Abstract
Cardiovascular diseases are common in patients with chronic obstructive pulmonary disease (COPD). Clot formation and resolution secondary to systemic inflammation may be a part of the explanation. The aim was to determine whether biomarkers of clot formation (products of von Willebrand Factor formation and activation) and clot resolution (product of fibrin degeneration) during COPD exacerbation predicted major cardiovascular events (MACE). The cohort was based on clinical data and biobank plasma samples from a trial including patients admitted with an acute exacerbation of COPD (CORTICO-COP). Neo-epitope biomarkers of formation and the activation of von Willebrand factor (VWF-N and V-WFA, respectively) and cross-linked fibrin degradation (X-FIB) were assessed using ELISAs in EDTA plasma at the time of acute admission, and analyzed for time-to-first MACE within 36 months, using multivariable Cox proportional hazards models. In total, 299/318 participants had samples available for analysis. The risk of MACE for patients in the upper quartile of each biomarker versus the lower quartile was: X-FIB: HR 0.98 (95% CI 0.65–1.48), VWF-N: HR 1.56 (95% CI 1.07–2.27), and VWF-A: HR 0.78 (95% CI 0.52–1.16). Thus, in COPD patients with an acute exacerbation, VWF-N was associated with future MACE and warrants further studies in a larger population.
1. Introduction
Chronic obstructive pulmonary disease (COPD) affects 210 million people worldwide, and causes 1.9 million deaths annually [1]. Cardiovascular diseases (CVDs) are common among COPD patients, with a high prevalence of congestive heart failure, coronary heart disease, peripheral vascular disease, arrhythmia, stroke, and unspecified cardiovascular disease [2]. CVDs are associated with an increased rate of hospitalization and mortality in COPD patients [3,4].
COPD is a heterogenous disease and is often associated with persistent systemic inflammation [5]. Systemic inflammation induces increased stress on the endothelium, leading to varying degrees of endothelial damage and dysfunction [6].
Damage of the endothelium leads to a healing process, where von Willebrand Factor (vWF) is an initiator via exposure to extracellular matrix proteins through the damaged endothelium, which leads to local platelet adhesion [7]. Initially, when vWF is formed, an n-terminal pro-peptide is released as a bi-product. Antibodies for this pro-peptide are available, making it possible to assess the quantity of vWF formed (as the biomarker VWF-N) [8]. During its activation, vWF unfolds, which reveals a cleavage-site for degradation by the metalloprotease ADAMTS13 (a disintegrin and metalloprotease with ThromboSpondin motif repeats 13) [9,10]. When vWF is cleaved by ADAMTS13 following activation, a neo-epitope is released, where targeted antibodies allow for the quantification of vWF activation (as the biomarker VWF-A) [8].
Patients with COPD have increased vWF levels and relative serum activity [11]. vWF-antigen levels have been associated with first-time coronary heart disease [12], all-cause mortality [13], and stroke [14]. Additionally, low levels of ADAMTS13 activity are associated with all-cause and cardiovascular mortality [13], as well as stroke [15]. In patients with chronic heart failure, a high ADAMTS13/vWF-ratio is associated with a lower risk of clinical events [16]. The neo-epitope markers VWF-N and VWF-A were analyzed in plasma samples from the ECLIPSE trial, and revealed an association of VWF-N with the chronic condition of emphysema, and VWF-A with prior exacerbations [17]. Additionally, in a dichotomized form, higher values of both vWF products were associated with an increase in all-cause mortality [17].
Clotting is a complex interplay between clot formation and clot resolution, with both processes happening simultaneously. Further, fibrin is cross-linked as the clot matures and stabilizes [18]. As part of the ongoing clot resolution, cross-linked fibrin is degraded, releasing bi-products, including X-FIB, which is a neo-epitope of the plasmin-mediated degradation of cross-linked fibrin [19]. Thus, both vWF and the products of fibrin degradation may help describe an ongoing process of endovascular damage, clot formation, and clot resolution in patients with COPD.
Several non-interventional studies have indicated an increased risk of cardio- and vascular events among persons with an increased plasma level of Vwf [12,20].
The marker of degradation of cross-linked fibrin (X-FIB) has previously been shown to be related to emphysema and dyspnea, and predict mortality in stable COPD patients [19]. D-dimer, another marker of fibrin degradation, has been related to a prognosis in both healthy individuals and patients with COPD [12,21,22,23]. Similarly, fibrinogen, the precursor of fibrin, has been approved as a biomarker to enrich drug trials with endpoints of acute exacerbation or mortality [24].
The above-mentioned properties of both clot formation and resolution, with vWF as an initiator of clot formation and X-FIB as a marker of clot resolution, led to the hypotheses that high plasma levels of these neo-epitope biomarkers reflecting vWF formation (VWF-N) and activation (VWF-A), and of fibrin clot resolution (X-FIB) at the time of acute exacerbation of COPD (AECOPD) were associated with future MACE. Additionally, we aimed to describe the behavior of each biomarker, comparing acute with stable phase values.
2. Materials and Methods
2.1. Patients
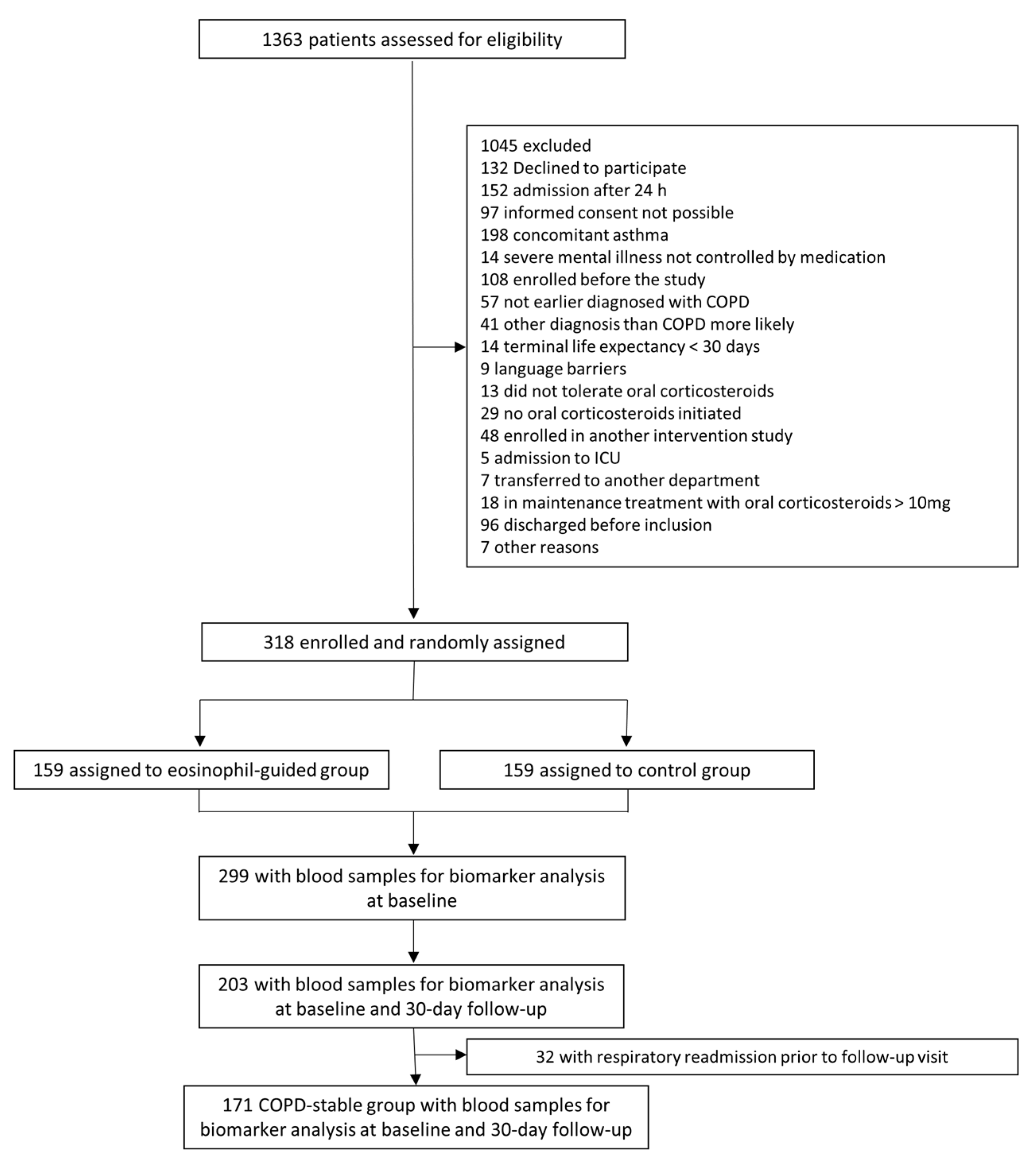
The study population originated from the randomized controlled trial, CORTICO-COP, as described in detail in a previous publication [25]. Briefly, patients presenting with an AECOPD within 24 h of hospital admission between August 2016 and September 2018 were eligible for the trial. Blood samples (full blood and plasma) from 318 patients with COPD hospitalized for an acute exacerbation (severe or very severe COPD GOLD (Global initiative for chronic Obstructive Lung Disease) stage C/D and age ≥ 40) were collected at time of hospitalization, and at 30 days follow-up for all patients. Of these, 299 patients had blood sampled for biomarker analysis at baseline; of these, 203 had blood sampled after 30 days. Patients were recruited at three different Respiratory Departments in the Capital Region of Denmark (Copenhagen University Hospital Herlev & Gentofte, Copenhagen University Hospital Bispebjerg, and Copenhagen University Hospital Hvidovre). A follow-up visit was performed 30 days after the onset of an exacerbation. In the present study, only participants who had the biomarkers in question measured at baseline were included (299 (94%) participants), see Figure 1.
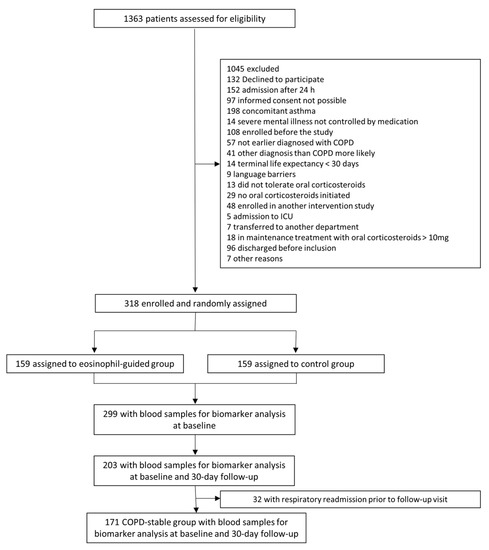
Figure 1.
Study flowchart.
2.2. Measurements
The Spirometry and Medical Research Council (MRC) Dyspnea Scale was performed at baseline and at the 30 day follow-up. Venous blood samples were collected at hospital admission and at the 30 day follow-up via venipuncture into vacutainer EDTA-coated tubes. Plasma was obtained via centrifugation at 2000× g for 10 min at 4 °C within 30 min of blood collection. EDTA plasma samples were stored in aliquots at –80 °C within 1 h and until analysed. Neo-epitopes of vWF formation (VWF-N), vWF activation (VWF-A), and clot resolution (X-FIB) were assessed in EDTA plasma using specific enzyme-linked immunosorbent assays (ELISAs) (Nordic Bioscience A/S, Herlev, Denmark), as previously described [8,26]. In brief, all assays employed mouse monoclonal antibodies specific for a neo-epitope: VWF-N quantifies the N-terminal of the vWF pro-peptide released upon formation using competitive ELISA [8], VWF-A quantifies the ADAMTS13-mediated degradation fragment of vWF using competitive ELISA [8], and X-FIB quantifies the plasmin-mediated degradation of cross-linked fibrin using sandwich ELISA [26].
2.3. Confounding Factors
Data on possible confounding factors were collected from patients at baseline. Data on previous ischemic heart disease and heart failure were supplied with lifetime data from registries. We identified the following potential confounding factors: Sex, age, smoking status (current, former, or never smoked), C-reactive protein, forced expiratory volume in one second (FEV1, % of predicted), patient-reported use of inhaled corticosteroids, ischemic heart disease, heart failure, atrial fibrillation/flutter, hypertension, hypercholesterolemia, peripheral vascular diseases, and diabetes mellitus type 2.
2.4. Outcome
Registry-based follow-up on MACE was performed during the 36 month follow-up after hospital admission for an AECOPD. Hospital-related events and deaths were respectively obtained by linking the CORTICO-COP data to the Danish National Patient Registry and the Danish Civil Registration System. MACE was defined as either death or any admission to hospital with one of the International Classification of Disease codes as the primary diagnosis (see Supplementary Table S1).
2.5. Reporting
Reporting was carried out in accordance with the STROBE (STrengthening the Reporting of Observational studies in Epidemiology) guidelines [27]. Prior to the conduction of this sub-study, a protocol was published online [28].
2.6. Statistical Analyses
Assuming an annual event rate of the primary outcome of 2% and a hazard ratio (HR) of 3 over 36 months follow-up, corresponding to a MACE rate of 6 vs. 18%, 326 participants were required.
Descriptive statistics was performed on the baseline data at the time of AECOPD. A comparison of baseline data was performed with a Chi-squared test if the expected observations were above five. When the expected observations were five or below, Fischer’s exact test was applied. For continuous data, the normally distributed data were compared with a t test, and not normally distributed data were compared with Mann–Whitney Wilcoxon test. Patients who did not experience a new AECOPD within 30 days formed the “COPD-stable group” at the 30 day follow-up. Patients in the COPD-stable group who had biomarkers at both index admission and follow-up was used to investigate the change in each biomarker between the acute and stable phases with a Wilcoxon Signed-Rank test, and their correlation was examined with Spearman’s correlation coefficient.
On the biomarkers in acute phase: Each of the three biomarkers were dichotomized at the upper quartile, allowing for a comparison of “high value groups” (upper quartile) with “low value groups” (three lowest quartiles) in a multivariable Cox proportional hazards regression analysis adjusted for the previously mentioned potential confounding factors.
Missing data was subject to Substantive Model Compatible Fully Conditional Specification multiple imputation [29] (for details, see Supplementary Document S1). For missing data, where the missing completely at random/missing at random assumption could be doubted, both the best-case and worst-case imputations were applied (see Supplementary Document S1 for further information).
Data management, descriptive statistics, and comparison of biomarkers were performed with Statistical Analysis Software 9.4 (SAS Institute, Cary, NC, USA). Multiple imputations and combinations of results were conducted in R 4.1.3 (R Foundation for Statistical Computing, Vienna, Austria) with the SMCFCS 1.6.1 and MITOOLS 2.4, packages, respectively. Crude Cox proportional hazards regression models were conducted using the SURVIVAL 3.3-1 package.
2.7. Model Control
Each biomarker was investigated for interaction with both known ischemic heart disease and known heart failure. Both best-case and worst-case imputations were performed as sensitivity analyses.
Proportional hazards assumption was tested as the interaction between exposure and time, and linearity was tested for the continuous covariates.
2.8. Explorative Analyses
Additional analyses were performed for the primary outcome, with the exclusion of all-cause mortality and with all-cause mortality as the sole outcome.
As an explorative analyses, each biomarker was investigated as a continuous variable. Further, each biomarker was investigated for an association with all parts of the composite outcome with a Chi-squared test (or Fischer’s exact test, if the expected observations were below five). When a significant association was identified, a multivariable Cox proportional hazards regression analysis was applied.
3. Results
Patient characteristics at baseline are shown in Table 1. In total, 299 (94%) patients had biomarkers assessed at baseline. For the assessment of biomarker stability between the acute and stable phases, 171 patients were stable at the 30 day follow-up, and had biomarkers accessible for comparison between baseline and the 30 day follow-up.

Table 1.
Baseline characteristics. MACE: Major cardiovascular event, n: Number, SD: Standard deviation, IQR: Interquartile range, FEV1: Forced expiratory Volume in 1 s, MRC: Medical research council dyspnea scale score. *: For ever-smokers.
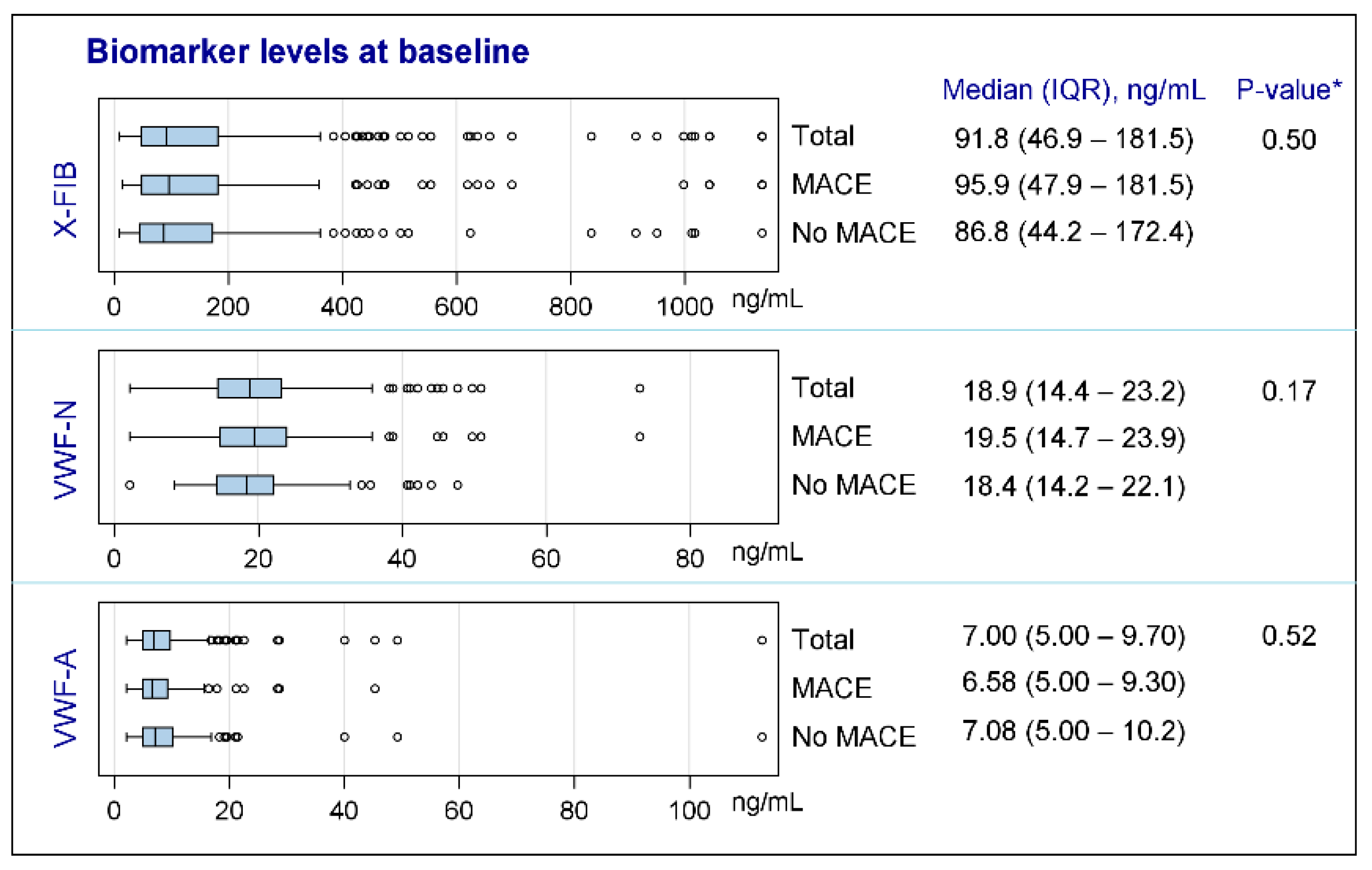
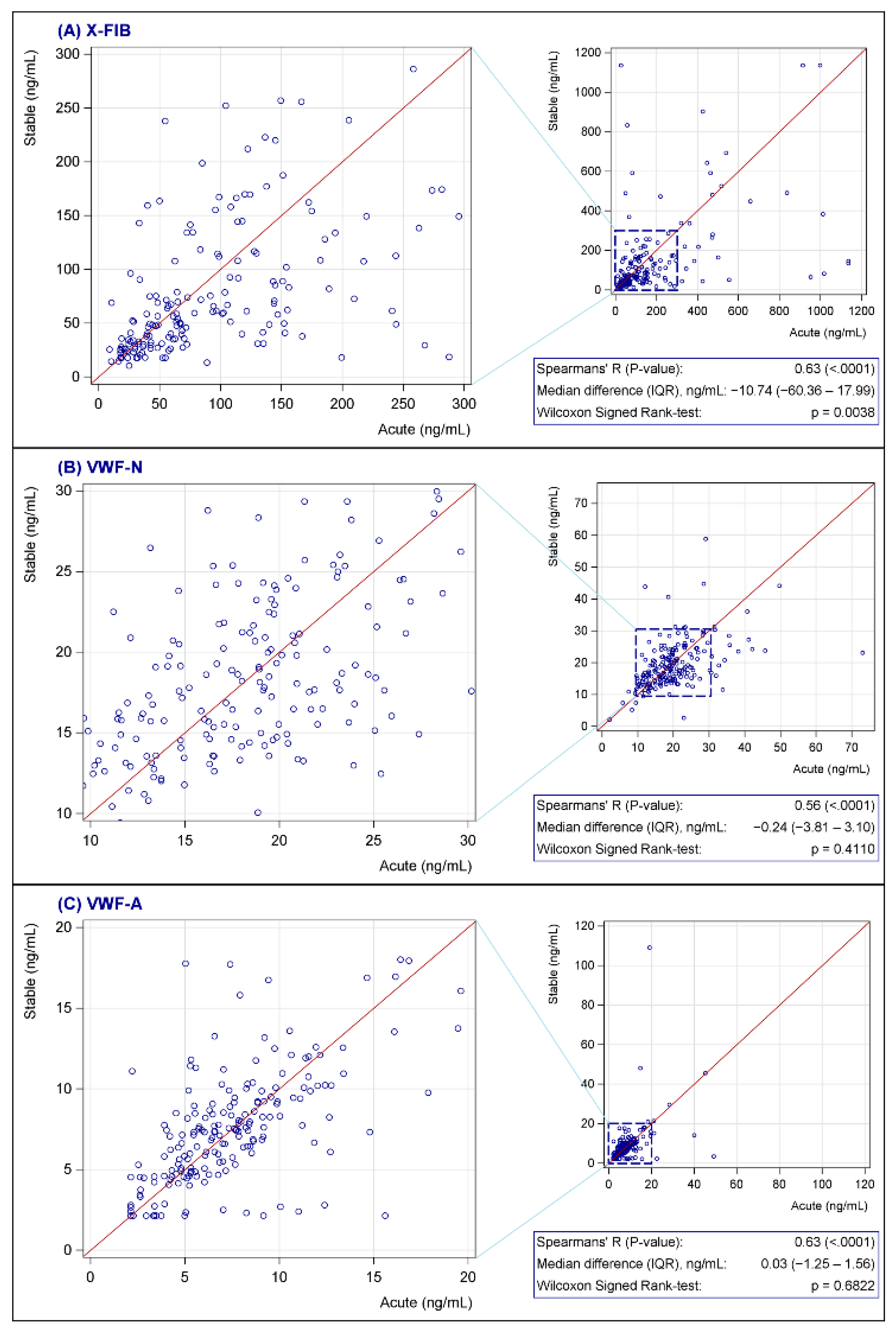
None of the biomarkers were normally distributed; see Figure 2 for characteristics of the biomarkers at baseline. Both vWF markers were generally unchanged from exacerbation to the stable phase (VWF-N: p = 0.41, VWF-A: p = 0.68), whereas there was a decrease in the stable phase for the clot resolution marker, X-FIB, with a median difference of 10.74 ng/mL (IQR: −60.36–17.99, p = 0.0038); Figure 3A–C. X-FIB and VWF-A had a strong correlation from the acute to stable phase, whereas VWF-N had a moderate correlation; Figure 3A–C.
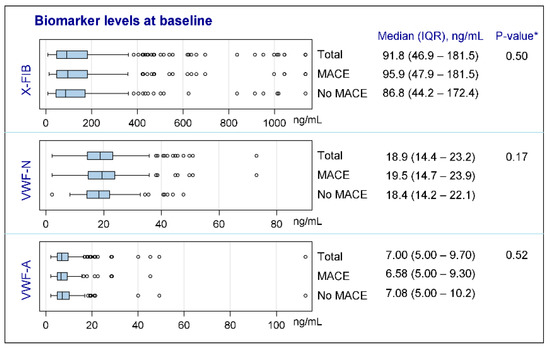
Figure 2.
Characteristics of biomarkers at baseline. X-FIB: Cross-linked fibrin degeneration. VWF-N: N-terminal of von Willebrand Factor formation. VWF-A: Neoepitope of ADAMTS13-mediated activation of von Willebrand Factor. IQR: Interquartile range. MACE: Major cardiovascular event. *: For comparison between ‘MACE’ and ‘No MACE’ subgroups.
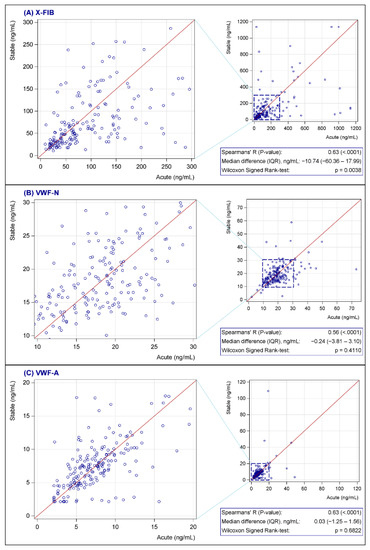
Figure 3.
(A–C) Change in X-FIB (A), VWF-N (B), and VWF-A (C) from admission to follow-up. X-FIB: Cross-linked fibrin degeneration. VWF-N: N-terminal of von Willebrand Factor formation. VWF-A: Neoepitope of ADAMTS13-mediated activation of von Willebrand Factor. IQR: Interquartile range.
In total, 149 (50%) of the eligible participants experienced MACE in the study period. Of these, seven experienced acute myocardial infarctions, and one, a stroke; 21 were admitted with heart failure and 120 died of all causes.
3.1. Primary Outcome
In the unadjusted Cox proportional hazards regression, no difference between high and low levels of acute-phase X-FIB (Hazard Ratio (HR): 1.10 (95% confidence interval (CI): 0.77–1.60), p = 0.60) nor VWF-A (HR: 0.85 (95% CI: 0.58–1.24), p = 0.41) on MACE was observed, while VWF-N was associated with MACE, HR: 1.41 (95% CI 1.00–2.00, p = 0.056).
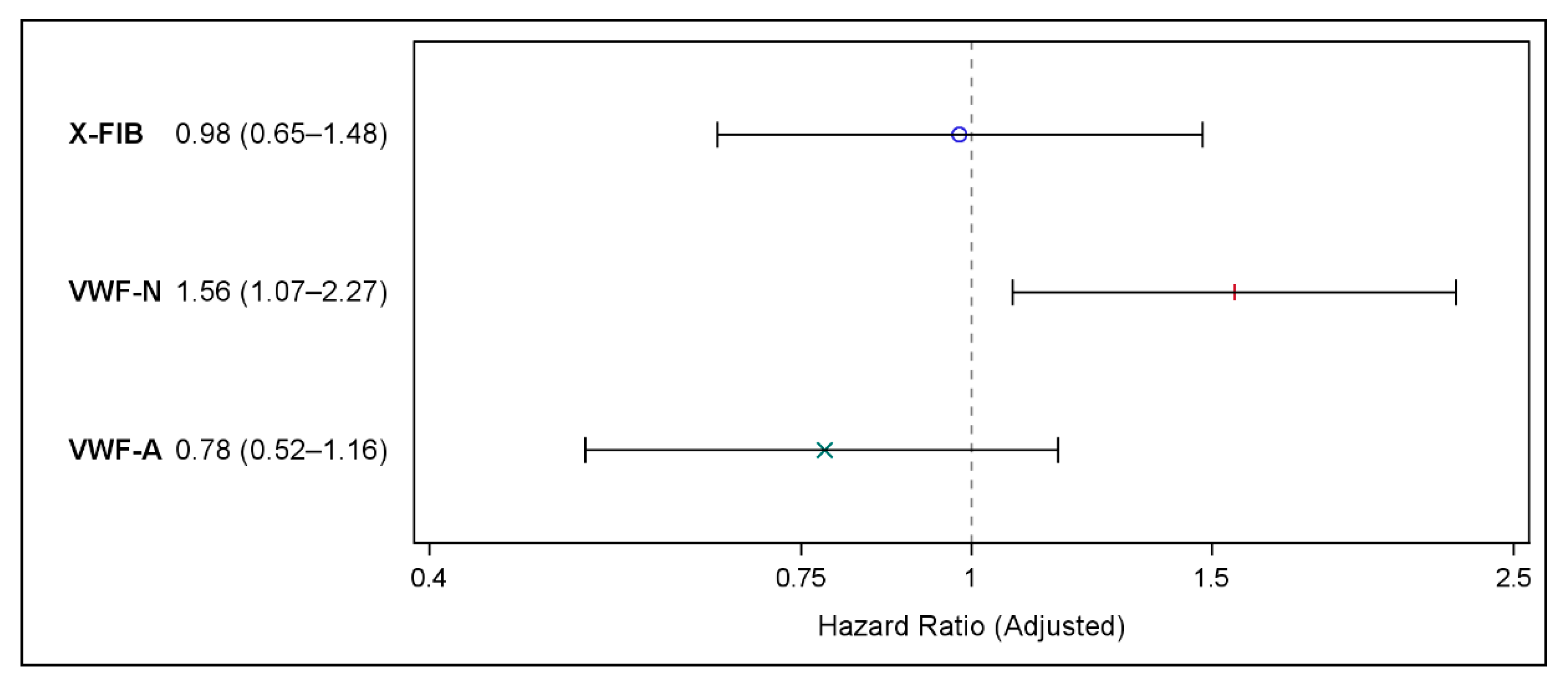
Adjustments for known and suspected confounding factors did not change our findings: Neither X-FIB nor VWF-A predicted future MACE, X-FIB (HR 0.98 (95% CI 0.65–1.48), p = 0.93), and VWF-A: (HR 0.78 (95% CI 0.52–1.16), p = 0.21), whereas VWF-N (HR 1.56 (95% CI 1.07–2.27), p = 0.02) significantly predicted future MACE (Figure 4).
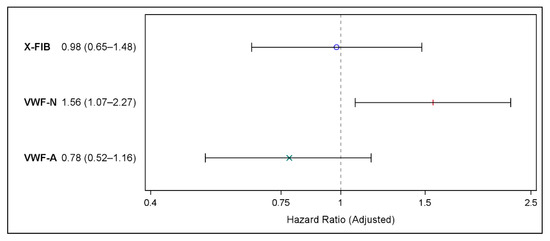
Figure 4.
Forest plot showing the results of the multivariable Cox proportional hazards regression on risk of major cardiovascular events for X-FIB, VWF-N, and VWF-A; Hazard Ratio (95% confidence interval). Adjusted for age, sex, forced expiratory volume in one second (% of predicted), former ischemic heart disease, former heart failure, atrial fibrillation/flutter, hypertension, hypercholesterolemia, peripheral vascular disease type 2 diabetes, smoking status, and ICS use. °: X-FIB HR estimate, |: VWF-N HR estimate, ×: VWF-A HR estimate.
Neither the worst-case nor the best-case imputations altered the results (see Supplementary Table S2 for complete results from all primary and secondary Cox proportional hazards regression models).
3.2. Secondary Outcome
None of the three biomarkers were associated with future MACE, when death was excluded from the composite outcome and included as a competing risk (X-FIB: HR 1.23 (95% CI 0.49–3.06, p = 0.66), VWF-N: HR 1.76 (95% CI 0.75–4.12, p = 0.19), and VWF-A: HR 0.66 (95% CI 0.24–1.81, p = 0.42).
3.3. Explorative Outcomes
Analyzing each biomarker as continuous variables did not alter the results. Each of the four composites of the main outcomes were tested against the dichotomized biomarkers. Of these, only VWF-A and heart failure had a significant association (p = 0.03); see Table 2. When using heart failure as an event in the multivariable Cox proportional hazards model (considering the other parts of the composite outcome as competing risks), VWF-A had a HR for heart failure of 0.16 (0.02–1.21, p = 0.08). No interaction between VWF-N and VWF-A was identified.
Table 2.
Distribution in outcome for high (upper quartile) versus low (lower three quartiles) levels of each biomarker.
Post-hoc explorative-adjusted Cox proportional hazards regressions were performed on each biomarker using all-cause mortality as outcome. Of these, VWF-N showed a significant association with all-cause mortality (HR 1.52 (95% CI 1.02–2.29), p = 0.04).
4. Discussion
We found that high levels of VWF-N, a biomarker of vWF formation, were strongly associated with an increased risk of MACE. However, neither a marker of clot resolution (X-FIB) or vWF activation (VWF-A) was associated with MACE. Our results were robust through unadjusted and adjusted analyses.
The two clot activation factors were largely unchanged in blood levels from the time when patients were admitted with an AECOPD to the time when they reached a stable phase, one month later.
We found an association with a higher risk of MACE for patients with high VWF-N levels, which was completely attenuated when all-cause mortality was excluded from the primary outcome. This is consistent with previous studies, where high levels of vWF-antigen were linked to an increased risk of MACE [20]. Accordingly, in an explorative analysis solely using all-cause mortality, we found an association between VWF-N and all-cause mortality, which is line with a previous study, where VWF-N was associated with all-cause mortality [17]. Our findings on the formation of von Willebrand factor (VWF-N) are in line with the previous findings of von Willebrand factor levels, where increased levels have been associated with all-cause mortality, stroke, and first-time coronary heart disease [12,13,14]. The probable mechanisms of this are both the underlying endothelial dysfunction, which may increase von Willebrand factor levels, as well as the thrombogenicity of von Willebrand factor itself [7,9]. VWF-A was not associated with any of our prespecified outcomes, but showed a near-significant association with heart failure. Subsequent multivariable Cox proportional hazards regression showed a trend towards a lower risk of heart failure admissions in patients with high VWF-A levels. A protective effect of increased activation of a clotting factor is contra-intuitive; rather, it seems more likely that patients with high VWF-A levels in this case are patients with a higher activity of the metalloprotease ADAMTS13, since ADAMTS13 activity is decreased in patients with congestive heart failure [16].
Our neutral finding regarding fibrin degradation contradicts the sparse available data. In the ECLIPSE trial, X-FIB had a predictive value for two-year mortality, with an adjusted HR of 1.48 [19]. Of note, the ECLIPSE study recruited COPD patients in a stable state, whereas our study was based on patients acutely admitted with an exacerbation of COPD, which we showed resulted in higher values of X-FIB. D-dimer, which is also a marker of fibrin degradation similar to X-FIB, has previously been shown to predict 1-year mortality when sampled upon admission for AECOPD and all-cause mortality in a cohort of healthy persons [22,23]. Additionally, high D-dimer levels have been shown to predict coronary heart disease in persons without prior coronary heart disease [12]. Due to few ischemic events in our study, we did not have sufficient power to investigate this outcome.
This study has several strengths; mainly that it was a multicenter prospective study. The population was well-defined, consisting of severe COPD patients with an acute admission due to AECOPD. The study was Good Clinical Practice monitored, and had complete data on the outcome. Furthermore, blood samples were standardized and performed during baseline and follow-up at three hospitals. Finally, missing data were sparse, and no data were missing regarding biomarkers or regarding the explored outcomes (0% missing).
Despite the above-mentioned strengths, our study has some limitations: First, according to the calculated sample size, the study did not reach sufficient numbers, although it was close. This may have decreased the sensitivity to detect signals, or in contrast, it may have increased the risk of false findings. Second, a small proportion of patients did not have spirometry performed. We tried to compensate for this by performing multiple imputations and best-case/worst-case analyses; the results were the same from these approaches, and given that the vast majority of patients had spirometry, we do not believe this to have influenced the main analyses decisively. Lastly, the descriptive statistics on the change in biomarkers from the acute to stable phase may be influenced by patients not completing follow-up. However, this did not affect the primary analyses of this study.
There is a need to understand the mechanisms of life-threatening events such as MACE in COPD patients, both for identifying patients at risk of these events, and to gain knowledge for how to treat these patients in the future, to protect against such events. Thus, while the direct clinical use of the marker is currently limited, it has the possibility to aid in the yet limited classification of COPD phenotypes, and to identify patients who need interventions to avoid MACE. Further, it highlights the importance of von Willebrand factor in COPD patients during acute admissions.
In conclusion, in this study, where we investigated the biomarkers of clot formation and resolution among severe COPD (GOLD 3–4) patients admitted with AECOPD, we confirmed an association between VWF-N and future MACE from other studies. This may help to explain the mechanism of increased cardiovascular events among persons with COPD exacerbation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biomedicines10082011/s1, Table S1: Definition of diagnoses in the registry-based data; Document S1: Handling of missing values; Table S2A–C: Complete information on all regression models.
Author Contributions
Conceptualization: J.-U.J., P.S., J.V. and T.W.; Methodology: P.K., J.M.B.S., C.S.U., J.J., C.P.R., S.R.R., D.J.L., S.G.J., T.W., A.G.M., M.M., T.L., E.B., R.F.-S., D.D.M., T.I., A.B., S.D.N., J.V., T.B.-S., M.K., J.-U.J. and P.S.; Software: P.K. and C.P.R.; Formal analysis: P.K. and C.P.R.; Investigation: J.M.B.S., C.S.U., J.J., S.R.R., D.J.L., T.L., M.K. and P.S.; Resources: J.M.B.S., S.R.R., D.J.L., M.K., P.S. and J.-U.J.; Data curation: P.K., C.P.R. and P.S.; Writing—original draft preparation: P.K., J.M.B.S., P.S. and J.-U.J.; Writing—review and editing: P.K., J.M.B.S., C.S.U., J.J., C.P.R., S.R.R., D.J.L., S.G.J., T.W., A.G.M., M.M., T.L., E.B., R.F.-S., D.D.M., T.I., A.B., S.D.N., J.V., T.B.-S., M.K., J.-U.J. and P.S.; Visualization: P.K.; Supervision: P.S. and J.-U.J.; Project administration: P.K., P.S. and J.-U.J.; Funding acquisition: P.S. and J.-U.J. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Danish Regions Medical Fund (grant no. 5894/16), the Danish Council for Independent Research (grant no. 6110-00268B), and the Novo Nordisk foundation (grant no. NNF20OC0060657). Author D.D.M. received personal funding from the Danish National Research Foundation (DNRF126).
Institutional Review Board Statement
The study was approved by the Ethics Committees of all participating sites (H-15012207), the Danish Medicines Agency (EudraCT no: 2015-003441-26) and the Danish Data Protection Agency (HGH-2015-038 and I-Suite number 04014). The study was registered at clinicaltrials.gov accessed on 1 October 2022 (NCT02857842).
Informed Consent Statement
Written informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data collected for the CORTICO-COP trial, including individual participant data and a data dictionary defining each field in the set, will be made available to others in form of deidentified participant data. Informed consent forms will not be available according to Danish legislation. These data will become available from 1 January 2023 upon request from investigators. Registry-based data will not be made available, due to Danish legislation.
Acknowledgments
CORTICO-COP was an investigator-initiated and driven study. No commercial sponsor had any involvement in the design and undertaking of the trial. The participating investigators in the Steering Committee have the right to the results. We thank the patients, relatives, clinical staff, and research staff at all trial sites, without whose support CORTICO-COP would not have been completed. A.M. and J.V. are supported by the NIHR Manchester Biomedical Research Centre.
Conflicts of Interest
Authors J.M.B.S., S.R.R., D.J.L. and M.K. are employees and shareholders of Nordic Bioscience. Outside the submitted work: C.S.U. has received grants from Sanofi, Boehringer Ingelheim, AstraZeneca and Novartis; speaker fees from Orion Pharma, AstraZeneca and TEVA; consulting fees from Chiesi, Orion Pharma, AstraZeneca, GSK and TEVA; and has been on advisory boards for Novartis, Sanofi, Glaxo-Smith Kline, Chiesi, AstraZeneca and Boehringer Ingelheim. M.M. has received a grant from Grifols and consulting fees from AstraZeneca, Atriva Therapeutics, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Bial, Gebro Pharma, CSL Behring, Laboratorios Esteve, Ferrer, Mereo Biopharma, Verona Pharma, Spin Therapeutics, pH Pharma, ONO Pharma, Palobiofarma SL, Takeda, Novartis, Sanofi and Grifols; speaker fees from AstraZeneca, GlaxoSmithKline, Boehringer Ingelheim, Chiesi, Cipla, Kamada, Takeda, Menarini, Rovi, Bial, Sandoz, Zambon, CSL Behring, Grifols and Novartis; support for attending meetings/travel from Novartis, Boehringer Ingelheim and Menarini; and participation on DSMB for Mereo. EB has received speaker fees from Boehringer Ingelheim and Chiesi, support for attending meetings/travel from Boehringer Ingelheim, and participation on DSMB or advisory board for Boehringer Ingelheim. AB is the secretary of Assembly 5, Airway diseases, Asthma, COPD and Chronic Cough; European Respiratory Society. J.V. has received a grant from Boehringer Ingelheim UK; consulting fees from AstraZeneca, ALK Abello, Boehringer Ingelheim, GSK, Novartis and TEVA; speaker fees from AstraZeneca, Boehringer Ingelheim, Chiesi, GSK and Novartis; and participation on DSMB/advisory boards for AstraZeneca and GSK. All other authors report no conflict of interest. TB-S received consulting fees from GSK and Sanofi Pasteur; received speaker payments from Bayer, Sanofi Pasteur and GSK; support for meetings/travel from AstraZeneca; and received equipment for his department from GE.
References
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Müllerova, H.; Agusti, A.; Erqou, S.; Mapel, D.W. Cardiovascular comorbidity in COPD: Systematic literature review. Chest 2013, 144, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, A.I.; Bartziokas, K.; Loukides, S.; Tsikrika, S.; Karakontaki, F.; Haniotou, A.; Papiris, S.; Stolz, D.; Kostikas, K. Cardiovascular comorbidities in hospitalised COPD patients: A determinant of future risk? Eur. Respir. J. 2015, 46, 846–849. [Google Scholar] [CrossRef]
- Terzano, C.; Conti, V.; Di Stefano, F.; Petroianni, A.; Ceccarelli, D.; Graziani, E.; Mariotta, S.; Ricci, A.; Vitarelli, A.; Puglisi, G.; et al. Comorbidity, hospitalization, and mortality in COPD: Results from a longitudinal study. Lung 2010, 188, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Agusti, A.; Edwards, L.D.; Rennard, S.I.; MacNee, W.; Tal-Singer, R.; Miller, B.E.; Vestbo, J.; Lomas, D.A.; Calverley, P.M.; Wouters, E.; et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: A novel phenotype. PLoS ONE 2012, 7, e37483. [Google Scholar] [CrossRef]
- Goedemans, L.; Bax, J.J.; Delgado, V. COPD and acute myocardial infarction. Eur. Respir. Rev. 2020, 29, 190139. [Google Scholar] [CrossRef] [PubMed]
- Mojzisch, A.; Brehm, M.A. The Manifold Cellular Functions of von Willebrand Factor. Cells 2021, 10, 2351. [Google Scholar] [CrossRef]
- Manon-Jensen, T. Initiation of the wound healing cascade in inflammatory bowel disease: Assessment of Von Willebrand factor ADAMTS-13 processing and formation in crohn’s disease. EC Gastroenterol. Dig. Syst. 2019, 6, 143–154. [Google Scholar]
- Pimanda, J.; Hogg, P. Control of von Willebrand factor multimer size and implications for disease. Blood Rev. 2002, 16, 185–192. [Google Scholar] [CrossRef]
- Reardon, B.; Pasalic, L.; Favaloro, E.J. The Intriguing Relationships of von Willebrand Factor, ADAMTS13 and Cardiac Disease. J. Cardiovasc. Dev. Dis 2021, 8, 115. [Google Scholar] [CrossRef]
- Bartholo, T.P.; Costa, C.H.; Rufino, R. Evaluation of von Willebrand factor in COPD patients. J. Bras. Pneumol. 2014, 40, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Thompson, A.; Aspelund, T.; Rumley, A.; Eiriksdottir, G.; Lowe, G.; Gudnason, V.; Di Angelantonio, E. Hemostatic factors and risk of coronary heart disease in general populations: New prospective study and updated meta-analyses. PLoS ONE 2013, 8, e55175. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, M.A.; Franco, O.H.; Ikram, M.A.; Hofman, A.; Kavousi, M.; de Maat, M.P.; Leebeek, F.W. Von Willebrand Factor, ADAMTS13, and the Risk of Mortality: The Rotterdam Study. Arter. Thromb. Vasc. Biol. 2016, 36, 2446–2451. [Google Scholar] [CrossRef]
- Wieberdink, R.G.; van Schie, M.C.; Koudstaal, P.J.; Hofman, A.; Witteman, J.C.; de Maat, M.P.; Leebeek, F.W.; Breteler, M.M. High von Willebrand factor levels increase the risk of stroke: The Rotterdam study. Stroke 2010, 41, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Sonneveld, M.A.; de Maat, M.P.; Portegies, M.L.; Kavousi, M.; Hofman, A.; Turecek, P.L.; Rottensteiner, H.; Scheiflinger, F.; Koudstaal, P.J.; Ikram, M.A.; et al. Low ADAMTS13 activity is associated with an increased risk of ischemic stroke. Blood 2015, 126, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Gombos, T.; Mako, V.; Cervenak, L.; Papassotiriou, J.; Kunde, J.; Harsfalvi, J.; Forhecz, Z.; Pozsonyi, Z.; Borgulya, G.; Janoskuti, L.; et al. Levels of von Willebrand factor antigen and von Willebrand factor cleaving protease (ADAMTS13) activity predict clinical events in chronic heart failure. Thromb. Haemost. 2009, 102, 573–580. [Google Scholar] [CrossRef]
- Langholm, L.L.; Ronnow, S.R.; Sand, J.M.B.; Leeming, D.J.; Tal-Singer, R.; Miller, B.E.; Vestbo, J.; Karsdal, M.A.; Manon-Jensen, T. Increased von Willebrand Factor Processing in COPD, Reflecting Lung Epithelium Damage, Is Associated with Emphysema, Exacerbations and Elevated Mortality Risk. Int. J. Chronic Obs. Pulm. Dis. 2020, 15, 543–552. [Google Scholar] [CrossRef]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef]
- Manon-Jensen, T.; Langholm, L.L.; Rønnow, S.R.; Karsdal, M.A.; Tal-Singer, R.; Vestbo, J.; Leeming, D.J.; Miller, B.E.; Bülow Sand, J.M. End-product of fibrinogen is elevated in emphysematous chronic obstructive pulmonary disease and is predictive of mortality in the ECLIPSE cohort. Respir. Med. 2019, 160, 105814. [Google Scholar] [CrossRef]
- Kovacevic, K.D.; Mayer, F.J.; Jilma, B.; Buchtele, N.; Obermayer, G.; Binder, C.J.; Blann, A.D.; Minar, E.; Schillinger, M.; Hoke, M. Von Willebrand factor antigen levels predict major adverse cardiovascular events in patients with carotid stenosis of the ICARAS study. Atherosclerosis 2019, 290, 31–36. [Google Scholar] [CrossRef]
- Cushman, M.; Folsom, A.R.; Wang, L.; Aleksic, N.; Rosamond, W.D.; Tracy, R.P.; Heckbert, S.R. Fibrin fragment D-dimer and the risk of future venous thrombosis. Blood 2003, 101, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Di Castelnuovo, A.; de Curtis, A.; Costanzo, S.; Persichillo, M.; Olivieri, M.; Zito, F.; Donati, M.B.; de Gaetano, G.; Iacoviello, L.; Investigators, M.-S.P. Association of D-dimer levels with all-cause mortality in a healthy adult population: Findings from the MOLI-SANI study. Haematologica 2013, 98, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wu, Y.; Zhou, Y.; Wu, Z.; Wei, L.; Li, Y.; Peng, G.; Liang, W.; Ran, P. Prognostic role of D-dimer for in-hospital and 1-year mortality in exacerbations of COPD. Int. J. Chronic Obs. Pulm. Dis. 2016, 11, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food & Drug Administration. Biomarkers Qualification Review for Plasma Fibrinogen. Available online: https://www.fda.gov/media/92567/download (accessed on 30 May 2022).
- Sivapalan, P.; Lapperre, T.S.; Janner, J.; Laub, R.R.; Moberg, M.; Bech, C.S.; Eklof, J.; Holm, F.S.; Armbruster, K.; Sivapalan, P.; et al. Eosinophil-guided corticosteroid therapy in patients admitted to hospital with COPD exacerbation (CORTICO-COP): A multicentre, randomised, controlled, open-label, non-inferiority trial. Lancet Respir. Med. 2019, 7, 699–709. [Google Scholar] [CrossRef]
- Sun, S.; Karsdal, M.A.; Mortensen, J.H.; Luo, Y.; Kjeldsen, J.; Krag, A.; Jensen, M.D.; Bay-Jensen, A.C.; Manon-Jensen, T. Serological Assessment of the Quality of Wound Healing Processes in Crohn’s Disease. J. Gastrointest. Liver Dis. 2019, 28, 175–182. [Google Scholar] [CrossRef]
- Von Elm, E.; Altman, D.G.; Egger, M.; Pocock, S.J.; Gotzsche, P.C.; Vandenbroucke, J.P.; Initiative, S. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: Guidelines for reporting observational studies. J. Clin. Epidemiol. 2008, 61, 344–349. [Google Scholar] [CrossRef]
- Peter Kamstrup, J.M.B.S.; Ulrik, C.; Janner, J.; Rønnow, S.R.; Leeming, D.J.; Jensen, S.G.; Wilcke, T.; Miravitlles, M.; Mathioudakis, A.; Lapperre, T.; et al. Protocol: Biomarkers of Endothelial Damage in Patients with COPD. A Substudy of the CORTICO-COP Randomized Controlled Trial. Available online: http://coptrin.dk/wp-content/uploads/2022/01/cortico-endotheldamage-sign-protokol.pdf (accessed on 10 June 2022).
- Bartlett, J.W.; Seaman, S.R.; White, I.R.; Carpenter, J.R. Multiple imputation of covariates by fully conditional specification: Accommodating the substantive model. Stat. Methods Med. Res. 2015, 24, 462–487. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).