
Pathophysiological Mechanisms of Antipsychotic-Induced Parkinsonism
 ,
,  ,
,  ,
,  , and
, and
Abstract
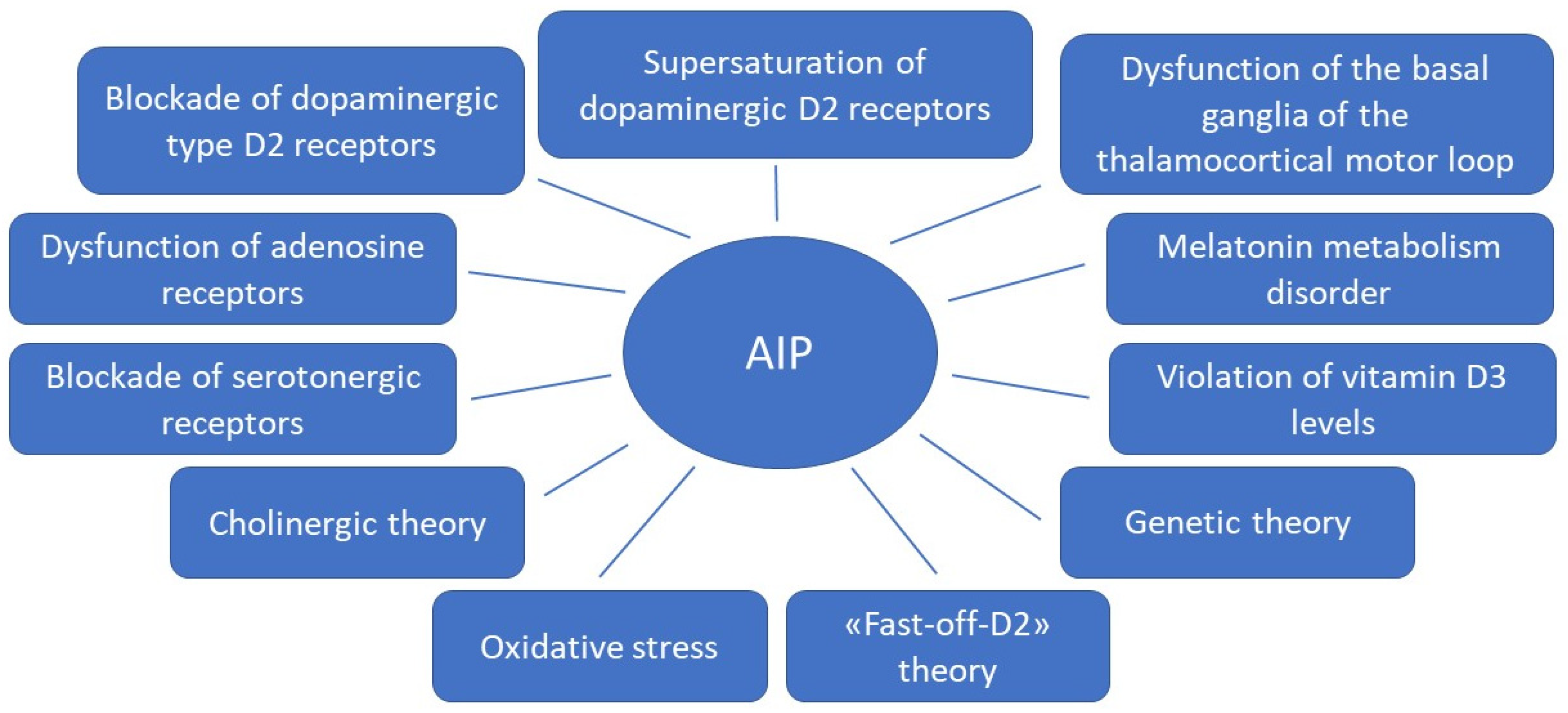
1. Introduction
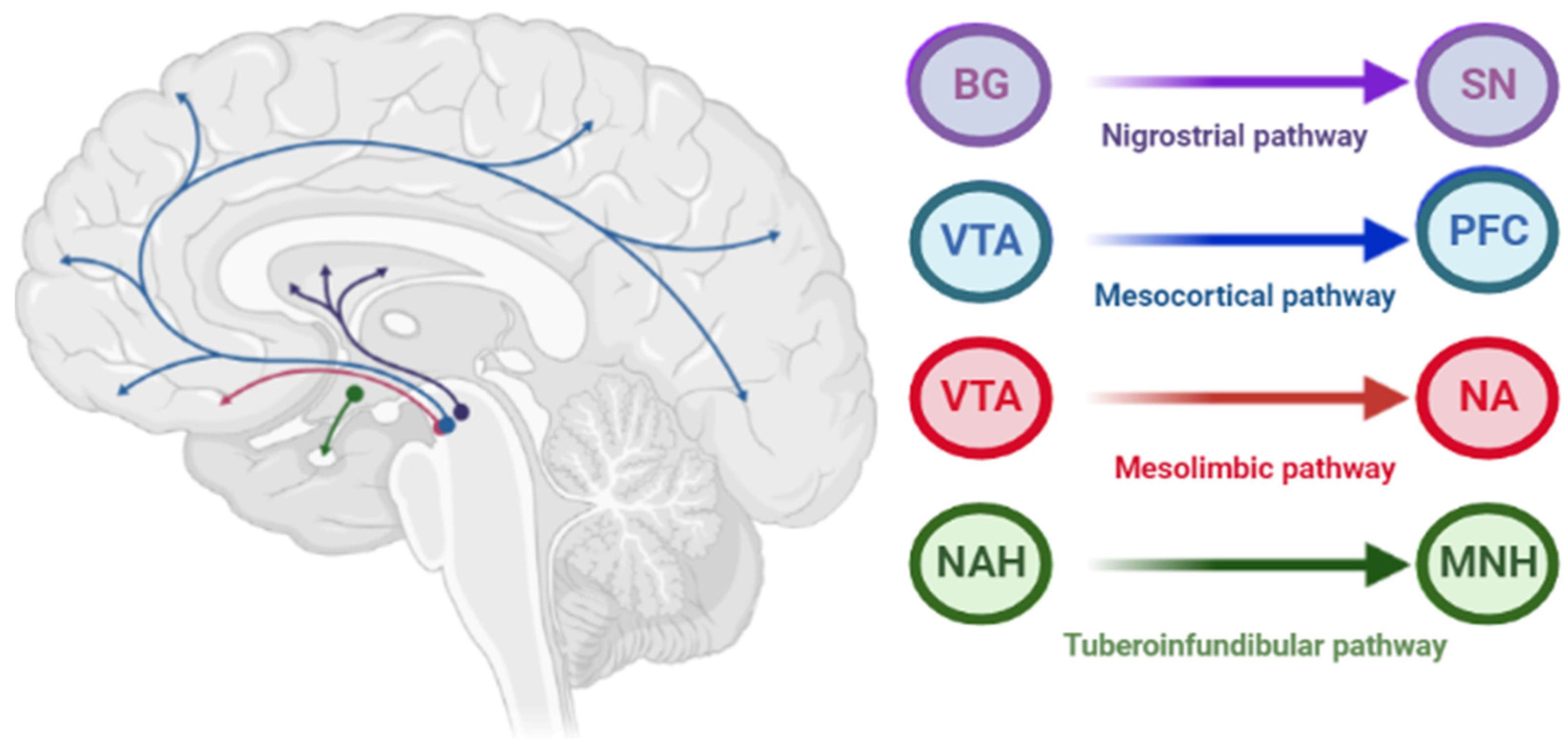
1.1. Dopamine D2 Type Receptors Blockade
1.2. Supersaturation (“Occupancies”) of Striatal Dopamine D2/3 Receptors
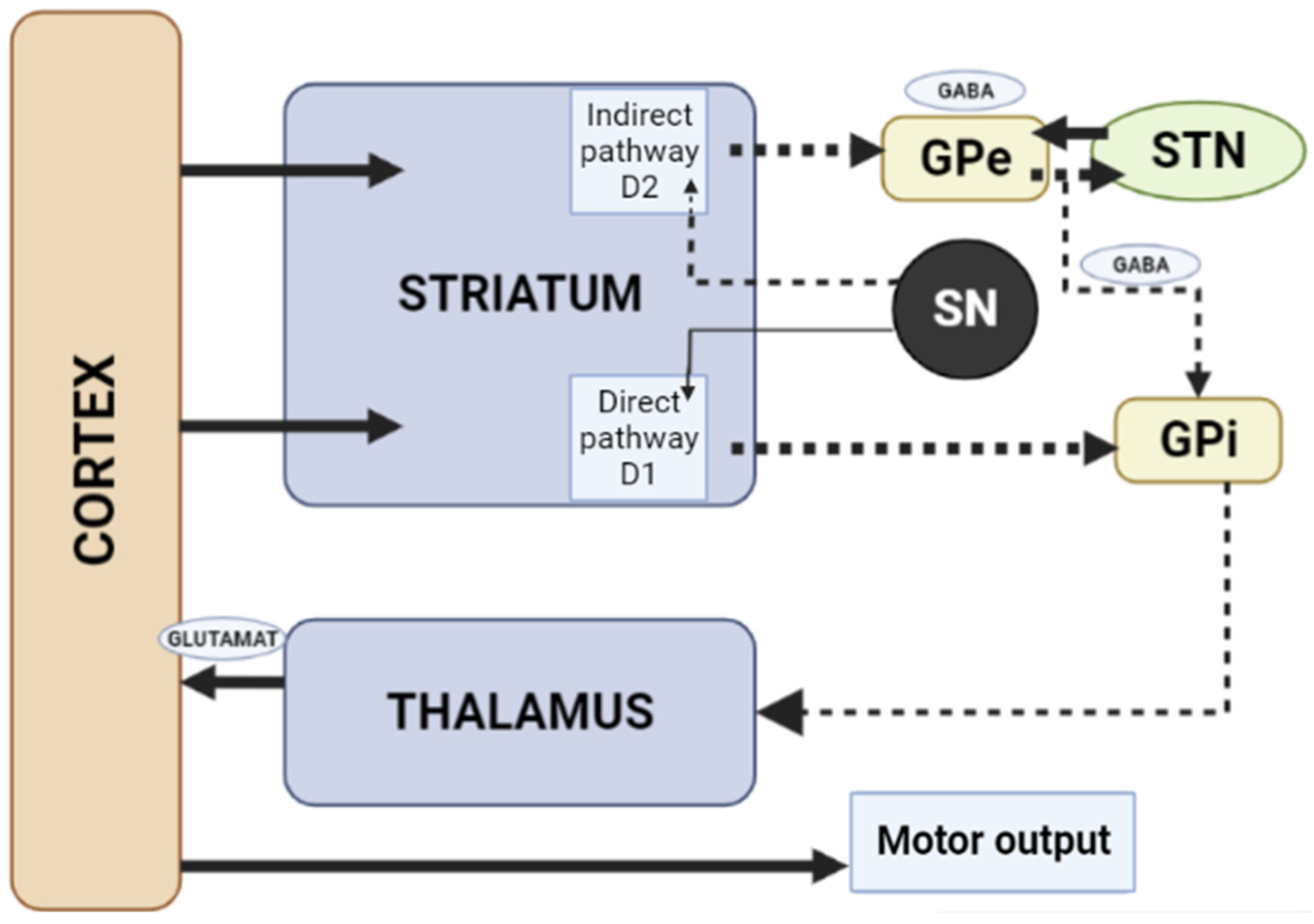
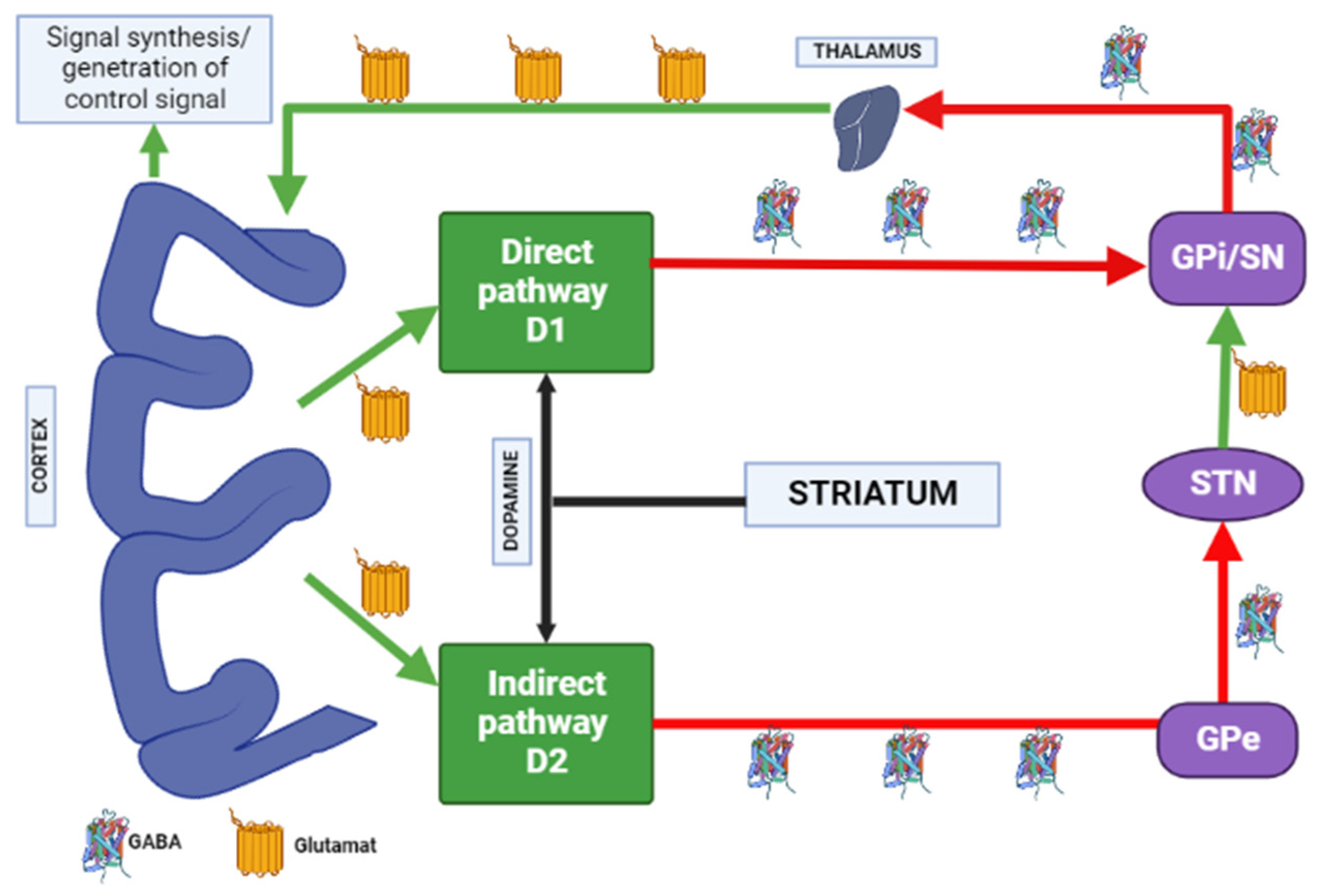
1.3. Influence of the Basal Ganglia of the Thalamocortical Motor Loop
1.4. Fast-off-D Theory
1.5. Role of Adenosine Receptors
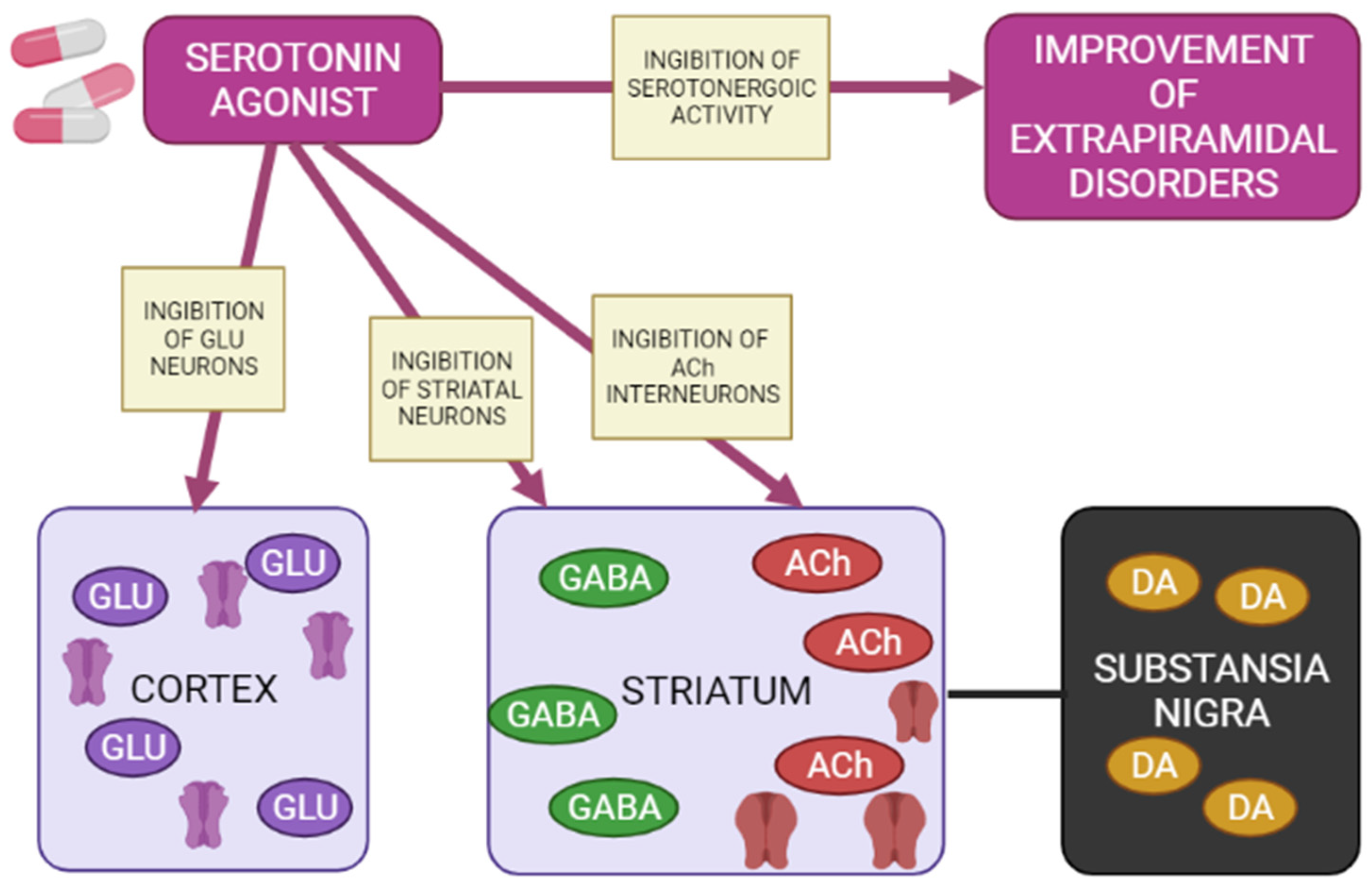
1.6. Blockade of the Serotonergic System
1.7. Cholinergic Theory
1.8. Melatonin Theory
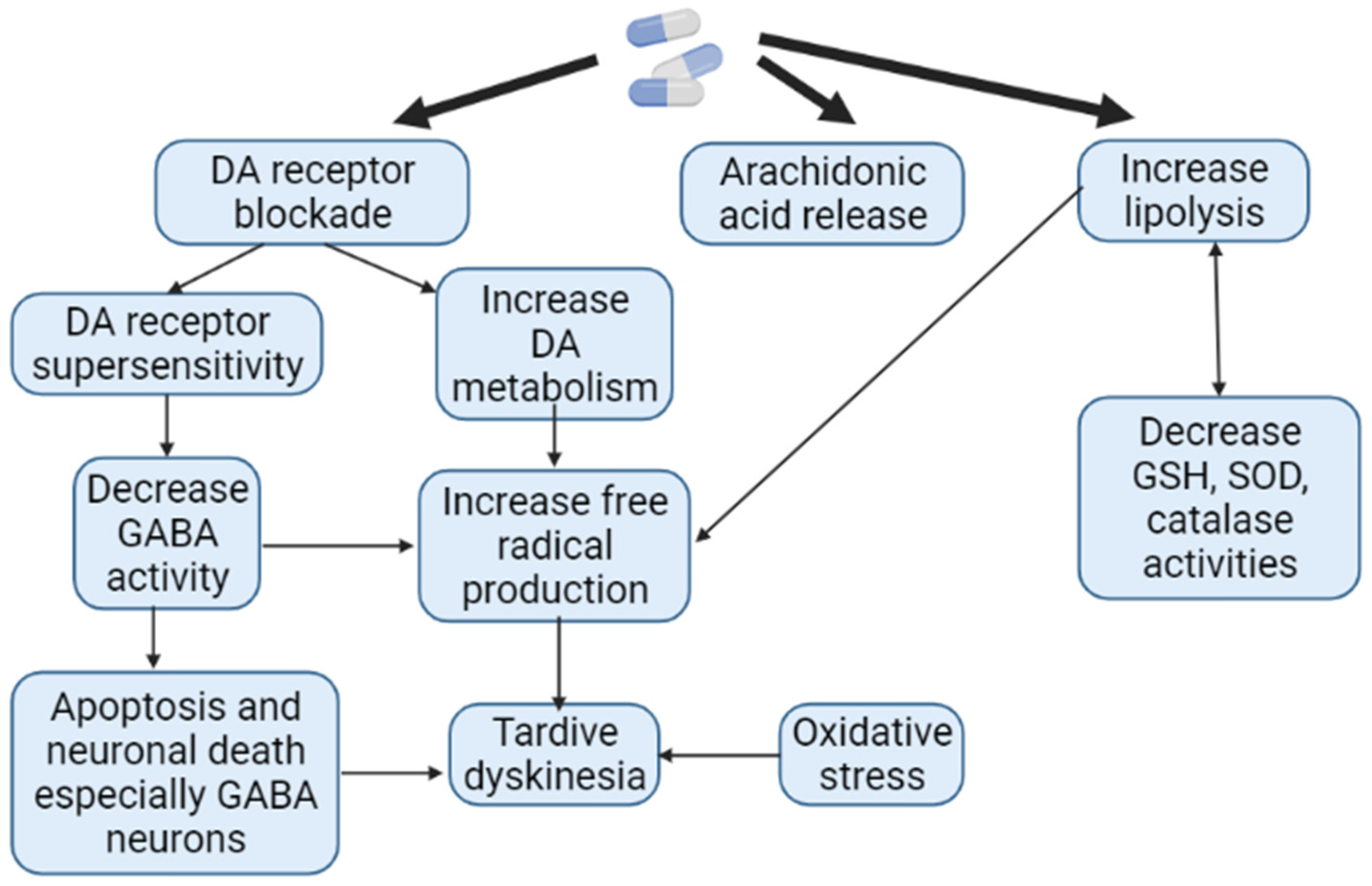
1.9. Theory of Oxidative Stress
1.10. Role of Vitamin D3
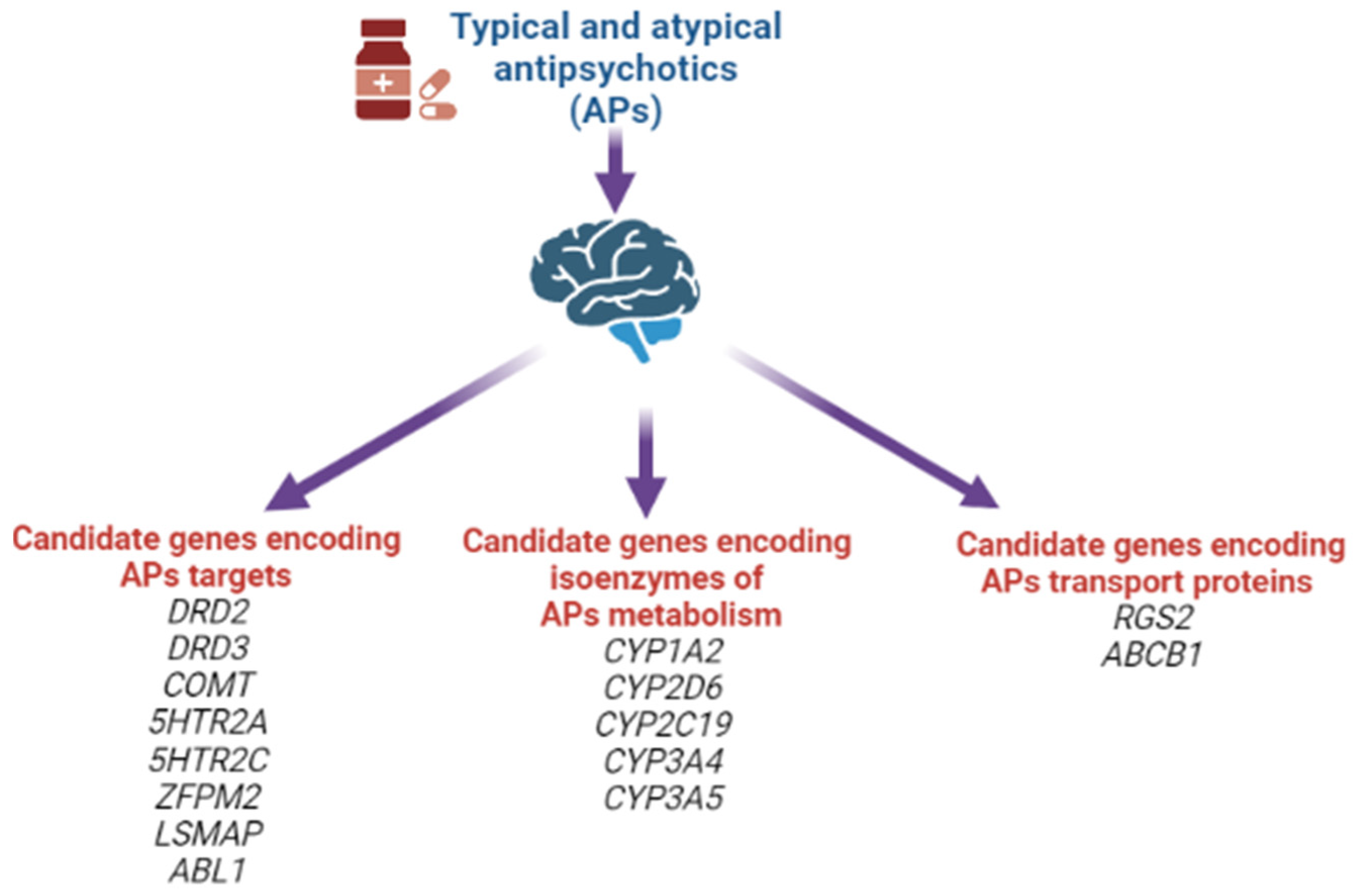
1.11. Genetic Theory
2. Discussion
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, L.G.; Jankovic, J. Neurologic approach to drug-induced movement disorders: A study of 125 patients. South. Med. J. 1990, 83, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Sethi, K.D. Movement disorders induced by dopamine blocking agents. Semin. Neurol. 2001, 21, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Esper, C.D.; Factor, S.A. Failure of recognition of drug-induced parkinsonism in the elderly. Mov. Disord. 2008, 23, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.W.; Chung, S.J. Drug-induced parkinsonism. J. Clin. Neurol. 2012, 8, 15–21. [Google Scholar] [CrossRef]
- Caroff, S.N.; Hurford, I.; Lybrand, J.; Campbell, E.C. Movement disorders induced by antipsychotic drugs: Implications of the CATIE schizophrenia trial. Neurol. Clin. 2011, 29, 127–148. [Google Scholar] [CrossRef]
- Shnayder, N.A.; Vaiman, E.E.; Neznanov, N.G.; Nasyrova, R.F. Pharmacogenetics of Antipsychotic-Induced Extrapyramidal Disorders; DEAN Publishing House: St. Petersburg, Russia, 2022; p. 288. [Google Scholar]
- Lauder, J.M.; Bloom, F.E. Ontogeny of monoamine neurons in the locus coeruleus, Raphe nuclei and substantia nigra of the rat. I. Cell differentiation. J. Comp. Neurol. 1974, 155, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524–532. [Google Scholar] [CrossRef]
- Lynd-Balta, E.; Haber, S.N. The organization of midbrain projections to the striatum in the primate: Sensorimotor-related striatum versus ventral striatum. Neuroscience. 1994, 59, 625–640. [Google Scholar] [CrossRef]
- Nyberg, S.; Dencker, S.J.; Malm, U.; Dahl, M.-L.; Svetnson, J.-O.; Halldin, C.; Naskashima, Y.; Farde, L. D(2)- and 5-Ht(2) receptor occupancy in high-dose neuroleptictreated patients. Int. J. Neuropsychopharmacol. 1998, 1, 95–101. [Google Scholar] [CrossRef]
- Crocker, A.D.; Hemsley, K.M. An animal model of extrapyramidal side effects induced by antipsychotic drugs: Relationship with D2 dopamine receptor occupancy. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2001, 25, 573–590. [Google Scholar] [CrossRef]
- Farde, L.; Nordstrom, A.L.; Wiesel, F.A.; Pauli, S.; Halldin, C.; Sedvall, G. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch. Gen. Psychiatry 1992, 49, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Scharrer, J.; Tatsch, K.; Schwarz, J.; Oertel, W.H.; Konjarczyk, M.; Albus, M. D2-dopamine receptor occupancy differs between patients with and without extrapyramidal side effects. Acta Psychiatr. Scand. 1994, 90, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Haddad, P.M.; Dursun, S.M. Neurological complications of psychiatric drugs: Clinical features and management. Hum. Psychopharmacol. 2008, 23 (Suppl. 1), S15–S26. [Google Scholar] [CrossRef] [PubMed]
- Margolese, H.C.; Chouinard, G.; Kolivakis, T.T.; Beauclair, L.; Miller, R. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 1: Pathophysiology and mechanisms of induction. Can. J. Psychiatry. 2005, 50, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Sebastiao, A.M.; Ribeiro, J.A. Adenosine receptors and the central nervous system. Handb. Exp. Pharmacol. 2009, 193, 471–534. [Google Scholar] [CrossRef]
- Grunder, G.; Carlsson, A.; Wong, D.F. Mechanism of new antipsychotic medications: Occupancy is not just antagonism. Arch. Gen. Psychiatry 2003, 60, 974–977. [Google Scholar] [CrossRef]
- Sharma, A.; Sorrell, J.H. Aripiprazole-induced parkinsonism. Int. Clin. Psychopharmacol. 2006, 21, 127–129. [Google Scholar] [CrossRef]
- Gunne, L.M.; Andrén, P.E. An animal model for coexisting tardive dyskinesia and tardive parkinsonism: A glutamate hypothesis for tardive dyskinesia. Clin. Neuropharmacol. 1993, 16, 90–95. [Google Scholar] [CrossRef]
- Snyder, S.; Greenberg, D.; Yamamura, H.I. Antischizophrenic drugs and brain cholinergic receptors. Affinity for muscarinic sites predicts extrapyramidal effects. Arch. Gen. Psychiatry. 1974, 31, 58–61. [Google Scholar] [CrossRef]
- Susatia, F.; Fernandez, H.H. Drug-induced parkinsonism. Curr. Treat. Options Neurol. 2009, 11, 162–169. [Google Scholar] [CrossRef]
- Ward, K.M.; Citrome, L. Antipsychotic-Related Movement Disorders: Drug-Induced Parkinsonism vs. Tardive Dyskinesia-Key Differences in Pathophysiology and Clinical Management. Neurol. Ther. 2018, 7, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Vaiman, E.E.; Shnayder, N.A.; Neznanov, N.G.; Nasyrova, R.F. Pathophysiological mechanisms underlying antipsychotic-induced tardive dyskinesia. Bull. Sib. Med. 2019, 18, 169–184. [Google Scholar] [CrossRef]
- Ossowska, K. Neuronal basis of neuroleptic-induced extrapyramidal side effects. Pol. J. Pharmacol. 2002, 54, 299–312. [Google Scholar] [PubMed]
- Powell, A.; Gallur, L.; Koopowitz, L.; Hayes, M.W. Parkinsonism in the psychiatric setting: An update on clinical differentiation and management. BMJ Neurol. Open 2020, 2, e000034. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S.; Seeman, P. Does fast dissociation from the dopamine D2 receptor explain the action of atypical antipsychotics? A new hypothesis. Am. J. Psychiatry 2001, 158, 360–369. [Google Scholar] [CrossRef]
- Bohlega, S.A.; Al-Foghom, N.B. Drug-induced Parkinson’s disease. A clinical review. Neurosci. J. 2013, 18, 215–221. [Google Scholar]
- Fuxe, K.; Ferré, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A2A and dopamine D2 heteromeric receptor complexes and their function. J Mol. Neurosci. 2005, 26, 209–220. [Google Scholar] [CrossRef]
- Kulisevsky, J.; Poyurovsky, M. Adenosine A2A-receptor antagonism and pathophysiology of Parkinson’s disease and drug-induced movement disorders. Eur. Neurol. 2012, 67, 4–11. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Hauser, R.; Morelli, M.; Fredholm, B.; Chen, J. Adenosine, adenosine A 2A antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 406–413. [Google Scholar] [CrossRef]
- Hettinger, B.D.; Lee, A.; Linden, J.; Rosin, D.L. Ultrastructural localization of adenosine A2A receptors suggests multiple celular sites for modulation of GABAergic neurons in rat striatum. J. Comp. Neurol. 2001, 431, 331–346. [Google Scholar] [CrossRef]
- Varty, G.B.; Hodgson, R.A.; Pond, A.J.; Grzelak, M.E.; Parker, E.M.; Hunter, J.C. The effects of adenosine A2A receptor antagonists on haloperidol-induced movement disorders in primates. Psychopharmacology 2008, 200, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E.; Ferre, S. Blocking striatal adenosine A 2A receptors: A new strategy for basal ganglia disorders. Recent Pat. CNS Drug Discov. 2007, 2, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Dayne, M.R.; Larson, G.; Orona, R.A.; Zahniser, N.R. Opposing actions of adenosine A2a and dopamine D2 receptor activation on GABA release in the basal ganglia: Evidence for an A2a/D2 receptor interaction in globus pallidus. Synapse 1996, 22, 132–138. [Google Scholar] [CrossRef]
- Ferre, S.; Fredholm, B.B.; Morelli, M.; Popoli, P. Adenosine-dopamine receptor- receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997, 20, 482–487. [Google Scholar] [CrossRef]
- Parsons, B.; Togasaki, D.M.; Kassir, S.; Przedborski, S. Neuroleptics up-regulate adenosine A 2a receptors in rat striatum: Im- 487 plications for the mechanism and the treatment of tardive dyskinesia. J. Neurochem. 1995, 65, 2057–2064. [Google Scholar] [CrossRef]
- Bishnoi, M.; Chopra, K.; Kulkarni, S.K. Involvement of adenosinergic receptor system in an animal model of tardive dyskinesia and associated behavioural, biochemical and neurochemical changes. Eur. J. Pharmcol. 2006, 552, 55–66. [Google Scholar] [CrossRef]
- Bishnoi, M.; Chopra, K.; Kulkarni, S.K. Theophylline, adenosine receptor antagonist prevents behavioral, biochemical and neurochemical changes associated with an animal model of tardive dyskinesia. Pharmacol. Rep. 2007, 59, 181–191. [Google Scholar]
- Wardas, J.; Konieczny, J.; Lorenc-Koci, E. SCH 58261, an A(2A) adenosine receptor antagonist, counteracts parkinsonian-like muscle rigidity in rats. Synapse 2001, 41, 160–171. [Google Scholar] [CrossRef]
- John, D.S.; Salamone, J.D.; Betz, A.J.; Ishiwari, K.; Felsted, J.; Madson, L.; Mirante, B.; Clark, K.; Font, L.; Korbey, S.; et al. Tremorolytic effects of adenosine A2A antagonists: Implications for parkinsonism. Front. Biosci. 2008, 13, 3594–3605. [Google Scholar] [CrossRef]
- Roth, B.L. Multiple serotonin receptors: Clinical and experimental aspects. Ann. Clin. Psychiatry. 1994, 6, 67–78. [Google Scholar] [CrossRef]
- Baumgarten, H.G.; Grozdanovic, Z. Psychopharmacology of central serotonergic systems. Pharmacopsychiatry 1995, 28 (Suppl. 2), 73–79. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y. Therapeutic role of 5-HT1A receptors in the treatment of schizophrenia and Parkinson’s disease. CNS Neurosci. Ther. 2011, 17, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef]
- Ohno, Y.; Tatara, A.; Shimizu, S.; Sasa, M. Management of Cognitive Impairments in Schizophrenia: The Therapeutic Role of 5-HT Receptors. In Schizophrenia Research: Recent Advances; Sumiyoshi, T., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2012; pp. 321–335. [Google Scholar]
- Ohno, Y.; Shimizu, S.; Imaki, J.; Masui, A.; Tatara, A. Management of Antipsychotic-Induced Extrapyramidal Motor Disorders: Regulatory Roles of the Serotonergic Nervous System. In Antipsychotic Drugs: Pharmacology, Side Effects and Abuse Prevention; Schwartz, T.L., Topel, M., Menga, J.L., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2013; pp. 219–234. [Google Scholar]
- Pucadyil, T.J.; Kalipatnapu, S.; Chattopadhyay, A. The serotonin1A receptor: A representative member of the serotonin receptor family. Cell. Mol. Neurobiol. 2005, 25, 553–580. [Google Scholar] [CrossRef] [PubMed]
- Neal-Beliveau, B.S.; Joyce, J.N.; Lucki, I. Serotonergic involvement in haloperidol-induced catalepsy. J. Pharmacol. Exp. Ther. 1993, 265, 207–217. [Google Scholar]
- Wadenberg, M.L.; Young, K.A.; Richter, J.T.; Hicks, P.B. Effects of local application of 5-hydroxytryptamine into the dorsal or median raphe nuclei on haloperidol-induced catalepsy in the rat. Neuropharmacology 1999, 38, 151–156. [Google Scholar] [CrossRef]
- Mignon, L.; Wolf, W.A. Postsynaptic 5-HT1A receptors mediate an increase in locomotor activity in the monoamine-depleted rat. Psychopharmacology 2002, 163, 85–94. [Google Scholar] [CrossRef]
- Ohno, Y.; Shimizu, S.; Imaki, J. Evaluation of the antibradykinetic actions of 5-HT1A agonists using the mouse pole test. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1302–1307. [Google Scholar] [CrossRef]
- Ohno, Y.; Shimizu, S.; Imaki, J. Anticataleptic 8-OH-DPAT preferentially counteracts with haloperidol-induced Fos expression in the dorsolateral striatum and the core region of the nucleus accumbens. Neuropharmacology 2008, 55, 717–723. [Google Scholar] [CrossRef]
- Ohno, Y.; Shimizu, S.; Imaki, J. Effects of tandospirone, a 5-HT1A agonistic anxiolytic agent, on haloperidol-induced catalepsy and forebrain Fos expression in mice. J. Pharmacol. Sci. 2009, 109, 593–599. [Google Scholar] [CrossRef]
- Shimizu, S.; Tatara, A.; Imaki, J.; Ohno, Y. Role of cortical and striatal 5-HT1A receptors in alleviating antipsychotic-induced extrapyramidal disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y. The mechanism of action of novel antipsychotic drugs. Schizophr. Bull. 1991, 17, 263–287. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Ishida-Tokuda, K.; Ishibashi, T. Potential role of 5-HT2 and D2 receptor interaction in the atypical antipsychotic action of the novel succimide derivative, perospirone. Pol. J. Pharmacol. 1997, 49, 213–219. [Google Scholar] [PubMed]
- Kapur, S.; Remington, G. Atypical antipsychotics: New directions and new challenges in the treatment of schizophrenia. Annu. Rev. Med. 2001, 52, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Livesey, M.R.; Cooper, M.A.; Deeb, T.Z. Structural determinants of Ca2+ permeability and conduction in the human 5-hydroxytryptamine type 3A receptor. J. Biol. Chem. 2008, 283, 19301–19313. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Kang, W.H.; Li, Q. Beneficial effects of ondansetron as an adjunct to haloperidol for chronic, treatment-resistant schizophrenia: A double-blind, randomize, placebo-controlled study. Schizophr. Res. 2006, 88, 102–110. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Mohammadi, N.; Noroozian, M.; Karamghadiri, N.; Ghoreishi, A.; Jamshidi, A.-H.; Forghani, S. Added ondansetron for stable schizophrenia: A double blind, placebo controlled trial. Schizophr. Res. 2009, 107, 206–212. [Google Scholar] [CrossRef]
- Bonsi, P.; Cuomo, D.; Ding, J.; Sciamanna, G.; Ulrich, S.; Tscherter, A.; Bernardi, G.; Surmeier, D.J.; Pisani, A. Endogenous serotonin excites striatal cholinergic interneurons via the activation of 5-HT2C, 5-HT6, and 5-HT7 serotonin receptors: Implications for extrapyramidal side effects of serotonin reuptake inhibitors. Neuropsychopharmacology 2007, 32, 1840–1854. [Google Scholar] [CrossRef]
- Ohno, Y.; Imaki, J.; Mae, Y. Serotonergic modulation of extrapyramidal motor disorders in mice and rats: Role of striatal 5-HT3 and 5-HT6 receptors. Neuropharmacology 2011, 60, 201–208. [Google Scholar] [CrossRef]
- Tatara, A.; Shimizu, S.; Shin, N.; Sato, M.; Sugiuchi, T.; Imalki, J.; Ohno, Y. Modulation of antipsychotic-induced extrapyramidal side effects by medications for mood disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 38, 252–259. [Google Scholar] [CrossRef]
- Ohno, Y.; Shimizu, S.; Tokudome, K. Pathophysiological roles of serotonergic system in regulating extrapyramidal motor functions. Biol. Pharm. Bull. 2013, 36, 1396–1400. [Google Scholar] [CrossRef] [PubMed]
- Ebmeier, K.P.; O’Brien, J.T.; Taylor, J.-P. Psychiatry of Parkinson’s Disease. Adv. Biol. Psychiatry 2012, 12, 49–60. [Google Scholar] [CrossRef]
- Naber, D.; Wirz-Justice, A.; Kafka, M.S. Seasonal variations in the endogenous rhythms of dopamine receptor binding in the rat striatum. Biol. Psychiatry 1981, 16, 831–839. [Google Scholar] [PubMed]
- Naber, D.; Wirz-Justice, A.; Kafka, M.S. Chronic fluphenazine treatment modifies circadian rhythms of neurotransmitter receptor binding in the rat. J. Neural Transm. 1982, 55, 277–288. [Google Scholar] [CrossRef]
- Brown, W.A.; Herz, L.R. Response to neuroleptic drugs as a device for classifying schizophrenia. Schizophr. Bull. 1989, 15, 123–129. [Google Scholar] [CrossRef][Green Version]
- Gaffori, O.; Geffard, M.; Van Ree, J.M. des-Tyr1-gamma-endorphin and haloperidol increase pineal gland melatonin levels in rats. Peptides 1983, 4, 393–395. [Google Scholar] [CrossRef]
- Smith, J.A.; Mee, T.J.X.; Barnes, J.D. Increased serum melatonin levels in chlorpromazine-treated psychiatric patients. J. Neural Transm. 1978, 13, 397. [Google Scholar]
- Horita, N.; Ishii, T.; Moroji, T. Effects of long-term administration of chlorpromazine on the pineal gland of rats. Acta Neuropathol. 1978, 42, 49–52. [Google Scholar] [CrossRef]
- Ayd, F.U. A survey of drug-induced extrapyramidal reactions. J. Am. Med. Assoc. 1961, 175, 1054–1060. [Google Scholar] [CrossRef]
- Sack, R.L.; Lewy, A.J.; Erb, D.L.; Vollmer, W.M.; Singer, C.M. Human melatonin production decreases with age. J. Pineal Res. 1986, 3, 379–388. [Google Scholar] [CrossRef]
- Miles, A.; Philbrick, D.R.S. Melatonin and psychiatry. Biol. Psychiatry 1988, 23, 405–425. [Google Scholar] [CrossRef]
- Trentini, G.P.; De Gaetani, C.F.; Criscuolo, M.; Migaldi, M.; Ferrari, G. Pineal Calcification in Different Physiopathological Conditions in Humans. In Fundamentals and Clinics in Pineal Research; Trentini, G.P., De Gaetani, C., Pevet, P., Eds.; Raven Press: New York, NY, USA, 1987; pp. 291–304. [Google Scholar]
- Sandyk, R.; Kay, S.R.; Gillman, M.A. The role of melatonin in the antipsychotic and motor-side effects of neuroleptics: A hypothesis. Int. J. Neurosci. 1992, 64, 203–207. [Google Scholar] [CrossRef]
- Zenkov, N.K.; Lankin, V.Z.; Men’shikova, E.B. Oxidative Stress: Biochemical and Pathophysiological Aspects; Nauka: Moscow, Russia, 2001; 343p. [Google Scholar]
- Dolgo-Saburov, V.B.; Dagaev, S.G.; Kubarskaya, L.G.; Solovjeva, N.E. The role of the nitric oxide generation system in neuro lepticinduced Parkinsonism. Dokl. Biol. Sci. 2012, 444, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, K.; Anemo, K.; Nakamura, K.; Fukunishi, I.; Igarashi, K. Analysis of the metabolism of haloperidol and its neurotoxic pyridinium metabolite in patients with drug-induced parkinsonism. Neuropsychobiology 2001, 44, 126–128. [Google Scholar] [CrossRef]
- Ulrich, S.; Sandmann, U.; Genz, A. Serum concentrations of haloperidol pyridinium metabolites and the relationship with tardive dyskinesia and parkinsonism: A cross-section study in psychiatric patients. Pharmacopsychiatry 2005, 38, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Usuki, E.; Bloomquist, J.R.; Freeborn, E.; Castagnoli, K.; Van Der Schyf, C.J.; Castagnoli, N. Metabolic studies on haloperidol and its tetrahydropyridinyl dehydrationproduct (HPTP) in C57BL/6 mouse brain preparations. Neurotox. Res. 2002, 4, 51–58. [Google Scholar] [CrossRef]
- Reinke, A.; Martins, M.R.; Lima, M.S.; Moreira, J.C.; Dal-Pizzol, F.; Quevedo, J. Haloperidol and clozapine, but not olanzapine, induces oxidative stress in rat brain. Neurosci. Lett. 2004, 372, 157–160. [Google Scholar] [CrossRef]
- Ukai, W.; Ozawa, H.; Tateno, M.; Hashimoto, E.; Saito, T. Neurotoxic potential of haloperidol in comparison with risperidone: Implication of Akt-mediated signal changes by haloperidol. J. Neural Transm. 2004, 111, 667–681. [Google Scholar] [CrossRef]
- Saldana, M.; Bonastre, M.; Aguilar, E.; Marin, C. Role of nigral NFkappaB p50 and p65 subunit expression in haloperidol-induced neurotoxicity and stereotyped behavior in rats. Eur. Neuropsychopharmacol. 2006, 16, 491–497. [Google Scholar] [CrossRef]
- Mena, M.A.; de Yébenes, J.G. Drug-induced parkinsonism. Expert Opin. Drug Saf. 2006, 5, 759–771. [Google Scholar] [CrossRef]
- Cui, X.; Pelekanos, M.; Liu, P.-Y.; Burne, T.H.J.; McGrath, J.J.; Eyles, D.W. The vitamin D receptor in dopamine neurons; its presence in human substantia nigra and its ontogenesis in rat midbrain. Neuroscience 2013, 236, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, Y.W.; Tang, Y.L.; Liu, X.; Jiang, J.-H.; Li, Q.-G.; Yuan, J.-Y. Vitamin D: Preventive and therapeutic potential in Parkinson’s disease. Curr. Drug Metab. 2013, 14, 989–993. [Google Scholar] [CrossRef]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yang, Y.F.; Luo, P.; Liu, W.; Dai, S.-H.; Zheng, X.-R.; Fei, Z.; Jiang, X.-F. Homer1 knockdown protects dopamine neurons through regulating calcium homeostasis in an in vitro model of Parkinson’s disease. Cell. Signal. 2013, 25, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Sandoval, O.; Carrillo-Reid, L.; Galarraga, E.; Tapia, D.; Mendoza, E.; Gomora, J.C.; Aceves, J.; Bargas, J. Bursting in substantia nigra pars reticulata neurons in vitro: Possible relevance for Parkinson disease. J. Neurophysiol. 2007, 98, 2311–2323. [Google Scholar] [CrossRef]
- Dontseva, E.A.; Trefilova, V.V.; Popova, T.E.; Petrova, M.M.; Al-Zamil, M. Perspectives of personalized approach to prevention and treatment of anticonvulsant-induced osteoporosis via action on vitamin D exchange and VDR expression. Pers. Psychiatry Neurol. 2021, 1, 46–62. [Google Scholar] [CrossRef]
- Blandini, F.; Porter, R.H.P.; Greenamyre, J.T. Glutamate and Parkinson’s disease. Mol. Neurobiol. 1996, 12, 73–94. [Google Scholar] [CrossRef]
- Hurley, M.J.; Dexter, D.T. Voltage-gated calcium channels and Parkinson’s disease. Pharmacol Ther. 2012, 133, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Ogundele, O.M.; Okunnuga, A.A.; Fabiyi, T.D.; Olajide, O.J.; Akinrinade, I.D.; Adeniyi, P.A.; Ojo, A.A. NMDA receptor inhibition and potentiation affects cellular process formation in melanocytes; a model for synaptic denervation in Parkinsonism. Metabol. Brain Dis. 2014, 29, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Ogundele, O.M.; Nanakumo, E.T.; Ishola, A.O.; Obende, O.M.; Enye, L.A.; Balogun, W.; Cobham, A.; Abdulbasit, A. NMDA R/+VDR pharmacological phenotype as a novel therapeutic target in relieving motor-cognitive impairments in Parkinsonism. Drug. Chem. Toxicol. 2015, 38, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Cazorla, M.; de Carvalho, F.D.; Chohan, M.O.; Shegda, M.; Chuhma, N.; Rayport, S.; Ahmari, S.E.; Moore, H.; Kellendonk, C. Dopamine D2 receptors regulate the anatomical and functional balance of basal ganglia circuitry. Neuron 2014, 81, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Bishnoi, M.; Chopra, K.; Kulkarni, S.K. Activation of striatal inflammatory mediators and caspase-3 is central to haloperidolinduced orofacial dyskinesia. Eur. J. Pharmacol. 2008, 590, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Voronkov, D.N.; Khudoerkov, R.M.; Dovedova, E.L. Changes in neuroglial interactions in the cerebral nigrostriatal structures in a model of dopamine system dysfunction. Zhurnal Nevrol. I Psikhiatrii Im. S.S. Korsakova 2013, 113, 47–51. [Google Scholar]
- Byron, K.Y.; Bitanihirwe, B.K.; Tsung-Ung, W.W. Oxidative Stress in Schizophrenia: An Integrated Approach. Neurosci. Biobehav. Rev. 2010, 35, 878–893. [Google Scholar]
- Drago, A.; Giegling, I.; Schäfer, M.; Hartmann, A.M.; Friedl, M.; Konte, B.; Möller, H.-J.; De Ronchi, D.; Stassen, H.H.; Serretti, A.; et al. AKAP13, CACNA1, GRIK4 and GRIA1 genetic variations may be associated with haloperidol efficacy during acute treatment. Eur. Neuropsychopharmacol. 2013, 23, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-M.; Wang, C.-Y.; Zheng, F.-C.; Gao, F.-F.; Chen, Y.-C.; Huang, Z.-Q.; Xia, Z.-Y.; Irwin, M.G.; Li, W.-Q.; Liu, X.-P.; et al. Effects of N-n-butyl haloperidol iodide on the rat myocardial sarcoplasmic re- 635 ticulum Ca(2+)-ATPase during ischemia/reperfusion. Biochem. Biophys. Res. Commun. 2012, 425, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Delotterie, D.; Ruiz, G.; Brocard, J.; Schweitzer, A.; Roucard, C.; Roche, Y.; Suaud-Chagny, M.-F.; Bressand, K.; Andrieux, A. Chronic administration of atypical antipsychotics improves behavioral and synaptic defects of STOP null mice. Psychopharmacology 2010, 208, 131–141. [Google Scholar] [CrossRef]
- Hasbi, A.; Fan, T.; Alijaniaram, M.; Nguyen, T.; Perreault, M.L.; O’Dowd, B.F.; George, S.R. Calcium signaling cascade links dopamine D1–D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc. Natl. Acad. Sci. USA 2009, 106, 21377–21382. [Google Scholar] [CrossRef]
- Villalba, R.M.; Smith, Y. Striatal Spine Plasticity in Parkinson’s Disease. Front. Neuroanat. 2010, 4, 133. [Google Scholar] [CrossRef]
- Petersen, M.S.; Bech, S.; Christiansen, D.H.; Schmedes, A.V.; Halling, J. The role of vitamin D levels and vitamin D receptor polymorphism on Parkinson’s disease in the Faroe Islands. Neurosci. Lett. 2014, 561, 74–79. [Google Scholar] [CrossRef]
- Peterson, A.L.; Murchison, C.; Zabetian, C.; Leverenz, J.B.; Watson, G.S.; Montine, T.; Carney, N.; Bowman, G.L.; Edwards, K.; Quinn, J.F. Memory, mood, and vitamin d in persons with Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.L. A review of vitamin D and Parkinson’s disease. Maturitas 2014, 78, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, N.C.; Kurland, A.A.; Kurland, L.T. Hereditary predisposition in drug-induced parkinsonism. Arch. Neurol. 1962, 6, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.A.; Haugland, G.; Craig, T.J. Neuroleptic use, parkinsonian symptoms, tardive dyskinesia, and associated factors in child and adolescent psychiatric patients. Am. J. Psychiatry. 1991, 148, 1322–1328. [Google Scholar] [CrossRef]
- Theodoulou, G.; Milner, G.; Jumaian, A. Neuroleptics and family history of Parkinson’s diseases: Case report. East. Mediterr. Health J. 2001, 7, 559–561. [Google Scholar] [CrossRef]
- Honer, W.G.; Kopala, L.C.; Rabinowitz, J. Extrapyramidal symptoms and signs in first-episode, antipsychotic exposed and non-exposed patients with schizophrenia or related psychotic illness. J. Psychopharmacol. 2005, 19, 277–285. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Taylor, T.J.; Venkatakrishnan, K.; Isin, E.M. Assessment of the contributions of CYP3A4 and CYP3A5 in the metabolism of the antipsychotic agent haloperidol to its potentially neurotoxic pyridinium metabolite and effect of antidepressants on the bioactivation pathway. Drug Metab. Dispos. 2003, 31, 243–249. [Google Scholar] [CrossRef]
- Vaiman, E.E.; Shnayder, N.A.; Neznanov, N.G.; Nasyrova, R.F. Candidate genes of the development of antipsychotic-induced parkinsonism in patients with schizophrenia. V.M. Bekhterev Rev. Psychiatry Med. Psychol. 2021, 55, 15–35. [Google Scholar] [CrossRef]
- Abdyrakhmanova, A.K.; Nasyrova, R.F. Pharmacogenetic testing of cytochrome P450 metabolizing enzymes in 28-year-old man with treatment-resistant schizophrenia. Pers. Psychiatry Neurol. 2022, 2, 81–88. [Google Scholar] [CrossRef]
- Vaiman, E.E.; Novitsky, M.A.; Nasyrova, R.F. Pharmacogenetics of chlorpromazine and its role in the development of antipsychotic-induced parkinsonism. Pers. Psychiatry Neurol. 2021, 1, 11–17. [Google Scholar] [CrossRef]
- Vaiman, E.E.; Shnayder, N.A.; Novitsky, M.A.; Dobrodeeva, V.S.; Goncharova, P.S.; Bochanova, E.N.; Sapronova, M.R.; Popova, T.E.; Tappakhov, A.A.; Nasyrova, R.F. Candidate genes encoding dopamine receptors as predictors of the risk of antipsychotic-induced parkinsonism and tardive dyskinesia in schizophrenic patients. Biomedicines 2021, 9, 879. [Google Scholar] [CrossRef] [PubMed]
- Vaiman, E.E.; Shnayder, N.A.; Neznanov, N.G.; Nasyrova, R.F. Antipsychotic-induced tardive dyskinesia as a serious adverse effect in the psychopharmacotherapy of schizophrenia. Neurol. Neuropsychiatry Psychosom. 2019, 11, 4–13. [Google Scholar] [CrossRef]
- Vaiman, E.E.; Shnayder, N.A.; Neznanov, N.G.; Nasyrova, R.F. Candidate genes involved in the development of antipsychotic-induced tardive dyskinesia in patients with schizophrenia. Neuromuscul. Dis. 2020, 10, 10–26. [Google Scholar] [CrossRef]
- Shireen, E. Experimental treatment of antipsychotic-induced movement disorders. J. Exp. Pharmacol. 2016, 8, 1–10. [Google Scholar] [CrossRef]
- Ivashchenko, D.V.; Buromskaya, N.I.; Shimanov, P.V.; Deitch, D.V.; Ryzhykova, K.A.; Grishina, A.; Shevchenko, Y.S.; Sychev, D.A. Pharmacogenetics biomarkers of antipsychotics’ safety in adolescents with acute psychotic episode. V.M. Bekhterev Rev. Psychiatry Med. Psychol. [CrossRef]
- Golimbet, V.E.; Golov, A.K.; Kondratyev, N.V. Post-GWAS era in genetics of schizophrenia. V.M. Bekhterev Rev. Psychiatry Med. Psychol. 2019, 6–7. [Google Scholar] [CrossRef]
- Abdyrakhmanova, A.K.; Shnayder, N.A.; Neznanov, N.G.; Nasyrova, R.F. Pharmacogenetics of quetiapine. Pers. Psychiatry Neurol. 2021, 1, 73–83. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antipsychotics of First Generation | Risk of Developing AIP | Antipsychotics of New Generations | Risk of Developing AIP |
|---|---|---|---|
| Haloperidol | +++ | Lurasidone | ++ |
| Pimozide | +++ | Olanzapine | ++ |
| Thiothixene | +++ | Paliperidone | ++ |
| Fluphenazine | +++ | Risperidone | ++ |
| Loxapin | ++ | Azenapine | + |
| Molindon | ++ | Aripiprazole | + |
| Perphenazine | ++ | Brexpiprazole | + |
| Trifluoperazine | ++ | Ziprasidone | + |
| Chlorpromazine | ++ | Iloperidone | + |
| Thioridazine | + | Karipazin | + |
| Quetiapine | + | ||
| Clozapine | + | ||
| Lumateperone | + | ||
| Pimavenzirin | + |
| Antipsychotics | Occupancy Percentage (%) | ||
|---|---|---|---|
| D2 Receptors | D3 Receptors | D4 Receptors | |
| Clozapine | 38–63 | 62 | 49–73 |
| Chlorpromazine | 78 | 62 | 17 |
| Haloperidol | 85 | 52 | 57 |
| Olanzapine | 43–89 | 10–55 | 27–80 |
| Risperidone | 63–89 | 25–61 | 22–55 |
| Quetiapine | 51 | 24 | 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaiman, E.E.; Shnayder, N.A.; Khasanova, A.K.; Strelnik, A.I.; Gayduk, A.J.; Al-Zamil, M.; Sapronova, M.R.; Zhukova, N.G.; Smirnova, D.A.; Nasyrova, R.F. Pathophysiological Mechanisms of Antipsychotic-Induced Parkinsonism. Biomedicines 2022, 10, 2010. https://doi.org/10.3390/biomedicines10082010
Vaiman EE, Shnayder NA, Khasanova AK, Strelnik AI, Gayduk AJ, Al-Zamil M, Sapronova MR, Zhukova NG, Smirnova DA, Nasyrova RF. Pathophysiological Mechanisms of Antipsychotic-Induced Parkinsonism. Biomedicines. 2022; 10(8):2010. https://doi.org/10.3390/biomedicines10082010
Chicago/Turabian StyleVaiman, Elena E., Natalia A. Shnayder, Aiperi K. Khasanova, Anna I. Strelnik, Arseny J. Gayduk, Mustafa Al-Zamil, Margarita R. Sapronova, Natalia G. Zhukova, Daria A. Smirnova, and Regina F. Nasyrova. 2022. "Pathophysiological Mechanisms of Antipsychotic-Induced Parkinsonism" Biomedicines 10, no. 8: 2010. https://doi.org/10.3390/biomedicines10082010
APA StyleVaiman, E. E., Shnayder, N. A., Khasanova, A. K., Strelnik, A. I., Gayduk, A. J., Al-Zamil, M., Sapronova, M. R., Zhukova, N. G., Smirnova, D. A., & Nasyrova, R. F. (2022). Pathophysiological Mechanisms of Antipsychotic-Induced Parkinsonism. Biomedicines, 10(8), 2010. https://doi.org/10.3390/biomedicines10082010