
Immunomodulatory Effects of Non-Thermal Plasma in a Model for Latent HIV-1 Infection: Implications for an HIV-1-Specific Immunotherapy

 , ,
, ,  and
and 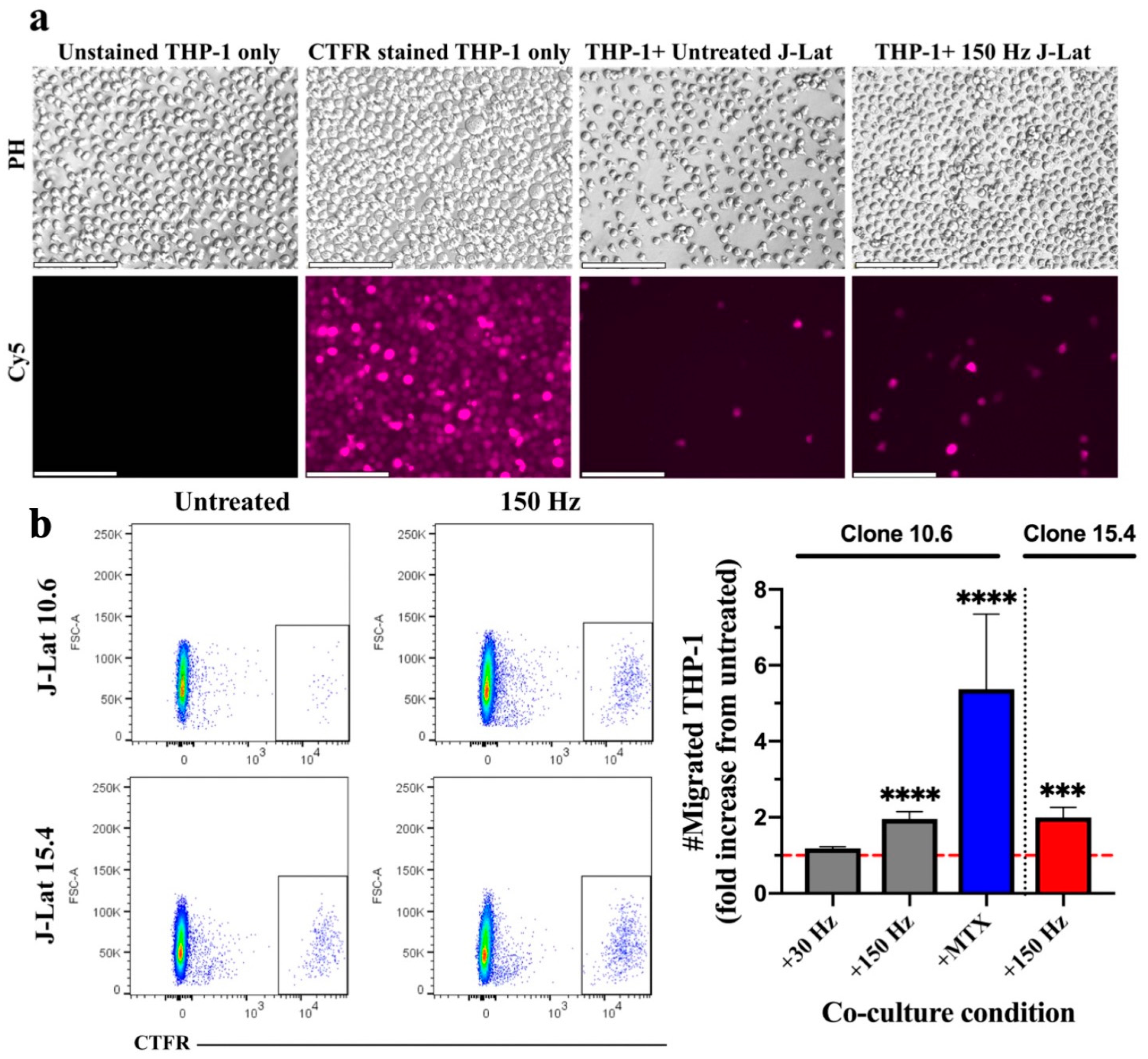{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Non-Thermal Plasma Application
2.3. Migration Assay
2.4. Phagocytosis and Maturation Assay
2.5. Cross-Presentation Assay
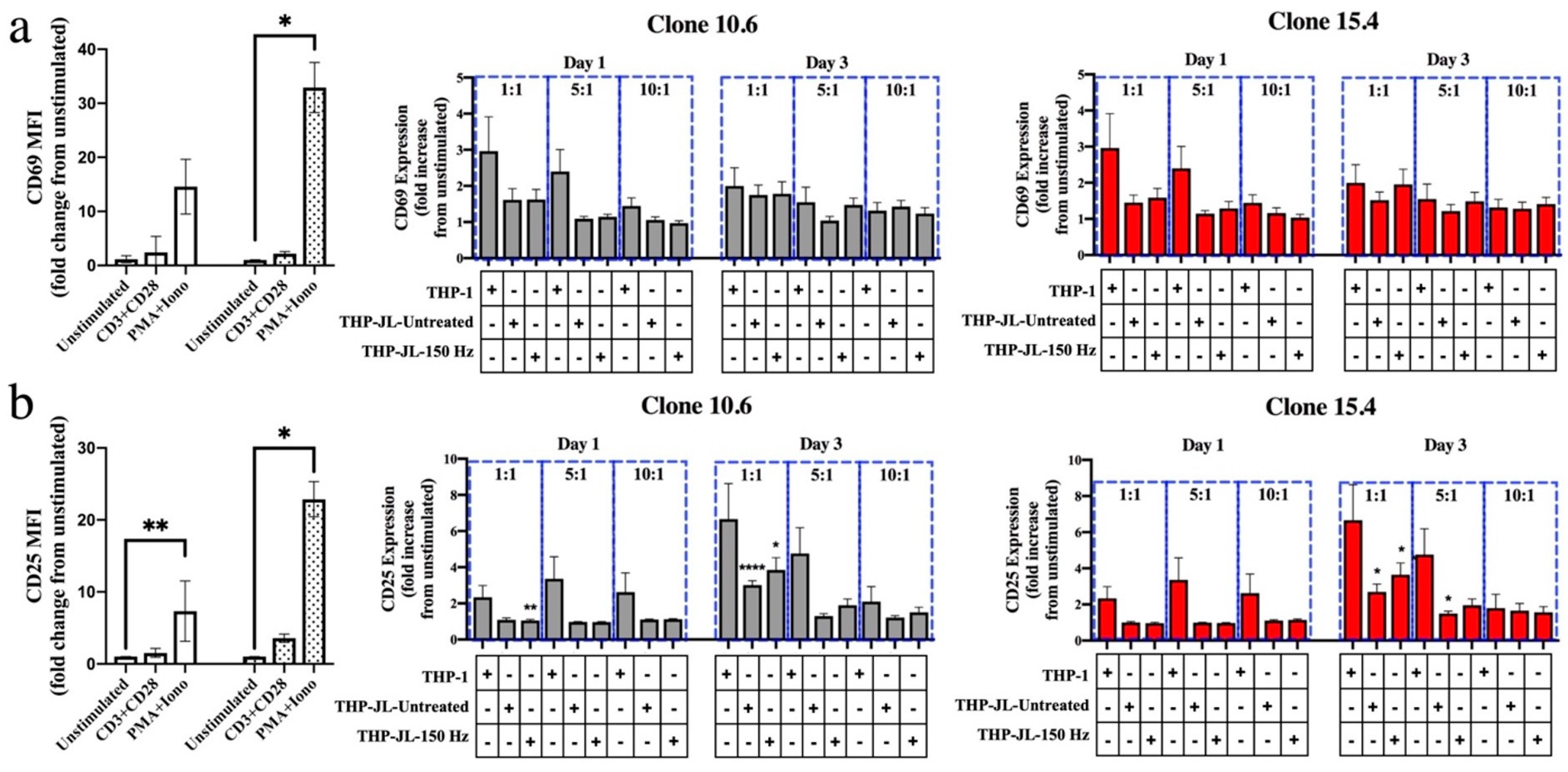
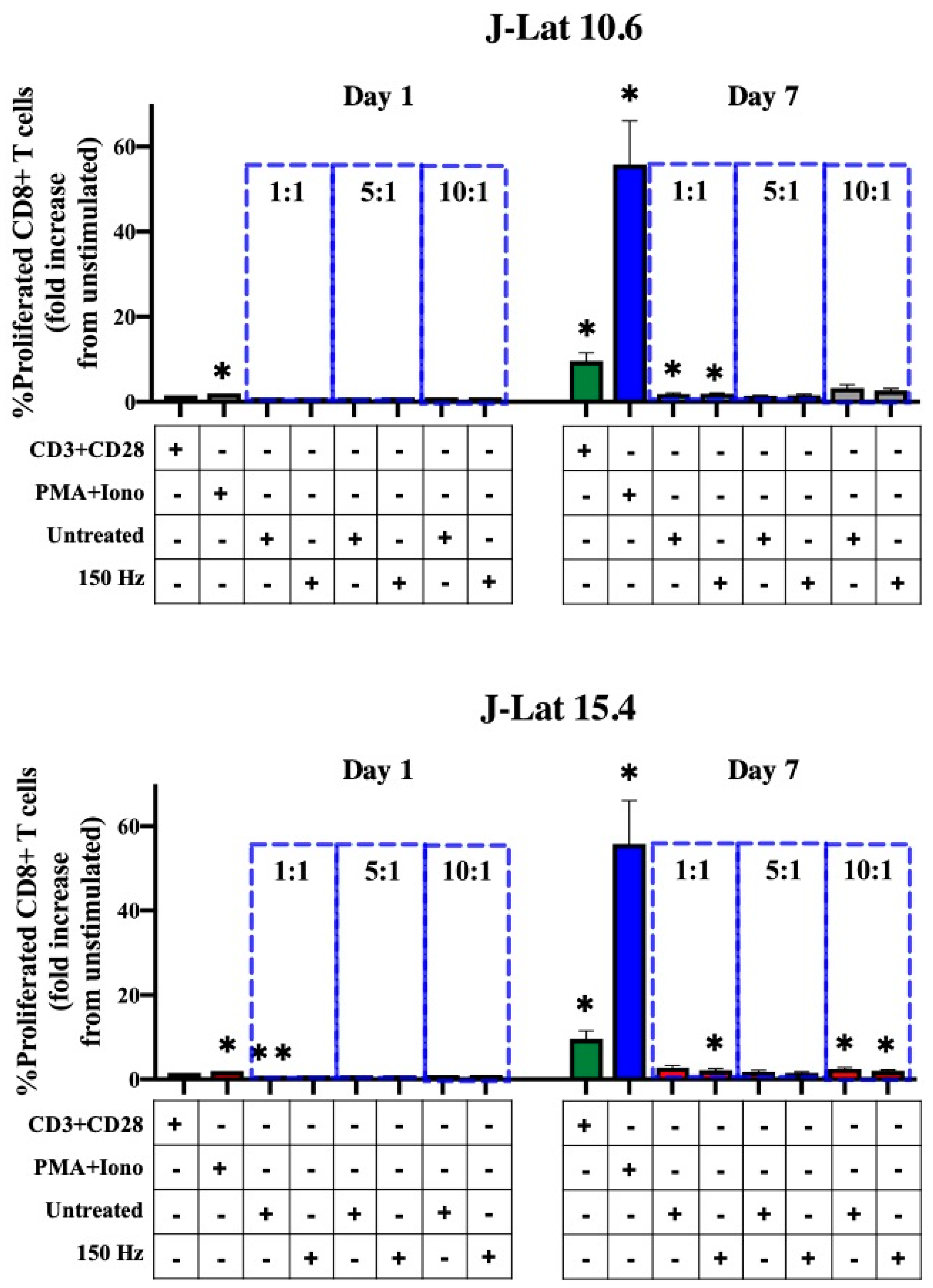
2.6. Mixed Lymphocyte Reaction (MLR)
2.7. Analysis of the Chromatin State at LTR Integration Sites
2.8. Statistical Analyses
3. Results
3.1. Effect of NTP on Migration of Monocytes toward NTP-Exposed J-Lat Clones
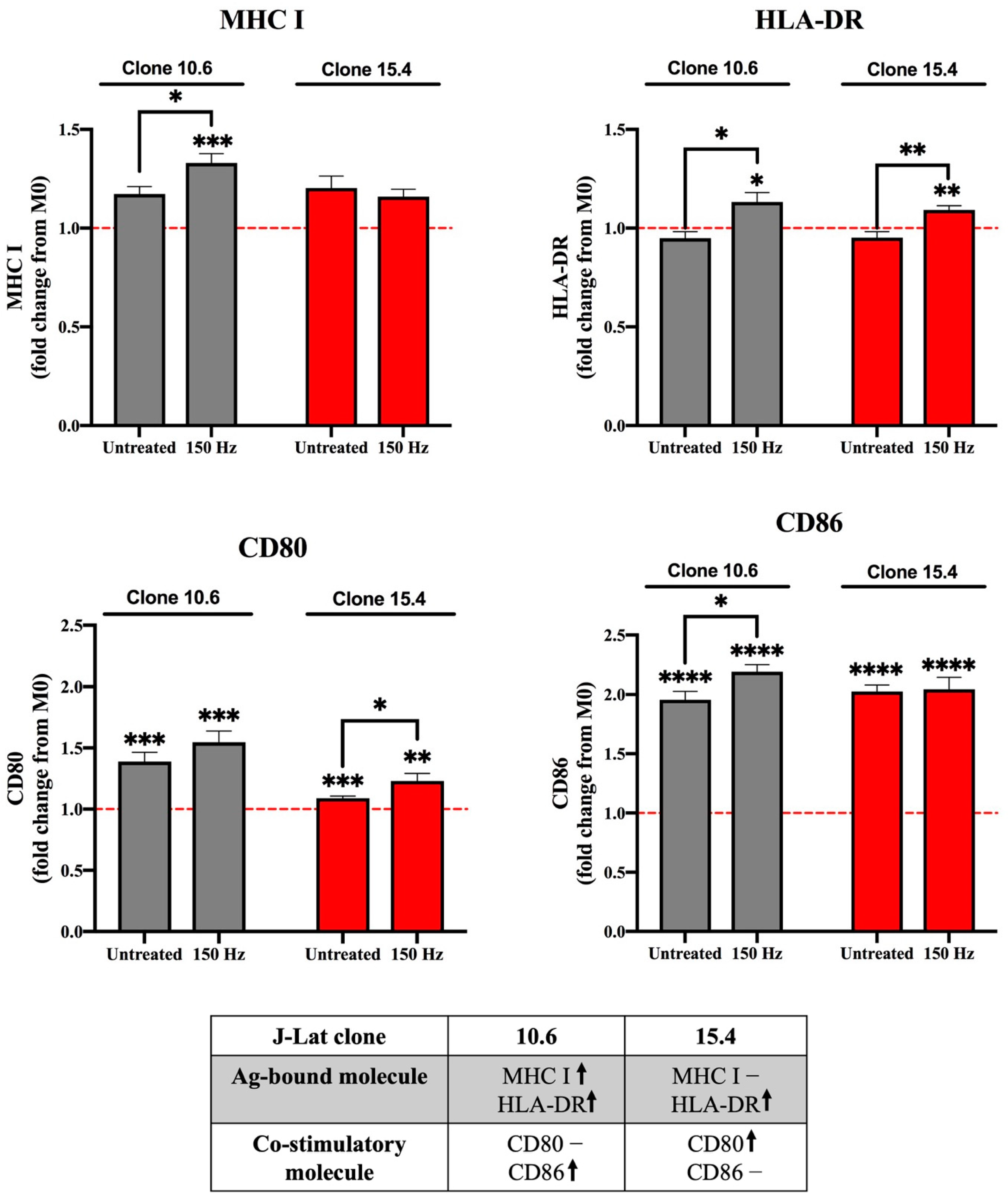
3.2. Effect of NTP on Macrophage Maturation Markers Following Uptake of NTP-Exposed Cells
3.3. Effect of NTP-Exposed J-Lat Cells on Antigen Cross-Presentation by Macrophages
3.4. Effect of NTP on Antigenicity of J-Lat Cells
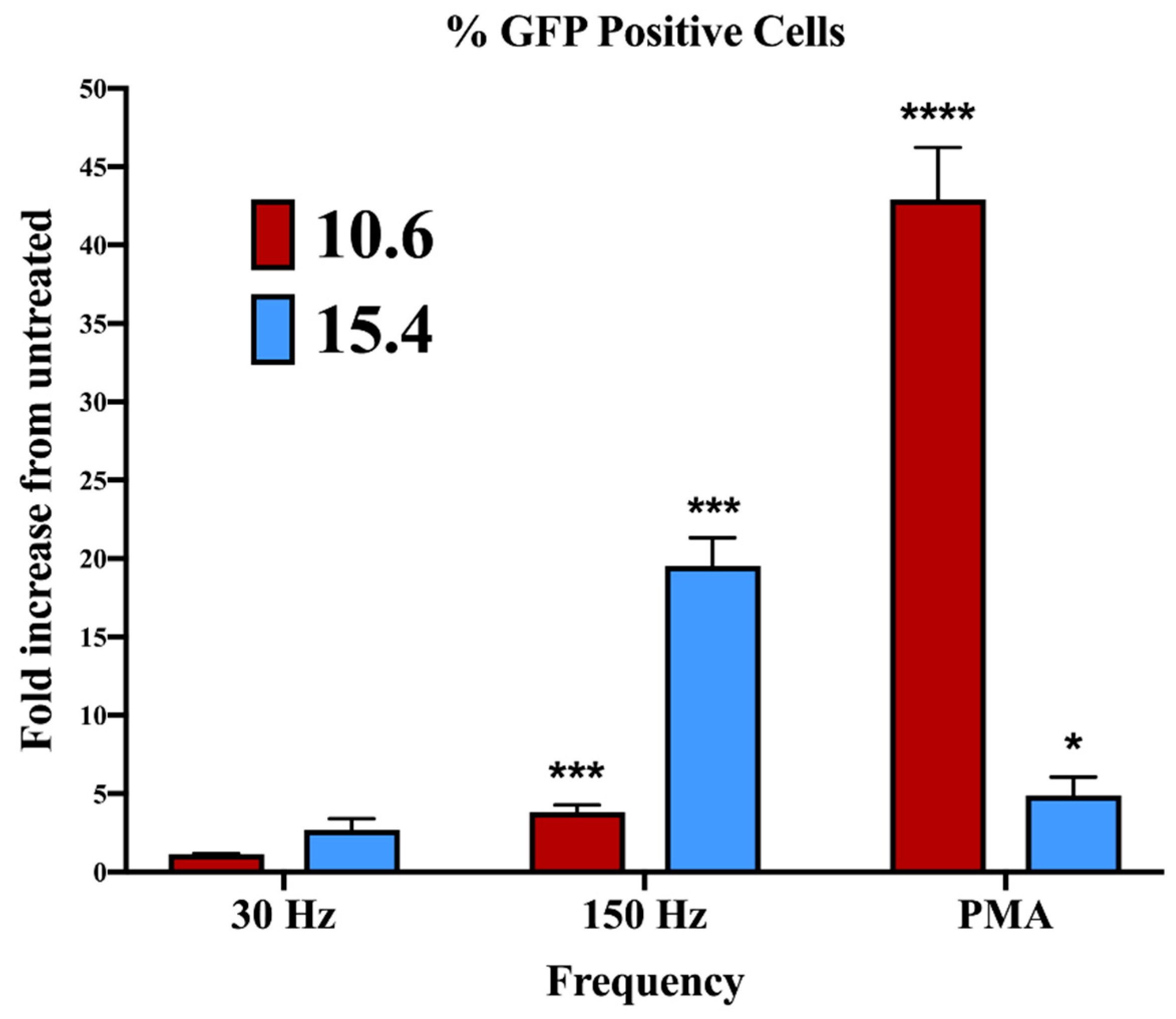
3.5. Effect of NTP on HIV-1 Gene Expression in J-Lat Clones 10.6 and 15.4
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 10 October 2022).
- Bushman, F.D.; Fujiwara, T.; Craigie, R. Retroviral DNA integration directed by HIV integration protein in vitro. Science 1990, 249, 1555–1558. [Google Scholar] [CrossRef] [PubMed]
- Lusic, M.; Siliciano, R.F. Nuclear landscape of HIV-1 infection and integration. Nat. Rev. Microbiol. 2017, 15, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Saag, M.S.; Benson, C.A.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Mugavero, M.J.; Sax, P.E.; Smith, D.M.; Thompson, M.A.; Buchbinder, S.P.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2018 Recommendations of the International Antiviral Society-USA Panel. JAMA 2018, 320, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, A.; Liu, Y.; Wigdahl, B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 2008, 6, 388–400. [Google Scholar] [CrossRef]
- Datta, P.K.; Kaminski, R.; Hu, W.; Pirrone, V.; Sullivan, N.T.; Nonnemacher, M.R.; Dampier, W.; Wigdahl, B.; Khalili, K. HIV-1 Latency and Eradication: Past, Present and Future. Curr. HIV Res. 2016, 14, 431–441. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Greene, W.C.; Peterlin, R.M. Global HIV/AIDS Medicine; Volberding, P.A., Ed.; Saunders: Philadelphia, PA, USA, 2008. [Google Scholar]
- Collini, P.; Noursadeghi, M.; Sabroe, I.; Miller, R.F.; Dockrell, D.H. Monocyte and macrophage dysfunction as a cause of HIV-1 induced dysfunction of innate immunity. Curr. Mol. Med. 2010, 10, 727–740. [Google Scholar] [CrossRef]
- Collins, D.R.; Gaiha, G.D.; Walker, B.D. CD8(+) T cells in HIV control, cure and prevention. Nat. Rev. Immunol. 2020, 20, 471–482. [Google Scholar] [CrossRef]
- Ward, A.R.; Mota, T.M.; Jones, R.B. Immunological approaches to HIV cure. Semin. Immunol. 2021, 51, 101412. [Google Scholar] [CrossRef]
- von Woedtke, T.; Laroussi, M.; Gherardi, M. Foundations of plasmas for medical applications. Plasma Sources Sci. Technol. 2022, 31, 054002. [Google Scholar] [CrossRef]
- Fridman, A. Plasma Chemistry; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Lin, A.; Truong, B.; Pappas, A.; Kirifides, L.; Oubarri, A.; Chen, S.; Lin, S.; Dobrynin, D.; Fridman, G.; Fridman, A.; et al. Uniform Nanosecond Pulsed Dielectric Barrier Discharge Plasma Enhances Anti-Tumor Effects by Induction of Immunogenic Cell Death in Tumors and Stimulation of Macrophages. Plasma Process. Polym. 2015, 12, 1392–1399. [Google Scholar] [CrossRef]
- Bekeschus, S.; Clemen, R.; Metelmann, H.-R. Potentiating anti-tumor immunity with physical plasma. Clin. Plasma Med. 2018, 12, 17–22. [Google Scholar] [CrossRef]
- Khalili, M.; Daniels, L.; Lin, A.; Krebs, F.C.; Snook, A.E.; Bekeschus, S.; Bowne, W.B.; Miller, V. Non-Thermal Plasma-Induced Immunogenic Cell Death in Cancer: A Topical Review. J. Phys. D Appl. Phys. 2019, 52, 423001. [Google Scholar] [CrossRef] [PubMed]
- Brulle, L.; Vandamme, M.; Ries, D.; Martel, E.; Robert, E.; Lerondel, S.; Trichet, V.; Richard, S.; Pouvesle, J.M.; Le Pape, A. Effects of a non thermal plasma treatment alone or in combination with gemcitabine in a MIA PaCa2-luc orthotopic pancreatic carcinoma model. PLoS ONE 2012, 7, e52653. [Google Scholar] [CrossRef] [PubMed]
- Freund, E.; Liedtke, K.R.; van der Linde, J.; Metelmann, H.R.; Heidecke, C.D.; Partecke, L.I.; Bekeschus, S. Physical plasma-treated saline promotes an immunogenic phenotype in CT26 colon cancer cells in vitro and in vivo. Sci. Rep. 2019, 9, 634. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, K.R.; Bekeschus, S.; Kaeding, A.; Hackbarth, C.; Kuehn, J.P.; Heidecke, C.D.; von Bernstorff, W.; von Woedtke, T.; Partecke, L.I. Non-thermal plasma-treated solution demonstrates antitumor activity against pancreatic cancer cells in vitro and in vivo. Sci. Rep. 2017, 7, 8319. [Google Scholar] [CrossRef]
- Lin, A.G.; Xiang, B.; Merlino, D.J.; Baybutt, T.R.; Sahu, J.; Fridman, A.; Snook, A.E.; Miller, V. Non-thermal plasma induces immunogenic cell death in vivo in murine CT26 colorectal tumors. Oncoimmunology 2018, 7, e1484978. [Google Scholar] [CrossRef]
- Smolkova, B.; Frtus, A.; Uzhytchak, M.; Lunova, M.; Kubinova, S.; Dejneka, A.; Lunov, O. Critical Analysis of Non-Thermal Plasma-Driven Modulation of Immune Cells from Clinical Perspective. Int. J. Mol. Sci. 2020, 21, 6226. [Google Scholar] [CrossRef]
- Lin, L.; Wang, L.; Liu, Y.; Xu, C.; Tu, Y.; Zhou, J. Nonthermal plasma inhibits tumor growth and proliferation and enhances the sensitivity to radiation in vitro and in vivo. Oncol. Rep. 2018, 40, 3405–3415. [Google Scholar] [CrossRef]
- Mohamed, H.; Clemen, R.; Freund, E.; Lackmann, J.W.; Wende, K.; Connors, J.; Haddad, E.K.; Dampier, W.; Wigdahl, B.; Miller, V.; et al. Non-thermal plasma modulates cellular markers associated with immunogenicity in a model of latent HIV-1 infection. PLoS ONE 2021, 16, e0247125. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef]
- Kerpedjiev, P.; Abdennur, N.; Lekschas, F.; McCallum, C.; Dinkla, K.; Strobelt, H.; Luber, J.M.; Ouellette, S.B.; Azhir, A.; Kumar, N.; et al. HiGlass: Web-based visual exploration and analysis of genome interaction maps. Genome Biol. 2018, 19, 125. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, W. Epilogos. Available online: https://epilogos.altius.org (accessed on 10 October 2022).
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D.; et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen 2019, 39, 12. [Google Scholar] [CrossRef]
- Mohamed, H.; Gebski, E.; Reyes, R.; Beane, S.; Wigdahl, B.; Krebs, F.C.; Stapelmann, K.; Miller, V. Differential Effect of Non-Thermal Plasma RONS on Two Human Leukemic Cell Populations. Cancers 2021, 13, 2437. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV latency: The road to an HIV cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef]
- Sherrill-Mix, S.; Lewinski, M.K.; Famiglietti, M.; Bosque, A.; Malani, N.; Ocwieja, K.E.; Berry, C.C.; Looney, D.; Shan, L.; Agosto, L.M.; et al. HIV latency and integration site placement in five cell-based models. Retrovirology 2013, 10, 90. [Google Scholar] [CrossRef]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef]
- Embgenbroich, M.; Burgdorf, S. Current Concepts of Antigen Cross-Presentation. Front. Immunol. 2018, 9, 1643. [Google Scholar] [CrossRef] [PubMed]
- Van Loenhout, J.; Flieswasser, T.; Freire Boullosa, L.; De Waele, J.; Van Audenaerde, J.; Marcq, E.; Jacobs, J.; Lin, A.; Lion, E.; Dewitte, H.; et al. Cold Atmospheric Plasma-Treated PBS Eliminates Immunosuppressive Pancreatic Stellate Cells and Induces Immunogenic Cell Death of Pancreatic Cancer Cells. Cancers 2019, 11, 1597. [Google Scholar] [CrossRef] [PubMed]
- Gulzar, N.; Copeland, K.F. CD8+ T-cells: Function and response to HIV infection. Curr. HIV Res. 2004, 2, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Maranon, C.; Desoutter, J.F.; Hoeffel, G.; Cohen, W.; Hanau, D.; Hosmalin, A. Dendritic cells cross-present HIV antigens from live as well as apoptotic infected CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 6092–6097. [Google Scholar] [CrossRef]
- Ferre, A.L.; Hunt, P.W.; Critchfield, J.W.; Young, D.H.; Morris, M.M.; Garcia, J.C.; Pollard, R.B.; Yee, H.F., Jr.; Martin, J.N.; Deeks, S.G.; et al. Mucosal immune responses to HIV-1 in elite controllers: A potential correlate of immune control. Blood 2009, 113, 3978–3989. [Google Scholar] [CrossRef]
- Way, K.J.; Chou, E.; King, G.L. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol. Sci. 2000, 21, 181–187. [Google Scholar] [CrossRef]
- Chung, C.H.; Allen, A.G.; Sullivan, N.T.; Atkins, A.; Nonnemacher, M.R.; Wigdahl, B.; Dampier, W. Computational Analysis Concerning the Impact of DNA Accessibility on CRISPR-Cas9 Cleavage Efficiency. Mol. Ther. 2020, 28, 19–28. [Google Scholar] [CrossRef]
- Sunshine, S.; Kirchner, R.; Amr, S.S.; Mansur, L.; Shakhbatyan, R.; Kim, M.; Bosque, A.; Siliciano, R.F.; Planelles, V.; Hofmann, O.; et al. HIV Integration Site Analysis of Cellular Models of HIV Latency with a Probe-Enriched Next-Generation Sequencing Assay. J. Virol. 2016, 90, 4511–4519. [Google Scholar] [CrossRef]
- Campbell, G.R.; Spector, S.A. Current strategies to induce selective killing of HIV-1-infected cells. J. Leukoc. Biol. 2022, 112, 1273–1284. [Google Scholar] [CrossRef]
- Dampier, W.; Nonnemacher, M.R.; Mell, J.; Earl, J.; Ehrlich, G.D.; Pirrone, V.; Aiamkitsumrit, B.; Zhong, W.; Kercher, K.; Passic, S.; et al. HIV-1 Genetic Variation Resulting in the Development of New Quasispecies Continues to Be Encountered in the Peripheral Blood of Well-Suppressed Patients. PLoS ONE 2016, 11, e0155382. [Google Scholar] [CrossRef]
- Wells, D.W.; Guo, S.; Shao, W.; Bale, M.J.; Coffin, J.M.; Hughes, S.H.; Wu, X. An analytical pipeline for identifying and mapping the integration sites of HIV and other retroviruses. BMC Genom. 2020, 21, 216. [Google Scholar] [CrossRef]
- Telwatte, S.; Moron-Lopez, S.; Aran, D.; Kim, P.; Hsieh, C.; Joshi, S.; Montano, M.; Greene, W.C.; Butte, A.J.; Wong, J.K.; et al. Heterogeneity in HIV and cellular transcription profiles in cell line models of latent and productive infection: Implications for HIV latency. Retrovirology 2019, 16, 32. [Google Scholar] [CrossRef]
- Park, J.; Suh, D.; Tang, T.; Lee, H.J.; Roe, J.S.; Kim, G.C.; Han, S.; Song, K. Non-thermal atmospheric pressure plasma induces epigenetic modifications that activate the expression of various cytokines and growth factors in human mesoderm-derived stem cells. Free Radic. Biol. Med. 2020, 148, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Staric, P.; Vogel-Mikus, K.; Mozetic, M.; Junkar, I. Effects of Nonthermal Plasma on Morphology, Genetics and Physiology of Seeds: A Review. Plants 2020, 9, 1736. [Google Scholar] [CrossRef] [PubMed]
- Mahdikia, H.; Saadati, F.; Freund, E.; Gaipl, U.S.; Majidzadeh, A.K.; Shokri, B.; Bekeschus, S. Gas plasma irradiation of breast cancers promotes immunogenicity, tumor reduction, and an abscopal effect in vivo. Oncoimmunology 2020, 10, 1859731. [Google Scholar] [CrossRef]
- Debaisieux, S.; Lachambre, S.; Gross, A.; Mettling, C.; Besteiro, S.; Yezid, H.; Henaff, D.; Chopard, C.; Mesnard, J.M.; Beaumelle, B. HIV-1 Tat inhibits phagocytosis by preventing the recruitment of Cdc42 to the phagocytic cup. Nat. Commun. 2015, 6, 6211. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.; Nayak, G.; Rendine, N.; Wigdahl, B.; Krebs, F.C.; Bruggeman, P.J.; Miller, V. Non-Thermal Plasma as a Novel Strategy for Treating or Preventing Viral Infection and Associated Disease. Front. Phys. 2021, 9, 286. [Google Scholar] [CrossRef]
- Goral, S. The three-signal hypothesis of lymphocyte activation/targets for immunosuppression. Dial. Transplant. 2011, 40, 14–16. [Google Scholar] [CrossRef]
- Henry, C.J.; Ornelles, D.A.; Mitchell, L.M.; Brzoza-Lewis, K.L.; Hiltbold, E.M. IL-12 produced by dendritic cells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J. Immunol. 2008, 181, 8576–8584. [Google Scholar] [CrossRef]
- Vacaflores, A.; Freedman, S.N.; Chapman, N.M.; Houtman, J.C. Pretreatment of activated human CD8 T cells with IL-12 leads to enhanced TCR-induced signaling and cytokine production. Mol. Immunol. 2017, 81, 1–15. [Google Scholar] [CrossRef]
- Rutishauser, R.L.; Trautmann, L. CD8 + T-cell responses in HIV controllers: Potential implications for novel HIV remission strategies. Curr. Opin. HIV AIDS 2022, 17, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, T.; Zhang, Y.; Luo, S.; Chen, H.; Chen, D.; Li, C.; Li, W. The reservoir of latent HIV. Front. Cell. Infect. Microbiol. 2022, 12, 945956. [Google Scholar] [CrossRef] [PubMed]
- Pardons, M.; Baxter, A.E.; Massanella, M.; Pagliuzza, A.; Fromentin, R.; Dufour, C.; Leyre, L.; Routy, J.P.; Kaufmann, D.E.; Chomont, N. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog. 2019, 15, e1007619. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.R.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Altfeld, M.; Gale, M., Jr. Innate immunity against HIV-1 infection. Nat. Immunol. 2015, 16, 554–562. [Google Scholar] [CrossRef]
- Kloverpris, H.N.; Jackson, A.; Handley, A.; Hayes, P.; Gilmour, J.; Riddell, L.; Chen, F.; Atkins, M.; Boffito, M.; Walker, B.D.; et al. Non-immunogenicity of overlapping gag peptides pulsed on autologous cells after vaccination of HIV infected individuals. PLoS ONE 2013, 8, e74389. [Google Scholar] [CrossRef]
- Garcia, F.; Climent, N.; Guardo, A.C.; Gil, C.; Leon, A.; Autran, B.; Lifson, J.D.; Martinez-Picado, J.; Dalmau, J.; Clotet, B.; et al. A dendritic cell-based vaccine elicits T cell responses associated with control of HIV-1 replication. Sci. Transl. Med. 2013, 5, 166ra162. [Google Scholar] [CrossRef]
- Cafaro, A.; Ensoli, B. HIV-1 therapeutic vaccines in clinical development to intensify or replace antiretroviral therapy: The promising results of the Tat vaccine. Expert Rev. Vaccines 2022, 21, 1243–1253. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, H.; Berman, R.; Connors, J.; Haddad, E.K.; Miller, V.; Nonnemacher, M.R.; Dampier, W.; Wigdahl, B.; Krebs, F.C. Immunomodulatory Effects of Non-Thermal Plasma in a Model for Latent HIV-1 Infection: Implications for an HIV-1-Specific Immunotherapy. Biomedicines 2023, 11, 122. https://doi.org/10.3390/biomedicines11010122
Mohamed H, Berman R, Connors J, Haddad EK, Miller V, Nonnemacher MR, Dampier W, Wigdahl B, Krebs FC. Immunomodulatory Effects of Non-Thermal Plasma in a Model for Latent HIV-1 Infection: Implications for an HIV-1-Specific Immunotherapy. Biomedicines. 2023; 11(1):122. https://doi.org/10.3390/biomedicines11010122
Chicago/Turabian StyleMohamed, Hager, Rachel Berman, Jennifer Connors, Elias K. Haddad, Vandana Miller, Michael R. Nonnemacher, Will Dampier, Brian Wigdahl, and Fred C. Krebs. 2023. "Immunomodulatory Effects of Non-Thermal Plasma in a Model for Latent HIV-1 Infection: Implications for an HIV-1-Specific Immunotherapy" Biomedicines 11, no. 1: 122. https://doi.org/10.3390/biomedicines11010122
APA StyleMohamed, H., Berman, R., Connors, J., Haddad, E. K., Miller, V., Nonnemacher, M. R., Dampier, W., Wigdahl, B., & Krebs, F. C. (2023). Immunomodulatory Effects of Non-Thermal Plasma in a Model for Latent HIV-1 Infection: Implications for an HIV-1-Specific Immunotherapy. Biomedicines, 11(1), 122. https://doi.org/10.3390/biomedicines11010122