023; 11(1):189. https://doi.org/10.3390/biomedicines11010189
Chicago/Turabian StyleSipos, Ferenc, and Györgyi Műzes. 2023. "Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment" Biomedicines 11, no. 1: 189. https://doi.org/10.3390/biomedicines11010189
APA StyleSipos, F., & Műzes, G. (2023). Cancer Stem Cell Relationship with Pro-Tumoral Inflammatory Microenvironment. Biomedicines, 11(1), 189. https://doi.org/10.3390/biomedicines11010189