
Wnt/β-Catenin Signaling and Resistance to Immune Checkpoint Inhibitors: From Non-Small-Cell Lung Cancer to Other Cancers

 ,
,
Abstract
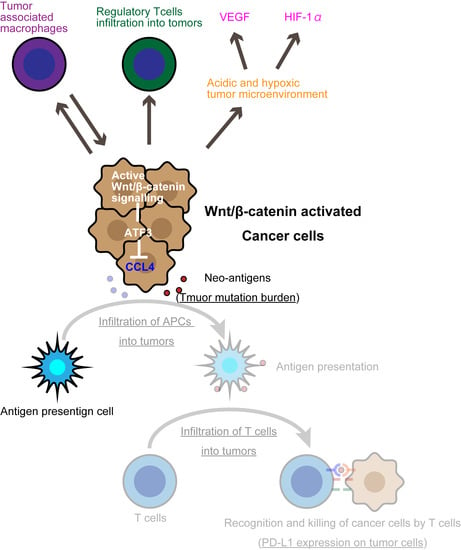
1. Introduction
2. Wnt Pathways in Cancer
2.1. Canonical Wnt Pathway
2.2. Non-Canonical Wnt Pathways
2.3. The Wnt/β-Catenin Pathway and Cancer
3. The Wnt/β-Catenin Pathway Is a Tumor-Intrinsic Mechanism of Resistance to Immune Checkpoint Inhibitors
3.1. Tumor-Intrinsic Mechanisms of Immune-Checkpoint-Inhibitor Resistance
3.2. Wnt/β-Catenin Induces T-Cell Exclusion
3.3. The Wnt/β-Catenin Pathway and Immunoediting in Cancer
4. Overcoming Immune-Checkpoint-Inhibitor Resistance with Wnt/β-Catenin Signaling
4.1. Combination Therapy with Chemotherapy and Immune Checkpoint Inhibitors
4.2. Neoadjuvant Treatment Strategies
4.3. Combination Therapy with Inhibitors of Wnt/β-Catenin Signaling and Immune Checkpoint Inhibitors
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Goldstraw, P.; Chansky, K.; Crowley, J.; Rami-Porta, R.; Asamura, H.; Eberhardt, W.E.E.; Nicholson, A.G.; Groome, P.; Mitchell, A.; Bolejack, V.; et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016, 11, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Gettinger, S.; Vokes, E.E.; Chow, L.Q.M.; Burgio, M.A.; de Castro Carpeno, J.; Pluzanski, A.; Arrieta, O.; Frontera, O.A.; Chiari, R.; et al. Five-Year Outcomes from the Randomized, Phase III Trials CheckMate 017 and 057: Nivolumab Versus Docetaxel in Previously Treated Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2021, 39, 723–733. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1–Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.-G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Barlesi, F.; West, H.; Ball, S.; Bordoni, R.; Cobo, M.; Longeras, P.D.; Goldschmidt, J., Jr.; Novello, S.; Orlandi, F.; et al. Atezolizumab Plus Chemotherapy for First-Line Treatment of Nonsquamous NSCLC: Results from the Randomized Phase 3 IMpower132 Trial. J. Thorac. Oncol. 2021, 16, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Ciuleanu, T.-E.; Cobo, M.; Schenker, M.; Zurawski, B.; Menezes, J.; Richardet, E.; Bennouna, J.; Felip, E.; Juan-Vidal, O.; et al. First-line nivolumab plus ipilimumab combined with two cycles of chemotherapy in patients with non-small-cell lung cancer (CheckMate 9LA): An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.L.; Cho, B.C.; Luft, A.; Alatorre-Alexander, J.; Geater, S.L.; Laktionov, K.; Kim, S.-W.; Ursol, G.; Hussein, M.; Lim, F.L.; et al. Durvalumab with or without Tremelimumab in Combination with Chemotherapy as First-Line Therapy for Metastatic Non–Small-Cell Lung Cancer: The Phase III POSEIDON Study. J. Clin. Oncol. 2022; ahead of print. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef]
- Galluzzi, L.; Spranger, S.; Fuchs, E.; López-Soto, A. WNT Signaling in Cancer Immunosurveillance. Trends Cell Biol. 2019, 29, 44–65. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Goldsberry, W.N.; Londoño, A.; Randall, T.D.; Norian, L.A.; Arend, R.C. A Review of the Role of Wnt in Cancer Immunomodulation. Cancers 2019, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, M.; Jho, E.-h. Wnt/β-catenin signalling: From plasma membrane to nucleus. Biochem. J. 2013, 450, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.; Luis, T.C.; Tiemessen, M.M. WNT signalling in the immune system: WNT is spreading its wings. Nat. Rev. Immunol. 2008, 8, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Ji, Y.; Restifo, N.P. Wnt/β-Catenin Signaling in T-Cell Immunity and Cancer Immunotherapy. Clin. Cancer Res. 2010, 16, 4695–4701. [Google Scholar] [CrossRef]
- Kusserow, A.; Pang, K.; Sturm, C.; Hrouda, M.; Lentfer, J.; Schmidt, H.A.; Technau, U.; von Haeseler, A.; Hobmayer, B.; Martindale, M.Q.; et al. Unexpected complexity of the Wnt gene family in a sea anemone. Nature 2005, 433, 156–160. [Google Scholar] [CrossRef]
- Du, S.J.; Purcell, S.M.; Christian, J.L.; McGrew, L.L.; Moon, R.T. Identification of distinct classes and functional domains of Wnts through expression of wild-type and chimeric proteins in Xenopus embryos. Mol. Cell Biol. 1995, 15, 2625–2634. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Masuda, T.; Ishitani, T. Context-dependent regulation of the β-catenin transcriptional complex supports diverse functions of Wnt/β-catenin signaling. J. Biochem. 2017, 161, 9–17. [Google Scholar] [CrossRef]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, Z.; Yang, P.; Zhang, L.; Fan, Y.; Li, Z. PKM2 depletion induces the compensation of glutaminolysis through β-catenin/c-Myc pathway in tumor cells. Cell. Signal. 2014, 26, 2397–2405. [Google Scholar] [CrossRef]
- Corda, G.; Sala, A. Non-canonical WNT/PCP signalling in cancer: Fzd6 takes centre stage. Oncogenesis 2017, 6, e364. [Google Scholar] [CrossRef]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, L.; Cui, B.; Chuang, H.-Y.; Yu, J.; Wang-Rodriguez, J.; Tang, L.; Chen, G.; Basak, G.W.; Kipps, T.J. ROR1 Is Expressed in Human Breast Cancer and Associated with Enhanced Tumor-Cell Growth. PLoS ONE 2012, 7, e31127. [Google Scholar] [CrossRef]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. 2011, 43, 745. [Google Scholar] [CrossRef]
- Rijsewijk, F.; van Deemter, L.; Wagenaar, E.; Sonnenberg, A.; Nusse, R. Transfection of the int-1 mammary oncogene in cuboidal RAC mammary cell line results in morphological transformation and tumorigenicity. EMBO J. 1987, 6, 127–131. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef]
- Nishisho, I.; Nakamura, Y.; Miyoshi, Y.; Miki, Y.; Ando, H.; Horii, A.; Koyama, K.; Utsunomiya, J.; Baba, S.; Hedge, P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 1991, 253, 665–669. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dong, X.; Mai, M.; Seelan, R.S.; Taniguchi, K.; Krishnadath, K.K.; Halling, K.C.; Cunningham, J.M.; Boardman, L.A.; Qian, C.; et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat. Genet. 2000, 26, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Guezguez, B.; Almakadi, M.; Benoit, Y.D.; Shapovalova, Z.; Rahmig, S.; Fiebig-Comyn, A.; Casado, F.L.; Tanasijevic, B.; Bresolin, S.; Masetti, R.; et al. GSK3 Deficiencies in Hematopoietic Stem Cells Initiate Pre-neoplastic State that Is Predictive of Clinical Outcomes of Human Acute Leukemia. Cancer Cell 2016, 29, 61–74. [Google Scholar] [CrossRef]
- Kim, Y.; Jin, D.; Lee, B.B.; Cho, E.Y.; Han, J.; Shim, Y.M.; Kim, H.K.; Kim, D.-H. Overexpression of β-Catenin and Cyclin D1 is Associated with Poor Overall Survival in Patients with Stage IA–IIA Squamous Cell Lung Cancer Irrespective of Adjuvant Chemotherapy. J. Thorac. Oncol. 2016, 11, 2193–2201. [Google Scholar] [CrossRef]
- Bodnar, L.; Stanczak, A.; Cierniak, S.; Smoter, M.; Cichowicz, M.; Kozlowski, W.; Szczylik, C.; Wieczorek, M.; Lamparska-Przybysz, M. Wnt/β-catenin pathway as a potential prognostic and predictive marker in patients with advanced ovarian cancer. J. Ovarian Res. 2014, 7, 16. [Google Scholar] [CrossRef]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, G.; Zhang, H.; Zhang, F.; Zhou, B.; Ning, F.; Wang, H.-S.; Cai, S.-H.; Du, J. Acquisition of epithelial–mesenchymal transition phenotype and cancer stem cell-like properties in cisplatin-resistant lung cancer cells through AKT/β-catenin/Snail signaling pathway. Eur. J. Pharmacol. 2014, 723, 156–166. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Menon, A.G.; Morreau, H.; Tollenaar, R.A.E.M.; Alphenaar, E.; van Puijenbroek, M.; Putter, H.; Janssen-van Rhijn, C.M.; van de Velde, C.J.H.; Fleuren, G.J.; Kuppen, P.J.K. Down-Regulation of HLA-A Expression Correlates with a Better Prognosis in Colorectal Cancer Patients. Lab. Investig. 2002, 82, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Becker, C.; Benner, A.; Woerner, S.M.; Gebert, J.; Ferrone, S.; von Knebel Doeberitz, M. Immunoselective Pressure and Human Leukocyte Antigen Class I Antigen Machinery Defects in Microsatellite Unstable Colorectal Cancers. Cancer Res. 2005, 65, 6418–6424. [Google Scholar] [CrossRef]
- Meissner, M.; Reichert, T.E.; Kunkel, M.; Gooding, W.; Whiteside, T.L.; Ferrone, S.; Seliger, B. Defects in the Human Leukocyte Antigen Class I Antigen Processing Machinery in Head and Neck Squamous Cell Carcinoma: Association with Clinical Outcome. Clin. Cancer Res. 2005, 11, 2552–2560. [Google Scholar] [CrossRef]
- Sucker, A.; Zhao, F.; Real, B.; Heeke, C.; Bielefeld, N.; Maβen, S.; Horn, S.; Moll, I.; Maltaner, R.; Horn, P.A.; et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin. Cancer Res. 2014, 20, 6593–6604. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef]
- Montesion, M.; Murugesan, K.; Jin, D.X.; Sharaf, R.; Sanchez, N.; Guria, A.; Minker, M.; Li, G.; Fisher, V.; Sokol, E.S.; et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discov. 2021, 11, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e9. [Google Scholar] [CrossRef] [PubMed]
- Sucker, A.; Zhao, F.; Pieper, N.; Heeke, C.; Maltaner, R.; Stadtler, N.; Real, B.; Bielefeld, N.; Howe, S.; Weide, B.; et al. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat. Commun. 2017, 8, 15440. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Leonardelli, S.; Sucker, A.; Schadendorf, D.; Griewank, K.G.; Paschen, A. Tumor CDKN2A-Associated JAK2 Loss and Susceptibility to Immunotherapy Resistance. J. Natl. Cancer Inst. 2018, 110, 677–681. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of essential genes for cancer immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef]
- Han, P.; Dai, Q.; Fan, L.; Lin, H.; Zhang, X.; Li, F.; Yang, X. Genome-Wide CRISPR Screening Identifies JAK1 Deficiency as a Mechanism of T-Cell Resistance. Front. Immunol. 2019, 10, 251. [Google Scholar] [CrossRef]
- Cha, Y.J.; Kim, H.R.; Lee, C.Y.; Cho, B.C.; Shim, H.S. Clinicopathological and prognostic significance of programmed cell death ligand-1 expression in lung adenocarcinoma and its relationship with p53 status. Lung Cancer 2016, 97, 73–80. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.E.; Hanke, M.L.; Kargl, J.; Metz, H.E.; MacPherson, D.; Houghton, A.M. Lung Cancer Subtypes Generate Unique Immune Responses. J. Immunol. 2016, 197, 4493–4503. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non–small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937. [Google Scholar] [CrossRef]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef]
- Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; Le Moulec, S.; et al. Hyperprogressive Disease in Patients with Advanced Non–Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol. 2018, 4, 1543–1552. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Yang, J.C.; Yu, H.; Kim, S.W.; Saka, H.; Horn, L.; Goto, K.; Ohe, Y.; Mann, H.; Thress, K.S.; et al. TATTON: A multi-arm, phase Ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in EGFR-mutant lung cancer. Ann. Oncol. 2020, 31, 507–516. [Google Scholar] [CrossRef]
- Pereira, F.; Ferreira, A.; Reis, C.A.; Sousa, M.J.; Oliveira, M.J.; Preto, A. KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications. Cells 2022, 11, 398. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]
- Kumagai, S.; Koyama, S.; Itahashi, K.; Tanegashima, T.; Lin, Y.T.; Togashi, Y.; Kamada, T.; Irie, T.; Okumura, G.; Kono, H.; et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell 2022, 40, 201–218.e9. [Google Scholar] [CrossRef]
- Pinyol, R.; Sia, D.; Llovet, J.M. Immune Exclusion-Wnt/CTNNB1 Class Predicts Resistance to Immunotherapies in HCC. Clin. Cancer Res. 2019, 25, 2021–2023. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and Comparative Genomic Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef]
- Sweis, R.F.; Spranger, S.; Bao, R.; Paner, G.P.; Stadler, W.M.; Steinberg, G.; Gajewski, T.F. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer Immunol. Res. 2016, 4, 563–568. [Google Scholar] [CrossRef]
- Sridharan, V.; Gjini, E.; Liao, X.; Chau, N.G.; Haddad, R.I.; Severgnini, M.; Hammerman, P.; El-Naggar, A.; Freeman, G.J.; Hodi, F.S.; et al. Immune Profiling of Adenoid Cystic Carcinoma: PD-L2 Expression and Associations with Tumor-Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 679–687. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938.e20. [Google Scholar] [CrossRef]
- Muto, S.; Ozaki, Y.; Yamaguchi, H.; Mine, H.; Takagi, H.; Watanabe, M.; Inoue, T.; Yamaura, T.; Fukuhara, M.; Okabe, N.; et al. Tumor β-catenin expression is associated with immune evasion in non-small cell lung cancer with high tumor mutation burden. Oncol. Lett. 2021, 21, 203. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Tanegashima, T.; Sato, E.; Irie, T.; Sai, A.; Itahashi, K.; Kumagai, S.; Tada, Y.; Togashi, Y.; Koyama, S.; et al. Highly immunogenic cancer cells require activation of the WNT pathway for immunological escape. Sci. Immunol. 2021, 6, eabc6424. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; El-Gamil, M.; Li, Y.F.; Kawakami, Y.; Loftus, D.; Appella, E.; Rosenberg, S.A. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 1996, 183, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Activating Mutations in β-Catenin in Colon Cancer Cells Alter Their Interaction with Macrophages; the Role of Snail. PLoS ONE 2012, 7, e45462. [Google Scholar] [CrossRef]
- Hong, Y.; Manoharan, I.; Suryawanshi, A.; Majumdar, T.; Angus-Hill, M.L.; Koni, P.A.; Manicassamy, B.; Mellor, A.L.; Munn, D.H.; Manicassamy, S. β-Catenin Promotes Regulatory T-cell Responses in Tumors by Inducing Vitamin A Metabolism in Dendritic Cells. Cancer Res. 2015, 75, 656–665. [Google Scholar] [CrossRef]
- Van Loosdregt, J.; Fleskens, V.; Tiemessen, M.M.; Mokry, M.; van Boxtel, R.; Meerding, J.; Pals, C.E.; Kurek, D.; Baert, M.R.; Delemarre, E.M.; et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity 2013, 39, 298–310. [Google Scholar] [CrossRef]
- Yaguchi, T.; Goto, Y.; Kido, K.; Mochimaru, H.; Sakurai, T.; Tsukamoto, N.; Kudo-Saito, C.; Fujita, T.; Sumimoto, H.; Kawakami, Y. Immune Suppression and Resistance Mediated by Constitutive Activation of Wnt/β-Catenin Signaling in Human Melanoma Cells. J. Immunol. 2012, 189, 2110. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9, e55185. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Ortenberg, R.; Varanasi, S.K.; Mangalhara, K.C.; Mardamshina, M.; Markovits, E.; Baruch, E.N.; Tripple, V.; Arama-Chayoth, M.; Greenberg, E.; et al. Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell 2019, 179, 236–250.e18. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Till, B.; Gao, Q. Chemotherapeutic agent-mediated elimination of myeloid-derived suppressor cells. Oncoimmunology 2017, 6, e1331807. [Google Scholar] [CrossRef]
- Roselli, M.; Cereda, V.; di Bari, M.G.; Formica, V.; Spila, A.; Jochems, C.; Farsaci, B.; Donahue, R.; Gulley, J.L.; Schlom, J.; et al. Effects of conventional therapeutic interventions on the number and function of regulatory T cells. Oncoimmunology 2013, 2, e27025. [Google Scholar] [CrossRef]
- Kalbasi, A.; June, C.H.; Haas, N.; Vapiwala, N. Radiation and immunotherapy: A synergistic combination. J. Clin. Investig. 2013, 123, 2756–2763. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Daguenet, E.; Louati, S.; Wozny, A.S.; Vial, N.; Gras, M.; Guy, J.B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magné, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef]
- De Castro, G.; Kudaba, I.; Wu, Y.-L.; Lopes, G.; Kowalski, D.M.; Turna, H.Z.; Caglevic, C.; Zhang, L.; Karaszewska, B.; Laktionov, K.K.; et al. Five-Year Outcomes with Pembrolizumab Versus Chemotherapy as First-Line Therapy in Patients with Non–Small-Cell Lung Cancer and Programmed Death Ligand-1 Tumor Proportion Score ≥ 1% in the KEYNOTE-042 Study. J. Clin. Oncol. 2022; ahead of print. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Five-Year Outcomes with Pembrolizumab Versus Chemotherapy for Metastatic Non–Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥50%. J. Clin. Oncol. 2021, 39, 2339–2349. [Google Scholar] [CrossRef]
- Rodríguez-Abreu, D.; Powell, S.F.; Hochmair, M.J.; Gadgeel, S.; Esteban, E.; Felip, E.; Speranza, G.; De Angelis, F.; Dómine, M.; Cheng, S.Y.; et al. Pemetrexed plus platinum with or without pembrolizumab in patients with previously untreated metastatic nonsquamous NSCLC: Protocol-specified final analysis from KEYNOTE-189. Ann. Oncol. 2021, 32, 881–895. [Google Scholar] [CrossRef]
- West, H.J.; McCleland, M.; Cappuzzo, F.; Reck, M.; Mok, T.S.K.; Jotte, R.M.; Nishio, M.; Kim, E.; Morris, S.; Zou, W.; et al. Clinical efficacy of atezolizumab plus bevacizumab and chemotherapy in KRAS-mutated non-small cell lung cancer with STK11, KEAP1, or TP53 comutations: Subgroup results from the phase III IMpower150 trial. J. Immunother. Cancer 2022, 10, e003027. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Smits, R.; Hao, H.; He, C. Wnt/β-Catenin Signaling in Liver Cancers. Cancers 2019, 11, 926. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Abbas, A.R.; de Galarreta, M.R.; Guan, Y.; Lu, S.; Koeppen, H.; Zhang, W.; Hsu, C.-H.; He, A.R.; Ryoo, B.-Y.; et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat. Med. 2022, 28, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Spicer, J.; Lu, S.; Provencio, M.; Mitsudomi, T.; Awad, M.M.; Felip, E.; Broderick, S.R.; Brahmer, J.R.; Swanson, S.J.; et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N. Engl. J. Med. 2022, 386, 1973–1985. [Google Scholar] [CrossRef] [PubMed]
- Provencio, M.; Nadal, E.; Insa, A.; García-Campelo, M.R.; Casal-Rubio, J.; Dómine, M.; Majem, M.; Rodríguez-Abreu, D.; Martínez-Martí, A.; De Castro Carpeño, J.; et al. Neoadjuvant chemotherapy and nivolumab in resectable non-small-cell lung cancer (NADIM): An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Wnt is back in drugmakers’ sights, but is it druggable? Nat. Biotechnol. 2018, 36, 1028–1029. [Google Scholar] [CrossRef]
- Ganesh, S.; Shui, X.; Craig, K.P.; Park, J.; Wang, W.; Brown, B.D.; Abrams, M.T. RNAi-Mediated β-Catenin Inhibition Promotes T Cell Infiltration and Antitumor Activity in Combination with Immune Checkpoint Blockade. Mol. Ther. 2018, 26, 2567–2579. [Google Scholar] [CrossRef]
- Ganesh, S.; Koser, M.L.; Cyr, W.A.; Chopda, G.R.; Tao, J.; Shui, X.; Ying, B.; Chen, D.; Pandya, P.; Chipumuro, E.; et al. Direct pharmacological inhibition of β-catenin by RNA interference in tumors of diverse origin. Mol. Cancer Ther. 2016, 15, 2143–2154. [Google Scholar] [CrossRef]
- Deldar Abad Paskeh, M.; Mirzaei, S.; Ashrafizadeh, M.; Zarrabi, A.; Sethi, G. Wnt/β-Catenin Signaling as a Driver of Hepatocellular Carcinoma Progression: An Emphasis on Molecular Pathways. J. Hepatocell. Carcinoma 2021, 8, 1415–1444. [Google Scholar] [CrossRef]
- Liu, Y.; Qi, X.; Donnelly, L.; Elghobashi-Meinhardt, N.; Long, T.; Zhou, R.W.; Sun, Y.; Wang, B.; Li, X. Mechanisms and inhibition of Porcupine-mediated Wnt acylation. Nature 2022, 607, 816–822. [Google Scholar] [CrossRef]
- Janku, F.; de Vos, F.; de Miguel, M.; Forde, P.; Ribas, A.; Nagasaka, M.; Argiles, G.; Arance, A.M.; Calvo, A.; Giannakis, M.; et al. Abstract CT034: Phase I study of WNT974 + spartalizumab in patients (pts) with advanced solid tumors. Cancer Res. 2020, 80, CT034. [Google Scholar] [CrossRef]
- Kagey, M.H.; He, X. Rationale for targeting the Wnt signalling modulator Dickkopf-1 for oncology. Br. J. Pharmacol. 2017, 174, 4637–4650. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.S.; Kagey, M.H.; Heath, H.; Schuerpf, F.; Rottman, J.B.; Newman, W. mDKN-01, a Novel Anti-DKK1 mAb, Enhances Innate Immune Responses in the Tumor Microenvironment. Mol. Cancer Res. 2021, 19, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Klempner, S.J.; Bendell, J.C.; Villaflor, V.M.; Tenner, L.L.; Stein, S.M.; Rottman, J.B.; Naik, G.S.; Sirard, C.A.; Kagey, M.H.; Chaney, M.F.; et al. Safety, Efficacy, and Biomarker Results from a Phase Ib Study of the Anti-DKK1 Antibody DKN-01 in Combination with Pembrolizumab in Advanced Esophagogastric Cancers. Mol. Cancer Ther. 2021, 20, 2240–2249. [Google Scholar] [CrossRef]

{kind=link}
| Gene | Genetic Aberration | Resulting Immune Editing | Reference |
|---|---|---|---|
| EGFR | Mutation | PD-L1↑, Tregs | [70,73] |
| Myeloid cells↑ | [72] | ||
| KRAS | Mutation | Myeloid cells↑, CD8 T cells↑, Tregs↑ | [72] |
| MYC | Amplification | Macrophages↑, NK cells↓, T cells↓ | [78] |
| Tregs↑, Lactic acid↑ | [79] | ||
| STK11/LKB1 | Loss | Neutrophils↑, T cells↓, PD-L1↓ | [80,81] |
| β-catenin target genes | Amplification | DC↓ | [82] |
| Drug | Target | ICI Agent | Cancer Type | Trial Phase | Clinical Trial |
|---|---|---|---|---|---|
| LGK974 | PORCN | PDR001 | Solid tumors | I | NCT01351103 |
| ETC-1922159 | PORCN | Pembrolizumab | Solid tumors | I | NCT02521844 |
| CGX1321 | PORCN | Pembrolizumab | Solid/GI tumors | I | NCT02675946 |
| RXC004 | PORCN | Nivolumab | Solid tumors | I | NCT03447470 |
| RXC004 | PORCN | Nivolumab | Colorectal cancer | II | NCT04907539 |
| DKN-01 | DKK1 | Pembrolizumab | Esophagogastric cancer | I | NCT02013154 |
| DKN-01 | DKK1 | Nivolumab | Biliary tract cancer | II | NCT04057365 |
| DKN-01 | DKK1 | Atezolizumab | Esophagogastric cancer | I/II | NCT04166721 |
| DKN-01 | DKK1 | Tiselizumab | Esophagogastric cancer | II | NCT04363801 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muto, S.; Enta, A.; Maruya, Y.; Inomata, S.; Yamaguchi, H.; Mine, H.; Takagi, H.; Ozaki, Y.; Watanabe, M.; Inoue, T.; et al. Wnt/β-Catenin Signaling and Resistance to Immune Checkpoint Inhibitors: From Non-Small-Cell Lung Cancer to Other Cancers. Biomedicines 2023, 11, 190. https://doi.org/10.3390/biomedicines11010190
Muto S, Enta A, Maruya Y, Inomata S, Yamaguchi H, Mine H, Takagi H, Ozaki Y, Watanabe M, Inoue T, et al. Wnt/β-Catenin Signaling and Resistance to Immune Checkpoint Inhibitors: From Non-Small-Cell Lung Cancer to Other Cancers. Biomedicines. 2023; 11(1):190. https://doi.org/10.3390/biomedicines11010190
Chicago/Turabian StyleMuto, Satoshi, Akio Enta, Yoshiyuki Maruya, Sho Inomata, Hikaru Yamaguchi, Hayato Mine, Hironori Takagi, Yuki Ozaki, Masayuki Watanabe, Takuya Inoue, and et al. 2023. "Wnt/β-Catenin Signaling and Resistance to Immune Checkpoint Inhibitors: From Non-Small-Cell Lung Cancer to Other Cancers" Biomedicines 11, no. 1: 190. https://doi.org/10.3390/biomedicines11010190
APA StyleMuto, S., Enta, A., Maruya, Y., Inomata, S., Yamaguchi, H., Mine, H., Takagi, H., Ozaki, Y., Watanabe, M., Inoue, T., Yamaura, T., Fukuhara, M., Okabe, N., Matsumura, Y., Hasegawa, T., Osugi, J., Hoshino, M., Higuchi, M., Shio, Y., ... Suzuki, H. (2023). Wnt/β-Catenin Signaling and Resistance to Immune Checkpoint Inhibitors: From Non-Small-Cell Lung Cancer to Other Cancers. Biomedicines, 11(1), 190. https://doi.org/10.3390/biomedicines11010190