
Protein Kinase CK2 and Its Potential Role as a Therapeutic Target in Huntington’s Disease


Abstract
1. Introduction
2. Current Therapeutic Approaches in HD
3. Post-Translational Modifications and Their Role in HTT Toxicity
3.1. Phosphorylation

{kind=link}
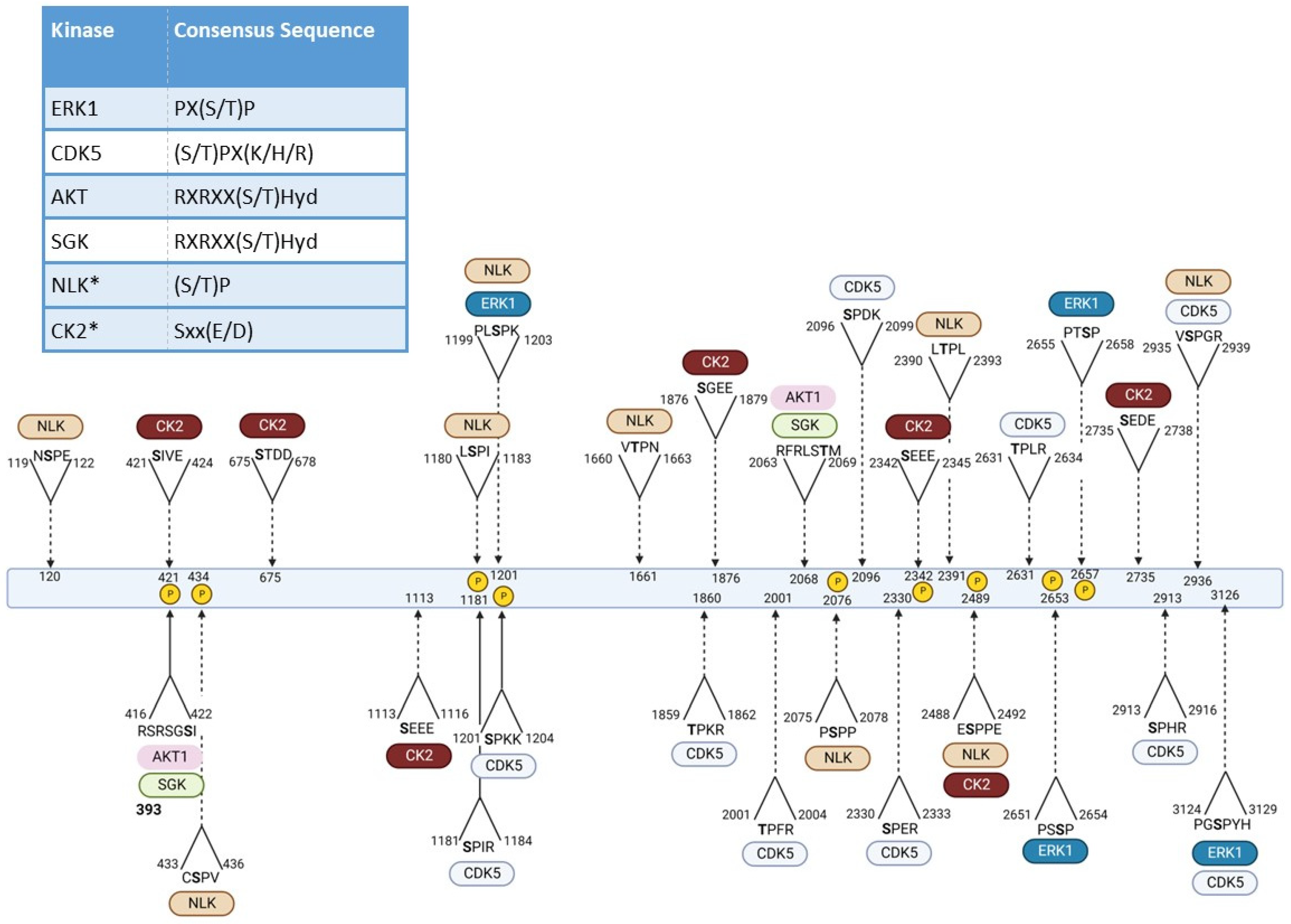{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Effect | Implicated Kinase |
|---|---|---|
| T3 | Effects mtHTT aggregation (conflicting reports enhance vs. prevention) [93,95] Reduces HTT mediated toxicity [93,95] | IKKβ [96] |
| S13 | Decrease mtHTT toxicity [71,87,97,98,99] Decrease HTT aggregates [71,94,97,99] Promote HTT clearance [94,99] Increases nuclear localization [71] | TBK1 [99] CK2 [71,98] IKKβ [94,100] |
| S16 | Decrease mtHTT toxicity [71,87,97,98] Decrease HTT aggregates [71,94,97] Promote HTT clearance [94] Increases nuclear localization [71] | CK2 [71,98] IKKβ [94] |
| S116 | Decrease mtHTT mediated cell death in HEK293 cells and mouse primary cortical and striatal neurons [72,101] | Not described |
| S120 | Reduce mtHTT levels in striatal and HEK293T cells [92] Influence neuropathology in HD mice [92] | NLK [92] |
| T271 | Phospho-null mutation at this site decreases mitochondrial potential in HD HEK293 cells [72] | Not described |
| S421 | Improves mitochondrial potential and form in hIPSCs [89] | AKT/SGK [102,103] |
| S431 | Lack of phosphorylation on mtHTT increases cell viability and decreases mtHTT accumulation [104] | Not described |
| S432 | Lack of phosphorylation on mtHTT decreases cell viability and increases mtHTT accumulation [104] | Not described |
| S434 | Reduces caspase-mediated HTT cleavage [105] | CDK5 [105] |
| S536 | Decreases HTT cleavage by Calpain and mtHTT toxicity [106] | Not described |
| S1181 | Protects against polyQ induced p53-mediated cell death in response to DNA damage [107] | CDK5 [107] |
| S1201 | Protects against polyQ induced p53-mediated cell death in response to DNA damage [107] | CDK5 [107] |
| S2076 | Not described | ERK1 [106] |
| S2116 | Impacts HTT structure and interaction with other proteins such as Polycomb Repressive complex 2 (PRC2) histone methyltransferase [108] | Not described |
| S2342 | Modulation of mitochondrial potential [72] | Not described |
| S2489 | Modulation of mitochondrial potential [72] | Not described |
| S2562 | Reduces mtHTT cellular toxicity [72] | Not described |
| S2653/2657 | Not described | ERK1 [106] |
3.2. Other PTMs
3.2.1. Acetylation
3.2.2. Palmitoylation
3.2.3. SUMOylation
3.2.4. Methylation
4. Kinases Involved in HTT Phosphorylation and Their Potential as Therapeutic Agents
4.1. IKKβ
4.2. TBK1
4.3. CDK5
4.4. NLK
4.5. CK2
5. Other Roles of Kinase CK2 in HD
5.1. CK2α’ Facilitates Protein Homeostasis in HD
5.2. CK2α’ Associates with Increased Striatal Inflammation in HD
5.3. CK2α’ Contributes to the Dysregulation of Expression and Function of Synaptic Genes in HD
5.4. CK2α’ Promotes Synucleinopathy in HD
6. CK2 in Other Neurodegenerative Disorders
6.1. Alzheimer’s Disease and Other Tauopathies
6.2. Parkinson’s Disease and Synucleinopathy
6.3. Amyotrophic Lateral Sclerosis
6.4. Spinocerebellar Ataxia
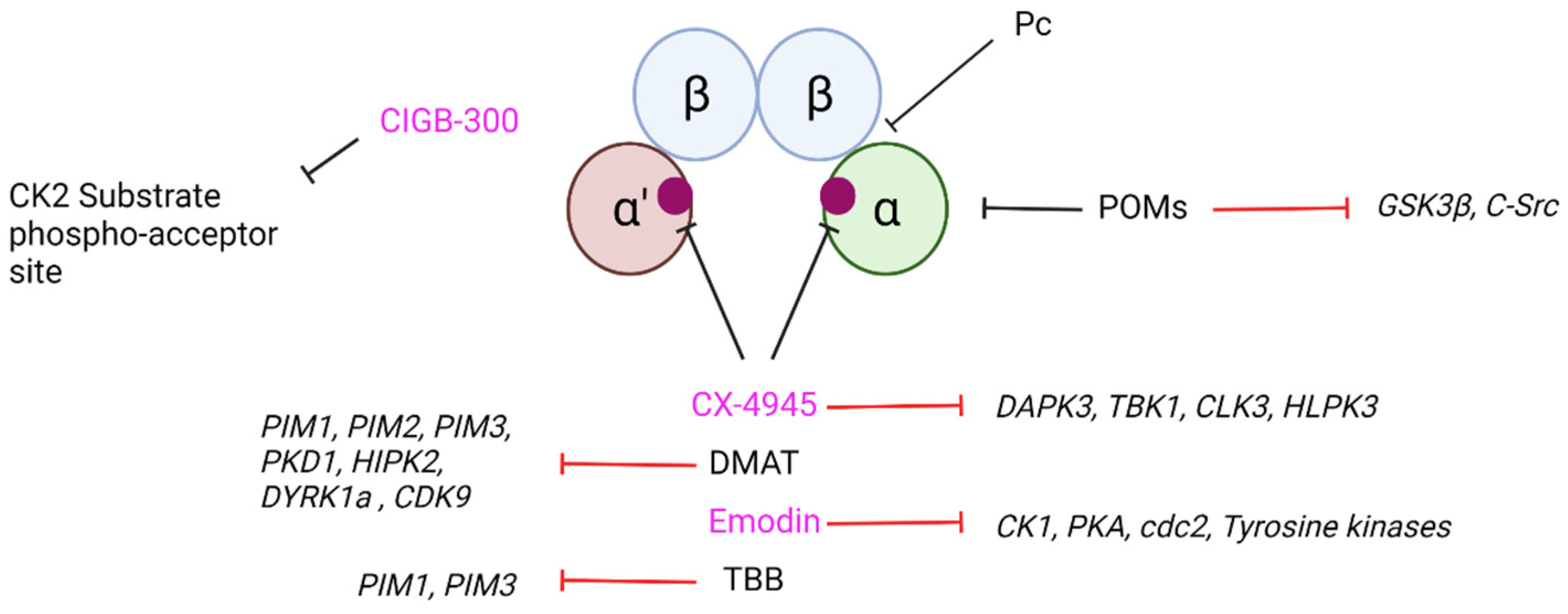
7. Current CK2 Inhibitory Strategies and Their Potential as Therapeutic Agents in Neurodegeneration
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The Huntington’s Disease Collaborative Research. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Roos, R.A.C. Huntington’s disease: A clinical review. Orphanet. J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef]
- Van Duijn, E.; Kingma, E.M.; Van Der Mast, R.C. Psychopathology in verified Huntington’s disease gene carriers. J. Neuropsychiatry Clin. Neurosci. 2007, 19, 441–448. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Han, I.; You, Y.; Kordower, J.H.; Brady, S.T.; Morfini, G.A. Differential vulnerability of neurons in Huntington’s disease: The role of cell type-specific features. J. Neurochem. 2010, 113, 1073–1091. [Google Scholar] [CrossRef]
- Albin, R.L.; Reiner, A.; Anderson, K.D.; Dure, L.S.; Handelin, B.; Balfour, R.; Whetsell, W.O.; Penney, J.B.; Young, A.B. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann. Neurol. 1992, 31, 425–430. [Google Scholar] [CrossRef]
- Petrasch-Parwez, E.; Nguyen, H.P.; Löbbecke-Schumacher, M.; Habbes, H.W.; Wieczorek, S.; Riess, O.; Andres, K.H.; Dermietzel, R.; Von Hörsten, S. Cellular and subcellular localization of Huntingtin [corrected] aggregates in the brain of a rat transgenic for Huntington disease. J. Comp. Neurol. 2007, 501, 716–730. [Google Scholar] [CrossRef]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein-protein interactions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef]
- Yao, J.; Ong, S.E.; Bajjalieh, S. Huntingtin is associated with cytomatrix proteins at the presynaptic terminal. Mol. Cell. Neurosci. 2014, 63, 96–100. [Google Scholar] [CrossRef]
- Mackenzie, K.D.; Lumsden, A.L.; Guo, F.; Duffield, M.D.; Chataway, T.; Lim, Y.; Zhou, X.F.; Keating, D.J. Huntingtin-associated protein-1 is a synapsin I-binding protein regulating synaptic vesicle exocytosis and synapsin I trafficking. J. Neurochem. 2016, 138, 710–721. [Google Scholar] [CrossRef]
- Parker, J.A.; Metzler, M.; Georgiou, J.; Mage, M.; Roder, J.C.; Rose, A.M.; Hayden, M.R.; Néri, C. Huntingtin-interacting protein 1 influences worm and mouse presynaptic function and protects Caenorhabditis elegans neurons against mutant polyglutamine toxicity. J. Neurosci. 2007, 27, 11056–11064. [Google Scholar] [CrossRef]
- Engelender, S.; Sharp, A.H.; Colomer, V.; Tokito, M.K.; Lanahan, A.; Worley, P.; Holzbaur, E.L.; Ross, C.A. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 1997, 6, 2205–2212. [Google Scholar] [CrossRef]
- Li, S.H.; Gutekunst, C.A.; Hersch, S.M.; Li, X.J. Interaction of huntingtin-associated protein with dynactin P150Glued. J. Neurosci. 1998, 18, 1261–1269. [Google Scholar] [CrossRef]
- McGuire, J.R.; Rong, J.; Li, S.H.; Li, X.J. Interaction of Huntingtin-associated protein-1 with kinesin light chain: Implications in intracellular trafficking in neurons. J. Biol. Chem. 2006, 281, 3552–3559. [Google Scholar] [CrossRef]
- Vitet, H.; Brandt, V.; Saudou, F. Traffic signaling: New functions of huntingtin and axonal transport in neurological disease. Curr. Opin. Neurobiol. 2020, 63, 122–130. [Google Scholar] [CrossRef]
- Twelvetrees, A.E.; Yuen, E.Y.; Arancibia-Carcamo, I.L.; MacAskill, A.F.; Rostaing, P.; Lumb, M.J.; Humbert, S.; Triller, A.; Saudou, F.; Yan, Z.; et al. Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron 2010, 65, 53–65. [Google Scholar] [CrossRef]
- Mandal, M.; Wei, J.; Zhong, P.; Cheng, J.; Duffney, L.J.; Liu, W.; Yuen, E.Y.; Twelvetrees, A.E.; Li, S.; Li, X.J.; et al. Impaired alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor trafficking and function by mutant huntingtin. J. Biol. Chem. 2011, 286, 33719–33728. [Google Scholar] [CrossRef]
- Savas, J.N.; Ma, B.; Deinhardt, K.; Culver, B.P.; Restituito, S.; Wu, L.; Belasco, J.G.; Chao, M.V.; Tanese, N. A role for huntington disease protein in dendritic RNA granules. J. Biol. Chem. 2010, 285, 13142–13153. [Google Scholar] [CrossRef]
- Eom, T.; Antar, L.N.; Singer, R.H.; Bassell, G.J. Localization of a beta-actin messenger ribonucleoprotein complex with zipcode-binding protein modulates the density of dendritic filopodia and filopodial synapses. J. Neurosci. 2003, 23, 10433–10444. [Google Scholar] [CrossRef]
- Huang, K.; Sanders, S.S.; Kang, R.; Carroll, J.B.; Sutton, L.; Wan, J.; Singaraja, R.; Young, F.B.; Liu, L.; El-Husseini, A.; et al. Wild-type HTT modulates the enzymatic activity of the neuronal palmitoyl transferase HIP14. Hum. Mol. Genet. 2011, 20, 3356–3365. [Google Scholar] [CrossRef]
- Parsons, M.P.; Kang, R.; Buren, C.; Dau, A.; Southwell, A.L.; Doty, C.N.; Sanders, S.S.; Hayden, M.R.; Raymond, L.A. Bidirectional control of postsynaptic density-95 (PSD-95) clustering by Huntingtin. J. Biol. Chem. 2014, 289, 3518–3528. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef]
- Martin, D.D.; Heit, R.J.; Yap, M.C.; Davidson, M.W.; Hayden, M.R.; Berthiaume, L.G. Identification of a post-translationally myristoylated autophagy-inducing domain released by caspase cleavage of huntingtin. Hum. Mol. Genet. 2014, 23, 3166–3179. [Google Scholar] [CrossRef]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef]
- Fox, L.M.; Kim, K.; Johnson, C.W.; Chen, S.; Croce, K.R.; Victor, M.B.; Eenjes, E.; Bosco, J.R.; Randolph, L.K.; Dragatsis, I.; et al. Huntington’s Disease Pathogenesis Is Modified In Vivo by Alfy/Wdfy3 and Selective Macroautophagy. Neuron 2020, 105, 813–821.e6. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef]
- Landles, C.; Bates, G.P. Huntingtin and the molecular pathogenesis of Huntington’s disease. Fourth in molecular medicine review series. EMBO Rep. 2004, 5, 958–963. [Google Scholar] [CrossRef]
- Benn, C.L.; Sun, T.; Sadri-Vakili, G.; McFarland, K.N.; DiRocco, D.P.; Yohrling, G.J.; Clark, T.W.; Bouzou, B.; Cha, J.H. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 2008, 28, 10720–10733. [Google Scholar] [CrossRef]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef]
- Seong, I.S.; Woda, J.M.; Song, J.J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.M.; Wheeler, V.C.; Walz, T.; et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010, 19, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.; Roos, R.A.; Howland, D.; et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Neueder, A.; Dumas, A.A.; Benjamin, A.C.; Bates, G.P. Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat. Commun. 2018, 9, 3955. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Trotta, C.R.; Narasimhan, J.; Wiedinger, K.J.; Li, W.; Effenberger, K.A.; Woll, M.G.; Jani, M.B.; Risher, N.; Yeh, S.; et al. Small molecule splicing modifiers with systemic HTT-lowering activity. Nat. Commun. 2021, 12, 7299. [Google Scholar] [CrossRef] [PubMed]
- Huntington’s Disease—Diagnosis and Treatment—Mayo Clinic. Available online: https://www.mayoclinic.org/diseases-conditions/huntingtons-disease/diagnosis-treatment/drc-20356122 (accessed on 1 June 2022).
- Scherman, D.; Jaudon, P.; Henry, J.P. Characterization of the monoamine carrier of chromaffin granule membrane by binding of [2-3H]dihydrotetrabenazine. Proc. Natl. Acad. Sci. USA 1983, 80, 584–588. [Google Scholar] [CrossRef]
- Frank, S. Tetrabenazine as anti-chorea therapy in Huntington disease: An open-label continuation study. Huntington Study Group/TETRA-HD Investigators. BMC Neurol. 2009, 9, 62. [Google Scholar] [CrossRef]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef]
- Gardner, J.; Fidler, B. Roche Revives a Closely Watched Huntington’s Disease Drug; Biopharma Dive. Industry Dive: Washington, DC, USA, 2022; Available online: https://www.biopharmadive.com/news/roche-ionis-start-new-huntington-trial-tominersen/617253/ (accessed on 1 June 2022).
- Gardner, J. Roche Stops Giving Huntington’s Disease Drug in Closely Watched Study; Biopharma Dive. Industry Dive: Washington, DC, USA, 2021; Available online: https://www.biopharmadive.com/news/roche-stop-dosing-huntington-tominersen-ionis/597171/ (accessed on 1 June 2022).
- Videnovic, A. Treatment of huntington disease. Curr. Treat. Options Neurol. 2013, 15, 424–438. [Google Scholar] [CrossRef]
- Beglinger, L.J.; Adams, W.H.; Langbehn, D.; Fiedorowicz, J.G.; Jorge, R.; Biglan, K.; Caviness, J.; Olson, B.; Robinson, R.G.; Kieburtz, K.; et al. Results of the citalopram to enhance cognition in Huntington disease trial. Mov. Disord. 2014, 29, 401–405. [Google Scholar] [CrossRef]
- Sohel, A.J.; Shutter, M.C.; Molla, M. Fluoxetine. In StatPearls [Internet]; StatPearls Publsihing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ranen, N.G.; Lipsey, J.R.; Treisman, G.; Ross, C.A. Sertraline in the treatment of severe aggressiveness in Huntington’s disease. J. Neuropsychiatry Clin. Neurosci. 1996, 8, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Sangkuhl, K.; Klein, T.E.; Altman, R.B. Selective serotonin reuptake inhibitors pathway. Pharm. Genom. 2009, 19, 907–909. [Google Scholar] [CrossRef] [PubMed]
- Holl, A.K.; Wilkinson, L.; Painold, A.; Holl, E.M.; Bonelli, R.M. Combating depression in Huntington’s disease: Effective antidepressive treatment with venlafaxine XR. Int. Clin. Psychopharmacol. 2010, 25, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Squitieri, F.; Cannella, M.; Porcellini, A.; Brusa, L.; Simonelli, M.; Ruggieri, S. Short-term effects of olanzapine in Huntington disease. Neuropsychiatry Neuropsychol. Behav. Neurol. 2001, 14, 69–72. [Google Scholar] [PubMed]
- Kristina, T.; Abdolreza, S. Olanzapine. In StatPearls [Internet]; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Koller, W.C.; Trimble, J. The gait abnormality of Huntington’s disease. Neurology 1985, 35, 1450–1454. [Google Scholar] [CrossRef]
- Girotti, F.; Carella, F.; Scigliano, G.; Grassi, M.P.; Soliveri, P.; Giovannini, P.; Parati, E.; Caraceni, T. Effect of neuroleptic treatment on involuntary movements and motor performances in Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 1984, 47, 848–852. [Google Scholar] [CrossRef]
- Rahman, S.; Marwaha, R. Haloperidol. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- de Bartolomeis, A.; Tomasetti, C.; Iasevoli, F. Update on the Mechanism of Action of Aripiprazole: Translational Insights into Antipsychotic Strategies Beyond Dopamine Receptor Antagonism. CNS Drugs 2015, 29, 773–799. [Google Scholar] [CrossRef]
- Brusa, L.; Orlacchio, A.; Moschella, V.; Iani, C.; Bernardi, G.; Mercuri, N.B. Treatment of the symptoms of Huntington’s disease: Preliminary results comparing aripiprazole and tetrabenazine. Mov. Disord. 2009, 24, 126–129. [Google Scholar] [CrossRef]
- Ciammola, A.; Sassone, J.; Colciago, C.; Mencacci, N.E.; Poletti, B.; Ciarmiello, A.; Squitieri, F.; Silani, V. Aripiprazole in the treatment of Huntington’s disease: A case series. Neuropsychiatr. Dis. Treat. 2009, 5, 1–4. [Google Scholar]
- Dallocchio, C.; Buffa, C.; Tinelli, C.; Mazzarello, P. Effectiveness of risperidone in Huntington chorea patients. J. Clin. Psychopharmacol. 1999, 19, 101–103. [Google Scholar] [CrossRef]
- McNeil, S.E.; Gibbons, J.R.R.; Cogburn, M. Risperidone. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Coppen, E.M.; Roos, R.A. Current Pharmacological Approaches to Reduce Chorea in Huntington’s Disease. Drugs 2017, 77, 29–46. [Google Scholar] [CrossRef]
- Haidary, H.A.; Padhy, R.K. Clozapine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Bonuccelli, U.; Ceravolo, R.; Maremmani, C.; Nuti, A.; Rossi, G.; Muratorio, A. Clozapine in Huntington’s chorea. Neurology 1994, 44, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Vallette, N.; Gosselin, O.; Kahn, J.P. Efficacy of clozapine in the course of Huntington chorea: Apropos of a clinical case. Encephale 2001, 27, 169–171. [Google Scholar] [PubMed]
- van Vugt, J.P.; Siesling, S.; Vergeer, M.; van der Velde, E.A.; Roos, R.A. Clozapine versus placebo in Huntington’s disease: A double blind randomised comparative study. J. Neurol. Neurosurg. Psychiatry 1997, 63, 35–39. [Google Scholar] [CrossRef]
- Patel, P.H.; Gupta, V. Rivastigmine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- de Tommaso, M.; Difruscolo, O.; Sciruicchio, V.; Specchio, N.; Livrea, P. Two years’ follow-up of rivastigmine treatment in Huntington disease. Clin. Neuropharmacol. 2007, 30, 43–46. [Google Scholar] [CrossRef]
- Gottlieb, L.; Guo, L.; Shorter, J.; Marmorstein, R. N-alpha-acetylation of Huntingtin protein increases its propensity to aggregate. J. Biol. Chem. 2021, 297, 101363. [Google Scholar] [CrossRef]
- Lemarié, F.L.; Caron, N.S.; Sanders, S.S.; Schmidt, M.E.; Nguyen, Y.T.N.; Ko, S.; Xu, X.; Pouladi, M.A.; Martin, D.D.O.; Hayden, M.R. Rescue of aberrant huntingtin palmitoylation ameliorates mutant huntingtin-induced toxicity. Neurobiol. Dis. 2021, 158, 105479. [Google Scholar] [CrossRef] [PubMed]
- Yanai, A.; Huang, K.; Kang, R.; Singaraja, R.R.; Arstikaitis, P.; Gan, L.; Orban, P.C.; Mullard, A.; Cowan, C.M.; Raymond, L.A.; et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat. Neurosci. 2006, 9, 824–831. [Google Scholar] [CrossRef]
- Soares, E.S.; Prediger, R.D.; Brocardo, P.S.; Cimarosti, H.I. SUMO-modifying Huntington’s disease. IBRO Neurosci. Rep. 2022, 12, 203–209. [Google Scholar] [CrossRef]
- Migazzi, A.; Scaramuzzino, C.; Anderson, E.N.; Tripathy, D.; Hernández, I.H.; Grant, R.A.; Roccuzzo, M.; Tosatto, L.; Virlogeux, A.; Zuccato, C.; et al. Huntingtin-mediated axonal transport requires arginine methylation by PRMT6. Cell Rep. 2021, 35, 108980. [Google Scholar] [CrossRef]
- Ratovitski, T.; Jiang, M.; O’Meally, R.N.; Rauniyar, P.; Chighladze, E.; Faragó, A.; Kamath, S.V.; Jin, J.; Shevelkin, A.V.; Cole, R.N.; et al. Interaction of huntingtin (HTT) with PRMTs and its subsequent arginine methylation affects HTT solubility, phase transition behavior and neuronal toxicity. Hum. Mol. Genet. 2022, 31, 1651–1672. [Google Scholar] [CrossRef] [PubMed]
- Atwal, R.S.; Desmond, C.R.; Caron, N.; Maiuri, T.; Xia, J.; Sipione, S.; Truant, R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 2011, 7, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Arbez, N.; Ratovitski, T.; Roby, E.; Chighladze, E.; Stewart, J.C.; Ren, M.; Wang, X.; Lavery, D.J.; Ross, C.A. Post-translational modifications clustering within proteolytic domains decrease mutant huntingtin toxicity. J. Biol. Chem. 2017, 292, 19238–19249. [Google Scholar] [CrossRef] [PubMed]
- Cariulo, C.; Azzollini, L.; Verani, M.; Martufi, P.; Boggio, R.; Chiki, A.; Deguire, S.M.; Cherubini, M.; Gines, S.; Marsh, J.L.; et al. Phosphorylation of huntingtin at residue T3 is decreased in Huntington’s disease and modulates mutant huntingtin protein conformation. Proc. Natl. Acad. Sci. USA 2017, 114, E10809–E10818. [Google Scholar] [CrossRef] [PubMed]
- Chiki, A.; DeGuire, S.M.; Ruggeri, F.S.; Sanfelice, D.; Ansaloni, A.; Wang, Z.M.; Cendrowska, U.; Burai, R.; Vieweg, S.; Pastore, A.; et al. Mutant Exon1 Huntingtin Aggregation is Regulated by T3 Phosphorylation-Induced Structural Changes and Crosstalk between T3 Phosphorylation and Acetylation at K6. Angew. Chem. Int. Ed. 2017, 56, 5202–5207. [Google Scholar] [CrossRef]
- Di Pardo, A.; Maglione, V.; Alpaugh, M.; Horkey, M.; Atwal, R.S.; Sassone, J.; Ciammola, A.; Steffan, J.S.; Fouad, K.; Truant, R.; et al. Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc. Natl. Acad. Sci. USA 2012, 109, 3528–3533. [Google Scholar] [CrossRef]
- Chaibva, M.; Jawahery, S.; Pilkington, A.W.; Arndt, J.R.; Sarver, O.; Valentine, S.; Matysiak, S.; Legleiter, J. Acetylation within the First 17 Residues of Huntingtin Exon 1 Alters Aggregation and Lipid Binding. Biophys. J. 2016, 111, 349–362. [Google Scholar] [CrossRef]
- Ratovitski, T.; O’Meally, R.N.; Jiang, M.; Chaerkady, R.; Chighladze, E.; Stewart, J.C.; Wang, X.; Arbez, N.; Roby, E.; Alexandris, A.; et al. Post-Translational Modifications (PTMs), Identified on Endogenous Huntingtin, Cluster within Proteolytic Domains between HEAT Repeats. J. Proteome Res. 2017, 16, 2692–2708. [Google Scholar] [CrossRef]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.Z.; Cattaneo, E.; et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef]
- Jeong, H.; Then, F.; Melia, T.J.; Mazzulli, J.R.; Cui, L.; Savas, J.N.; Voisine, C.; Paganetti, P.; Tanese, N.; Hart, A.C.; et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009, 137, 60–72. [Google Scholar] [CrossRef]
- Lontay, B.; Kiss, A.; Virág, L.; Tar, K. How Do Post-Translational Modifications Influence the Pathomechanistic Landscape of Huntington’s Disease? A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 4282. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, T.; Woloshansky, T.; Xia, J.; Truant, R. The huntingtin N17 domain is a multifunctional CRM1 and Ran-dependent nuclear and cilial export signal. Hum. Mol. Genet. 2013, 22, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Li, A.; Holmes, B.B.; Marasa, J.C.; Diamond, M.I. An N-terminal nuclear export signal regulates trafficking and aggregation of Huntingtin (Htt) protein exon 1. J. Biol. Chem. 2013, 288, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bertolotti, A. Critical role of the proline-rich region in Huntingtin for aggregation and cytotoxicity in yeast. J. Biol. Chem. 2006, 281, 35608–35615. [Google Scholar] [CrossRef]
- Gao, Y.G.; Yan, X.Z.; Song, A.X.; Chang, Y.G.; Gao, X.C.; Jiang, N.; Zhang, Q.; Hu, H.Y. Structural insights into the specific binding of huntingtin proline-rich region with the SH3 and WW domains. Structure 2006, 14, 1755–1765. [Google Scholar] [CrossRef]
- Southwell, A.L.; Khoshnan, A.; Dunn, D.E.; Bugg, C.W.; Lo, D.C.; Patterson, P.H. Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity. J. Neurosci. 2008, 28, 9013–9020. [Google Scholar] [CrossRef]
- Chatterjee, M.; Steffan, J.S.; Lukacsovich, T.; Marsh, J.L.; Agrawal, N. Serine residues 13 and 16 are key modulators of mutant huntingtin induced toxicity in Drosophila. Exp. Neurol. 2021, 338, 113463. [Google Scholar] [CrossRef]
- Warby, S.C.; Chan, E.Y.; Metzler, M.; Gan, L.; Singaraja, R.R.; Crocker, S.F.; Robertson, H.A.; Hayden, M.R. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum. Mol. Genet. 2005, 14, 1569–1577. [Google Scholar] [CrossRef]
- Xu, X.; Ng, B.; Sim, B.; Radulescu, C.I.; Yusof, N.A.B.M.; Goh, W.I.; Lin, S.; Lim, J.S.Y.; Cha, Y.; Kusko, R.; et al. pS421 huntingtin modulates mitochondrial phenotypes and confers neuroprotection in an HD hiPSC model. Cell Death Dis. 2020, 11, 809. [Google Scholar] [CrossRef]
- Zala, D.; Colin, E.; Rangone, H.; Liot, G.; Humbert, S.; Saudou, F. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum. Mol. Genet. 2008, 17, 3837–3846. [Google Scholar] [CrossRef] [PubMed]
- Kratter, I.H.; Zahed, H.; Lau, A.; Tsvetkov, A.S.; Daub, A.C.; Weiberth, K.F.; Gu, X.; Saudou, F.; Humbert, S.; Yang, X.W.; et al. Serine 421 regulates mutant huntingtin toxicity and clearance in mice. J. Clin. Investig. 2016, 126, 3585–3597. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, X.; Liu, H.; LeBron, J.; Alexandris, A.; Peng, Q.; Gu, H.; Yang, F.; Li, Y.; Wang, R.; et al. Nemo-like kinase reduces mutant huntingtin levels and mitigates Huntington’s disease. Hum. Mol. Genet. 2020, 29, 1340–1352. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.T.; Steffan, J.S.; Guerrero, C.M.; Khashwji, H.; Lukacsovich, T.; Simmons, D.; Purcell, J.M.; Menhaji, K.; Zhu, Y.Z.; Green, K.; et al. Phosphorylation of threonine 3: Implications for Huntingtin aggregation and neurotoxicity. J. Biol. Chem. 2009, 284, 29427–29436. [Google Scholar] [CrossRef]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vincente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Branco-Santos, J.; Herrera, F.; Poças, G.M.; Pires-Afonso, Y.; Giorgini, F.; Domingos, P.M.; Outeiro, T.F. Protein phosphatase 1 regulates huntingtin exon 1 aggregation and toxicity. Hum. Mol. Genet. 2017, 26, 3763–3775. [Google Scholar] [CrossRef]
- Bustamante, M.B.; Ansaloni, A.; Pedersen, J.F.; Azzollini, L.; Cariulo, C.; Wang, Z.M.; Petricca, L.; Verani, M.; Puglisi, F.; Park, H.; et al. Detection of huntingtin exon 1 phosphorylation by Phos-Tag SDS-PAGE: Predominant phosphorylation on threonine 3 and regulation by IKKβ. Biochem. Biophys. Res. Commun. 2015, 463, 1317–1322. [Google Scholar] [CrossRef]
- Gu, X.; Greiner, E.R.; Mishra, R.; Kodali, R.; Osmand, A.; Finkbeiner, S.; Steffan, J.S.; Thompson, L.M.; Wetzel, R.; Yang, X.W. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 2009, 64, 828–840. [Google Scholar] [CrossRef]
- Bowie, L.E.; Maiuri, T.; Alpaugh, M.; Gabriel, M.; Arbez, N.; Galleguillos, D.; Hung, C.L.K.; Patel, S.; Xia, J.; Hertz, N.T.; et al. N6-Furfuryladenine is protective in Huntington’s disease models by signaling huntingtin phosphorylation. Proc. Natl. Acad. Sci. USA 2018, 115, E7081–E7090. [Google Scholar] [CrossRef]
- Hegde, R.N.; Chiki, A.; Petricca, L.; Martufi, P.; Arbez, N.; Mouchiroud, L.; Auwerx, J.; Landles, C.; Bates, G.P.; Singh-Bains, M.K.; et al. TBK1 phosphorylates mutant Huntingtin and suppresses its aggregation and toxicity in Huntington’s disease models. EMBO J. 2020, 39, e104671. [Google Scholar] [CrossRef]
- Ochaba, J.; Fote, G.; Kachemov, M.; Thein, S.; Yeung, S.Y.; Lau, A.L.; Hernandez, S.; Lim, R.G.; Casale, M.; Neel, M.J.; et al. IKKβ slows Huntington’s disease progression in R6/1 mice. Proc. Natl. Acad. Sci. USA 2019, 116, 10952–10961. [Google Scholar] [CrossRef] [PubMed]
- Watkin, E.E.; Arbez, N.; Waldron-Roby, E.; O’Meally, R.; Ratovitski, T.; Cole, R.N.; Ross, C.A. Phosphorylation of mutant huntingtin at serine 116 modulates neuronal toxicity. PLoS ONE 2014, 9, e88284. [Google Scholar] [CrossRef] [PubMed]
- Humbert, S.; Bryson, E.A.; Cordelières, F.P.; Connors, N.C.; Datta, S.R.; Finkbeiner, S.; Greenberg, M.E.; Saudou, F. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev. Cell 2002, 2, 831–837. [Google Scholar] [CrossRef]
- Rangone, H.; Poizat, G.; Troncoso, J.; Ross, C.A.; MacDonald, M.E.; Saudou, F.; Humbert, S. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur. J. Neurosci. 2004, 19, 273–279. [Google Scholar] [CrossRef]
- Dong, G.; Callegari, E.; Gloeckner, C.J.; Ueffing, M.; Wang, H. Mass spectrometric identification of novel posttranslational modification sites in Huntingtin. Proteomics 2012, 12, 2060–2064. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Vacher, C.; Davies, J.E.; Rubinsztein, D.C. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: Implications for mutant huntingtin toxicity. J. Cell Biol. 2005, 169, 647–656. [Google Scholar] [CrossRef]
- Schilling, B.; Gafni, J.; Torcassi, C.; Cong, X.; Row, R.H.; LaFevre-Bernt, M.A.; Cusack, M.P.; Ratovitski, T.; Hirschhorn, R.; Ross, C.A.; et al. Huntingtin phosphorylation sites mapped by mass spectrometry. Modulation of cleavage and toxicity. J. Biol. Chem. 2006, 281, 23686–23697. [Google Scholar] [CrossRef]
- Anne, S.L.; Saudou, F.; Humbert, S. Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J. Neurosci. 2007, 27, 7318–7328. [Google Scholar] [CrossRef]
- Jung, T.; Shin, B.; Tamo, G.; Kim, H.; Vijayvargia, R.; Leitner, A.; Marcaida, M.J.; Astorga-Wells, J.; Jung, R.; Aebersold, R.; et al. The Polyglutamine Expansion at the N-Terminal of Huntingtin Protein Modulates the Dynamic Configuration and Phosphorylation of the C-Terminal HEAT Domain. Structure 2020, 28, 1035–1050.e1038. [Google Scholar] [CrossRef]
- DeGuire, S.M.; Ruggeri, F.S.; Fares, M.B.; Chiki, A.; Cendrowska, U.; Dietler, G.; Lashuel, H.A. N-terminal Huntingtin (Htt) phosphorylation is a molecular switch regulating Htt aggregation, helical conformation, internalization, and nuclear targeting. J. Biol. Chem. 2018, 293, 18540–18558. [Google Scholar] [CrossRef]
- Chiki, A.; Ricci, J.; Hegde, R.; Abriata, L.A.; Reif, A.; Boudeffa, D.; Lashuel, H.A. Site-Specific Phosphorylation of Huntingtin Exon 1 Recombinant Proteins Enabled by the Discovery of Novel Kinases. Chembiochem 2021, 22, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Pardo, R.; Colin, E.; Régulier, E.; Aebischer, P.; Déglon, N.; Humbert, S.; Saudou, F. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J. Neurosci. 2006, 26, 1635–1645. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Metzler, M.; Gan, L.; Mazarei, G.; Graham, R.K.; Liu, L.; Bissada, N.; Lu, G.; Leavitt, B.R.; Hayden, M.R. Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A. J. Neurosci. 2010, 30, 14318–14329. [Google Scholar] [CrossRef] [PubMed]
- Hecklau, K.; Mueller, S.; Koch, S.P.; Mehkary, M.H.; Kilic, B.; Harms, C.; Boehm-Sturm, P.; Yildirim, F. The Effects of Selective Inhibition of Histone Deacetylase 1 and 3 in Huntington’s Disease Mice. Front. Mol. Neurosci. 2021, 14, 616886. [Google Scholar] [CrossRef] [PubMed]
- Singaraja, R.R.; Huang, K.; Sanders, S.S.; Milnerwood, A.J.; Hines, R.; Lerch, J.P.; Franciosi, S.; Drisdel, R.C.; Vaid, K.; Young, F.B.; et al. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum. Mol. Genet. 2011, 20, 3899–3909. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Gareau, J.R.; Ochaba, J.; Song, W.; Raskó, T.; Reverter, D.; Lee, J.; Monteys, A.M.; Pallos, J.; Mee, L.; et al. SUMO-2 and PIAS1 modulate insoluble mutant huntingtin protein accumulation. Cell Rep. 2013, 4, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009, 324, 1327–1330. [Google Scholar] [CrossRef]
- Ochaba, J.; Monteys, A.M.; O’Rourke, J.G.; Reidling, J.C.; Steffan, J.S.; Davidson, B.L.; Thompson, L.M. PIAS1 Regulates Mutant Huntingtin Accumulation and Huntington’s Disease-Associated Phenotypes In Vivo. Neuron 2016, 90, 507–520. [Google Scholar] [CrossRef]
- Ramírez-Jarquín, U.N.; Sharma, M.; Zhou, W.; Shahani, N.; Subramaniam, S. Deletion of SUMO1 attenuates behavioral and anatomical deficits by regulating autophagic activities in Huntington disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2107187119. [Google Scholar] [CrossRef]
- Sedighi, F.; Adegbuyiro, A.; Legleiter, J. SUMOylation Prevents Huntingtin Fibrillization and Localization onto Lipid Membranes. ACS Chem. Neurosci. 2020, 11, 328–343. [Google Scholar] [CrossRef]
- Carlson, S.M.; Chouinard, C.R.; Labadorf, A.; Lam, C.J.; Schmelzle, K.; Fraenkel, E.; White, F.M. Large-scale discovery of ERK2 substrates identifies ERK-mediated transcriptional regulation by ETV3. Sci. Signal. 2011, 4, rs11. [Google Scholar] [CrossRef] [PubMed]
- Bórquez, D.A.; Olmos, C.; Álvarez, S.; Di Genova, A.; Maass, A.; González-Billault, C. Bioinformatic survey for new physiological substrates of Cyclin-dependent kinase 5. Genomics 2013, 101, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Caudwell, F.B.; Andjelkovic, M.; Hemmings, B.A.; Cohen, P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996, 399, 333–338. [Google Scholar] [CrossRef]
- Della-Morte, D.; Pastore, D.; Capuani, B.; Pacifici, F.; Lauro, D. SGK-1 (Serum- and Glucocorticoid-Inducible Kinase-1). In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Ishitani, T.; Ishitani, S. NLK. In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer: Cham, Switzerland, 2018. [Google Scholar]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef]
- Hornbeck, P.; Zhang, B.; Murray, B.; Kornhauser, J.; Latham, V.; Skrzypek, E. Phosphositeplus 2014 mutations Ptms and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- St-Denis, N.; Gabriel, M.; Turowec, J.P.; Gloor, G.B.; Li, S.S.; Gingras, A.C.; Litchfield, D.W. Systematic investigation of hierarchical phosphorylation by protein kinase CK2. J. Proteom. 2015, 118, 49–62. [Google Scholar] [CrossRef]
- Stothard, P. The Sequence Manipulation Suite: JavaScript Programs for Analyzing and Formatting Protein and DNA Sequences. BioTechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef]
- Fan, M.M.; Zhang, H.; Hayden, M.R.; Pelech, S.L.; Raymond, L.A. Protective up-regulation of CK2 by mutant huntingtin in cells co-expressing NMDA receptors. J. Neurochem. 2008, 104, 790–805. [Google Scholar] [CrossRef]
- Alvarez-Periel, E.; Puigdellívol, M.; Brito, V.; Plattner, F.; Bibb, J.A.; Alberch, J.; Ginés, S. Cdk5 Contributes to Huntington’s Disease Learning and Memory Deficits via Modulation of Brain Region-Specific Substrates. Mol. Neurobiol. 2018, 55, 6250–6268. [Google Scholar] [CrossRef]
- Daams, R.; Massoumi, R. Nemo-Like Kinase in Development and Diseases: Insights from Mouse Studies. Int. J. Mol. Sci 2020, 21, 9203. [Google Scholar] [CrossRef]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Neef, D.W.; Jaeger, A.M.; Cabiscol, E.; McKinstry, S.U.; Doss, A.; Aballay, A.; Lo, D.C.; Akimov, S.S.; et al. Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington’s disease. Nat. Commun. 2017, 8, 14405. [Google Scholar] [CrossRef]
- Yu, D.; Zarate, N.; White, A.; Coates, D.; Tsai, W.; Nanclares, C.; Cuccu, F.; Yue, J.S.; Brown, T.G.; Mansky, R.H.; et al. CK2 alpha prime and alpha-synuclein pathogenic functional interaction mediates synaptic dysregulation in huntington’s disease. Acta Neuropathol. Commun. 2022, 10, 83. [Google Scholar] [CrossRef]
- Mikula, M.; Hanusek, K.; Paziewska, A.; Dzwonek, A.; Rubel, T.; Bomsztyk, K.; Ostrowski, J. Halogenated imidazole derivatives block RNA polymerase II elongation along mitogen inducible genes. BMC Mol. Biol. 2010, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Zandomeni, R.; Bunick, D.; Ackerman, S.; Mittleman, B.; Weinmann, R. Mechanism of action of DRB. III. Effect on specific in vitro initiation of transcription. J. Mol. Biol. 1983, 167, 561–574. [Google Scholar] [CrossRef]
- Dubois, M.F.; Bellier, S.; Seo, S.J.; Bensaude, O. Phosphorylation of the RNA polymerase II largest subunit during heat shock and inhibition of transcription in HeLa cells. J. Cell Physiol. 1994, 158, 417–426. [Google Scholar] [CrossRef]
- Pagano, M.A.; Bain, J.; Kazimierczuk, Z.; Sarno, S.; Ruzzene, M.; Di Maira, G.; Elliott, M.; Orzeszko, A.; Cozza, G.; Meggio, F.; et al. The selectivity of inhibitors of protein kinase CK2: An update. Biochem. J. 2008, 415, 353–365. [Google Scholar] [CrossRef]
- Bian, Y.; Ye, M.; Wang, C.; Cheng, K.; Song, C.; Dong, M.; Pan, Y.; Qin, H.; Zou, H. Global screening of CK2 kinase substrates by an integrated phosphoproteomics workflow. Sci. Rep. 2013, 3, 3460. [Google Scholar] [CrossRef]
- Franchin, C.; Borgo, C.; Cesaro, L.; Zaramella, S.; Vilardell, J.; Salvi, M.; Arrigoni, G.; Pinna, L.A. Re-evaluation of protein kinase CK2 pleiotropy: New insights provided by a phosphoproteomics analysis of CK2 knockout cells. Cell. Mol. Life Sci. 2018, 75, 2011–2026. [Google Scholar] [CrossRef]
- Perez, D.I.; Gil, C.; Martinez, A. Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med. Res. Rev. 2011, 31, 924–954. [Google Scholar] [CrossRef] [PubMed]
- Rusin, S.F.; Adamo, M.E.; Kettenbach, A.N. Identification of Candidate Casein Kinase 2 Substrates in Mitosis by Quantitative Phosphoproteomics. Front. Cell Dev. Biol. 2017, 5, 97. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.Y.; Dominguez, I.; Toselli, P.; Landesman-Bollag, E.; O’Brien, C.; Seldin, D.C. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol. Cell Biol. 2008, 28, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Toselli, P.A.; Russell, L.D.; Seldin, D.C. Globozoospermia in mice lacking the casein kinase II alpha’ catalytic subunit. Nat. Genet. 1999, 23, 118–121. [Google Scholar] [CrossRef]
- Sarno, S.; Salvi, M.; Battistutta, R.; Zanotti, G.; Pinna, L.A. Features and potentials of ATP-site directed CK2 inhibitors. Biochim. Biophys. Acta 2005, 1754, 263–270. [Google Scholar] [CrossRef]
- Roffey, S.E.; Litchfield, D.W. CK2 Regulation: Perspectives in 2021. Biomedicines 2021, 9, 1361. [Google Scholar] [CrossRef]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef]
- Labbadia, J.; Cunliffe, H.; Weiss, A.; Katsyuba, E.; Sathasivam, K.; Seredenina, T.; Woodman, B.; Moussaoui, S.; Frentzel, S.; Luthi-Carter, R.; et al. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J. Clin. Investig. 2011, 121, 3306–3319. [Google Scholar] [CrossRef]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef]
- Chafekar, S.M.; Duennwald, M.L. Impaired heat shock response in cells expressing full-length polyglutamine-expanded huntingtin. PLoS ONE 2012, 7, e37929. [Google Scholar] [CrossRef]
- Maheshwari, M.; Bhutani, S.; Das, A.; Mukherjee, R.; Sharma, A.; Kino, Y.; Nukina, N.; Jana, N.R. Dexamethasone induces heat shock response and slows down disease progression in mouse and fly models of Huntington’s disease. Hum. Mol. Genet. 2014, 23, 2737–2751. [Google Scholar] [CrossRef] [PubMed]
- Zarate, N.; Intihar, T.A.; Yu, D.; Sawyer, J.; Tsai, W.; Syed, M.; Carlson, L.; Gomez-Pastor, R. Heat Shock Factor 1 Directly Regulates Postsynaptic Scaffolding PSD95 in Aging and Huntington’s Disease and Influences Striatal Synaptic Density. Int. J. Mol. Sci. 2021, 22, 13113. [Google Scholar] [CrossRef] [PubMed]
- Burchfiel, E.T.; Vihervaara, A.; Guertin, M.J.; Gomez-Pastor, R.; Thiele, D.J. Comparative interactomes of HSF1 in stress and disease reveal a role for CTCF in HSF1-mediated gene regulation. J. Biol. Chem. 2020, 296, 100097. [Google Scholar] [CrossRef] [PubMed]
- Knauf, U.; Newton, E.M.; Kyriakis, J.; Kingston, R.E. Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev. 1996, 10, 2782–2793. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Zhong, R.; Soncin, F.; Stevenson, M.A.; Calderwood, S.K. Transcriptional activity of heat shock factor 1 at 37 degrees C is repressed through phosphorylation on two distinct serine residues by glycogen synthase kinase 3 and protein kinases Calpha and Czeta. J. Biol. Chem. 1998, 273, 18640–18646. [Google Scholar] [CrossRef]
- Hietakangas, V.; Ahlskog, J.K.; Jakobsson, A.M.; Hellesuo, M.; Sahlberg, N.M.; Holmberg, C.I.; Mikhailov, A.; Palvimo, J.J.; Pirkkala, L.; Sistonen, L. Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol. Cell Biol. 2003, 23, 2953–2968. [Google Scholar] [CrossRef]
- Batista-Nascimento, L.; Neef, D.W.; Liu, P.C.; Rodrigues-Pousada, C.; Thiele, D.J. Deciphering human heat shock transcription factor 1 regulation via post-translationa-l modification in yeast. PLoS ONE 2011, 6, e15976. [Google Scholar] [CrossRef]
- Riva, L.; Koeva, M.; Yildirim, F.; Pirhaji, L.; Dinesh, D.; Mazor, T.; Duennwald, M.L.; Fraenkel, E. Poly-glutamine expanded huntingtin dramatically alters the genome wide binding of HSF1. J. Huntingtons Dis. 2012, 1, 33–45. [Google Scholar] [CrossRef]
- Singh, N.N.; Ramji, D.P. Protein kinase CK2, an important regulator of the inflammatory response? J. Mol. Med. 2008, 86, 887–897. [Google Scholar] [CrossRef]
- Gibson, S.A.; Benveniste, E.N. Protein Kinase CK2: An Emerging Regulator of Immunity. Trends Immunol. 2018, 39, 82–85. [Google Scholar] [CrossRef]
- Dominguez, I.; Sonenshein, G.E.; Seldin, D.C. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: Linking development and cancer. Cell. Mol. Life Sci. 2009, 66, 1850–1857. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, A.F.; Morrema, T.H.; Gerritsen, W.H.; van Haastert, E.S.; Snkhchyan, H.; Hilhorst, R.; Rozemuller, A.J.; Scheltens, P.; van der Vies, S.M.; Hoozemans, J.J. Increased occurrence of protein kinase CK2 in astrocytes in Alzheimer’s disease pathology. J. Neuroinflamm. 2016, 13, 4. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Byrne, L.M.; McColgan, P.; Robertson, N.; Tabrizi, S.J.; Zetterberg, H.; Wild, E.J. Cerebrospinal Fluid Inflammatory Biomarkers Reflect Clinical Severity in Huntington’s Disease. PLoS ONE 2016, 11, e0163479. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M. Excitotoxic injury of the neostriatum: A model for Huntington’s disease. Trends Neurosci. 1990, 13, 286–289. [Google Scholar] [CrossRef]
- Chung, H.J.; Huang, Y.H.; Lau, L.F.; Huganir, R.L. Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J. Neurosci. 2004, 24, 10248–10259. [Google Scholar] [CrossRef]
- Soto, D.; Pancetti, F.; Marengo, J.J.; Sandoval, M.; Sandoval, R.; Orrego, F.; Wyneken, U. Protein kinase CK2 in postsynaptic densities: Phosphorylation of PSD-95/SAP90 and NMDA receptor regulation. Biochem. Biophys. Res. Commun. 2004, 322, 542–550. [Google Scholar] [CrossRef]
- Sanz-Clemente, A.; Matta, J.A.; Isaac, J.T.; Roche, K.W. Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 2010, 67, 984–996. [Google Scholar] [CrossRef]
- Sanz-Clemente, A.; Gray, J.A.; Ogilvie, K.A.; Nicoll, R.A.; Roche, K.W. Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep. 2013, 3, 607–614. [Google Scholar] [CrossRef]
- Marshall, C.A.; McBride, J.D.; Changolkar, L.; Riddle, D.M.; Trojanowski, J.Q.; Lee, V.M. Inhibition of CK2 mitigates Alzheimer’s tau pathology by preventing NR2B synaptic mislocalization. Acta Neuropathol. Commun. 2022, 10, 30. [Google Scholar] [CrossRef]
- Kimura, R.; Matsuki, N. Protein kinase CK2 modulates synaptic plasticity by modification of synaptic NMDA receptors in the hippocampus. J. Physiol. 2008, 586, 3195–3206. [Google Scholar] [CrossRef]
- Charles, V.; Mezey, E.; Reddy, P.H.; Dehejia, A.; Young, T.A.; Polymeropoulos, M.H.; Brownstein, M.J.; Tagle, D.A. Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci. Lett. 2000, 289, 29–32. [Google Scholar] [CrossRef]
- Tomás-Zapico, C.; Díez-Zaera, M.; Ferrer, I.; Gómez-Ramos, P.; Morán, M.A.; Miras-Portugal, M.T.; Díaz-Hernández, M.; Lucas, J.J. α-Synuclein accumulates in huntingtin inclusions but forms independent filaments and its deficiency attenuates early phenotype in a mouse model of Huntington’s disease. Hum. Mol. Genet. 2012, 21, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.; Outeiro, T.F. α-Synuclein modifies huntingtin aggregation in living cells. FEBS Lett. 2012, 586, 7–12. [Google Scholar] [CrossRef]
- Breza, M.; Emmanouilidou, E.; Leandrou, E.; Kartanou, C.; Bougea, A.; Panas, M.; Stefanis, L.; Karadima, G.; Vekrellis, K.; Koutsis, G. Elevated Serum α-Synuclein Levels in Huntington’s Disease Patients. Neuroscience 2020, 431, 34–39. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—lessons and emerging principles. Mol. Neurodegener 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- St-Amour, I.; Turgeon, A.; Goupil, C.; Planel, E.; Hébert, S.S. Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease. Acta Neuropathol. 2018, 135, 249–265. [Google Scholar] [CrossRef]
- Corrochano, S.; Renna, M.; Carter, S.; Chrobot, N.; Kent, R.; Stewart, M.; Cooper, J.; Brown, S.D.M.; Rubinsztein, D.C.; Acevedo-Arozena, A. Article Navigation α-Synuclein levels modulate Huntington’s disease in mice. Hum. Mol. Genet. 2012, 21, 485–494. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Lee, G.; Tanaka, M.; Park, K.; Lee, S.S.; Kim, Y.M.; Junn, E.; Lee, S.H.; Mouradian, M.M. Casein kinase II-mediated phosphorylation regulates alpha-synuclein/synphilin-1 interaction and inclusion body formation. J. Biol. Chem. 2004, 279, 6834–6839. [Google Scholar] [CrossRef]
- Oueslati, A. Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade? J. Parkinsons Dis. 2016, 6, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Pinho, R.; Paiva, I.; Jercic, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Garcia-Esparcia, P.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Piqué, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2019, 28, 31–50. [Google Scholar] [CrossRef] [PubMed]
- Schell, H.; Hasegawa, T.; Neumann, M.; Kahle, P.J. Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience 2009, 160, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Sugeno, N.; Takeda, A.; Hasegawa, T.; Kobayashi, M.; Kikuchi, A.; Mori, F.; Wakabayashi, K.; Itoyama, Y. Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 2008, 283, 23179–23188. [Google Scholar] [CrossRef]
- Ishii, A.; Nonaka, T.; Taniguchi, S.; Saito, T.; Arai, T.; Mann, D.; Iwatsubo, T.; Hisanaga, S.; Goedert, M.; Hasegawa, M. Casein kinase 2 is the major enzyme in brain that phosphorylates Ser129 of human alpha-synuclein: Implication for alpha-synucleinopathies. FEBS Lett. 2007, 581, 4711–4717. [Google Scholar] [CrossRef]
- Wakamatsu, M.; Ishii, A.; Ukai, Y.; Sakagami, J.; Iwata, S.; Ono, M.; Matsumoto, K.; Nakamura, A.; Tada, N.; Kobayashi, K.; et al. Accumulation of phosphorylated alpha-synuclein in dopaminergic neurons of transgenic mice that express human alpha-synuclein. J. Neurosci. Res. 2007, 85, 1819–1825. [Google Scholar] [CrossRef]
- Masliah, E.; Iimoto, D.S.; Mallory, M.; Albright, T.; Hansen, L.; Saitoh, T. Casein kinase II alteration precedes tau accumulation in tangle formation. Am. J. Pathol. 1992, 140, 263–268. [Google Scholar]
- Yadikar, H.; Torres, I.; Aiello, G.; Kurup, M.; Yang, Z.; Lin, F.; Kobeissy, F.; Yost, R.; Wang, K.K. Screening of tau protein kinase inhibitors in a tauopathy-relevant cell-based model of tau hyperphosphorylation and oligomerization. PLoS ONE 2020, 15, e0224952. [Google Scholar] [CrossRef]
- Zhang, Q.; Xia, Y.; Wang, Y.; Shentu, Y.; Zeng, K.; Mahaman, Y.A.R.; Huang, F.; Wu, M.; Ke, D.; Wang, Q.; et al. CK2 Phosphorylating I. Front. Mol. Neurosci. 2018, 11, 146. [Google Scholar] [CrossRef]
- Fernández-Nogales, M.; Cabrera, J.R.; Santos-Galindo, M.; Hoozemans, J.J.; Ferrer, I.; Rozemuller, A.J.; Hernández, F.; Avila, J.; Lucas, J.J. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 2014, 20, 881–885. [Google Scholar] [CrossRef]
- Vuono, R.; Winder-Rhodes, S.; de Silva, R.; Cisbani, G.; Drouin-Ouellet, J.; Spillantini, M.G.; Cicchetti, F.; Barker, R.A.; REGISTRY Investigators of the European Huntington’s Disease Network. The role of tau in the pathological process and clinical expression of Huntington’s disease. Brain 2015, 138, 1907–1918. [Google Scholar] [CrossRef]
- Liu, P.; Smith, B.R.; Huang, E.S.; Mahesh, A.; Vonsattel, J.P.G.; Petersen, A.J.; Gomez-Pastor, R.; Ashe, K.H. A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington’s disease patients. Acta Neuropathol. Commun. 2019, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Pimenova, A.A.; Thathiah, A.; De Strooper, B.; Tesseur, I. Regulation of amyloid precursor protein processing by serotonin signaling. PLoS ONE 2014, 9, e87014. [Google Scholar] [CrossRef]
- Lenzken, S.C.; Stanga, S.; Lanni, C.; De Leonardis, F.; Govoni, S.; Racchi, M. Recruitment of casein kinase 2 is involved in AbetaPP processing following cholinergic stimulation. J. Alzheimers Dis. 2010, 20, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Sung, C.C.; Yu, P.; Li, J.; Chung, K.K.K. S-Nitrosylation of G protein-coupled receptor kinase 6 and Casein kinase 2 alpha modulates their kinase activity toward alpha-synuclein phosphorylation in an animal model of Parkinson’s disease. PLoS ONE 2020, 15, e0232019. [Google Scholar] [CrossRef]
- Lin, K.D.; Yang, C.Y.; Lee, M.Y.; Ho, S.C.; Liu, C.K.; Shin, S.J. Statin therapy prevents the onset of Parkinson disease in patients with diabetes. Ann. Neurol. 2016, 80, 532–540. [Google Scholar] [CrossRef]
- Lin, C.H.; Chang, C.H.; Tai, C.H.; Cheng, M.F.; Chen, Y.C.; Chao, Y.T.; Huang, T.L.; Yen, R.F.; Wu, R.M. A Double-Blind, Randomized, Controlled Trial of Lovastatin in Early-Stage Parkinson’s Disease. Mov. Disord. 2021, 36, 1229–1237. [Google Scholar] [CrossRef]
- Kawada, T. Risk reduction of Parkinson disease by statin therapy in patients with diabetes. Ann. Neurol. 2017, 81, 157. [Google Scholar] [CrossRef]
- Dai, L.; Wang, J.; He, M.; Xiong, M.; Tian, Y.; Liu, C.; Zhang, Z. Lovastatin Alleviates α-Synuclein Aggregation and Phosphorylation in Cellular Models of Synucleinopathy. Front. Mol. Neurosci. 2021, 14, 682320. [Google Scholar] [CrossRef]
- Cortés, M.; Malave, L.; Castello, J.; Flajolet, M.; Cenci, M.A.; Friedman, E.; Rebholz, H. CK2 Oppositely Modulates l-DOPA-Induced Dyskinesia via Striatal Projection Neurons Expressing D1 or D2 Receptors. J. Neurosci. 2017, 37, 11930–11946. [Google Scholar] [CrossRef]
- Li, H.Y.; Yeh, P.A.; Chiu, H.C.; Tang, C.Y.; Tu, B.P. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS ONE 2011, 6, e23075. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, Y.; Zhang, Y.; Davis, M.; Lin, W.L.; Cook, C.; Dunmore, J.; Tay, W.; Menkosky, K.; Cao, X.; Petrucelli, L.; et al. Casein kinase II induced polymerization of soluble TDP-43 into filaments is inhibited by heat shock proteins. PLoS ONE 2014, 9, e90452. [Google Scholar] [CrossRef] [PubMed]
- Eck, R.J.; Kraemer, B.C.; Liachko, N.F. Regulation of TDP-43 phosphorylation in aging and disease. Geroscience 2021, 43, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Breuer, P.; Schmitt, I.; Walter, J.; Evert, B.O.; Wüllner, U. CK2-dependent phosphorylation determines cellular localization and stability of ataxin-3. Hum. Mol. Genet. 2009, 18, 3334–3343. [Google Scholar] [CrossRef]
- Pastori, V.; Sangalli, E.; Coccetti, P.; Pozzi, C.; Nonnis, S.; Tedeschi, G.; Fusi, P. CK2 and GSK3 phosphorylation on S29 controls wild-type ATXN3 nuclear uptake. Biochim. Biophys. Acta 2010, 1802, 583–592. [Google Scholar] [CrossRef]
- Chen, Y.S.; Hong, Z.X.; Lin, S.Z.; Harn, H.J. Identifying Therapeutic Targets for Spinocerebellar Ataxia Type 3/Machado-Joseph Disease through Integration of Pathological Biomarkers and Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 3063. [Google Scholar] [CrossRef]
- Laudet, B.; Barette, C.; Dulery, V.; Renaudet, O.; Dumy, P.; Metz, A.; Prudent, R.; Deshiere, A.; Dideberg, O.; Filhol, O.; et al. Structure-based design of small peptide inhibitors of protein kinase CK2 subunit interaction. Biochem. J. 2007, 408, 363–373. [Google Scholar] [CrossRef]
- Prudent, R.; Moucadel, V.; Laudet, B.; Barette, C.; Lafanechère, L.; Hasenknopf, B.; Li, J.; Bareyt, S.; Lacôte, E.; Thorimbert, S.; et al. Identification of polyoxometalates as nanomolar noncompetitive inhibitors of protein kinase CK2. Chem. Biol. 2008, 15, 683–692. [Google Scholar] [CrossRef]
- Perea, S.E.; Baladrón, I.; Valenzuela, C.; Perera, Y. CIGB-300: A peptide-based drug that impairs the Protein Kinase CK2-mediated phosphorylation. Semin. Oncol. 2018, 45, 58–67. [Google Scholar] [CrossRef]
- Janeczko, M.; Masłyk, M.; Kubiński, K.; Golczyk, H. Emodin, a natural inhibitor of protein kinase CK2, suppresses growth, hyphal development, and biofilm formation of Candida albicans. Yeast 2017, 34, 253–265. [Google Scholar] [CrossRef]
- Castello, J.; Ragnauth, A.; Friedman, E.; Rebholz, H. CK2-An Emerging Target for Neurological and Psychiatric Disorders. Pharmaceuticals 2017, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Font, L.; Villamañan, L.; Arias-Ramos, N.; Vilardell, J.; Plana, M.; Ruzzene, M.; Pinna, L.A.; Itarte, E.; Arús, C.; Candiota, A.P. Targeting Protein Kinase CK2: Evaluating CX-4945 Potential for GL261 Glioblastoma Therapy in Immunocompetent Mice. Pharmaceuticals 2017, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; McFarland, B.C.; Drygin, D.; Yu, H.; Bellis, S.L.; Kim, H.; Bredel, M.; Benveniste, E.N. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin. Cancer Res. 2013, 19, 6484–6494. [Google Scholar] [CrossRef] [PubMed]
- Pierre, F.; Chua, P.C.; O’Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef]
- Kim, H.; Choi, K.; Kang, H.; Lee, S.Y.; Chi, S.W.; Lee, M.S.; Song, J.; Im, D.; Choi, Y.; Cho, S. Identification of a novel function of CX-4945 as a splicing regulator. PLoS ONE 2014, 9, e94978. [Google Scholar] [CrossRef]
- Kim, H.; Lee, K.S.; Kim, A.K.; Choi, M.; Choi, K.; Kang, M.; Chi, S.W.; Lee, M.S.; Lee, J.S.; Lee, S.Y.; et al. A chemical with proven clinical safety rescues Down-syndrome-related phenotypes in through DYRK1A inhibition. Dis. Model. Mech. 2016, 9, 839–848. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef]
- Yim, H.; Lee, Y.H.; Lee, C.H.; Lee, S.K. Emodin, an anthraquinone derivative isolated from the rhizomes of Rheum palmatum, selectively inhibits the activity of casein kinase II as a competitive inhibitor. Planta Med. 1999, 65, 9–13. [Google Scholar] [CrossRef]
- Battistutta, R.; Sarno, S.; De Moliner, E.; Papinutto, E.; Zanotti, G.; Pinna, L.A. The replacement of ATP by the competitive inhibitor emodin induces conformational modifications in the catalytic site of protein kinase CK2. J. Biol. Chem. 2000, 275, 29618–29622. [Google Scholar] [CrossRef]
- Sarno, S.; Moro, S.; Meggio, F.; Zagotto, G.; Dal Ben, D.; Ghisellini, P.; Battistutta, R.; Zanotti, G.; Pinna, L.A. Toward the rational design of protein kinase casein kinase-2 inhibitors. Pharmacol. Ther. 2002, 93, 159–168. [Google Scholar] [CrossRef]





| Agent | Target and Purpose | References |
|---|---|---|
| Citalopram (Celexa) Flouexetine (Prozac) Setraline (Zoloft) | Inhibitors of serotonin transporter SLC6A4 to treat depressive and aggressive symptoms | [42,43,44,45,46] |
| Venlafaxine (Effexor) | Sequential inhibitor of serotonin and norepinephrine reuptake transporters to treat depressive symptoms | [42,47] |
| Olanzapine (Zyprexa) Haloperidol (Haldol) Aripiprazole (Abilify) Risperidone (Risperdal) | D2 receptor antagonists used for the treatment of chorea | [42,48,49,50,51,52,53,54,55,56,57,58] |
| Clozapine (Clozaril) | Antagonist of the D1 and D4 dopamine receptors and 5-HT2A and 5-HT2C serotonin receptors used for treatment of chorea | [42,58,59,60,61,62] |
| Tetrabenzine (Xenazine) | Inhibits transport of monoamines via binding to type-2 vesicular monoamine transporters (VMAT2) used for the treatment of chorea | [37,42,58] |
| Rivastigmine (Exelon) | Inhibits activity of the cholinesterase enzymes, acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) used to treat cognitive deficits | [42,63,64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, A.; McGlone, A.; Gomez-Pastor, R. Protein Kinase CK2 and Its Potential Role as a Therapeutic Target in Huntington’s Disease. Biomedicines 2022, 10, 1979. https://doi.org/10.3390/biomedicines10081979
White A, McGlone A, Gomez-Pastor R. Protein Kinase CK2 and Its Potential Role as a Therapeutic Target in Huntington’s Disease. Biomedicines. 2022; 10(8):1979. https://doi.org/10.3390/biomedicines10081979
Chicago/Turabian StyleWhite, Angel, Anna McGlone, and Rocio Gomez-Pastor. 2022. "Protein Kinase CK2 and Its Potential Role as a Therapeutic Target in Huntington’s Disease" Biomedicines 10, no. 8: 1979. https://doi.org/10.3390/biomedicines10081979
APA StyleWhite, A., McGlone, A., & Gomez-Pastor, R. (2022). Protein Kinase CK2 and Its Potential Role as a Therapeutic Target in Huntington’s Disease. Biomedicines, 10(8), 1979. https://doi.org/10.3390/biomedicines10081979