

Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options
 ,
,  , ,
, , 
 and
and
Abstract
1. Introduction
2. Pathophysiology of CMD in Diabetes
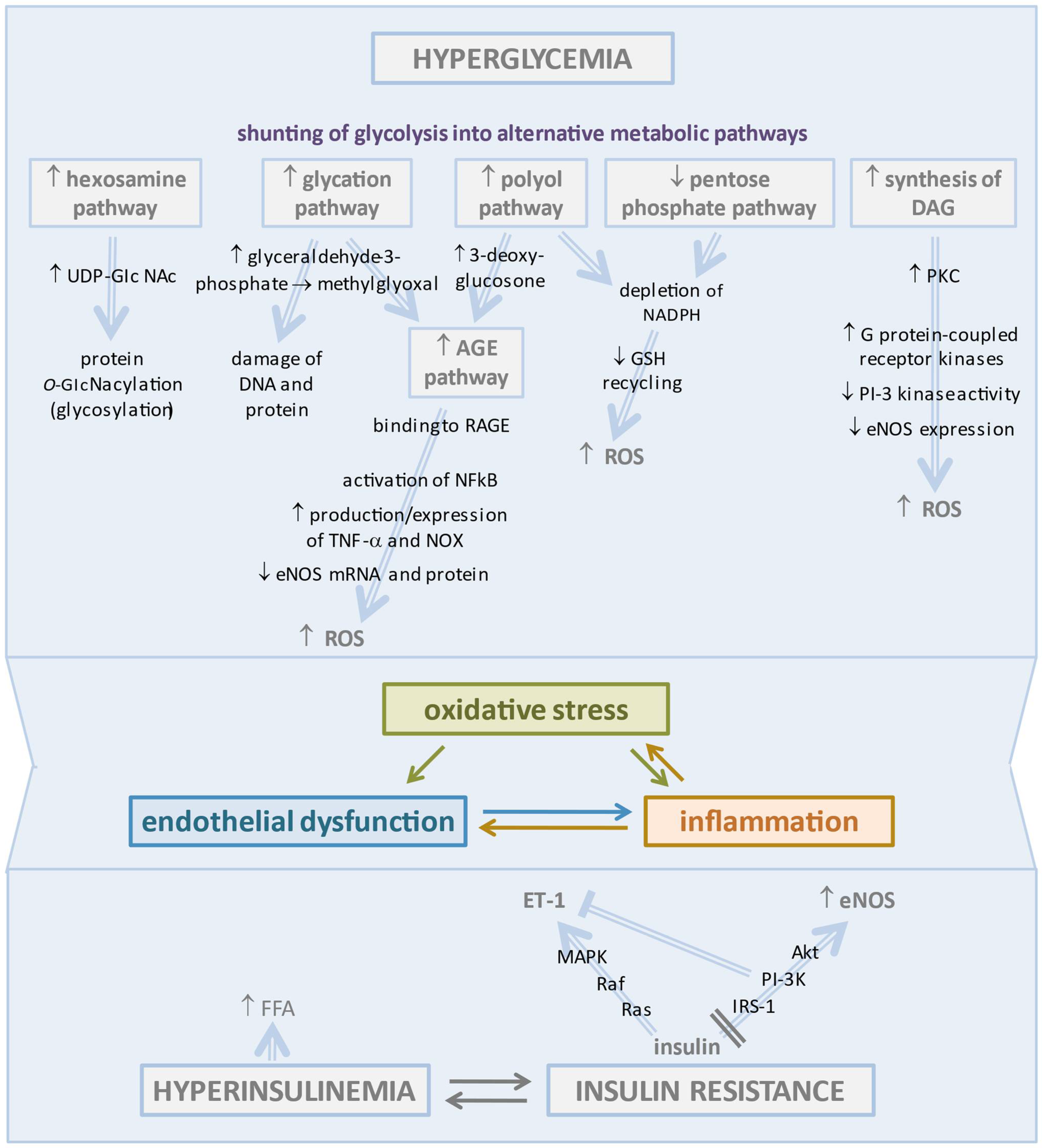
2.1. Hyperglycemia-Induced Changes in Vascular Endothelium
2.2. Insulin Resistance in the Pathogenesis of CMD
2.3. Decreased NO Bioavailability in Diabetes
2.4. Role of Mitochondrial Dysfunction in Diabetic CMD
2.5. Oxidative Stress and Inflammation in Diabetic CMD
2.6. Remodeling of Coronaric Microvessel and Myocardium in Diabetes
2.7. Other Mechanisms of CMD: Hemodynamic Forces, Epigenetics and microRNAs
3. Therapeutic Management of CMD in Diabetes
3.1. Correction of CV Risk Factors
3.2. Pharmacological Options
4. Glucagon like Peptide-1 (GLP-1) and GLP-1 Receptor Agonists (GLP-1 RAs)
4.1. CV Benefits by GLP-1 RAs
4.1.1. Correction of Traditional Risk Factors for CVD
4.1.2. Direct Anti-Atherosclerotic Effects
4.2. Effects of GLP-1 RA on Coronary Microvasculature
4.2.1. Preclinical Studies
4.2.2. Human Studies

{kind=link}
| Preclinical Studies | ||||||
|---|---|---|---|---|---|---|
| Experimental Model | Treatment | Duration of Treatment | Effects | Suggested Mediating Mechanisms | Ref. | |
| STZ-induced diabetic rats | exenatide or vildagliptin | 12 weeks | protection of endothelial barrier function by both drugs (at transmission electron microscopy) | [226] | ||
| HG-cultured CMECs | Incubation with GLP-1 | ↓ ROS production and apoptotic index | inhibition of Rho though a cAMP/PKA-mediated pathway | [226] | ||
| H/R injuried CMECs | liraglutide 12 h before the induction of hypoxia | suppression of XO-mediated ROS release | stimulation of PI3K/Akt/surviving pathways | [229] | ||
| Zucker obese rats at high Na+ diet | liraglutide | 8 weeks | restoration of ET-1/NO balance (↓ nitrosative stress and proinflamm./profibrotic markers) | [230] | ||
| STZ-induced diabetic rats | liraglutide | 6 weeks | ↓ TNF-α ↑ VEGF and α-smooth muscle actin | [232] | ||
| swine model of ventricular fibrillation | GLP-1 | 4-h infusion 1 min after resuscitation | ↑ adenosine-stimulated CFR (by intracoronary Doppler flow) | [234] | ||
| swine model of ventricular fibrillation | GLP-1 | 4-h infusion 1 min after resuscitation | preserved endothelial function ↓ of oxidative stress (↓ 8-iso-PGF2α) | [236] | ||
| Clinical Studies | ||||||
| Experimental model | Treatment | Duration of treatment | Effects | Suggested mediating mechanisms | Diagnostic modality | Ref. |
| 15 obese people | GLP-1 +/− euglyc. insulin clamp | 150-min infusion ≥120 min | ↑ MBF by ~40% Independently of insulin | preserved recruitment of coronary microvasculature | myocardial contrast echocardiography | [221] |
| 8 non-insulin treated T2DM pts without CAD | exenatide | IV infusion | ↑ MBF by 24% (no change of myocardial myocardial glucose uptake) | direct action on a myocardial receptor (?) | 13N-ammonia PET with hyperinsulinemic clamp | [237] |
| 26 healty young people | GLP-1 | 150-min infusion | ↑ MBF by 47% | recruitment of coronary microvasculature | myocardial contrast echocardiography | [239] |
| 12 normo-tolerant obese people | GLP-1 + sitagliptin | 120-min infusion | no variation of CFVR | no direct effect of intact GLP-1 on coronary microvessels | TTDE | [240] |
| 21 patients awaiting PCI for stable angina | GLP-1 (n.10) or saline (n. 11) | post-PCI vasodilation of coronary microvessels | direct actions on cardiomyocytes | pressure-flow wire | [241] | |
| 41 patients awaiting PCI for stable angina | GLP-1 (n. 10) saline (n. 11) GLP-1 + Theophylline (n. 10) Theophylline (n. 10) | acute infusion post successful PCI | post-PCI vasodilation of coronary microvessels | adenosine-independent mechanism | pressure-flow wire | [243] |
| STEMI patients treated with PCI | liraglutide | 7 days (started 30 m’ before PCI) | tendency for a lower rate of no-reflow | TTDE | [244] | |
| 20 T2DM patients without heart disease | liraglutide | 10 weeks | borderline improvement of CFVR | TTDE with dipyridamole stress | [245] | |
| 31 newly diagnosed T2DM subjects | exenatide | 12 weeks | improved CFVR ↓ serum ICAM and VCAM | activation of AMPK/PI3K/Akt pathway in a GLP-1R/cAMP dependent manner (observed in exenatide-treated HUVEC) | TTDE | [246] |
| 36 non-diabetic patients with stable HFrEF | liraglutide (n. 18) or placebo (n. 18) | 24 weeks | no variation in MBF or MBF reserve | 15O-H2O PET | [247] | |
5. SGLT2 Inhibitors (SGLT2-Is)
5.1. CV Benefits by SGLT2-Is
5.2. Effects of SGLT2-Is on Coronary Microvasculature
5.2.1. Preclinical Studies
5.2.2. Clinical Studies
| Preclinical Studies | |||||
|---|---|---|---|---|---|
| Experimental Model | Treatment | Duration of Treatment | Effects | Suggested Mediating Mechanisms | Ref. |
| co-culture model of human CMECs and adult rat cardiomyocytes stimulated by TNF-α | empagliflozin | correction of TNF-α-mediated impairment of CMEC cardiomyocyte interaction | reduction of mitochondrial ROS level, prevention of cytoplasmic ROS buildup and restoration of NO bioavailability | [274] | |
| co-culture model of human CMECs and adult rat cardiomyocytes exposed at uremic serum | empagliflozin | restoration of NO level in ECs | reversion of mitochondrial fission, responsible of ↑ ROS production and intracellular accumulation | [279] | |
| mice with metabolic syndrome and pre/early diabetes | empagliflozin | 10 weeks | ↑ CFVR, no difference in CD31 staining, and ↑ L-arginine and ↑ L-arginine/ADMA | NO-dependent improvement of endothelial function | [269] |
| STZ-induced diabetic mice | empagliflozin or an inhibitor of mitochondrial fission | 20 weeks | preserved cardiac microvascular barrier, sustained eNOS phosphorylation and endothelium-dependent relaxation, and increased microvessel density | ↓ mitochondrial fission through ↓ activation of Drp1 | [283] |
| mice subjected to myocardial I/R injury and CMECs from mice after myocardial I/R injury | in vivo pre-treatment with empagliflozin | 7 days | normalized mitochondrial fission/fusion, ↓ endothelial oxidative stress and hampered mitochondrial apoptotic signaling | ↑ FUNDC1-dependent mitophagy through AMPKα1 activation and ULK1 phosphorylation | [285] |
| mice model of HF (transv. aortic constriction) | empagliflozin | 2 weeks | improved capillary rarefaction and endothelial apoptosis | ↑ AKT/eNOS/NO pathway in ECs | [293] |
| yocardium from a rat model of HFpEF | empagliflozin | in vivo acute treatment | improved endothelial function by reduced oxidative stress and inflammation | reversed repression of NO–sGC–cGMP–PKG pathway and its downstream targets | [296] |
| Dahl salt-sensitive rats as a model of HFpEF | dapagliflozin | 6 weeks | ↓ endothelial inflammation/dysfunction | [297] | |
| Clinical Studies | |||||
| Subjects | Treatment | Duration of Treatment | Effects | Diagnostic Modality | Ref. |
| 13 T2DM people or placebo | empagliflozin | 4 weeks | ↓ MBF by 13% and no variation of adenosine stress MBF or MFR | 15O-H2O PET/CT | [299] |
| 90 T2DM patients with known CVD or high CV risk | empagliflozin or placebo | 13 weeks | no change in MFR | 82Rb-PET/CT | [300] |
| 19 T2DM patients | empagliflozin | 12 weeks | no improvement in CFVR | TTDE with adenosine stress | [301] |
| 160 T2DM patients | add-on to metformin of GLP-1RA, SGLT2 Is, or both, vs. insulin | 12 months | increase of endothelial glycocalyx thickness | measurements of perfused boundary region of sublingual arterial microvessels | [304] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Barrett, E.J.; Liu, Z.; Khamaisi, M.; King, G.L.; Klein, R.; Klein, B.E.K.; Hughes, T.M.; Craft, S.; Freedman, B.I.; Bowden, D.W.; et al. Diabetic Microvascular Disease: An Endocrine Society Scientific Statement. J. Clin. Endocrinol. Metab. 2017, 102, 4343–4410. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Control and Complications Trial Research Group; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865, Erratum in Lancet 1998, 352, 1558. [Google Scholar] [CrossRef]
- Eppens, M.C.; Craig, M.E.; Cusumano, J.; Hing, S.; Chan, A.K.; Howard, N.J.; Silink, M.; Donaghue, K.C. Prevalence of diabetes complications in adolescents with type 2 compared with type 1 diabetes. Diabetes Care 2006, 29, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Dabelea, D.; Stafford, J.M.; Mayer-Davis, E.J.; D’Agostino, R.; Dolan, L.; Imperatore, G.; Linder, B.; Lawrence, J.M.; Marcovina, S.M.; Mottl, A.K.; et al. Association of Type 1 Diabetes vs Type 2 Diabetes Diagnosed During Childhood and Adolescence with Complications During Teenage Years and Young Adulthood. JAMA 2017, 317, 825–835. [Google Scholar] [CrossRef]
- Ojaimi, E.; Nguyen, T.T.; Klein, R.; Islam, F.M.A.; Cotch, M.F.; Klein, B.E.; Wang, J.-J.; Wong, T.Y. Retinopathy Signs in People without Diabetes: The Multi-Ethnic Study of Atherosclerosis. Ophthalmology 2011, 118, 656–662. [Google Scholar] [CrossRef]
- Keenan, H.A.; Costacou, T.; Sun, J.K.; Doria, A.; Cavellerano, J.; Coney, J.; Orchard, T.J.; Aiello, L.P.; King, G.L. Clinical factors associated with resistance to microvascular complications in diabetic patients of extreme disease duration: The 50-year medalist study. Diabetes Care 2007, 30, 1995–1997. [Google Scholar] [CrossRef]
- Sun, J.K.; Keenan, H.A.; Cavallerano, J.D.; Asztalos, B.F.; Schaefer, E.J.; Sell, D.R.; Strauch, C.M.; Monnier, V.M.; Doria, A.; Aiello, L.P.; et al. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: The joslin 50-year medalist study. Diabetes Care 2011, 34, 968–974. [Google Scholar] [CrossRef]
- Horton, W.B.; Barrett, E.J. Microvascular Dysfunction in Diabetes Mellitus and Cardiometabolic Disease. Endocr. Rev. 2020, 42, 29–55. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Gelso, A.; Bono, V.; Costagliola, C.; Marfella, R.; Sardu, C.; Rinaldi, L.; Galiero, R.; Acierno, C.; et al. Telemedicine for screening diabetic retinopathy: The NO BLIND Italian multicenter study. Diabetes Metab. Res. Rev. 2018, 35, e3113. [Google Scholar] [CrossRef]
- Galiero, R.; Pafundi, P.C.; Nevola, R.; Rinaldi, L.; Acierno, C.; Caturano, A.; Salvatore, T.; Adinolfi, L.E.; Costagliola, C.; Sasso, F.C. The Importance of Telemedicine during COVID-19 Pandemic: A Focus on Diabetic Retinopathy. J. Diabetes Res. 2020, 2020, 9036847. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; De Nicola, L.; Carbonara, O.; Nasti, R.; Minutolo, R.; Salvatore, T.; Conte, G.; Torella, R. Cardiovascular Risk Factors and Disease Management in Type 2 Diabetic Patients with Diabetic Nephropathy. Diabetes Care 2006, 29, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Galiero, R.; Ricciardi, D.; Pafundi, P.C.; Todisco, V.; Tedeschi, G.; Cirillo, G.; Sasso, F.C. Whole plantar nerve conduction study: A new tool for early diagnosis of peripheral diabetic neuropathy. Diabetes Res. Clin. Pract. 2021, 176, 108856. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Salvatore, T.; Tranchino, G.; Cozzolino, D.; Caruso, A.A.; Persico, M.; Gentile, S.; Torella, D.; Torella, R. Cochlear dysfunction in type 2 diabetes: A complication independent of neuropathy and acute hyperglycemia. Metabolism 1999, 48, 1346–1350. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Simeon, V.; De Nicola, L.; Chiodini, P.; Galiero, R.; Rinaldi, L.; Nevola, R.; Salvatore, T.; Sardu, C.; et al. Efficacy and durability of multifactorial intervention on mortality and MACEs: A randomized clinical trial in type-2 diabetic kidney disease. Cardiovasc. Diabetol. 2021, 20, 145. [Google Scholar] [CrossRef]
- Marino, F.; Scalise, M.; Salerno, N.; Salerno, L.; Molinaro, C.; Cappetta, D.; Torella, M.; Greco, M.; Foti, D.; Sasso, F.C.; et al. Diabetes-Induced Cellular Senescence and Senescence-Associated Secretory Phenotype Impair Cardiac Regeneration and Function Independently of Age. Diabetes 2022, 71, 1081–1098. [Google Scholar] [CrossRef] [PubMed]
- Nitenberg, A.; Valensi, P.; Sachs, R.; Cosson, E.; Attali, J.-R.; Antony, I. Prognostic Value of Epicardial Coronary Artery Constriction to the Cold Pressor Test in Type 2 Diabetic Patients with Angiographically Normal Coronary Arteries and No Other Major Coronary Risk Factors. Diabetes Care 2004, 27, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Cortigiani, L.; Rigo, F.; Gherardi, S.; Galderisi, M.; Bovenzi, F.; Sicari, R. Prognostic Meaning of Coronary Microvascular Disease in Type 2 Diabetes Mellitus: A Transthoracic Doppler Echocardiographic Study. J. Am. Soc. Echocardiogr. 2014, 27, 742–748. [Google Scholar] [CrossRef]
- Murthy, V.L.; Naya, M.; Foster, C.R.; Gaber, M.; Hainer, J.; Klein, J.; Dorbala, S.; Blankstein, R.; Di Carli, M.F. Association Between Coronary Vascular Dysfunction and Cardiac Mortality in Patients with and Without Diabetes Mellitus. Circulation 2012, 126, 1858–1868. [Google Scholar] [CrossRef]
- Osborne, M.T.; Bajaj, N.S.; Taqueti, V.R.; Gupta, A.; Bravo, P.E.; Hainer, J.; Bibbo, C.F.; Dorbala, S.; Blankstein, R.; Di Carli, M.F. Coronary Microvascular Dysfunction Identifies Patients at High Risk of Adverse Events Across Cardiometabolic Diseases. J. Am. Coll. Cardiol. 2017, 70, 2835–2837. [Google Scholar] [CrossRef]
- Taqueti, V.R.; Solomon, S.D.; Shah, A.M.; Desai, A.S.; Groarke, J.D.; Osborne, M.T.; Hainer, J.; Bibbo, C.F.; Dorbala, S.; Blankstein, R.; et al. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur. Heart J. 2018, 39, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Indorkar, R.; Kwong, R.Y.; Romano, S.; White, B.E.; Chia, R.C.; Trybula, M.; Evans, K.; Shenoy, C.; Farzaneh-Far, A. Global Coronary Flow Reserve Measured During Stress Cardiac Magnetic Resonance Imaging Is an Independent Predictor of Adverse Cardiovascular Events. JACC Cardiovasc. Imaging 2019, 12, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Nitenberg, A.; Valensi, P.; Sachs, R.; Dali, M.; Aptecar, E.; Attali, J.-R. Impairment of Coronary Vascular Reserve and ACh-Induced Coronary Vasodilation in Diabetic Patients with Angiographically Normal Coronary Arteries and Normal Left Ventricular Systolic Function. Diabetes 1993, 42, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Nitenberg, A.; Ledoux, S.; Valensi, P.; Sachs, R.; Attali, J.-R.; Antony, I. Impairment of Coronary Microvascular Dilation in Response to Cold Pressor–Induced Sympathetic Stimulation in Type 2 Diabetic Patients with Abnormal Stress Thallium Imaging. Diabetes 2001, 50, 1180–1185. [Google Scholar] [CrossRef][Green Version]
- Labazi, H.; Trask, A.J. Coronary microvascular disease as an early culprit in the pathophysiology of diabetes and metabolic syndrome. Pharmacol. Res. 2017, 123, 114–121. [Google Scholar] [CrossRef]
- Sabe, S.A.; Feng, J.; Sellke, F.W.; Abid, M.R. Mechanisms and clinical implications of endothelium-dependent vasomotor dysfunction in coronary microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H819–H841. [Google Scholar] [CrossRef]
- Kaiser, N.; Sasson, S.; Feener, E.P.; Boukobza-Vardi, N.; Higashi, S.; Moller, D.E.; Davidheiser, S.; Przybylski, R.J.; King, G.L. Differential Regulation of Glucose Transport and Transporters by Glucose in Vascular Endothelial and Smooth Muscle Cells. Diabetes 1993, 42, 80–89. [Google Scholar] [CrossRef]
- Mandarino, L.J.; Finlayson, J.; Hassell, J.R. High glucose downregulates glucose transport activity in retinal capillary pericytes but not endothelial cells. Investig. Ophthalmol. Vis. Sci. 1994, 35, 964–972. [Google Scholar]
- Yokoyama, I.; Momomura, S.-I.; Ohtake, T.; Yonekura, K.; Nishikawa, J.; Sasaki, Y.; Omata, M. Reduced Myocardial Flow Reserve in Non–Insulin-Dependent Diabetes Mellitus. J. Am. Coll. Cardiol. 1997, 30, 1472–1477. [Google Scholar] [CrossRef]
- Feng, J.; Chu, L.M.; Dobrilovic, N.; Liu, Y.; Singh, A.K.; Sellke, F.W. Decreased coronary microvascular reactivity after cardioplegic arrest in patients with uncontrolled diabetes mellitus. Surgery 2012, 152, 262–269. [Google Scholar] [CrossRef]
- Pan, M.; Han, Y.; Basu, A.; Dai, A.; Si, R.; Willson, C.; Balistrieri, A.; Scott, B.T.; Makino, A. Overexpression of hexokinase 2 reduces mitochondrial calcium overload in coronary endothelial cells of type 2 diabetic mice. Am. J. Physiol. Physiol. 2018, 314, C732–C740. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, J.; Toan, S.; Muid, D.; Li, R.; Chang, X.; Zhou, H. Molecular mechanisms of coronary microvascular endothelial dys-function in diabetes mellitus: Focus on mitochondrial quality surveillance. Angiogenesis 2022, 25, 307–329. [Google Scholar] [CrossRef] [PubMed]
- Picchi, A.; Capobianco, S.; Qiu, T.; Focardi, M.; Zou, X.; Cao, J.M.; Zhang, C. Coronary microvascular dysfunction in diabetes mellitus: A review. World J. Cardiol. 2010, 2, 377–390. [Google Scholar] [CrossRef]
- Thornalley, P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems-role in ageing and disease. Drug Metabol. Drug Interact. 2008, 23, 125–150. [Google Scholar] [CrossRef]
- Matafome, P.; Sena, C.; Seiça, R. Methylglyoxal, obesity, and diabetes. Endocrine 2013, 43, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, H.; Schmidt, A.M.; Zhang, C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in Type 2 diabetic mice. Am. J. Physiol. Circ. Physiol. 2008, 295, H491–H498. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Ren, L.; Wei, Q.; Shao, H.; Chen, L.; Liu, N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc. Diabetol. 2017, 16, 1–12. [Google Scholar] [CrossRef]
- Luo, B.; Soesanto, Y.; McClain, D.A. Protein Modification by O-Linked GlcNAc Reduces Angiogenesis by Inhibiting Akt Activity in Endothelial Cells. Arter. Thromb. Vasc. Biol. 2008, 28, 651–657. [Google Scholar] [CrossRef]
- Bolanle, I.O.; Palmer, T.M. Targeting Protein O-GlcNAcylation, a Link between Type 2 Diabetes Mellitus and Inflammatory Disease. Cells 2022, 11, 705. [Google Scholar] [CrossRef]
- Leopold, J.A.; Cap, A.; Scribner, A.W.; Stanton, R.C.; Loscalzo, J. Glucose-6-phosphate dehydrogenase deficiency promotes endothelial oxidant stress and decreases endothelial nitric oxide bioavailability. FASEB J. 2001, 15, 1771–1773. [Google Scholar] [CrossRef]
- Das Evcimen, N.; King, G.L. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res. 2007, 55, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Tickerhoof, M.M.; Farrell, P.A.; Korzick, D.H. Alterations in rat coronary vasoreactivity and vascular protein kinase C isoforms in Type 1 diabetes. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2694–H2703. [Google Scholar] [CrossRef] [PubMed]
- Kuboki, K.; Jiang, Z.Y.; Takahara, N.; Ha, S.W.; Igarashi, M.; Yamauchi, T.; Feener, E.P.; Herbert, T.P.; Rhodes, C.J.; King, G.L. Regulation of Endothelial Constitutive Nitric Oxide Synthase Gene Expression in Endothelial Cells and In Vivo: A specific vascular action of insulin. Circulation 2000, 101, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Kobayashi, T.; Matsumoto, T.; Kamata, K. Dysfunction of endothelium-dependent relaxation to insulin via PKC-mediated GRK2/Akt activation in aortas of ob/ob mice. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H571–H583. [Google Scholar] [CrossRef] [PubMed]
- Thengchaisri, N.; Hein, T.W.; Ren, Y.; Kuo, L. Activation of Coronary Arteriolar PKCβ2 Impairs Endothelial NO-Mediated Vasodilation: Role of JNK/Rho Kinase Signaling and Xanthine Oxidase Activation. Int. J. Mol. Sci. 2021, 22, 9763. [Google Scholar] [CrossRef]
- Yuan, S.Y.; Ustinova, E.E.; Wu, M.H.; Tinsley, J.H.; Xu, W.; Korompai, F.L.; Taulman, A.C. Protein Kinase C Activation Contributes to Microvascular Barrier Dysfunction in the Heart at Early Stages of Diabetes. Circ. Res. 2000, 87, 412–417. [Google Scholar] [CrossRef]
- Haidari, M.; Zhang, W.; Willerson, J.T.; Dixon, R.A. Disruption of endothelial adherens junctions by high glucose is mediated by protein kinase C-β-dependent vascular endothelial cadherin tyrosine phosphorylation. Cardiovasc. Diabetol. 2014, 13, 105, Erratum in Cardiovasc. Diabetol. 2017, 16, 136. [Google Scholar] [CrossRef]
- Shah, M.S.; Brownlee, M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ. Res. 2016, 118, 1808–1829. [Google Scholar] [CrossRef]
- Wasserman, D.H.; Wang, T.; Brown, N.J. The Vasculature in Prediabetes. Circ. Res. 2018, 122, 1135–1150. [Google Scholar] [CrossRef]
- Liu, Z. Insulin at physiological concentrations increases microvascular perfusion in human myocardium. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1250–E1255. [Google Scholar] [CrossRef]
- Muniyappa, R.; Iantorno, M.; Quon, M.J. An Integrated View of Insulin Resistance and Endothelial Dysfunction. Endocrinol. Metab. Clin. North Am. 2008, 37, 685–711. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, L.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Morone, M.V.; Silvestri, C.F.; Giordano, M.; Salvatore, T.; Sasso, F.C. Mechanisms of Non-Alcoholic Fatty Liver Disease in the Metabolic Syndrome. A Narrative Review. Antioxidants 2021, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Aglitti, A.; Bucci, T.; Caruso, R.; Salvatore, T.; Sasso, F.C.; Tripodi, M.F.; Persico, M. Liver biopsy in type 2 diabetes mellitus: Steatohepatitis represents the sole feature of liver damage. PLoS ONE 2017, 12, e0178473. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Petta, S.; Fracanzani, A.L.; Nevola, R.; Coppola, C.; Narciso, V.; Rinaldi, L.; Calvaruso, V.; Pafundi, P.C.; Lombardi, R.; et al. Reduced incidence of type 2 diabetes in patients with chronic hepatitis C virus infection cleared by direct-acting antiviral therapy: A prospective study. Diabetes Obes. Metab. 2020, 22, 2408–2416. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; Pafundi, P.C.; Caturano, A.; Galiero, R.; Vetrano, E.; Nevola, R.; Petta, S.; Fracanzani, A.L.; Coppola, C.; Di Marco, V.; et al. Impact of direct acting antivirals (DAAs) on cardiovascular events in HCV cohort with pre-diabetes. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 2345–2353. [Google Scholar] [CrossRef]
- Adinolfi, L.E.; Petta, S.; Fracanzani, A.L.; Coppola, C.; Narciso, V.; Nevola, R.; Rinaldi, L.; Calvaruso, V.; Staiano, L.; Di Marco, V.; et al. Impact of hepatitis C virus clearance by direct-acting antiviral treatment on the incidence of major cardiovascular events: A prospective multicentre study. Atherosclerosis 2020, 296, 40–47. [Google Scholar] [CrossRef]
- Rinaldi, L.; Perrella, A.; Guarino, M.; De Luca, M.; Piai, G.; Coppola, N.; Pafundi, P.C.; Ciardiello, F.; Fasano, M.; Martinelli, E.; et al. Incidence and risk factors of early HCC occurrence in HCV patients treated with direct acting antivirals: A prospective multicentre study. J. Transl. Med. 2019, 17, 292. [Google Scholar] [CrossRef]
- Jagasia, D.; Whiting, J.M.; Concato, J.; Pfau, S.; McNulty, P.H. Effect of non-insulin-dependent diabetes mellitus on myocardial insulin responsiveness in patients with ischemic heart disease. Circulation 2001, 103, 1734–1739. [Google Scholar] [CrossRef]
- Sundell, J.; Laine, H.; Luotolahti, M.; Kalliokoski, K.; Raitakari, O.; Nuutila, P.; Knuuti, J. Obesity Affects Myocardial Vasoreactivity and Coronary Flow Response to Insulin. Obes. Res. 2002, 10, 617–624. [Google Scholar] [CrossRef]
- Laine, H.; Nuutila, P.; Luotolahti, M.; Meyer, C.; Elomaa, T.; Koskinen, P.; Rönnemaa, T.; Knuuti, J. Insulin-Induced Increment of Coronary Flow Reserve Is Not Abolished by Dexamethasone in Healthy Young Men1. J. Clin. Endocrinol. Metab. 2000, 85, 1868–1873. [Google Scholar] [CrossRef]
- Sundell, J.; Nuutila, P.; Laine, H.; Luotolahti, M.; Kalliokoski, K.; Raitakari, O.; Knuuti, J. Dose-Dependent Vasodilating Effects of Insulin on Adenosine-Stimulated Myocardial Blood Flow. Diabetes 2002, 51, 1125–1130. [Google Scholar] [CrossRef]
- Laine, H.; Sundell, J.; Nuutila, P.; Raitakari, O.T.; Luotolahti, M.; Rönnemaa, T.; Elomaa, T.; Koskinen, P.; Knuuti, J. Insulin induced increase in coronary flow reserve is abolished by dexamethasone in young men with uncomplicated type 1 diabetes. Heart 2004, 2004 90, 270–276. [Google Scholar] [CrossRef][Green Version]
- Sundell, J.; Laine, H.; Nuutila, P.; Rönnemaa, T.; Luotolahti, M.; Raitakari, O.; Knuuti, J. The effects of insulin and short-term hyper-glycaemia on myocardial blood flow in young men with uncomplicated Type I diabetes. Diabetologia 2002, 45, 775–782. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, J.; Jahn, L.A.; Fowler, D.E.; Barrett, E.J.; Cao, W.; Liu, Z. Free Fatty Acids Induce Insulin Resistance in Both Cardiac and Skeletal Muscle Microvasculature in Humans. J. Clin. Endocrinol. Metab. 2011, 96, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Picchi, A.; Gao, X.; Belmadani, S.; Potter, B.J.; Focardi, M.; Chilian, W.M.; Zhang, C. Tumor Necrosis Factor-α Induces Endothelial Dysfunction in the Prediabetic Metabolic Syndrome. Circ. Res. 2006, 99, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Boodhwani, M.; Sodha, N.R.; Mieno, S.; Xu, S.-H.; Feng, J.; Ramlawi, B.; Clements, R.T.; Sellke, F.W. Functional, Cellular, and Molecular Characterization of the Angiogenic Response to Chronic Myocardial Ischemia in Diabetes. Circulation 2007, 116, I31–I37. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cosentino, F.; Lüscher, T.F. Endothelial dysfunction in diabetes mellitus. J. Cardiovasc. Pharmacol. 1998, 32, S54–S61. [Google Scholar]
- Kibel, A.; Selthofer-Relatic, K.; Drenjancevic, I.; Bacun, T.; Bosnjak, I.; Kibel, D.; Gros, M. Coronary microvascular dysfunction in diabetes mellitus. J. Int. Med Res. 2017, 45, 1901–1929. [Google Scholar] [CrossRef]
- Bucala, R.; Tracey, K.J.; Cerami, A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J. Clin. Investig. 1991, 87, 432–438. [Google Scholar] [CrossRef]
- Christ, M.; Bauersachs, J.; Liebetrau, C.; Heck, M.; Günther, A.; Wehling, M. Glucose Increases Endothelial-Dependent Superoxide Formation in Coronary Arteries by NAD(P)H Oxidase Activation: Attenuation by the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor atorvastatin. Diabetes 2002, 51, 2648–2652. [Google Scholar] [CrossRef]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Ito, A.; Asagami, T.; Tsao, P.S.; Adimoolam, S.; Kimoto, M.; Tsuji, H.; Reaven, G.M.; Cooke, J.P. Impaired Nitric Oxide Synthase Pathway in Diabetes Mellitus: Role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation 2002, 106, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Elms, S.C.; Toque, H.A.; Rojas, M.; Xu, Z.; Caldwell, R.W.; Caldwell, R.B. The role of arginase I in diabetes-induced retinal vascular dysfunction in mouse and rat models of diabetes. Diabetologia 2012, 56, 654–662. [Google Scholar] [CrossRef]
- Toque, H.A.; Nunes, K.P.; Yao, L.; Xu, Z.; Kondrikov, D.; Su, Y.; Webb, R.C.; Caldwell, R.B.; Caldwell, R.W. Akita Spontaneously Type 1 Diabetic Mice Exhibit Elevated Vascular Arginase and Impaired Vascular Endothelial and Nitrergic Function. PLoS ONE 2013, 8, e72277. [Google Scholar] [CrossRef]
- Knapp, M.; Tu, X.; Wu, R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol. Sin. 2019, 40, 1–8. [Google Scholar] [CrossRef]
- Muzaffar, S.; Shukla, N.; Bond, M.; Sala-Newby, G.B.; Newby, A.C.; Angelini, G.D.; Jeremy, J.Y. Superoxide from NADPH oxidase upregulates type 5 phosphodiesterase in human vascular smooth muscle cells: Inhibition with iloprost and NONOate. J. Cereb. Blood Flow Metab. 2008, 155, 847–856. [Google Scholar] [CrossRef]
- Muzaffar, S.; Jeremy, J.Y.; Angelini, G.D.; Shukla, N. NADPH oxidase 4 mediates upregulation of type 4 phosphodiesterases in human endothelial cells. J. Cell. Physiol. 2012, 227, 1941–1950. [Google Scholar] [CrossRef]
- Csanyi, G.; Lepran, I.; Flesch, T.; Telegdy, G.; Szabo, G.; Mezei, Z. Lack of endothelium-derived hyperpolarizing factor (EDHF) up-regulation in endothelial dysfunction in aorta in diabetic rats. Pharmacol. Rep. 2007, 59, 447–455. [Google Scholar] [PubMed]
- Grassi, G.; Seravalle, G.; Quarti-Trevano, F.; Scopelliti, F.; Dell’Oro, R.; Bolla, G.; Mancia, G. Excessive Sympathetic Activation in Heart Failure With Obesity and Metabolic Syndrome: Characteristics and mechanisms. Hypertension 2007, 49, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Kachur, S.; Morera, R.; De Schutter, A.; Lavie, C.J. Cardiovascular Risk in Patients with Prehypertension and the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 15. [Google Scholar] [CrossRef]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered Mitochondrial Dynamics Contributes to Endothelial Dysfunction in Diabetes Mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Pangare, M.; Makino, A. Mitochondrial function in vascular endothelial cell in diabetes. J. Smooth Muscle Res. 2012, 48, 1–26. [Google Scholar] [CrossRef]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengellér, Z.; Szabó, C.; Brownlee, M. Inhibition of GAPDH activity by poly (ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef]
- Bakuy, V.; Unal, O.; Gursoy, M.; Kunt, A.; Ozisik, K.; Sargon, M.; Emir, M.; Sener, E. Electron Microscopic Evaluation of Internal Thoracic Artery Endothelial Morphology in Diabetic Coronary Bypass Patients. Ann. Thorac. Surg. 2014, 97, 851–857. [Google Scholar] [CrossRef]
- Makino, A.; Scott, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794. [Google Scholar] [CrossRef]
- Masi, S.; Rizzoni, D.; Taddei, S.; Widmer, R.J.; Montezano, A.C.; Lüscher, T.F.; Schiffrin, E.L.; Touyz, R.M.; Paneni, F.; Lerman, A.; et al. Assessment and pathophysiology of microvascular disease: Recent progress and clinical implications. Eur. Heart J. 2020, 42, 2590–2604. [Google Scholar] [CrossRef]
- Lee, S.; Park, Y.; Zhang, C. Exercise Training Prevents Coronary Endothelial Dysfunction in Type 2 Diabetic Mice. Am. J. Biomed. Sci. 2011, 3, 241–252. [Google Scholar] [CrossRef]
- Zhang, C.; Park, Y.; Picchi, A.; Potter, B.J. Maturation-induces endothelial dysfunction via vascular inflammation in diabetic mice. Basic Res. Cardiol. 2008, 103, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Capobianco, S.; Gao, X.; Falck, J.R.; Dellsperger, K.C.; Zhang, C. Role of EDHF in type 2 diabetes-induced endothelial dysfunction. Am. J. Physiol. Circ. Physiol. 2008, 295, H1982–H1988. [Google Scholar] [CrossRef]
- Liu, Y.; Bubolz, A.H.; Mendoza, S.; Zhang, D.X.; Gutterman, D.D. H2O2 Is the Transferrable Factor Mediating Flow-Induced Dilation in Human Coronary Arterioles. Circ. Res. 2011, 108, 566–573. [Google Scholar] [CrossRef]
- Shafique, E.; Choy, W.C.; Liu, Y.; Feng, J.; Cordeiro, B.; Lyra, A.; Arafah, M.; Yassin-Kassab, A.; Zanetti, A.V.; Clements, R.T.; et al. Oxidative stress improves coronary endothelial function through activation of the pro-survival kinase AMPK. Aging 2013, 5, 515–530. [Google Scholar] [CrossRef]
- Feng, J.; Damrauer, S.M.; Lee, M.; Sellke, F.W.; Ferran, C.; Abid, M.R. Endothelium-Dependent Coronary Vasodilatation Requires NADPH Oxidase-Derived Reactive Oxygen Species. Arter. Thromb. Vasc. Biol. 2010, 30, 1703–1710. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Del Buono, M.G.; Montone, R.A.; Camilli, M.; Carbone, S.; Narula, J.; Lavie, C.J.; Niccoli, G.; Crea, F. Coronary Microvascular Dysfunction Across the Spectrum of Cardiovascular Diseases: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2021, 78, 1352–1371. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pagano, P.J. Microvascular NADPH oxidase in health and disease. Free Radic. Biol. Med. 2017, 109, 33–47. [Google Scholar] [CrossRef]
- Magenta, A.; Greco, S.; Capogrossi, M.C.; Gaetano, C.; Martelli, F. Nitric Oxide, Oxidative Stress, andp66ShcInterplay in Diabetic Endothelial Dysfunction. BioMed. Res. Int. 2014, 2014, 193095. [Google Scholar] [CrossRef]
- Qamirani, E.; Ren, Y.; Kuo, L.; Hein, T.W. C-Reactive Protein Inhibits Endothelium-Dependent NO-Mediated Dilation in Coronary Arterioles by Activating p38 Kinase and NAD(P)H Oxidase. Arter. Thromb. Vasc. Biol. 2005, 25, 995–1001. [Google Scholar] [CrossRef]
- Hein, T.W.; Qamirani, E.; Ren, Y.; Kuo, L. C-reactive protein impairs coronary arteriolar dilation to prostacyclin synthase activation: Role of peroxynitrite. J. Mol. Cell. Cardiol. 2009, 47, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; You, B.; Chen, S.-P.; Liao, J.K.; Sun, J. Tumor Necrosis Factor-α Downregulates Endothelial Nitric Oxide Synthase mRNA Stability via Translation Elongation Factor 1-α 1. Circ. Res. 2008, 103, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hein, T.W.; Wang, W.; Ren, Y.; Shipley, R.D.; Kuo, L. Activation of JNK and xanthine oxidase by TNF-α impairs nitric oxide-mediated dilation of coronary arterioles. J. Mol. Cell. Cardiol. 2006, 40, 247–257. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, X.; Potter, B.J.; Wang, W.; Kuo, L.; Michael, L.; Bagby, G.J.; Chilian, W.M. TNF-α Contributes to Endothelial Dysfunction in Ischemia/Reperfusion Injury. Arter. Thromb. Vasc. Biol. 2006, 26, 475–480. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Zhang, H.; Hill, M.A.; Zhang, C.; Park, Y. Interaction of IL-6 and TNF-α contributes to endothelial dysfunction in type 2 diabetic mouse hearts. PLoS ONE 2017, 12, e0187189. [Google Scholar] [CrossRef]
- de Martin, R.; Hoeth, M.; Hofer-Warbinek, R.; Schmid, J.A. The Transcription Factor NF-κB and the Regulation of Vascular Cell Function. Arter. Thromb. Vasc. Biol. 2000, 20, e83–e88. [Google Scholar] [CrossRef]
- Sezer, M.; Kocaaga, M.; Aslanger, E.; Atici, A.; Demirkiran, A.; Bugra, Z.; Umman, S.; Umman, B. Bimodal Pattern of Coronary Microvascular Involvement in Diabetes Mellitus. J. Am. Heart Assoc. 2016, 5, e003995, Erratum in: J. Am. Heart Assoc. 2017, 6, e002163. [Google Scholar] [CrossRef]
- Godo, S.; Suda, A.; Takahashi, J.; Yasuda, S.; Shimokawa, H. Coronary Microvascular Dysfunction. Arter. Thromb. Vasc. Biol. 2021, 41, 1625–1637. [Google Scholar] [CrossRef]
- Padro, T.; Manfrini, O.; Bugiardini, R.; Canty, J.; Cenko, E.; De Luca, G.; Duncker, D.J.; Eringa, E.C.; Koller, A.; Tousoulis, D.; et al. ESC Working Group on Coronary Pathophysiology and Microcirculation position paper on ‘coronary microvascular dysfunction in cardiovascular disease’. Cardiovasc. Res. 2020, 116, 741–755. [Google Scholar] [CrossRef]
- Hinkel, R.; Howe, A.; Renner, S.; Ng, J.; Lee, S.; Klett, K.; Kaczmarek, V.; Moretti, A.; Laugwitz, K.-L.; Skroblin, P.; et al. Diabetes Mellitus–Induced Microvascular Destabilization in the Myocardium. J. Am. Coll. Cardiol. 2017, 69, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Myers, P.R.; Tanner, M.A. Vascular Endothelial Cell Regulation of Extracellular Matrix Collagen: Role of nitric oxide. Arter. Thromb. Vasc. Biol. 1998, 18, 717–722. [Google Scholar] [CrossRef]
- O’Riordan, E.; Mendelev, N.; Patschan, S.; Patschan, D.; Eskander, J.; Cohen-Gould, L.; Chander, P.; Goligorsky, M.S. Chronic NOS inhibition actuates endothelial-mesenchymal transformation. Am. J. Physiol. Circ. Physiol. 2007, 292, H285–H294. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Van Linthout, S. New Insights in (Inter)Cellular Mechanisms by Heart Failure with Preserved Ejection Fraction. Curr. Heart Fail. Rep. 2014, 11, 436–444. [Google Scholar] [CrossRef]
- Virdis, A.; Neves, M.F.; Amiri, F.; Touyz, R.M.; Schiffrin, E.L. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J. Hypertens. 2004, 22, 535–542. [Google Scholar] [CrossRef]
- Cho, Y.-E.; Basu, A.; Dai, A.; Heldak, M.; Makino, A. Coronary endothelial dysfunction and mitochondrial reactive oxygen species in type 2 diabetic mice. Am. J. Physiol. Cell Physiol. 2013, 305, C1033–C1040. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, L.; Zhang, M.; Hu, J.; Wang, T.; Duan, Y.; Man, W.; Wu, B.; Feng, J.; Sun, L.; et al. Mst1 inhibits CMECs autophagy and participates in the development of diabetic coronary microvascular dysfunction. Sci. Rep. 2016, 6, 34199. [Google Scholar] [CrossRef]
- Wynne, B.M.; Chiao, C.-W.; Webb, R.C. Vascular smooth muscle cell signaling mechanisms for contraction to angiotensin II and endothelin-1. J. Am. Soc. Hypertens. 2009, 3, 84–95. [Google Scholar] [CrossRef]
- Husarek, K.E.; Katz, P.S.; Trask, A.J.; Galantowicz, M.L.; Cismowski, M.J.; Lucchesi, P.A. The angiotensin receptor blocker losartan reduces coronary arteriole remodeling in type 2 diabetic mice. Vasc. Pharmacol. 2015, 76, 28–36. [Google Scholar] [CrossRef]
- Katz, P.S.; Trask, A.J.; Souza-Smith, F.M.; Hutchinson, K.R.; Galantowicz, M.L.; Lord, K.C.; Stewart, J.A., Jr.; Cismowski, M.J.; Varner, K.J.; Lucchesi, P.A. Coronary arterioles in type 2 diabetic (db/db) mice undergo a distinct pattern of remodeling associated with decreased vessel stiffness. Basic Res. Cardiol. 2011, 106, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Trask, A.J.; Katz, P.S.; Kelly, A.P.; Galantowicz, M.L.; Cismowski, M.J.; West, T.A.; Neeb, Z.P.; Berwick, Z.C.; Goodwill, A.; Alloosh, M.; et al. Dynamic micro- and macrovascular remodeling in coronary circulation of obese Ossabaw pigs with metabolic syndrome. J. Appl. Physiol. 2012, 113, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Trask, A.J.; Delbin, M.A.; Katz, P.S.; Zanesco, A.; Lucchesi, P.A. Differential coronary resistance microvessel remodeling between type 1 and type 2 diabetic mice: Impact of exercise training. Vasc. Pharmacol. 2012, 57, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Lipes, M.A.; Galderisi, A. Cardiac Autoimmunity as a Novel Biomarker, Mediator, and Therapeutic Target of Heart Disease in Type 1 Diabetes. Curr. Diabetes Rep. 2015, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, M.; Nádasy, G.L.; Dörnyei, G.; Szénási, A.; Koller, A. Remodeling of Wall Mechanics and the Myogenic Mechanism of Rat Intramural Coronary Arterioles in Response to a Short-Term Daily Exercise Program: Role of Endothelial Factors. J. Vasc. Res. 2018, 55, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Kushibiki, M.; Yamada, M.; Oikawa, K.; Tomita, H.; Osanai, T.; Okumura, K. Aldosterone causes vasoconstriction in coronary arterioles of rats via angiotensin II type-1 receptor: Influence of hypertension. Eur. J. Pharmacol. 2007, 572, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Rein, K.M.; Rusch, N.J. Distinct endothelial impairment in coronary microvessels from hypertensive Dahl rats. Hypertension 1998, 1, 328–334. [Google Scholar] [CrossRef]
- Siasos, G.; Tsigkou, V.; Zaromytidou, M.; Sara, J.D.; Varshney, A.; Coskun, A.U.; Lerman, A.; Stone, P.H. Role of local coronary blood flow patterns and shear stress on the development of microvascular and epicardial endothelial dysfunction and coronary plaque. Curr. Opin. Cardiol. 2018, 33, 638–644. [Google Scholar] [CrossRef]
- Costantino, S.; Paneni, F.; Virdis, A.; Hussain, S.; Mohammed, S.A.; Capretti, G.; Akhmedov, A.; Dalgaard, K.; Chiandotto, S.; Pospisilik, J.A.; et al. Interplay among H3K9-editing enzymes SUV39H1, JMJD2C and SRC-1 drives p66Shc transcription and vascular oxidative stress in obesity. Eur. Heart J. 2017, 40, 383–391. [Google Scholar] [CrossRef]
- Costantino, S.; Paneni, F.; Mitchell, K.; Mohammed, S.A.; Hussain, S.; Gkolfos, C.; Berrino, L.; Volpe, M.; Schwarzwald, C.; Lüscher, T.F.; et al. Hyperglycaemia-induced epigenetic changes drive persistent cardiac dysfunction via the adaptor p66Shc. Int. J. Cardiol. 2018, 268, 179–186. [Google Scholar] [CrossRef]
- Paneni, F.; Mocharla, P.; Akhmedov, A.; Costantino, S.; Osto, E.; Volpe, M.; Lüscher, T.F.; Cosentino, F. Gene Silencing of the Mitochondrial Adaptor p66Shc Suppresses Vascular Hyperglycemic Memory in Diabetes. Circ. Res. 2012, 111, 278–289. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, X.; Icli, B.; Feinberg, M.W. Emerging Roles for MicroRNAs in Diabetic Microvascular Disease: Novel Targets for Therapy. Endocr. Rev. 2017, 38, 145–168. [Google Scholar] [CrossRef]
- Samak, M.; Kaltenborn, D.; Kues, A.; Le Noble, F.; Hinkel, R.; Germena, G. Micro-RNA 92a as a Therapeutic Target for Cardiac Microvascular Dysfunction in Diabetes. Biomedicines 2021, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Song, Q.; Hu, C.; Da, X.; Yu, Y.; He, Z.; Xu, C.; Chen, Q.; Wang, Q.K. Endothelial cell metabolic memory causes cardiovascular dysfunction in diabetes. Cardiovasc. Res. 2021, 118, 196–211. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.P.; Kirkeeide, R.L.; Gould, K.L. Is Discordance of Coronary Flow Reserve and Fractional Flow Reserve Due to Methodology or Clinically Relevant Coronary Pathophysiology? JACC Cardiovasc. Imaging 2012, 5, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Shin, D.; Lee, J.M.; van de Hoef, T.P.; Hong, D.; Choi, K.H.; Hwang, D.; Boerhout, C.K.M.; de Waard, G.A.; Jung, J.; et al. Clinical Relevance of Ischemia with Nonobstructive Coronary Arteries According to Coronary Microvascular Dysfunction. J. Am. Heart Assoc. 2022, 11, e025171. [Google Scholar] [CrossRef]
- Sara, J.D.; Taher, R.; Kolluri, N.; Vella, A.; Lerman, L.O.; Lerman, A. Coronary microvascular dysfunction is associated with poor glycemic control amongst female diabetics with chest pain and non-obstructive coronary artery disease. Cardiovasc. Diabetol. 2019, 18, 22. [Google Scholar] [CrossRef]
- Jaffe, R.; Charron, T.; Puley, G.; Dick, A.; Strauss, B.H. Microvascular Obstruction and the No-Reflow Phenomenon After Percutaneous Coronary Intervention. Circulation 2008, 117, 3152–3156. [Google Scholar] [CrossRef]
- Kali, A.; Cokic, I.; Tang, R.; Dohnalkova, A.; Kovarik, L.; Yang, H.-J.; Kumar, A.; Prato, F.S.; Wood, J.C.; Underhill, D.; et al. Persistent Microvascular Obstruction After Myocardial Infarction Culminates in the Confluence of Ferric Iron Oxide Crystals, Proinflammatory Burden, and Adverse Remodeling. Circ. Cardiovasc. Imaging 2016, 9, e004996. [Google Scholar] [CrossRef]
- De Waha, S.; Patel, M.R.; Granger, C.B.; Ohman, E.M.; Maehara, A.; Eitel, I.; Ben-Yehuda, O.; Jenkins, P.; Thiele, H.; Stone, G.W. Relationship between microvascular obstruction and adverse events following primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: An individual patient data pooled analysis from seven randomized trials. Eur. Heart J. 2017, 38, 3502–3510. [Google Scholar] [CrossRef]
- Montone, R.A.; Camilli, M.; Del Buono, M.G.; Meucci, M.C.; Gurgoglione, F.; Russo, M.; Crea, F.; Niccoli, G. “No-reflow”: Update su diagnosi, fisiopatologia e strategie terapeutiche [No-reflow: Update on diagnosis, pathophysiology and therapeutic strategies]. G Ital. Cardiol. 2020, 21, 4S–14S. (In Italian) [Google Scholar] [CrossRef]
- Iwakura, K.; Ito, H.; Ikushima, M.; Kawano, S.; Okamura, A.; Asano, K.; Kuroda, T.; Tanaka, K.; Masuyama, T.; Hori, M.; et al. Association between hyperglycemia and the no-reflow phenomenon in patients with acute myocardial infarction. J. Am. Coll. Cardiol. 2003, 41, 1–7. [Google Scholar] [CrossRef]
- Liu, F.; Huang, R.; Li, Y.; Zhao, S.; Gong, Y.; Xu, Z. In-Hospital Peak Glycemia in Predicting No-Reflow Phenomenon in Diabetic Patients with STEMI Treated with Primary Percutaneous Coronary Intervention. J. Diabetes Res. 2021, 2021, 6683937. [Google Scholar] [CrossRef]
- Zhao, S.-R.; Huang, R.; Liu, F.; Li, Y.; Gong, Y.; Xing, J. Study on Correlation between Type 2 Diabetes and No-Reflow after PCI. Dis. Markers 2022, 2022, 7319277. [Google Scholar] [CrossRef]
- Dryer, K.; Gajjar, M.; Narang, N.; Lee, M.; Paul, J.; Shah, A.P.; Nathan, S.; Butler, J.; Davidson, C.J.; Fearon, W.F.; et al. Coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Am. J. Physiol. Circ. Physiol. 2018, 314, H1033–H1042. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Camici, P.G.; Tschöpe, C.; Di Carli, M.F.; Rimoldi, O.; Van Linthout, S. Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc. Res. 2020, 116, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Tromp, J.; Lim, S.L.; Tay, W.T.; Teng, T.-H.K.; Chandramouli, C.; Ouwerkerk, W.; Wander, G.S.; Sawhney, J.P.; Yap, J.; MacDonald, M.R.; et al. Microvascular Disease in Patients with Diabetes with Heart Failure and Reduced Ejection Versus Preserved Ejection Fraction. Diabetes Care 2019, 42, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Taqueti, V.R.; Di Carli, M.F. Coronary Microvascular Disease Pathogenic Mechanisms and Therapeutic Options: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 2625–2641. [Google Scholar] [CrossRef]
- Badimon, L.; Bugiardini, R.; Cenko, E.; Cubedo, J.; Dorobantu, M.; Duncker, D.J.; Estruch, R.; Milicic, D.; Tousoulis, D.; Vasiljevic, Z.; et al. Position paper of the European Society of Cardiology–working group of coronary pathophysiology and microcirculation: Obesity and heart disease. Eur. Heart J. 2017, 38, 1951–1958. [Google Scholar] [CrossRef]
- Mavri, A.; Poredoš, P.; Suran, D.; Gaborit, B.; Juhan-Vague, I.; Poredoš, P. Effect of diet-induced weight loss on endothelial dysfunction: Early improvement after the first week of dieting. Heart Vessel. 2011, 26, 31–38. [Google Scholar] [CrossRef]
- Csipo, T.; Fulop, G.A.; Lipecz, A.; Tarantini, S.; Kiss, T.; Balasubramanian, P.; Csiszar, A.; Ungvari, Z.; Yabluchanskiy, A. Short-term weight loss reverses obesity-induced microvascular endothelial dysfunction. GeroScience 2018, 40, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.H.; Pedersen, L.R.; Jürs, A.; Snoer, M.; Haugaard, S.B.; Prescott, E. A randomised trial comparing the effect of exercise training and weight loss on microvascular function in coronary artery disease. Int. J. Cardiol. 2015, 185, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, N.S.; Osborne, M.T.; Gupta, A.; Tavakkoli, A.; Bravo, P.E.; Vita, T.; Bibbo, C.F.; Hainer, J.; Dorbala, S.; Blankstein, R.; et al. Coronary Microvascular Dysfunction and Cardiovascular Risk in Obese Patients. J. Am. Coll. Cardiol. 2018, 72, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Nerla, R.; Tarzia, P.; Sestito, A.; Di Monaco, A.; Infusino, F.; Matera, D.; Greco, F.; Tacchino, R.M.; Lanza, G.A.; Crea, F. Effect of bariatric surgery on peripheral flow-mediated dilation and coronary microvascular function. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 626–634. [Google Scholar] [CrossRef]
- Quercioli, A.; Montecucco, F.; Pataky, Z.; Thomas, A.; Ambrosio, G.; Staub, C.; Di Marzo, V.; Ratib, O.; Mach, F.; Golay, A.; et al. Improvement in coronary circulatory function in morbidly obese individuals after gastric bypass-induced weight loss: Relation to alterations in endocannabinoids and adipocytokines. Eur. Heart J. 2013, 34, 2063–2073. [Google Scholar] [CrossRef]
- Guethlin, M.; Kasel, A.M.; Coppenrath, K.; Ziegler, S.; Delius, W.; Schwaiger, M. Delayed response of myocardial flow reserve to lipid-lowering therapy with Fluvastatin. Circulation 1999, 99, 475–481. [Google Scholar] [CrossRef]
- Solberg, O.G.; Stavem, K.; Ragnarsson, A.; Beitnes, J.O.; Skårdal, R.; Seljeflot, I.; Ueland, T.; Aukrust, P.; Gullestad, L.; Aaberge, L. Index of microvascular resistance to assess the effect of rosuvastatin on microvascular function in women with chest pain and no obstructive coronary artery disease: A double-blind randomized study. Catheter. Cardiovasc. Interv. 2019, 94, 660–668. [Google Scholar] [CrossRef]
- Baan, J., Jr.; Chang, P.C.; Vermeij, P.; Pfaffendorf, M.; van Zwieten, P.A. Effects of losartan on vasoconstrictor responses to angiotensin II in the forearm vascular bed of healthy volunteers. Cardiovasc. Res. 1996, 32, 973–979, Erratum in Cardiovasc. Res. 1996, 32, 1167. [Google Scholar] [CrossRef]
- Del Buono, M.G.; Iannaccone, G.; Scacciavillani, R.; Carbone, S.; Camilli, M.; Niccoli, G.; Borlaug, B.A.; Lavie, C.J.; Arena, R.; Crea, F.; et al. Heart failure with preserved ejection fraction diagnosis and treatment: An updated review of the evidence. Prog. Cardiovasc. Dis. 2020, 63, 570–584. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Junbo Ge, D.P.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin–Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef]
- Merz, C.N.B.; Handberg, E.M.; Shufelt, C.L.; Mehta, P.K.; Minissian, M.B.; Wei, J.; Thomson, L.E.; Berman, D.S.; Shaw, L.J.; Petersen, J.; et al. A randomized, placebo-controlled trial of late Na current inhibition (ranolazine) in coronary microvascular dysfunction (CMD): Impact on angina and myocardial perfusion reserve. Eur. Heart J. 2015, 37, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.R.; Cheezum, M.K.; Veeranna, V.; Horgan, S.J.; Taqueti, V.R.; Murthy, V.L.; Foster, C.; Hainer, J.; Daniels, K.M.; Rivero, J.; et al. Ranolazine in Symptomatic Diabetic Patients Without Obstructive Coronary Artery Disease: Impact on Microvascular and Diastolic Function. J. Am. Heart Assoc. 2017, 6, e005027. [Google Scholar] [CrossRef]
- Zhu, H.; Xu, X.; Fang, X.; Zheng, J.; Zhao, Q.; Chen, T.; Huang, J. Effects of the Antianginal Drugs Ranolazine, Nicorandil, and Ivabradine on Coronary Microvascular Function in Patients with Nonobstructive Coronary Artery Disease: A Meta-analysis of Randomized Controlled Trials. Clin. Ther. 2019, 41, 2137–2152.e12. [Google Scholar] [CrossRef]
- Vazzana, N.; Ranalli, P.; Cuccurullo, C.; Davì, G. Diabetes mellitus and thrombosis. Thromb. Res. 2012, 129, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association 10. Cardiovascular disease and risk management: Standards of medical care in diabetes—2019. Diabetes Care 2019, 42, S103–S123. [Google Scholar] [CrossRef]
- Capodanno, D.; Angiolillo, D.J. Antithrombotic Therapy for Atherosclerotic Cardiovascular Disease Risk Mitigation in Patients with Coronary Artery Disease and Diabetes Mellitus. Circulation 2020, 142, 2172–2188. [Google Scholar] [CrossRef]
- Moses, J.W.; Mehran, R.; Dangas, G.D.; Kobayashi, Y.; Lansky, A.J.; Mintz, G.S.; Aymong, E.D.; Fahy, M.; Stone, G.W.; Leon, M.B. Short- and long-term results after multivessel stenting in diabetic patients. J. Am. Coll. Cardiol. 2004, 43, 1348–1354. [Google Scholar] [CrossRef]
- Donahoe, S.M.; Stewart, G.C.; McCabe, C.H.; Mohanavelu, S.; Murphy, S.A.; Cannon, C.P.; Antman, E.M. Diabetes and Mortality Following Acute Coronary Syndromes. JAMA 2007, 298, 765–775. [Google Scholar] [CrossRef]
- Cattaneo, M.; Schulz, R.; Nylander, S. Adenosine-Mediated Effects of Ticagrelor: Evidence and potential clinical relevance. J. Am. Coll. Cardiol. 2014, 63, 2503–2509. [Google Scholar] [CrossRef] [PubMed]
- Echavarría-Pinto, M.; Gonzalo, N.; Ibañez, B.; Petraco, R.; Jimenez-Quevedo, P.; Sen, S.; Nijjer, S.; Tarkin, J.; Alfonso, F.; Núñez-Gil, I.J.; et al. Low Coronary Microcirculatory Resistance Associated with Profound Hypotension During Intravenous Adenosine Infusion: Implications for the functional assessment of coronary stenoses. Circ. Cardiovasc. Interv. 2014, 7, 35–42. [Google Scholar] [CrossRef]
- Liu, Y.; Ding, L.-Y.; Li, X.-Z. Therapy with ticagrelor for ST-elevated acute coronary syndrome accompanied by diabetes mellitus. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Caturano, A.; Vetrano, E.; Aprea, C.; Albanese, G.; Di Martino, A.; Ricozzi, C.; et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines 2020, 9, 3. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Morgillo, F.; Di Liello, R.; Galiero, R.; Nevola, R.; Marfella, R.; Monaco, L.; Rinaldi, L.; Adinolfi, L.E.; et al. Metformin: An old drug against old age and associated morbidities. Diabetes Res. Clin. Pract. 2020, 160, 108025. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of Incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Sherman, S.I.; Gorelick, F.S.; Bergenstal, R.M.; Sherwin, R.S.; Buse, J.B. Incretin-Based Therapies for the Treatment of Type 2 Diabetes: Evaluation of the Risks and Benefits. Diabetes Care 2010, 33, 428–433. [Google Scholar] [CrossRef]
- Nachawi, N.; Rao, P.P.; Makin, V. The role of GLP-1 receptor agonists in managing type 2 diabetes. Cleve. Clin. J. Med. 2022, 89, 457–464. [Google Scholar] [CrossRef]
- Huthmacher, J.A.; Meier, J.J.; Nauck, M.A. Efficacy and Safety of Short- and Long-Acting Glucagon-Like Peptide 1 Receptor Agonists on a Background of Basal Insulin in Type 2 Diabetes: A Meta-analysis. Diabetes Care 2020, 43, 2303–2312. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.E.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D'Agostino, R.B.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): A double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785, Erratum in Lancet Diabetes Endocrinol. 2020, 8, e2. [Google Scholar] [CrossRef]
- Ferrannini, G.; Gerstein, H.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Dyal, L.; Lakshmanan, M.; Mellbin, L.; Probstfield, J.; Riddle, M.C.; et al. Similar cardiovascular outcomes in patients with diabetes and established or high risk for coronary vascular disease treated with dulaglutide with and without baseline metformin. Eur. Heart J. 2020, 42, 2565–2573. [Google Scholar] [CrossRef]
- Crowley, M.J.; McGuire, D.K.; Alexopoulos, A.-S.; Jensen, T.J.; Rasmussen, S.; Saevereid, H.A.; Verma, S.; Buse, J.B. Effects of Liraglutide on Cardiovascular Outcomes in Type 2 Diabetes Patients with and Without Baseline Metformin Use: Post Hoc Analyses of the LEADER Trial. Diabetes Care 2020, 43, e108–e110. [Google Scholar] [CrossRef]
- Boyle, J.G.; Livingstone, R.; Petrie, J.R. Cardiovascular benefits of GLP-1 agonists in type 2 diabetes: A comparative review. Clin. Sci. 2018, 132, 1699–1709. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee 10. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes—2022. Diabetes Care 2021, 45, S144–S174, Erratum in Diabetes Care 2022, 45, 2178–2181. [Google Scholar] [CrossRef]
- Sun, F.; Wu, S.; Guo, S.; Yu, K.; Yang, Z.; Li, L.; Zhang, Y.; Quan, X.; Ji, L.; Zhan, S. Impact of GLP-1 receptor agonists on blood pressure, heart rate and hypertension among patients with type 2 diabetes: A systematic review and network meta-analysis. Diabetes Res. Clin. Pr. 2015, 110, 26–37. [Google Scholar] [CrossRef]
- Rizzo, M.; Rizvi, A.A.; Patti, A.M.; Nikolic, D.; Giglio, R.V.; Castellino, G.; Volti, G.L.; Caprio, M.; Montalto, G.; Provenzano, V.; et al. Liraglutide improves metabolic parameters and carotid intima-media thickness in diabetic patients with the metabolic syndrome: An 18-month prospective study. Cardiovasc. Diabetol. 2016, 15, 162. [Google Scholar] [CrossRef]
- Wilding, J.P.H.; Batterham, R.L.; Calanna, S.; Davies, M.; Van Gaal, L.F.; Lingvay, I.; McGowan, B.M.; Rosenstock, J.; Tran, M.T.D.; Wadden, T.A.; et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N. Engl. J. Med. 2021, 384, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Zeng, H.; Huang, M.; Fu, W.; Chen, Z. Efficacy and safety of once-weekly semaglutide in adults with overweight or obesity: A meta-analysis. Endocrine 2022, 75, 718–724. [Google Scholar] [CrossRef]
- Taha, M.B.; Yahya, T.; Satish, P.; Laird, R.; Agatston, A.S.; Cainzos-Achirica, M.; Patel, K.V.; Nasir, K. Glucagon-Like Peptide 1 Receptor Agonists: A Medication for Obesity Management. Curr. Atheroscler. Rep. 2022, 24, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.; Finlayson, G.; Axelsen, M.; Flint, A.; Gibbons, C.; Kvist, T.; Hjerpsted, J.B. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes. Metab. 2017, 19, 1242–1251. [Google Scholar] [CrossRef]
- Horowitz, M.; Flint, A.; Jones, K.L.; Hindsberger, C.; Rasmussen, M.F.; Kapitza, C.; Doran, S.; Jax, T.; Zdravkovic, M.; Chapman, I.M. Effect of the once-daily human GLP-1 analogue liraglutide on appetite, energy intake, energy expenditure and gastric emptying in type 2 diabetes. Diabetes Res. Clin. Pr. 2012, 97, 258–266. [Google Scholar] [CrossRef]
- Edwards, C.M.B.; Stanley, S.A.; Davis, R.; Brynes, A.E.; Frost, G.S.; Seal, L.J.; Ghatei, M.A.; Bloom, S.R. Exendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteers. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E155–E161. [Google Scholar] [CrossRef] [PubMed]
- van Bloemendaal, L.; IJzerman, R.G.; Kulve, J.S.T.; Barkhof, F.; Konrad, R.J.; Drent, M.L.; Veltman, D.J.; Diamant, M. GLP-1 receptor activation modulates appetite- and reward-related brain areas in humans. Diabetes 2014, 63, 4186–4196. [Google Scholar] [CrossRef] [PubMed]
- Coveleskie, K.; Kilpatrick, L.A.; Gupta, A.; Stains, J.; Connolly, L.; Labus, J.S.; SanMiguel, C.; Mayer, E.A. The effect of the GLP-1 analogue Exenatide on functional connectivity within an NTS-based network in women with and without obesity. Obes. Sci. Pract. 2017, 3, 434–445. [Google Scholar] [CrossRef]
- Secher, A.; Jelsing, J.; Baquero, A.F.; Hecksher-Sørensen, J.; Cowley, M.A.; Dalbøge, L.S.; Hansen, G.; Grove, K.L.; Pyke, C.; Raun, K.; et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J. Clin. Investig. 2014, 124, 4473–4488. [Google Scholar] [CrossRef]
- Beiroa, D.; Imbernon, M.; Gallego, R.; Senra, A.; Herranz, D.; Villarroya, F.; Serrano, M.; Fernø, J.; Salvador, J.; Escalada, J.; et al. GLP-1 Agonism Stimulates Brown Adipose Tissue Thermogenesis and Browning Through Hypothalamic AMPK. Diabetes 2014, 63, 3346–3358. [Google Scholar] [CrossRef]
- Gallego-Colon, E.; Wojakowski, W.; Francuz, T. Incretin drugs as modulators of atherosclerosis. Atherosclerosis 2018, 278, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Colon, E.; Kłych-Ratuszny, A.; Kosowska, A.; Garczorz, W.; Aghdam, M.R.F.; Wozniak, M.; Francuz, T. Exenatide modulates metalloproteinase expression in human cardiac smooth muscle cells via the inhibition of Akt signaling pathway. Pharmacol. Rep. 2018, 70, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, K.; Kyotani, Y.; Zhao, J.; Ito, S.; Ozawa, K.; Bolstad, F.A.; Yoshizumi, M. Exendin-4 Prevents Vascular Smooth Muscle Cell Proliferation and Migration by Angiotensin II via the Inhibition of ERK1/2 and JNK Signaling Pathways. PLoS ONE 2015, 10, e0137960. [Google Scholar] [CrossRef] [PubMed]
- Jojima, T.; Uchida, K.; Akimoto, K.; Tomotsune, T.; Yanagi, K.; Iijima, T.; Suzuki, K.; Kasai, K.; Aso, Y. Liraglutide, a GLP-1 receptor agonist, inhibits vascular smooth muscle cell proliferation by enhancing AMP-activated protein kinase and cell cycle regulation, and delays atherosclerosis in ApoE deficient mice. Atherosclerosis 2017, 261, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, Y.; Sato, K.; Watanabe, T.; Nohtomi, K.; Terasaki, M.; Nagashima, M.; Hirano, T. A glucagon-like peptide-1 analog liraglutide suppresses macrophage foam cell formation and atherosclerosis. Peptides 2014, 54, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Garczorz, W.; Gallego-Colon, E.; Kosowska, A.; Kłych-Ratuszny, A.; Woźniak, M.; Marcol, W.; Niesner, K.J.; Francuz, T. Exenatide exhibits anti-inflammatory properties and modulates endothelial response to tumor necrosis factor α-mediated activation. Cardiovasc. Ther. 2017, 36, e12317. [Google Scholar] [CrossRef]
- Forst, T.; Weber, M.M.; Pfützner, A. Cardiovascular Benefits of GLP-1-BasedTherapies in Patients with Diabetes Mellitus Type 2: Effects on Endothelial and Vascular Dysfunction beyond Glycemic Control. Exp. Diabetes Res. 2012, 2012, 635472. [Google Scholar] [CrossRef]
- Oeseburg, H.; de Boer, R.A.; Buikema, H.; van der Harst, P.; van Gilst, W.H.; Silljé, H.H. Glucagon-Like Peptide 1 Prevents Reactive Oxygen Species–Induced Endothelial Cell Senescence through the Activation of Protein Kinase A. Arter. Thromb. Vasc. Biol. 2010, 30, 1407–1414. [Google Scholar] [CrossRef]
- Batchuluun, B.; Inoguchi, T.; Sonoda, N.; Sasaki, S.; Inoue, T.; Fujimura, Y.; Miura, D.; Takayanagi, R. Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis 2014, 232, 156–164. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, S.; Chen, X.; Wang, Z.; Wang, X.; Zhou, Q.; Fang, W.; Zheng, C. Liraglutide prevents high glucose induced HUVECs dysfunction via inhibition of PINK1/Parkin-dependent mitophagy. Mol. Cell. Endocrinol. 2022, 545, 111560. [Google Scholar] [CrossRef]
- Shiraki, A.; Oyama, J.-I.; Komoda, H.; Asaka, M.; Komatsu, A.; Sakuma, M.; Kodama, K.; Sakamoto, Y.; Kotooka, N.; Hirase, T.; et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-α-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis 2012, 221, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Li, Y.; Ou, D.; Yang, Q. The GLP-1 receptor agonist liraglutide protects against oxidized LDL-induced endothelial inflammation and dysfunction via KLF2. IUBMB Life 2019, 71, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.; Liu, H.; Welungoda, I.; Hu, Y.; Widdop, R.E.; Knudsen, L.B.; Simpson, R.W.; Dear, A.E. A GLP-1 receptor agonist liraglutide inhibits endothelial cell dysfunction and vascular adhesion molecule expression in an ApoE-/- mouse model. Diabetes Vasc. Dis. Res. 2011, 8, 117–124, Erratum in Diabetes Vasc. Dis. Res. 2012, 9, 79. [Google Scholar] [CrossRef]
- Tang, Z.; Liu, L.; Guo, Y.; Deng, G.; Chen, M.; Wei, J. Exendin-4 reverses endothelial dysfunction in mice fed a high-cholesterol diet by a GTP cyclohydrolase-1/tetrahydrobiopterin pathway. Mol. Med. Rep. 2018, 18, 3350–3358. [Google Scholar] [CrossRef] [PubMed]
- Koska, J.; Sands, M.; Burciu, C.; D’Souza, K.M.; Raravikar, K.; Liu, J.; Truran, S.; Franco, D.A.; Schwartz, E.A.; Schwenke, D.C.; et al. Exenatide Protects Against Glucose- and Lipid-Induced Endothelial Dysfunction: Evidence for Direct Vasodilation Effect of GLP-1 Receptor Agonists in Humans. Diabetes 2015, 64, 2624–2635. [Google Scholar] [CrossRef] [PubMed]
- Bretón-Romero, R.; Weisbrod, R.M.; Feng, B.; Holbrook, M.; Ko, D.; Stathos, M.M.; Zhang, J.; Fetterman, J.L.; Hamburg, N.M. Liraglutide Treatment Reduces Endothelial Endoplasmic Reticulum Stress and Insulin Resistance in Patients with Diabetes Mellitus. J. Am. Heart Assoc. 2018, 7, e009379. [Google Scholar] [CrossRef]
- Nyström, T.; Gutniak, M.K.; Zhang, Q.; Zhang, F.; Holst, J.J.; Ahrén, B.; Sjöholm, A. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am. J. Physiol. Metab. 2004, 287, E1209–E1215. [Google Scholar] [CrossRef]
- Okada, K.; Kotani, K.; Yagyu, H.; Ando, A.; Osuga, J.-I.; Ishibashi, S. Effects of treatment with liraglutide on oxidative stress and cardiac natriuretic peptide levels in patients with type 2 diabetes mellitus. Endocrine 2014, 47, 962–964. [Google Scholar] [CrossRef]
- Rizzo, M.; Abate, N.; Chandalia, M.; Rizvi, A.A.; Giglio, R.V.; Nikolic, D.; Gammazza, A.M.; Barbagallo, I.; Isenovic, E.R.; Banach, M.; et al. Liraglutide Reduces Oxidative Stress and Restores Heme Oxygenase-1 and Ghrelin Levels in Patients with Type 2 Diabetes: A Prospective Pilot Study. J. Clin. Endocrinol. Metab. 2015, 100, 603–606. [Google Scholar] [CrossRef]
- Koska, J. Incretins and Preservation of Endothelial Function. Cardiovasc. Hematol. Agents Med. Chem. 2012, 10, 295–308. [Google Scholar] [CrossRef]
- Wang, N.; Tan, A.W.K.; Jahn, L.A.; Hartline, L.; Patrie, J.T.; Lin, S.; Barrett, E.J.; Aylor, K.W.; Liu, Z. Vasodilatory Actions of Glucagon-Like Peptide 1 Are Preserved in Skeletal and Cardiac Muscle Microvasculature but Not in Conduit Artery in Obese Humans with Vascular Insulin Resistance. Diabetes Care 2020, 43, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Richards, P.; Parker, H.E.; Adriaenssens, A.E.; Hodgson, J.M.; Cork, S.C.; Trapp, S.; Gribble, F.M.; Reimann, F. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes 2014, 63, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Pyke, C.; Heller, R.S.; Kirk, R.K.; Ørskov, C.; Reedtz-Runge, S.; Kaastrup, P.; Hvelplund, A.; Bardram, L.; Calatayud, D.; Knudsen, L.B. GLP-1 Receptor Localization in Monkey and Human Tissue: Novel Distribution Revealed with Extensively Validated Monoclonal Antibody. Endocrinology 2014, 155, 1280–1290. [Google Scholar] [CrossRef]
- Ban, K.; Noyan-Ashraf, M.H.; Hoefer, J.; Bolz, S.-S.; Drucker, D.J.; Husain, M. Cardioprotective and Vasodilatory Actions of Glucagon-Like Peptide 1 Receptor Are Mediated through Both Glucagon-Like Peptide 1 Receptor–Dependent and –Independent Pathways. Circulation 2008, 117, 2340–2350, Erratum in Circulation 2008, 118, e81. [Google Scholar] [CrossRef]
- Cai, X.; She, M.; Xu, M.; Chen, H.; Li, J.; Chen, X.; Zheng, D.; Liu, J.; Chen, S.; Zhu, J.; et al. GLP-1 treatment protects endothelial cells from oxidative stress-induced autophagy and endothelial dysfunction. Int. J. Biol. Sci. 2018, 14, 1696–1708. [Google Scholar] [CrossRef]
- Wang, D.; Luo, P.; Wang, Y.; Li, W.; Wang, C.; Sun, D.; Zhang, R.; Su, T.; Ma, X.; Zeng, C.; et al. Glucagon-like peptide-1 protects against cardiac microvascular injury in diabetes via a cAMP/PKA/Rho-dependent mechanism. Diabetes 2013, 62, 1697–1708. [Google Scholar] [CrossRef]
- Sinning, C.; Westermann, D.; Clemmensen, P. Oxidative stress in ischemia and reperfusion: Current concepts, novel ideas and future perspectives. Biomarkers Med. 2017, 11, 11031-1040. [Google Scholar] [CrossRef] [PubMed]
- Jarasch, E.-D.; Grund, C.; Bruder, G.; Heid, H.W.; Keenan, T.W.; Franke, W.W. Localization of xanthine oxidase in mammary-gland epithelium and capillary endothelium. Cell 1981, 25, 67–82. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, H.; Wu, W.; Shi, C.; Hu, S.; Yin, T.; Ma, Q.; Han, T.; Zhang, Y.; Tian, F.; et al. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca2+–XO–ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic. Biol. Med. 2016, 95, 278–292. [Google Scholar] [CrossRef]
- Sukumaran, V.; Tsuchimochi, H.; Sonobe, T.; Waddingham, M.T.; Shirai, M.; Pearson, J.T. Liraglutide treatment improves the coronary microcirculation in insulin resistant Zucker obese rats on a high salt diet. Cardiovasc. Diabetol. 2020, 19, 24. [Google Scholar] [CrossRef]
- Mendes-Junior, L.G.; Freitas-Lima, L.C.; Oliveira, J.R.; Melo, M.B.; Feltenberger, J.D.; Brandi, I.V.; Carvalho, B.M.A.; Guimarães, A.L.S.; De Paula, A.M.B.; D’Angelis, C.E.M.; et al. The usefulness of short-term high-fat/high salt diet as a model of metabolic syndrome in mice. Life Sci. 2018, 209, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Soliman, N.G.A.; Abdel-Hamid, A.A.; El-Hawwary, A.A.; Ellakkany, A. Impact of liraglutide on microcirculation in experimental diabetic cardiomyopathy. Acta Histochem. 2020, 122, 151533. [Google Scholar] [CrossRef] [PubMed]
- Kern, K.B.; Zuercher, M.; Cragun, D.; Ly, S.; Quash, J.; Bhartia, S.; Hilwig, R.W.; Berg, R.A.; Ewy, G.A. Myocardial microcirculatory dysfunction after prolonged ventricular fibrillation and resuscitation. Crit. Care Med. 2008, 36, S418–S421. [Google Scholar] [CrossRef] [PubMed]
- Dokken, B.B.; Hilwig, W.R.; Teachey, M.K.; Panchal, R.A.; Hubner, K.; Allen, D.; Rogers, D.C.; Kern, K.B. Glucagon-like peptide-1 (GLP-1) attenuates post-resuscitation myocardial microcirculatory dysfunction. Resuscitation 2010, 81, 755–760. [Google Scholar] [CrossRef]
- Biaggioni, I. Clinical and molecular pharmacologic characteristics of adenosine-induced vasodilation. Clin. Pharmacol. Ther. 2004, 75, 137–139. [Google Scholar] [CrossRef]
- Dokken, B.B.; Piermarini, C.V.; Teachey, M.K.; Gura, M.T.; Dameff, C.J.; Heller, B.D.; Krate, J.; Asghar, A.M.; Querin, L.; Mitchell, J.L.; et al. Glucagon-like peptide-1 preserves coronary microvascular endothelial function after cardiac arrest and resuscitation: Potential antioxidant effects. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H538–H546, Erratum in Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1861. [Google Scholar] [CrossRef]
- Gejl, M.; Søndergaard, H.M.; Stecher, C.; Bibby, B.M.; Moller, N.; Bøtker, H.E.; Hansen, S.B.; Gjedde, A.; Rungby, J.; Brock, B. Exenatide Alters Myocardial Glucose Transport and Uptake Depending on Insulin Resistance and Increases Myocardial Blood Flow in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, E1165–E1169. [Google Scholar] [CrossRef]
- Gill, A.; Hoogwerf, B.J.; Burger, J.; Bruce, S.; MacConell, L.; Yan, P.; Braun, D.; Giaconia, J.; Malone, J. Effect of exenatide on heart rate and blood pressure in subjects with type 2 diabetes mellitus: A double-blind, placebo-controlled, randomized pilot study. Cardiovasc. Diabetol. 2010, 9, 6. [Google Scholar] [CrossRef]
- Subaran, S.C.; Sauder, M.A.; Chai, W.; Jahn, L.A.; Fowler, D.E.; Aylor, K.W.; Basu, A.; Liu, Z. GLP-1 at physiological concentrations recruits skeletal and cardiac muscle microvasculature in healthy humans. Clin. Sci. 2014, 127, 163–170. [Google Scholar] [CrossRef]
- Nilsson, M.; Bové, K.B.; Suhrs, E.; Hermann, T.; Madsbad, S.; Holst, J.J.; Prescott, E.; Zander, M. The effect of DPP-4-protected GLP-1 (7–36) on coronary microvascular function in obese adults. IJC Heart Vasc. 2019, 22, 139–144. [Google Scholar] [CrossRef]
- Clarke, S.J.; Giblett, J.P.; Yang, L.L.; Hubsch, A.; Zhao, T.; Aetesam-Ur-Rahman, M.; West, N.E.J.; O'Sullivan, M.; Figg, N.; Bennett, M.; et al. GLP-1 Is a Coronary Artery Vasodilator in Humans. J. Am. Heart Assoc. 2018, 7, e010321. [Google Scholar] [CrossRef] [PubMed]
- Westerhof, N.; Boer, C.; Lamberts, R.R.; Sipkema, P. Cross-Talk Between Cardiac Muscle and Coronary Vasculature. Physiol. Rev. 2006, 86, 1263–1308. [Google Scholar] [CrossRef] [PubMed]
- Aetesam-Ur-Rahman, M.; Giblett, J.P.; Khialani, B.; Kyranis, S.; Clarke, S.J.; Zhao, T.X.; Braganza, D.M.; Clarke, S.C.; West, N.E.J.; Bennett, M.R.; et al. GLP-1 vasodilatation in humans with coronary artery disease is not adenosine mediated. BMC Cardiovasc. Disord. 2021, 21, 223. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.R.; Hu, S.Y.; Chen, Y.D.; Zhang, Y.; Qian, G.; Wang, J.; Yang, J.J.; Wang, Z.F.; Tian, F.; Ning, Q.X. Effects of liraglutide on left ventricular function in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am. Heart J. 2015, 170, 845–854. [Google Scholar] [CrossRef]
- Faber, R.; Zander, M.; Pena, A.; Michelsen, M.M.; Mygind, N.D.; Prescott, E. Effect of the glucagon-like peptide-1 analogue liraglutide on coronary microvascular function in patients with type 2 diabetes—A randomized, single-blinded, cross-over pilot study. Cardiovasc. Diabetol. 2015, 14, 41. [Google Scholar] [CrossRef]
- Wei, R.; Ma, S.; Wang, C.; Ke, J.; Yang, J.; Li, W.; Liu, Y.; Hou, W.; Feng, X.; Wang, G.; et al. Exenatide exerts direct protective effects on endothelial cells through the AMPK/Akt/eNOS pathway in a GLP-1 receptor-dependent manner. Am. J. Physiol. Metab. 2016, 310, E947–E957. [Google Scholar] [CrossRef]
- Nielsen, R.; Jorsal, A.; Iversen, P.; Tolbod, L.P.; Bouchelouche, K.; Sørensen, J.; Harms, H.J.; Flyvbjerg, A.; Tarnow, L.; Kistorp, C.; et al. Effect of liraglutide on myocardial glucose uptake and blood flow in stable chronic heart failure patients: A double-blind, randomized, placebo-controlled LIVE sub-study. J. Nucl. Cardiol. 2019, 26, 585–597. [Google Scholar] [CrossRef]
- Sano, R.; Shinozaki, Y.; Ohta, T. Sodium–glucose cotransporters: Functional properties and pharmaceutical potential. J. Diabetes Investig. 2020, 11, 770–782. [Google Scholar] [CrossRef]
- Kshirsagar, R.P.; Kulkarni, A.A.; Chouthe, R.S.; Pathan, S.K.; Une, H.D.; Reddy, G.B.; Diwan, P.V.; Ansari, S.A.; Sangshetti, J.N. SGLT inhibitors as antidiabetic agents: A comprehensive review. RSC Adv. 2020, 10, 1733–1756. [Google Scholar] [CrossRef]
- Thomas, M.C.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Salvatore, T.; Galiero, R.; Caturano, A.; Rinaldi, L.; Di Martino, A.; Albanese, G.; Di Salvo, J.; Epifani, R.; Marfella, R.; Docimo, G.; et al. An Overview of the Cardiorenal Protective Mechanisms of SGLT2 Inhibitors. Int. J. Mol. Sci. 2022, 23, 3651. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726, Erratum in Eur. Heart J. 2021, 42, 4901. [Google Scholar] [CrossRef]
- Sternlicht, H.; Bakris, G.L. Blood Pressure Lowering and Sodium-Glucose Co-transporter 2 Inhibitors (SGLT2is): More Than Osmotic Diuresis. Curr. Hypertens. Rep. 2019, 21, 12. [Google Scholar] [CrossRef]
- Rajeev, S.P.; Cuthbertson, D.; Wilding, J.P.H. Energy balance and metabolic changes with sodium-glucose co-transporter 2 inhibition. Diabetes Obes. Metab. 2015, 18, 125–134. [Google Scholar] [CrossRef]
- Lee, P.C.; Ganguly, S.; Goh, S.-Y. Weight loss associated with sodium-glucose cotransporter-2 inhibition: A review of evidence and underlying mechanisms. Obes. Rev. 2018, 19, 1630–1641. [Google Scholar] [CrossRef]
- McMurray, J. EMPA-REG—The “diuretic hypothesis”. J. Diabetes Its Complicat. 2015, 30, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Batzias, K.; Antonopoulos, A.; Oikonomou, E.; Siasos, G.; Bletsa, E.; Stampouloglou, P.K.; Mistakidi, C.-V.; Noutsou, M.; Katsiki, N.; Karopoulos, P.; et al. Effects of Newer Antidiabetic Drugs on Endothelial Function and Arterial Stiffness: A Systematic Review and Meta-Analysis. J. Diabetes Res. 2018, 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Caturano, A.; Galiero, R.; Di Martino, A.; Albanese, G.; Vetrano, E.; Sardu, C.; Marfella, R.; Rinaldi, L.; Sasso, F.C. Cardiovascular Benefits from Gliflozins: Effects on Endothelial Function. Biomedicines 2021, 9, 1356. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Williams, S.; Ho, S.; Loraine, H.; Hagan, D.; Whaley, J.M.; Feder, J.N. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010, 1, 57–92. [Google Scholar] [CrossRef]
- Maejima, Y. SGLT2 Inhibitors Play a Salutary Role in Heart Failure via Modulation of the Mitochondrial Function. Front. Cardiovasc. Med. 2020, 6, 186. [Google Scholar] [CrossRef]
- Mancini, S.J.; Boyd, D.; Katwan, O.J.; Strembitska, A.; Almabrouk, T.A.; Kennedy, S.; Palmer, T.M.; Salt, I.P. Canagliflozin inhibits inter-leukin-1β-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP-activated protein ki-nase-dependent and independent mechanisms. Sci. Rep. 2018, 8, 5276. [Google Scholar] [CrossRef]
- El-Daly, M.; Venu, V.K.P.; Saifeddine, M.; Mihara, K.; Kang, S.; Fedak, P.W.; Alston, L.A.; Hirota, S.A.; Ding, H.; Triggle, C.R.; et al. Hyperglycaemic impairment of PAR2-mediated vasodilation: Prevention by inhibition of aortic endothelial sodium-glucose-co-Transporter-2 and minimizing oxidative stress. Vasc. Pharmacol. 2018, 109, 56–71. [Google Scholar] [CrossRef]
- Adingupu, D.D.; Göpel, S.O.; Grönros, J.; Behrendt, M.; Sotak, M.; Miliotis, T.; Dahlqvist, U.; Gan, L.M.; Jönsson-Rylander, A.C. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob-/-mice. Cardiovasc. Diabetol. 2019, 18, 16. [Google Scholar] [CrossRef]
- Westergren, H.U.; Grönros, J.; Heinonen, S.E.; Miliotis, T.; Jennbacken, K.; Sabirsh, A.; Ericsson, A.; Jönsson-Rylander, A.-C.; Svedlund, S.; Gan, L.-M. Impaired Coronary and Renal Vascular Function in Spontaneously Type 2 Diabetic Leptin-Deficient Mice. PLoS ONE 2015, 10, e0130648. [Google Scholar] [CrossRef]
- Ashikhmina, E.A.; Schaff, H.V.; Suri, R.M.; Enriquez-Sarano, M.; Abel, M.D. Left ventricular remodeling early after correction of mitral regurgitation: Maintenance of stroke volume with decreased systolic indexes. J. Thorac. Cardiovasc. Surg. 2010, 140, 1300–1305. [Google Scholar] [CrossRef][Green Version]
- Gan, L.M.; Svedlund, S.; Wittfeldt, A.; Eklund, C.; Gao, S.; Matejka, G.; Jeppsson, A.; Albertsson, P.; Omerovic, E.; Lerman, A. Incremental Value of Transthoracic Doppler Echocardiography-Assessed Coronary Flow Reserve in Patients With Suspected Myocardial Ischemia Undergoing Myocardial Perfusion Scintigraphy. J. Am. Heart Assoc. 2017, 6, e004875. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.l.; Naya, M.; Foster, C.R.; Hainer, J.; Gaber, M.; Di Carli, G.; Blankstein, R.; Dorbala, S.; Sitek, A.; Pencina, M.J.; et al. Improved Cardiac Risk Assessment with Noninvasive Measures of Coronary Flow Reserve. Circulation 2011, 124, 2215–2224. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.D.C.A.; Lax, A.; Vicente, A.H.; Guillen, E.S.; Hernandez-Martinez, A.; Del Palacio, M.J.F.; Bayes-Genis, A.; Figal, D.A.P. Empagliflozin improves post-infarction cardiac remodeling through GTP enzyme cyclohydrolase 1 and irrespective of diabetes status. Sci. Rep. 2020, 10, 13553, Erratum in Sci. Rep. 2020, 10, 17266. [Google Scholar] [CrossRef]
- Juni, R.P.; Kuster, D.W.D.; Goebel, M.; Helmes, M.; Musters, R.J.; van der Velden, J.; Koolwijk, P.; Paulus, W.J.; van Hinsbergh, V.W. Cardiac Microvascular Endothelial Enhancement of Cardiomyocyte Function Is Impaired by Inflammation and Restored by Empagliflozin. JACC Basic Transl. Sci. 2019, 4, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; Van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell. Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef]
- Takimoto, E. Cyclic GMP-Dependent Signaling in Cardiac Myocytes. Circ. J. 2012, 76, 1819–1825. [Google Scholar] [CrossRef]
- Rush, C.J.; Berry, C.; Oldroyd, K.G.; Rocchiccioli, J.P.; Lindsay, M.M.; Touyz, R.M.; Murphy, C.L.; Ford, T.J.; Sidik, N.; McEntegart, M.B.; et al. Prevalence of Coronary Artery Disease and Coronary Microvascular Dysfunction in Patients with Heart Failure with Preserved Ejection Fraction. JAMA Cardiol. 2021, 6, 1130–1143. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Juni, R.P.; Al-Shama, R.; Kuster, D.W.D.; van der Velden, J.; Hamer, H.M.; Vervloet, M.G.; Eringa, E.C.; Koolwijk, P.; van Hinsbergh, V.W. Empagliflozin restores chronic kidney disease–induced impairment of endothelial regulation of cardiomyocyte relaxation and contraction. Kidney Int. 2020, 99, 1088–1101. [Google Scholar] [CrossRef]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The Cardiovascular Effect of the Uremic Solute Indole-3 Acetic Acid. J. Am. Soc. Nephrol. 2014, 26, 876–887. [Google Scholar] [CrossRef]
- Verkaik, M.; Juni, R.P.; Van Loon, E.P.M.; Van Poelgeest, E.M.; Kwekkeboom, R.F.J.; Gam, Z.; Richards, W.G.; Ter Wee, P.M.; Hoenderop, J.G.J.; Eringa, E.; et al. FGF23 impairs peripheral microvascular function in renal failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1414–H1424. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2017, 15, 335–346. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia–reperfusion injury. Acta Pharm. Sin. B 2020, 10, 1866–1879. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Guo, Z.; Chang, X.; Li, Z.; Wu, F.; He, J.; Cao, T.; Wang, K.; Shi, N.; Zhou, H.; et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 2022, 52, 102288. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, J.; Zhu, P.; Zhu, H.; Toan, S.; Hu, S.; Ren, J.; Chen, Y. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic Res. Cardiol. 2018, 113, 23. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef]
- Wu, H.; Ye, M.; Liu, D.; Yang, J.; Ding, J.W.; Zhang, J.; Wang, X.A.; Dong, W.S.; Fan, Z.X.; Yang, J. UCP2 protect the heart from myocardial ischemia/reperfusion injury via induction of mitochondrial autophagy. J. Cell. Biochem. 2019, 120, 15455–15466. [Google Scholar] [CrossRef]
- Wang, J.; Toan, S.; Zhou, H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis 2020, 23, 299–314. [Google Scholar] [CrossRef]
- Lim, V.G.; Bell, R.M.; Arjun, S.; Kolatsi-Joannou, M.; Long, D.A.; Yellon, D.M. SGLT2 Inhibitor, Canagliflozin, Attenuates Myocardial Infarction in the Diabetic and Nondiabetic Heart. JACC Basic Transl. Sci. 2019, 4, 15–26. [Google Scholar] [CrossRef]
- Lu, Q.; Liu, J.; Li, X.; Sun, X.; Zhang, J.; Ren, D.; Tong, N.; Li, J. Empagliflozin attenuates ischemia and reperfusion injury through LKB1/AMPK signaling pathway. Mol. Cell. Endocrinol. 2019, 501, 110642. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-W.; Que, J.-Q.; Liu, S.; Huang, K.-Y.; Qian, L.; Weng, Y.-B.; Rong, F.-N.; Wang, L.; Zhou, Y.-Y.; Xue, Y.-J.; et al. Sodium-Glucose Co-transporter-2 Inhibitor of Dapagliflozin Attenuates Myocardial Ischemia/Reperfusion Injury by Limiting NLRP3 Inflammasome Activation and Modulating Autophagy. Front. Cardiovasc. Med. 2022, 8, 68214. [Google Scholar] [CrossRef] [PubMed]
- Nakao, M.; Shimizu, I.; Katsuumi, G.; Yoshida, Y.; Suda, M.; Hayashi, Y.; Ikegami, R.; Hsiao, Y.T.; Okuda, S.; Soga, T.; et al. Em-pagliflozin maintains capillarization and improves cardiac function in a murine model of left ventricular pressure overload. Sci. Rep. 2021, 11, 18384. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nat. 2007, 446, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T.; Toko, H.; Okada, S.; Ikeda, H.; Yasuda, N.; Tateno, K.; Moriya, J.; Yokoyama, M.; Nojima, A.; et al. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J. Clin. Investig. 2010, 120, 1506–1514. [Google Scholar] [CrossRef]
- Kolijn, D.; Pabel, S.; Tian, Y.; Lódi, M.; Herwig, M.; Carrizzo, A.; Zhazykbayeva, S.; Kovács, Á.; Fülöp, G.; Falcão-Pires, I.; et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc. Res. 2020, 117, 495–507. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Ciuffreda, L.P.; Coppini, R.; Cozzolino, A.; Miccichè, A.; Dell'Aversana, C.; D’Amario, D.; Cianflone, E.; Scavone, C.; et al. Amelioration of diastolic dysfunction by dapagliflozin in a non-diabetic model involves coronary endothelium. Pharmacol. Res. 2020, 157, 104781. [Google Scholar] [CrossRef]
- Li, X.; Römer, G.; Kerindongo, R.P.; Hermanides, J.; Albrecht, M.; Hollmann, M.W.; Zuurbier, C.J.; Preckel, B.; Weber, N.C. Sodium Glucose Co-Transporter 2 Inhibitors Ameliorate Endothelium Barrier Dysfunction Induced by Cyclic Stretch through Inhibition of Reactive Oxygen Species. Int. J. Mol. Sci. 2021, 22, 6044. [Google Scholar] [CrossRef]
- Lauritsen, K.M.; Nielsen, B.R.; Tolbod, L.P.; Johannsen, M.; Hansen, J.; Hansen, T.K.; Wiggers, H.; Møller, N.; Gormsen, L.C.; Søndergaard, E. SGLT2 Inhibition Does Not Affect Myocardial Fatty Acid Oxidation or Uptake, but Reduces Myocardial Glucose Uptake and Blood Flow in Individuals With Type 2 Diabetes: A Randomized Double-Blind, Placebo-Controlled Crossover Trial. Diabetes 2020, 70, 800–808. [Google Scholar] [CrossRef]
- Jürgens, M.; Schou, M.; Hasbak, P.; Kjær, A.; Wolsk, E.; Zerahn, B.; Wiberg, M.; Brandt-Jacobsen, N.H.; Gæde, P.; Rossing, P.; et al. Effects of Empagliflozin on Myocardial Flow Reserve in Patients with Type 2 Diabetes Mellitus: The SIMPLE Trial. J. Am. Heart Assoc. 2021, 10, e020418. [Google Scholar] [CrossRef]
- Suhrs, H.E.; Nilsson, M.; Bové, K.B.; Zander, M.; Prescott, E. Effect of empagliflozin on coronary microvascular function in patients with type 2 diabetes mellitus–A randomized, placebo-controlled cross-over study. PLoS ONE 2022, 17, e0263481. [Google Scholar] [CrossRef] [PubMed]
- Löffler, A.I.; Bourque, J.M. Coronary Microvascular Dysfunction, Microvascular Angina, and Management. Curr. Cardiol. Rep. 2016, 8, 1. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; DeMets, D.L.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Langkilde, A.M.; Martinez, F.A.; Bengtsson, O.; Ponikowski, P.; Sabatine, M.S.; et al. A trial to evaluate the effect of the sodium–glucose co-transporter 2 inhibitor dapagliflozin on morbidity and mortality in patients with heart failure and reduced left ventricular ejection fraction (DAPA-HF). Eur. J. Heart Fail. 2019, 21, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Pavlidis, G.; Thymis, J.; Birba, D.; Kalogeris, A.; Kousathana, F.; Kountouri, A.; Balampanis, K.; Parissis, J.; Andreadou, I.; et al. Effects of Glucagon-Like Peptide-1 Receptor Agonists, Sodium-Glucose Cotransporter-2 Inhibitors, and Their Combination on Endothelial Glycocalyx, Arterial Function, and Myocardial Work Index in Patients With Type 2 Diabetes Mellitus After 12-Month Treatment. J. Am. Heart Assoc. 2020, 9, e015716. [Google Scholar] [CrossRef] [PubMed]
- Lekakis, J.; Abraham, P.; Balbarini, A.; Blann, A.; Boulanger, C.M.; Cockcroft, J.; Cosentino, F.; Deanfield, J.; Gallino, A.; Ikonomidis, I.; et al. Methods for evaluating endothelial function: A position statement from the European Society of Cardiology Working Group on Peripheral Circulation. Eur. J. Cardiovasc. Prev. Rehabil. 2011, 18, 775–789. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvatore, T.; Galiero, R.; Caturano, A.; Vetrano, E.; Loffredo, G.; Rinaldi, L.; Catalini, C.; Gjeloshi, K.; Albanese, G.; Di Martino, A.; et al. Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options. Biomedicines 2022, 10, 2274. https://doi.org/10.3390/biomedicines10092274
Salvatore T, Galiero R, Caturano A, Vetrano E, Loffredo G, Rinaldi L, Catalini C, Gjeloshi K, Albanese G, Di Martino A, et al. Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options. Biomedicines. 2022; 10(9):2274. https://doi.org/10.3390/biomedicines10092274
Chicago/Turabian StyleSalvatore, Teresa, Raffaele Galiero, Alfredo Caturano, Erica Vetrano, Giuseppe Loffredo, Luca Rinaldi, Christian Catalini, Klodian Gjeloshi, Gaetana Albanese, Anna Di Martino, and et al. 2022. "Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options" Biomedicines 10, no. 9: 2274. https://doi.org/10.3390/biomedicines10092274
APA StyleSalvatore, T., Galiero, R., Caturano, A., Vetrano, E., Loffredo, G., Rinaldi, L., Catalini, C., Gjeloshi, K., Albanese, G., Di Martino, A., Docimo, G., Sardu, C., Marfella, R., & Sasso, F. C. (2022). Coronary Microvascular Dysfunction in Diabetes Mellitus: Pathogenetic Mechanisms and Potential Therapeutic Options. Biomedicines, 10(9), 2274. https://doi.org/10.3390/biomedicines10092274