
Impact of Non-Invasive Physical Plasma on Heat Shock Protein Functionality in Eukaryotic Cells


Abstract
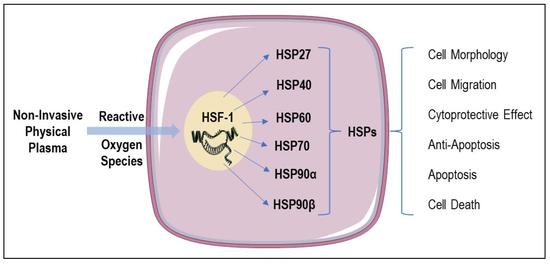
1. Introduction
2. HSP27
3. HSP40
4. HSP60
5. HSP70
6. HSP90
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Isbary, G.; Shimizu, T.; Li, Y.F.; Stolz, W.; Thomas, H.M.; Morfill, G.E.; Zimmermann, J.L. Cold atmospheric plasma devices for medical issues. Expert Rev. Med. Devices 2013, 10, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.G.; Kroesen, G.; Morfill, G.; Nosenko, T.; Shimizu, T.; van Dijk, J.; Zimmermann, J.L. Plasma medicine: An introductory review. New J. Phys. 2009, 11, 115012. [Google Scholar] [CrossRef]
- Kletschkus, K.; Haralambiev, L.; Mustea, A.; Bekeschus, S.; Stope, M.B. Review of innovative physical therapy methods: Introduction to the principles of cold physical plasma. In Vivo 2020, 34, 3103–3107. [Google Scholar] [CrossRef]
- Laroussi, M. Sterilization of contaminated matter with an atmospheric pressure plasma. IEEE Trans. Plasma Sci. 1996, 24, 1188–1191. [Google Scholar] [CrossRef]
- Kramer, A.; Bekeschus, S.; Matthes, R.; Bender, C.; Stope, M.B.; Napp, M.; Lademann, O.; Lademann, J.; Weltmann, K.-D.; Schauer, F. Cold physical plasmas in the field of hygiene—Relevance, significance, and future applications. Plasma Process. Polym. 2015, 12, 1410–1422. [Google Scholar] [CrossRef]
- Daeschlein, G.; Scholz, S.; Arnold, A.; von Podewils, S.; Haase, H.; Emmert, S.; Woedtke, T.; von Weltmann, K.-D.; Jünger, M. In vitro susceptibility of important skin and wound pathogens against low temperature atmospheric pressure plasma jet (APPJ) and dielectric barrier discharge plasma (DBD). Plasma Process Polym. 2012, 9, 380–389. [Google Scholar] [CrossRef]
- Von Woedtke, T.; Schmidt, A.; Bekeschus, S.; Wende, K.; Weltmann, K.D. Plasma medicine: A field of applied redox biology. In Vivo 2019, 33, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, T.; Semmler, M.L.; Schafer, M.; Bekeschus, S.; Emmert, S.; Boeckmann, L. Plasma medicine: Applications of cold atmospheric pressure plasma in dermatology. Oxid. Med. Cell Longev. 2019, 3, 3873928. [Google Scholar] [CrossRef]
- Chuangsuwanich, A.; Assadamongkol, T.; Boonyawan, D. The healing effect of low-temperature atmospheric-pressure plasma in pressure ulcer: A randomized controlled trial. Int. J. Low Extrem. Wounds 2016, 15, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, C.; Kluschke, F.; Patzelt, A.; Vandersee, S.; Czaika, V.A.; Richter, H.; Bob, A.; Hutten, J.; Painsi, C.; Huge, R.; et al. Clinical use of cold atmospheric pressure argon plasma in chronic leg ulcers: A pilot study. J. Wound Care 2015, 196, 198–200, 202–203. [Google Scholar] [CrossRef]
- Von Woedtke, T.; Reuter, S.; Masur, K.; Weltmann, K.-D. Plasmas for medicine. Phys. Rep. 2013, 530, 291–320. [Google Scholar] [CrossRef]
- Haertel, B.; Eiden, K.; Deuter, A.; Wende, K.; von Woedtke, T.; Lindequist, U. Differential effect of non-thermal atmospheric-pressure plasma on angiogenesis. Lett. Appl. NanoBioSci. 2014, 3, 159–166. [Google Scholar]
- Schmidt-Bleker, A.; Winter, J.; Bösel, A.; Reuter, S.; Weltmann, K.D. On the plasma chemistry of a cold atmospheric argon plasma jet with shielding gas device. Plasma Sources Sci. Technol. 2016, 25, 015005. [Google Scholar] [CrossRef]
- Arndt, S.; Unger, P.; Wacker, E.; Shimizu, T.; Heinlin, J.; Li, Y.F.; Thomas, H.M.; Morfill, G.E.; Zimmermann, J.L.; Bosserhoff, A.K.; et al. Cold atmospheric plasma (CAP) changes gene expression of key molecules of the wound healing machinery and improves wound healing in vitro and in vivo. PLoS ONE 2013, 8, e79325. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; von Woedtke, T.; Kramer, A.; Weltmann, K.D.; Masur, K. Cold physical plasma treatment alters redox balance in human immune cells. Plasma Med. 2013, 4, 267–278. [Google Scholar] [CrossRef]
- Jablonowski, H.; von Woedtke, T. Research on plasma medicine-relevant plasma-liquid interaction: What happened in the past five years? Clin. Plasma Med. 2015, 2, 42–52. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Friedman, G.; Fridman, A.; Clyne, A.M. Endothelial cell proliferation is enhanced by low dose non-thermal plasma through fibroblast growth factor-2 release. Ann. Biomed Eng. 2010, 38, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.; Nitsch, A.; Erb, H.H.H.; Egger, E.K.; Haralambiev, L.; Eggers, B.; Kramer, F.-J.; Weiss, M.; Mustea, A.; Stope, M.B. Non-invasive physical plasma enhances the membrane permeability to low molecular weight compounds and subsequently leads to the loss of cellular ATP and the devitalization of epithelial cancer cells. Appl. Sci. 2021, 21, 9801. [Google Scholar] [CrossRef]
- Haertel, B.; von Woedtke, T.; Weltmann, K.D.; Lindequist, U. Non-thermal atmospheric-pressure plasma possible application in wound healing. Biomol. Ther. 2014, 22, 477–490. [Google Scholar] [CrossRef]
- Stope, M.B. Plasma oncology—Physical plasma as innovative tumor therapy. J. Cancer Biol. 2020, 2, 53–56. [Google Scholar]
- Weiss, M.; Gumbel, D.; Hanschmann, E.M.; Mandelkow, R.; Gelbrich, N.; Zimmermann, U.; Walther, R.; Ekkernkamp, A.; Sckell, A.; Kramer, A.; et al. Cold atmospheric plasma treatment induces anti-proliferative effects in prostate cancer cells by redox and apoptotic signaling pathways. PLoS ONE 2015, 10, e0130350. [Google Scholar] [CrossRef] [PubMed]
- Marzi, J.; Stope, M.B.; Henes, M.; Koch, A.; Wenzel, T.; Holl, M.; Layland, S.L.; Neis, F.; Bosmuller, H.; Ruoff, F.; et al. Noninvasive physical plasma as innovative and tissue-preserving therapy for women positive for cervical intraepithelial neoplasia. Cancers 2022, 14, 1933. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Stope, M.B.; Koensgen, D.; Burchardt, M.; Concin, N.; Zygmunt, M.; Mustea, A. Jump in the fire—Heat shock proteins and their impact on ovarian cancer therapy. Crit. Rev. Oncol. Hematol. 2016, 97, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.J.; Prince, T.L.; Okusha, Y.; Bunch, H.; Calderwood, S.K. Heat shock proteins in cell signaling and cancer. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119187. [Google Scholar] [CrossRef] [PubMed]
- Stope, M.B.; Weiss, M.; Preuss, M.; Streitborger, A.; Ritter, C.A.; Zimmermann, U.; Walther, R.; Burchardt, M. Immediate and transient phosphorylation of the heat shock protein 27 initiates chemoresistance in prostate cancer cells. Oncol. Rep. 2014, 32, 2380–2386. [Google Scholar] [CrossRef]
- Rocchi, P.; So, A.; Kojima, S.; Signaevsky, M.; Beraldi, E.; Fazli, L.; Hurtado-Coll, A.; Yamanaka, K.; Gleave, M. Heat shock protein 27 increases after androgen ablation and plays a cytoprotective role in hormone-refractory prostate cancer. Cancer Res. 2004, 64, 6595–6602. [Google Scholar] [CrossRef]
- Grossebrummel, H.; Peter, T.; Mandelkow, R.; Weiss, M.; Muzzio, D.; Zimmermann, U.; Walther, R.; Jensen, F.; Knabbe, C.; Zygmunt, M.; et al. Cytochrome P450 17A1 inhibitor abiraterone attenuates cellular growth of prostate cancer cells independently from androgen receptor signaling by modulation of oncogenic and apoptotic pathways. Int. J. Oncol. 2016, 48, 793–800. [Google Scholar] [CrossRef]
- Banerji, U.; Sain, N.; Sharp, S.Y.; Valenti, M.; Asad, Y.; Ruddle, R.; Raynaud, F.; Walton, M.; Eccles, S.A.; Judson, I.; et al. An in vitro and in vivo study of the combination of the heat shock protein inhibitor 17-allylamino-17-demethoxygeldanamycin and carboplatin in human ovarian cancer models. Cancer Chemother. Pharmacol. 2008, 62, 769–778. [Google Scholar] [CrossRef]
- Brunnert, D.; Langer, C.; Zimmermann, L.; Bargou, R.C.; Burchardt, M.; Chatterjee, M.; Stope, M.B. The heat shock protein 70 inhibitor VER155008 suppresses the expression of HSP27, HOP and HSP90beta and the androgen receptor, induces apoptosis, and attenuates prostate cancer cell growth. J. Cell. Biochem. 2020, 121, 407–417. [Google Scholar] [CrossRef]
- Song, T.F.; Zhang, Z.F.; Liu, L.; Yang, T.; Jiang, J.; Li, P. Small interfering RNA-mediated silencing of heat shock protein 27 (HSP27) Increases chemosensitivity to paclitaxel by increasing production of reactive oxygen species in human ovarian cancer cells (HO8910). J. Int. Med. Res. 2009, 37, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Buzzard, K.A.; Giaccia, A.J.; Killender, M.; Anderson, R.L. Heat shock protein 72 modulates pathways of stress-induced apoptosis. J. Biol. Chem. 1998, 273, 17147–17153. [Google Scholar] [CrossRef] [PubMed]
- Abazid, A.; Martin, B.; Choinowski, A.; McNeill, R.V.; Brandenburg, L.O.; Ziegler, P.; Zimmermann, U.; Burchardt, M.; Erb, H.; Stope, M.B. The androgen receptor antagonist enzalutamide induces apoptosis, dysregulates the heat shock protein system, and diminishes the androgen receptor and estrogen receptor beta1 expression in prostate cancer cells. J. Cell. Biochem. 2019, 120, 16711–16722. [Google Scholar] [CrossRef] [PubMed]
- Gabai, V.L.; Kabakov, A.E. Rise in heat-shock protein level confers tolerance to energy deprivation. FEBS Lett. 1993, 327, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Preville, X.; Salvemini, F.; Giraud, S.; Chaufour, S.; Paul, C.; Stepien, G.; Ursini, M.V.; Arrigo, A.P. Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp. Cell Res. 1999, 247, 61–78. [Google Scholar] [CrossRef]
- Benjamin, I.J.; McMillan, D.R. Stress (heat shock) proteins: Molecular chaperones in cardiovascular biology and disease. Circ. Res. 1998, 83, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Polla, B.S.; Cossarizza, A. Stress proteins in inflammation. EXS 1996, 77, 375–391. [Google Scholar]
- Wong, H.R. Potential protective role of the heat shock response in sepsis. New Horiz. 1998, 6, 194–200. [Google Scholar]
- Gowda, A.; Yang, C.; Asimakis, G.K.; Rastegar, S.; Motamedi, M. Heat shock improves recovery and provides protection against global ischemia after hypothermic storage. Ann. Thorac. Surg. 1998, 66, 1991–1997. [Google Scholar] [CrossRef]
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 2, 185–194. [Google Scholar] [CrossRef]
- Shevtsov, M.; Multhoff, G. Heat Shock Protein-Peptide and HSP-Based Immunotherapies for the Treatment of Cancer. Front. Immunol. 2016, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Hagymasi, A.T.; Dempsey, J.P.; Srivastava, P.K. Heat-Shock Proteins. Curr. Protoc. 2022, 2, e592. [Google Scholar] [CrossRef] [PubMed]
- van Eden, W. Immune tolerance therapies for autoimmune diseases based on heat shock protein T-cell epitopes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160531. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Deng, G.; Peng, X.; Xu, X.; Liu, L.; Peng, J.; Ma, Y.; Zhang, P.; Wen, A.; Wang, Y.; et al. Intelligent photothermal dendritic cells restart the cancer immunity cycle through enhanced immunogenic cell death. Biomaterials 2021, 279, 121228. [Google Scholar] [CrossRef] [PubMed]
- Gulic, T.; Laskarin, G.; Glavan, L.; Grubic Kezele, T.; Haller, H.; Rukavina, D. Human Decidual CD1a(+) Dendritic Cells Undergo Functional Maturation Program Mediated by Gp96. Int. J. Mol. Sci. 2023, 24, 2278. [Google Scholar] [CrossRef]
- Spierings, J.; van Eden, W. Heat shock proteins and their immunomodulatory role in inflammatory arthritis. Rheumatology 2017, 56, 198–208. [Google Scholar] [CrossRef]
- Zininga, T.; Ramatsui, L.; Shonhai, A. Heat Shock Proteins as Immunomodulants. Molecules 2018, 23, 2846. [Google Scholar] [CrossRef]
- Duchesne, C.; Frescaline, N.; Blaise, O.; Lataillade, J.J.; Banzet, S.; Dussurget, O.; Rousseau, A. Cold Atmospheric Plasma Promotes Killing of Staphylococcus aureus by Macrophages. mSphere 2021, 6, e0021721. [Google Scholar] [CrossRef]
- Kaushik, N.K.; Kaushik, N.; Adhikari, M.; Ghimire, B.; Linh, N.N.; Mishra, Y.K.; Lee, S.J.; Choi, E.H. Preventing the Solid Cancer Progression via Release of Anticancer-Cytokines in Co-Culture with Cold Plasma-Stimulated Macrophages. Cancers 2019, 11, 842. [Google Scholar] [CrossRef]
- Bekeschus, S.; Winterbourn, C.C.; Kolata, J.; Masur, K.; Hasse, S.; Broker, B.M.; Parker, H.A. Neutrophil extracellular trap formation is elicited in response to cold physical plasma. J. Leukoc. Biol. 2016, 100, 791–799. [Google Scholar] [CrossRef]
- Haralambiev, L.; Wien, L.; Gelbrich, N.; Kramer, A.; Mustea, A.; Burchardt, M.; Ekkernkamp, A.; Stope, M.B.; Gumbel, D. Effects of Cold Atmospheric Plasma on the Expression of Chemokines, Growth Factors, TNF Superfamily Members, Interleukins, and Cytokines in Human Osteosarcoma Cells. Anticancer Res. 2019, 39, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Arndt, S.; Landthaler, M.; Zimmermann, J.L.; Unger, P.; Wacker, E.; Shimizu, T.; Li, Y.F.; Morfill, G.E.; Bosserhoff, A.K.; Karrer, S. Effects of cold atmospheric plasma (CAP) on ss-defensins, inflammatory cytokines, and apoptosis-related molecules in keratinocytes in vitro and in vivo. PLoS ONE 2015, 10, e0120041. [Google Scholar] [CrossRef] [PubMed]
- Turrini, E.; Laurita, R.; Stancampiano, A.; Catanzaro, E.; Calcabrini, C.; Maffei, F.; Gherardi, M.; Colombo, V.; Fimognari, C. Cold Atmospheric Plasma Induces Apoptosis and Oxidative Stress Pathway Regulation in T-Lymphoblastoid Leukemia Cells. Oxid. Med. Cell Longev. 2017, 2017, 4271065. [Google Scholar] [CrossRef]
- Haertel, B.; Volkmann, F.; von Woedtke, T.; Lindequist, U. Differential sensitivity of lymphocyte subpopulations to non-thermal atmospheric-pressure plasma. Immunobiology 2012, 217, 628–633. [Google Scholar] [CrossRef]
- Stratmann, B.; Costea, T.C.; Nolte, C.; Hiller, J.; Schmidt, J.; Reindel, J.; Masur, K.; Motz, W.; Timm, J.; Kerner, W.; et al. Effect of Cold Atmospheric Plasma Therapy vs Standard Therapy Placebo on Wound Healing in Patients with Diabetic Foot Ulcers: A Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e2010411. [Google Scholar] [CrossRef]
- Abbasi, E.; Mehrabadi, J.F.; Nourani, M.; Namini, Y.N.; Mohammadi, S.; Esmaeili, D.; Abbasi, A. Evaluation of cold atmospheric-pressure plasma against burn wound infections and gene silencing. Iran. J. Microbiol. 2021, 13, 544–552. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef] [PubMed]
- Dimauro, I.; Mercatelli, N.; Caporossi, D. Exercise-induced ROS in heat shock proteins response. Free. Radic. Biol. Med. 2016, 98, 46–55. [Google Scholar] [CrossRef]
- Schmidt, A.; Bekeschus, S.; Wende, K.; Vollmar, B.; von Woedtke, T. A cold plasma jet accelerates wound healing in a murine model of full-thickness skin wounds. Exp. Dermatol. 2017, 26, 156–162. [Google Scholar] [CrossRef]
- Tornin, J.; Mateu-Sanz, M.; Rodriguez, A.; Labay, C.; Rodriguez, R.; Canal, C. Pyruvate plays a main role in the antitumoral selectivity of cold atmospheric plasma in osteosarcoma. Sci. Rep. 2019, 9, 10681. [Google Scholar] [CrossRef]
- Tabuchi, Y.; Uchiyama, H.; Zhao, Q.L.; Yunoki, T.; Andocs, G.; Nojima, N.; Takeda, K.; Ishikawa, K.; Hori, M.; Kondo, T. Effects of nitrogen on the apoptosis of and changes in gene expression in human lymphoma U937 cells exposed to argon-based cold atmospheric pressure plasma. Int. J. Mol. Med. 2016, 37, 1706–1714. [Google Scholar] [CrossRef] [PubMed]
- Bolouki, N.; Hsu, Y.N.; Hsiao, Y.C.; Jheng, P.R.; Hsieh, J.H.; Chen, H.L.; Mansel, B.W.; Yeh, Y.Y.; Chen, Y.H.; Lu, C.X.; et al. Cold atmospheric plasma physically reinforced substances of platelets-laden photothermal-responsive methylcellulose complex restores burn wounds. Int. J. Biol. Macromol. 2021, 192, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; Lippert, M.; Diepold, K.; Chiosis, G.; Seufferlein, T.; Azoitei, N. Physical plasma-triggered ROS induces tumor cell death upon cleavage of HSP90 chaperone. Sci. Rep. 2019, 9, 4112. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Chuang, E.Y.; Jheng, P.R.; Hao, P.C.; Hsieh, J.H.; Chen, H.L.; Mansel, B.W.; Yeh, Y.Y.; Lu, C.X.; Lee, J.W.; et al. Cold-atmospheric plasma augments functionalities of hybrid polymeric carriers regenerating chronic wounds: In vivo experiments. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 131, 112488. [Google Scholar] [CrossRef] [PubMed]
- Bundscherer, L.; Wende, K.; Ottmuller, K.; Barton, A.; Schmidt, A.; Bekeschus, S.; Hasse, S.; Weltmann, K.D.; Masur, K.; Lindequist, U. Impact of non-thermal plasma treatment on MAPK signaling pathways of human immune cell lines. Immunobiology 2013, 218, 1248–1255. [Google Scholar] [CrossRef]
- Singer, D.; Ressel, V.; Stope, M.B.; Bekeschus, S. Heat shock protein 27 affects myeloid cell activation and interaction with prostate cancer cells. Biomedicines 2022, 10, 2192. [Google Scholar] [CrossRef]
- Singer, D.; Wulff, C.P.; Stope, M.B.; Bekeschus, S. Extracellular heat shock protein 27 is released by plasma-treated ovarian cancer cells and affects THP-1 monocyte activity. Plasma 2022, 5, 569–578. [Google Scholar] [CrossRef]
- Stock, A.D.; Spallone, P.A.; Dennis, T.R.; Netski, D.; Morris, C.A.; Mervis, C.B.; Hobart, H.H. Heat shock protein 27 gene: Chromosomal and molecular location and relationship to Williams syndrome. Am. J. Med. Genet. A 2003, 120A, 320–325. [Google Scholar] [CrossRef]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting Heat shock protein 27 in cancer: A druggable target for cancer treatment? Cancers 2019, 11, 1195. [Google Scholar] [CrossRef]
- Kocabiyik, S. Essential structural and functional features of small heat shock proteins in molecular chaperoning process. Protein Pept. Lett. 2009, 16, 613–622. [Google Scholar] [CrossRef]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat shock proteins 27 and 70: Anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006, 5, 2592–2601. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.P. Structure-functions of HspB1 (Hsp27). Methods Mol. Biol. 2011, 787, 105–119. [Google Scholar] [PubMed]
- Haslbeck, M.; Franzmann, T.; Weinfurtner, D.; Buchner, J. Some like it hot: The structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 2005, 12, 842–846. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.T.; Bortolus, M.; Koteiche, H.A.; McHaourab, H.S. Sequence, structure, and dynamic determinants of Hsp27 (HspB1) equilibrium dissociation are encoded by the N-terminal domain. Biochemistry 2012, 51, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.; Napoli, V.; Mazurkie, A.; Stafford, W.F.; Graceffa, P. Phosphorylation dependence of hsp27 multimeric size and molecular chaperone function. J. Biol. Chem. 2009, 284, 18801–18807. [Google Scholar] [CrossRef]
- Gusev, N.B.; Bogatcheva, N.V.; Marston, S.B. Structure and properties of small heat shock proteins (sHsp) and their interaction with cytoskeleton proteins. Biochemistry 2002, 67, 511–519. [Google Scholar]
- Mounier, N.; Arrigo, A.P. Actin cytoskeleton and small heat shock proteins: How do they interact? Cell Stress Chaperones 2002, 7, 167–176. [Google Scholar] [CrossRef]
- Ange, M.; Castanares-Zapatero, D.; De Poortere, J.; Dufeys, C.; Courtoy, G.E.; Bouzin, C.; Quarck, R.; Bertrand, L.; Beauloye, C.; Horman, S. Alpha1AMP-activated protein kinase protects against lipopolysaccharide-induced endothelial barrier disruption via junctional reinforcement and activation of the p38 MAPK/HSP27 pathway. Int. J. Mol. Sci. 2020, 21, 5581. [Google Scholar] [CrossRef]
- Wang, W.; Weng, J.; Yu, L.; Huang, Q.; Jiang, Y.; Guo, X. Role of TLR4-p38 MAPK-Hsp27 signal pathway in LPS-induced pulmonary epithelial hyperpermeability. BMC Pulm. Med. 2018, 18, 178. [Google Scholar] [CrossRef]
- Clements, R.T.; Feng, J.; Cordeiro, B.; Bianchi, C.; Sellke, F.W. p38 MAPK-dependent small HSP27 and alphaB-crystallin phosphorylation in regulation of myocardial function following cardioplegic arrest. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1669–H1677. [Google Scholar] [CrossRef][Green Version]
- Konishi, H.; Matsuzaki, H.; Tanaka, M.; Takemura, Y.; Kuroda, S.; Ono, Y.; Kikkawa, U. Activation of protein kinase B (Akt/RAC-protein kinase) by cellular stress and its association with heat shock protein Hsp27. FEBS Lett. 1997, 410, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Santell, L.; Bartfeld, N.S.; Levin, E.G. Identification of a protein transiently phosphorylated by activators of endothelial cell function as the heat-shock protein HSP27: A possible role for protein kinase C. Biochem. J. 1992, 284 Pt 3, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Gao, Y.; Zhang, L.; Weng, Z.; Zhang, F.; Hu, X.; Wang, S.; Vosler, P.; Cao, G.; Sun, D.; et al. Phosphorylation of HSP27 by protein kinase D is essential for mediating neuroprotection against ischemic neuronal injury. J. Neurosci. 2012, 32, 2667–2682. [Google Scholar] [CrossRef] [PubMed]
- Evans, I.M.; Britton, G.; Zachary, I.C. Vascular endothelial growth factor induces heat shock protein (HSP) 27 serine 82 phosphorylation and endothelial tubulogenesis via protein kinase D and independent of p38 kinase. Cell Signal. 2008, 20, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Hotta, K.; Hara, A.; Hirade, K.; Ito, H.; Kato, K.; Kozawa, O. TNF-alpha decreases hsp 27 in human blood mononuclear cells: Involvement of protein kinase c. Life Sci. 2006, 80, 181–186. [Google Scholar] [CrossRef]
- Hayashi, K.; Takai, S.; Matsushima-Nishiwaki, R.; Hanai, Y.; Kato, K.; Tokuda, H.; Kozawa, O. (−)-Epigallocatechin gallate reduces transforming growth factor beta-stimulated HSP27 induction through the suppression of stress-activated protein kinase/c-Jun N-terminal kinase in osteoblasts. Life Sci. 2008, 82, 1012–1017. [Google Scholar] [CrossRef]
- Kwon, S.M.; Kim, S.A.; Fujii, S.; Maeda, H.; Ahn, S.G.; Yoon, J.H. Transforming growth factor beta1 promotes migration of human periodontal ligament cells through heat shock protein 27 phosphorylation. Biol. Pharm. Bull. 2011, 34, 486–489. [Google Scholar] [CrossRef][Green Version]
- Geier, A.; Hemi, R.; Haimsohn, M.; Beery, R.; Karasik, A. Phosphorylation of a 27-kDa protein correlates with survival of protein-synthesis-inhibited MCF-7 cells. In Vitro Cell Dev. Biol. Anim. 1997, 33, 129–136. [Google Scholar] [CrossRef]
- Vahidinia, Z.; Mahdavi, E.; Talaei, S.A.; Naderian, H.; Tamtaji, A.; Haddad Kashani, H.; Beyer, C.; Azami Tameh, A. The effect of female sex hormones on Hsp27 phosphorylation and histological changes in prefrontal cortex after tMCAO. Pathol. Res. Pract. 2021, 221, 153415. [Google Scholar] [CrossRef]
- Tabibzadeh, S.; Broome, J. Heat shock proteins in human endometrium throughout the menstrual cycle. Infect. Dis. Obstet. Gynecol. 1999, 7, 5–9. [Google Scholar] [CrossRef]
- Bi, X.; Jiang, B.; Zhou, J.; Luo, L.; Yin, Z. Phosphorylated Hsp27 prevents LPS-induced excessive inflammation in THP-1 cells via suppressing ROS-mediated upregulation of CBP. Cell Biol. Int. 2020, 44, 253–267. [Google Scholar] [CrossRef]
- Nahomi, R.B.; Palmer, A.; Green, K.M.; Fort, P.E.; Nagaraj, R.H. Pro-inflammatory cytokines downregulate Hsp27 and cause apoptosis of human retinal capillary endothelial cells. Biochim. Biophys. Acta 2014, 1842, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; von Woedtke, T.; Bekeschus, S. Periodic exposure of keratinocytes to cold physical plasma: An in vitro model for redox-related diseases of the skin. Oxid. Med. Cell. Longev. 2016, 2016, 9816072. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Bekeschus, S.; Jarick, K.; Hasse, S.; von Woedtke, T.; Wende, K. Cold physical plasma modulates p53 and mitogen-activated protein kinase signaling in keratinocytes. Oxid. Med. Cell Longev. 2019, 2019, 7017363. [Google Scholar] [CrossRef] [PubMed]
- Landry, S.J. Structure and energetics of an allele-specific genetic interaction between dnaJ and dnaK: Correlation of nuclear magnetic resonance chemical shift perturbations in the J-domain of Hsp40/DnaJ with binding affinity for the ATPase domain of Hsp70/DnaK. Biochemistry 2003, 42, 4926–4936. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Kaida, A.; Yamamoto, S.; Parrales, A.; Young, E.D.; Ranjan, A.; Alalem, M.A.; Morita, K.I.; Oikawa, Y.; Harada, H.; Ikeda, T.; et al. DNAJA1 promotes cancer metastasis through interaction with mutant p53. Oncogene 2021, 40, 5013–5025. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Gotoh, T.; Terada, K.; Oyadomari, S.; Mori, M. hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ. 2004, 11, 390–402. [Google Scholar] [CrossRef]
- Beere, H.M. “The stress of dying”: The role of heat shock proteins in the regulation of apoptosis. J. Cell Sci. 2004, 117, 2641–2651. [Google Scholar] [CrossRef]
- Kabbage, M.; Dickman, M.B. The BAG proteins: A ubiquitous family of chaperone regulators. Cell Mol. Life Sci. 2008, 65, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.; Fernandez-Guerra, P. Disease-associated mutations in the HSPD1 gene encoding the large subunit of the mitochondrial HSP60/HSP10 chaperonin complex. Front Mol. Biosci. 2016, 3, 49. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.A.; Newman, S.M.; Hallberg, R.L.; Slaughter, C.A.; Perlman, P.S.; Butow, R.A. In organello formaldehyde crosslinking of proteins to mtDNA: Identification of bifunctional proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 7772–7777. [Google Scholar] [CrossRef]
- Kaufman, B.A.; Kolesar, J.E.; Perlman, P.S.; Butow, R.A. A function for the mitochondrial chaperonin Hsp60 in the structure and transmission of mitochondrial DNA nucleoids in Saccharomyces cerevisiae. J. Cell Biol. 2003, 163, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Bigman, L.S.; Horovitz, A. Reconciling the controversy regarding the functional importance of bullet- and football-shaped GroE complexes. J. Biol. Chem. 2019, 294, 13527–13529. [Google Scholar] [CrossRef] [PubMed]
- Erb, H.H.H.; Streitborger, A.; Mustea, A.; Stope, M.B. Physiological and genetically engineered expression modulation methods do not affect cellular levels of the heat shock protein HSP60 in prostate cancer cells. In Vivo 2022, 36, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Rottach, A.M.; Ahrend, H.; Martin, B.; Walther, R.; Zimmermann, U.; Burchardt, M.; Stope, M.B. Cabazitaxel inhibits prostate cancer cell growth by inhibition of androgen receptor and heat shock protein expression. World J. Urol. 2019, 37, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Cappello, F.; Conway de Macario, E.; Marasa, L.; Zummo, G.; Macario, A.J. Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biol. Ther. 2008, 7, 801–809. [Google Scholar] [CrossRef]
- Itoh, H.; Komatsuda, A.; Ohtani, H.; Wakui, H.; Imai, H.; Sawada, K.; Otaka, M.; Ogura, M.; Suzuki, A.; Hamada, F. Mammalian HSP60 is quickly sorted into the mitochondria under conditions of dehydration. Eur. J. Biochem. 2002, 269, 5931–5938. [Google Scholar] [CrossRef]
- Bhatt, J.M.; Enriquez, A.S.; Wang, J.; Rojo, H.M.; Molugu, S.K.; Hildenbrand, Z.L.; Bernal, R.A. Single-ring intermediates are essential for some chaperonins. Front. Mol. Biosci. 2018, 5, 42. [Google Scholar] [CrossRef]
- Weiss, C.; Jebara, F.; Nisemblat, S.; Azem, A. Dynamic complexes in the chaperonin-mediated protein folding cycle. Front. Mol. Biosci. 2016, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, A.; Fujimoto, M.; Tan, K.; Kurashima, A.; Srivastava, P.; Okada, M.; Takii, R.; Nakai, A. HSF1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio 2020, 10, 1135–1148. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Fujimoto, M.; Takii, R.; Takaki, E.; Hayashida, N.; Nakai, A. Mitochondrial SSBP1 protects cells from proteotoxic stresses by potentiating stress-induced HSF1 transcriptional activity. Nat. Commun. 2015, 6, 6580. [Google Scholar] [CrossRef] [PubMed]
- Chandra, D.; Choy, G.; Tang, D.G. Cytosolic accumulation of HSP60 during apoptosis with or without apparent mitochondrial release: Evidence that its pro-apoptotic or pro-survival functions involve differential interactions with caspase-3. J. Biol. Chem. 2007, 282, 31289–31301. [Google Scholar] [CrossRef] [PubMed]
- Samali, A.; Cai, J.; Zhivotovsky, B.; Jones, D.P.; Orrenius, S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J. 1999, 18, 2040–2048. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yeh, C.T. Functional compartmentalization of HSP60-survivin interaction between mitochondria and cytosol in cancer cells. Cells 2019, 9, 23. [Google Scholar] [CrossRef]
- Shan, Y.X.; Liu, T.J.; Su, H.F.; Samsamshariat, A.; Mestril, R.; Wang, P.H. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J. Mol. Cell Cardiol. 2003, 35, 1135–1143. [Google Scholar] [CrossRef]
- Song, E.; Tang, S.; Xu, J.; Yin, B.; Bao, E.; Hartung, J. Lenti-siRNA Hsp60 promote bax in mitochondria and induces apoptosis during heat stress. Biochem. Biophys. Res. Commun. 2016, 481, 125–131. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Dohi, T.; Kang, B.H.; Altieri, D.C. Hsp60 regulation of tumor cell apoptosis. J. Biol. Chem. 2008, 283, 5188–5194. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Siegelin, M.D.; Dohi, T.; Altieri, D.C. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 2010, 70, 8988–8993. [Google Scholar] [CrossRef]
- Hallberg, E.M.; Shu, Y.; Hallberg, R.L. Loss of mitochondrial hsp60 function: Nonequivalent effects on matrix-targeted and intermembrane-targeted proteins. Mol. Cell Biol. 1993, 13, 3050–3057. [Google Scholar]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef] [PubMed]
- Faust, O.; Rosenzweig, R. Structural and biochemical properties of Hsp40/Hsp70 chaperone system. Adv. Exp. Med. Biol. 2020, 1243, 3–20. [Google Scholar] [PubMed]
- Faust, O.; Abayev-Avraham, M.; Wentink, A.S.; Maurer, M.; Nillegoda, N.B.; London, N.; Bukau, B.; Rosenzweig, R. HSP40 proteins use class-specific regulation to drive HSP70 functional diversity. Nature 2020, 587, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lee, J.J.; Seo, J.S. HSP70 deficiency results in activation of c-Jun N-terminal kinase, extracellular signal-regulated kinase, and caspase-3 in hyperosmolarity-induced apoptosis. J. Biol. Chem. 2005, 280, 6634–6641. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Newton, A.C. The turn motif is a phosphorylation switch that regulates the binding of Hsp70 to protein kinase C. J. Biol. Chem. 2002, 277, 31585–31592. [Google Scholar] [CrossRef]
- Stankiewicz, A.R.; Lachapelle, G.; Foo, C.P.; Radicioni, S.M.; Mosser, D.D. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J. Biol. Chem. 2005, 280, 38729–38739. [Google Scholar] [CrossRef]
- Matsumori, Y.; Northington, F.J.; Hong, S.M.; Kayama, T.; Sheldon, R.A.; Vexler, Z.S.; Ferriero, D.M.; Weinstein, P.R.; Liu, J. Reduction of caspase-8 and -9 cleavage is associated with increased c-FLIP and increased binding of Apaf-1 and Hsp70 after neonatal hypoxic/ischemic injury in mice overexpressing Hsp70. Stroke 2006, 37, 507–512. [Google Scholar] [CrossRef]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar] [CrossRef]
- Cyr, D.M.; Ramos, C.H. Specification of Hsp70 function by Type I and Type II Hsp40. Subcell. Biochem. 2015, 78, 91–102. [Google Scholar] [PubMed]
- Attri, P.; Park, J.H.; Ali, A.; Choi, E.H. How does plasma activated media treatment differ from direct cold plasma treatment? Anticancer Agents Med. Chem. 2018, 18, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Kletschkus, K.; Haralambiev, L.; Nitsch, A.; Pfister, F.; Klinkmann, G.; Kramer, A.; Bekeschus, S.; Mustea, A.; Stope, M.B. The application of a low-temperature physical plasma device operating under atmospheric pressure leads to the production of toxic NO2. Anticancer Res. 2020, 40, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, J.; Zheng, X.; Patel, N.; Feder, Z.A.; Anandhakumar, J.; Valerius, K.; Gross, D.S.; Khalil, A.S.; Pincus, D. Hsf1 and Hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response. eLife 2018, 7, 31668. [Google Scholar] [CrossRef]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 family: Structure, regulation, function, and implications in health and disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Birbo, B.; Madu, E.E.; Madu, C.O.; Jain, A.; Lu, Y. Role of HSP90 in cancer. Int. J. Mol. Sci. 2021, 22, 10317. [Google Scholar] [CrossRef]
- Prodromou, C. Mechanisms of Hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef]
- Workman, P. Reflections and outlook on targeting HSP90, HSP70 and HSF1 in cancer: A personal perspective. Adv. Exp. Med. Biol. 2020, 1243, 163–179. [Google Scholar]
- Yang, S.; Zhao, T.; Ma, A.; Huang, Z.; Yang, J.; Yuan, C.; Guo, X.; Zhu, C. Heat stress-induced HSP90 expression is dependent on ERK and HSF1 activation in turbot (Scophthalmus maximus) kidney cells. Cell Stress Chaperones 2021, 26, 173–185. [Google Scholar] [CrossRef]
- Pincus, D. Regulation of Hsf1 and the Heat Shock Response. Adv. Exp. Med. Biol. 2020, 1243, 41–50. [Google Scholar]
- Zhang, H.; Shao, S.; Zeng, Y.; Wang, X.; Qin, Y.; Ren, Q.; Xiang, S.; Wang, Y.; Xiao, J.; Sun, Y. Reversible phase separation of HSF1 is required for an acute transcriptional response during heat shock. Nat. Cell Biol. 2022, 24, 340–352. [Google Scholar] [CrossRef]
- Ahn, S.G.; Thiele, D.J. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003, 17, 516–528. [Google Scholar] [CrossRef]
- Liebmann, J.; Scherer, J.; Bibinov, N.; Rajasekaran, P.; Kovacs, R.; Gesche, R.; Awakowicz, P.; Kolb-Bachofen, V. Biological effects of nitric oxide generated by an atmospheric pressure gas-plasma on human skin cells. Nitric Oxide 2011, 24, 8–16. [Google Scholar] [CrossRef]
- Manucha, W.; Valles, P.G. Cytoprotective role of nitric oxide associated with Hsp70 expression in neonatal obstructive nephropathy. Nitric Oxide 2008, 18, 204–215. [Google Scholar] [CrossRef]
- Nash, S.; Johnstone, J.; Rahman, M.S. Elevated temperature attenuates ovarian functions and induces apoptosis and oxidative stress in the American oyster, Crassostrea virginica: Potential mechanisms and signaling pathways. Cell Stress Chaperones 2019, 24, 957–967. [Google Scholar] [CrossRef]
- Fucarino, A.; Pitruzzella, A. Role of HSP60/HSP10 in Lung Cancer: Simple Biomarkers or Leading Actors? J. Oncol. 2020, 2020, 4701868. [Google Scholar] [CrossRef]
- Yadav, K.; Yadav, A.; Vashistha, P.; Pandey, V.P.; Dwivedi, U.N. Protein Misfolding Diseases and Therapeutic Approaches. Curr. Protein Pept. Sci. 2019, 20, 1226–1245. [Google Scholar] [CrossRef]
- David, S.; Bucchieri, F.; Corrao, S.; Czarnecka, A.M.; Campanella, C.; Farina, F.; Peri, G.; Tomasello, G.; Sciume, C.; Modica, G.; et al. Hsp10: Anatomic distribution, functions, and involvement in human disease. Front. Biosci. 2013, 5, 768–778. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Technology | Plasma Source | Voltage | Frequency | Airflow | Reference |
|---|---|---|---|---|---|
| Jet | Argon | 2–6 kV | N/A | 5 L/min | [59] |
| Jet | Helium | N/A | N/A | 1/3/5 L/min | [60] |
| Jet | Argon + N2 | 18 kV | 20 kHz | 2 L/min | [61] |
| Jet | Argon | 5 kV | 10 kHz | 3 L/min | [62] |
| Jet | Argon | N/A | N/A | N/A | [63] |
| DBD-based volume NIPP | Argon | 7 kV | 10 kHz | 3 L/min | [64] |
| Jet | Argon | 2–6 kV | 1.1 MHz | 3 L/min | [65] |
| Jet | N/A | N/A | N/A | N/A | [66] |
| Jet | Argon | N/A | 1 MHz | 4 L/min | [67] |
| Technology | Model | HSP | Methodology | Reference |
|---|---|---|---|---|
| Jet | HaCat Cells | HSP27 | WB | [59] |
| Jet | SaOS-2 Cells | HSP60 | WB | [60] |
| Jet | U937 Cells | HSP40/70 | PCR | [61] |
| Jet | Wistar Rat | IF | [62] | |
| Jet | MDA-MB-s31 | HSP90 | WB | [63] |
| DBD-based volume CAP | Human Wound Skin | IF | [64] | |
| Jet | THP-1 Cells | HSP27 | WB | [65] |
| Jet | LNCaP/PC-3 Cells | HSP27 | ELISA | [66] |
| Jet | OVCAR3 Cells | HSP27 | ELISA | [67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Abazid, A.; Badendieck, S.; Mustea, A.; Stope, M.B. Impact of Non-Invasive Physical Plasma on Heat Shock Protein Functionality in Eukaryotic Cells. Biomedicines 2023, 11, 1471. https://doi.org/10.3390/biomedicines11051471
Wang Y, Abazid A, Badendieck S, Mustea A, Stope MB. Impact of Non-Invasive Physical Plasma on Heat Shock Protein Functionality in Eukaryotic Cells. Biomedicines. 2023; 11(5):1471. https://doi.org/10.3390/biomedicines11051471
Chicago/Turabian StyleWang, Yanqing, Alexander Abazid, Steffen Badendieck, Alexander Mustea, and Matthias B. Stope. 2023. "Impact of Non-Invasive Physical Plasma on Heat Shock Protein Functionality in Eukaryotic Cells" Biomedicines 11, no. 5: 1471. https://doi.org/10.3390/biomedicines11051471
APA StyleWang, Y., Abazid, A., Badendieck, S., Mustea, A., & Stope, M. B. (2023). Impact of Non-Invasive Physical Plasma on Heat Shock Protein Functionality in Eukaryotic Cells. Biomedicines, 11(5), 1471. https://doi.org/10.3390/biomedicines11051471