
Postnatal Conditional Deletion of Bcl11b in Striatal Projection Neurons Mimics the Transcriptional Signature of Huntington’s Disease

 , , , , and
, , , , and 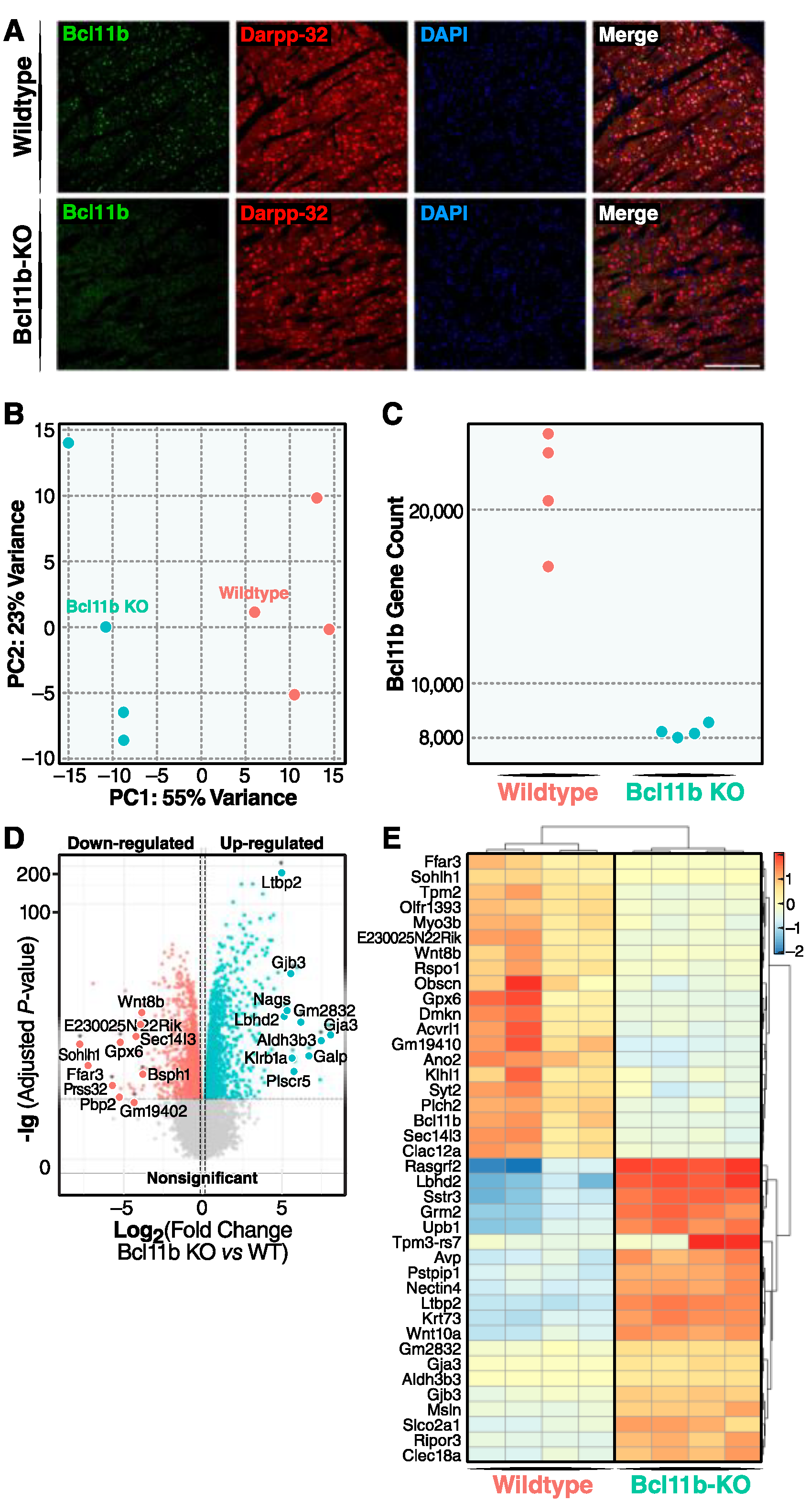{kind=link}
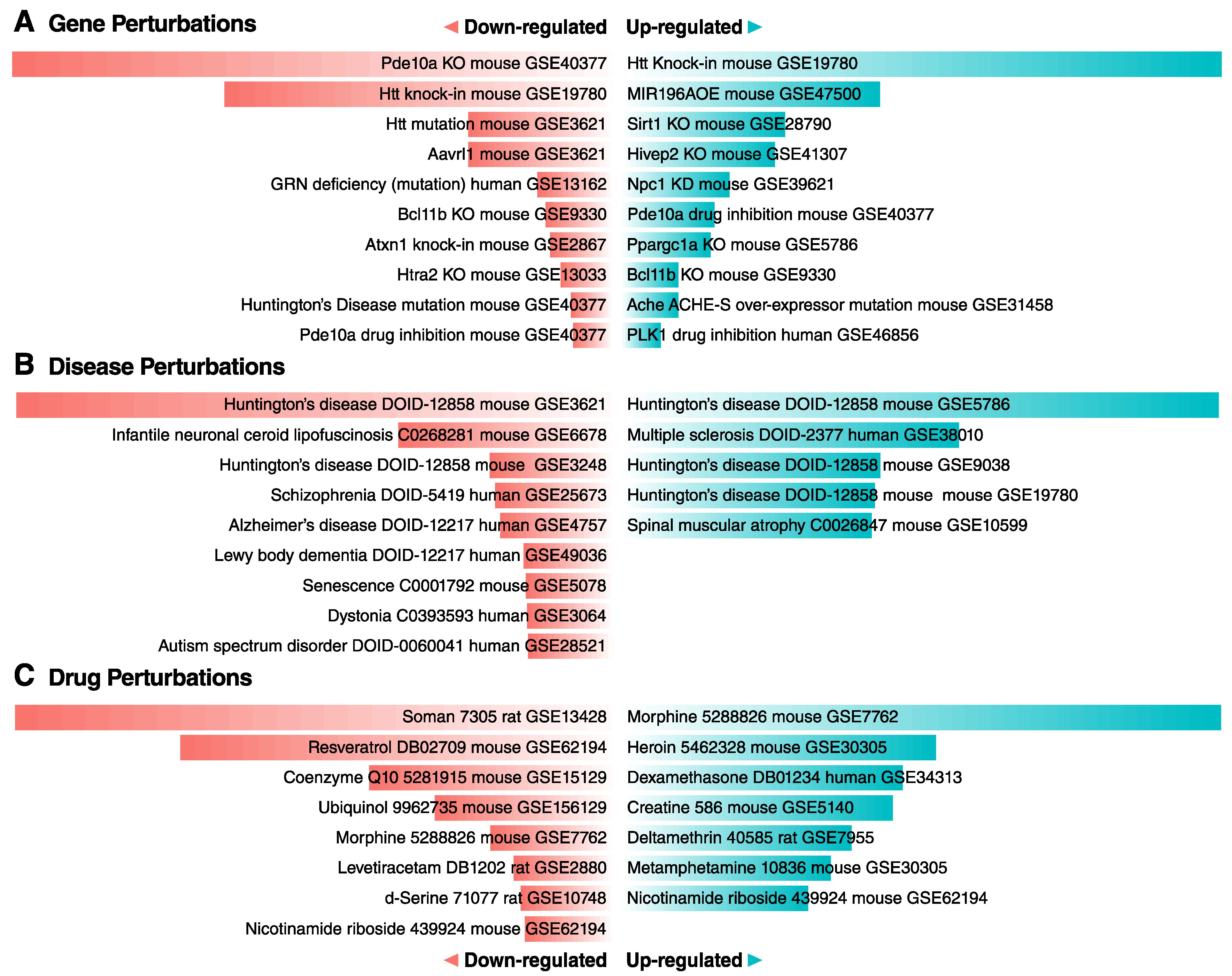{kind=link}
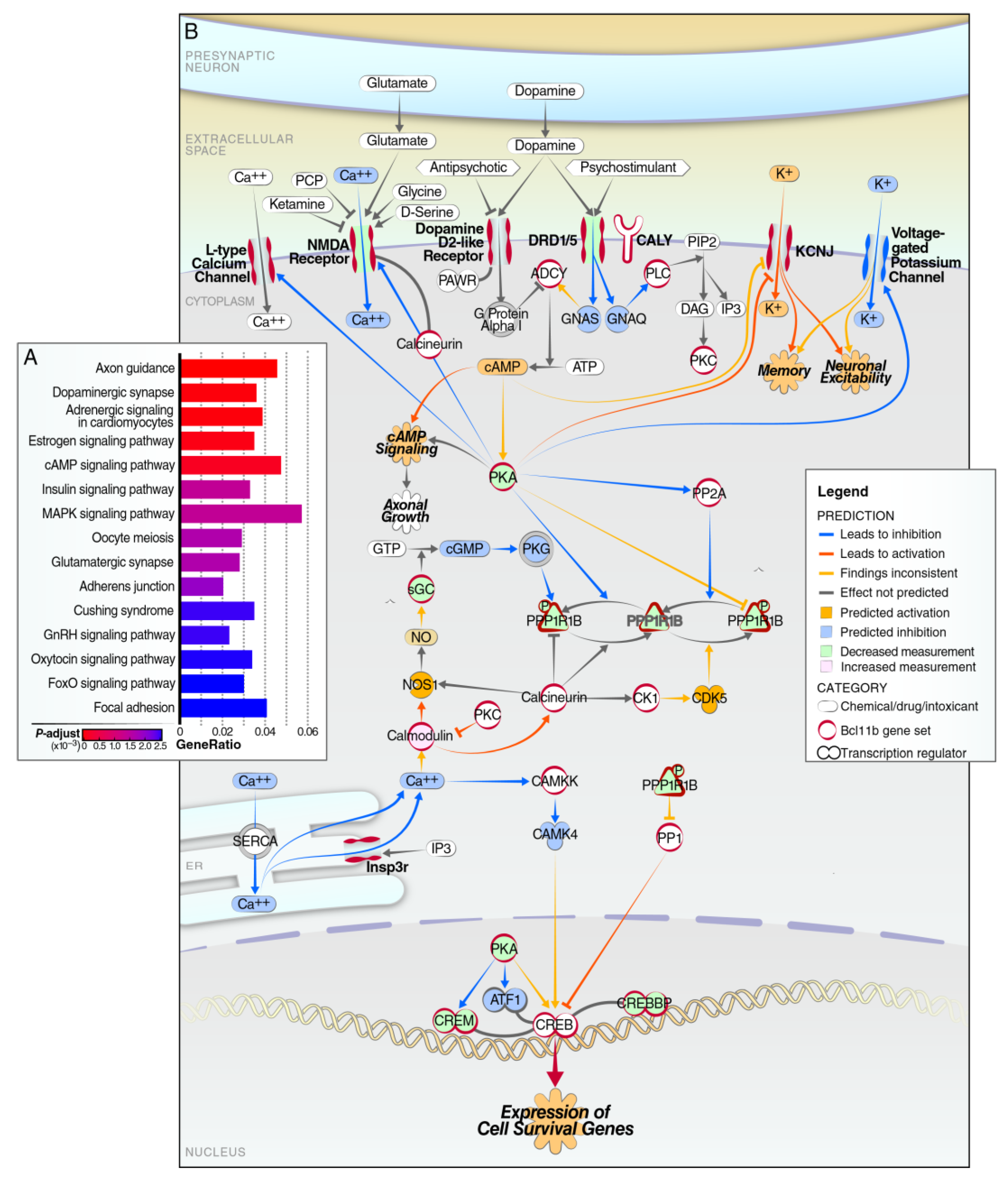{kind=link}
{kind=link}
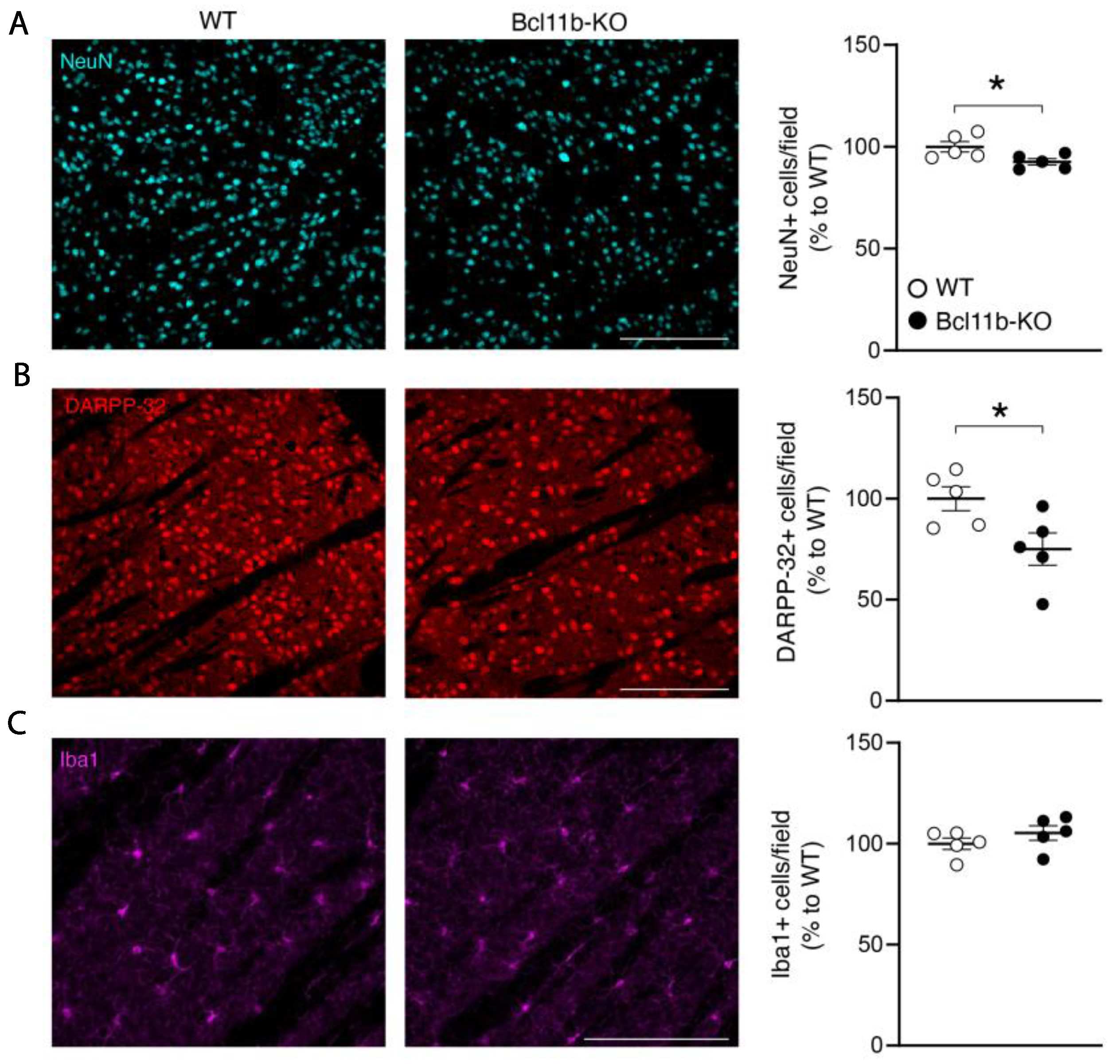{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
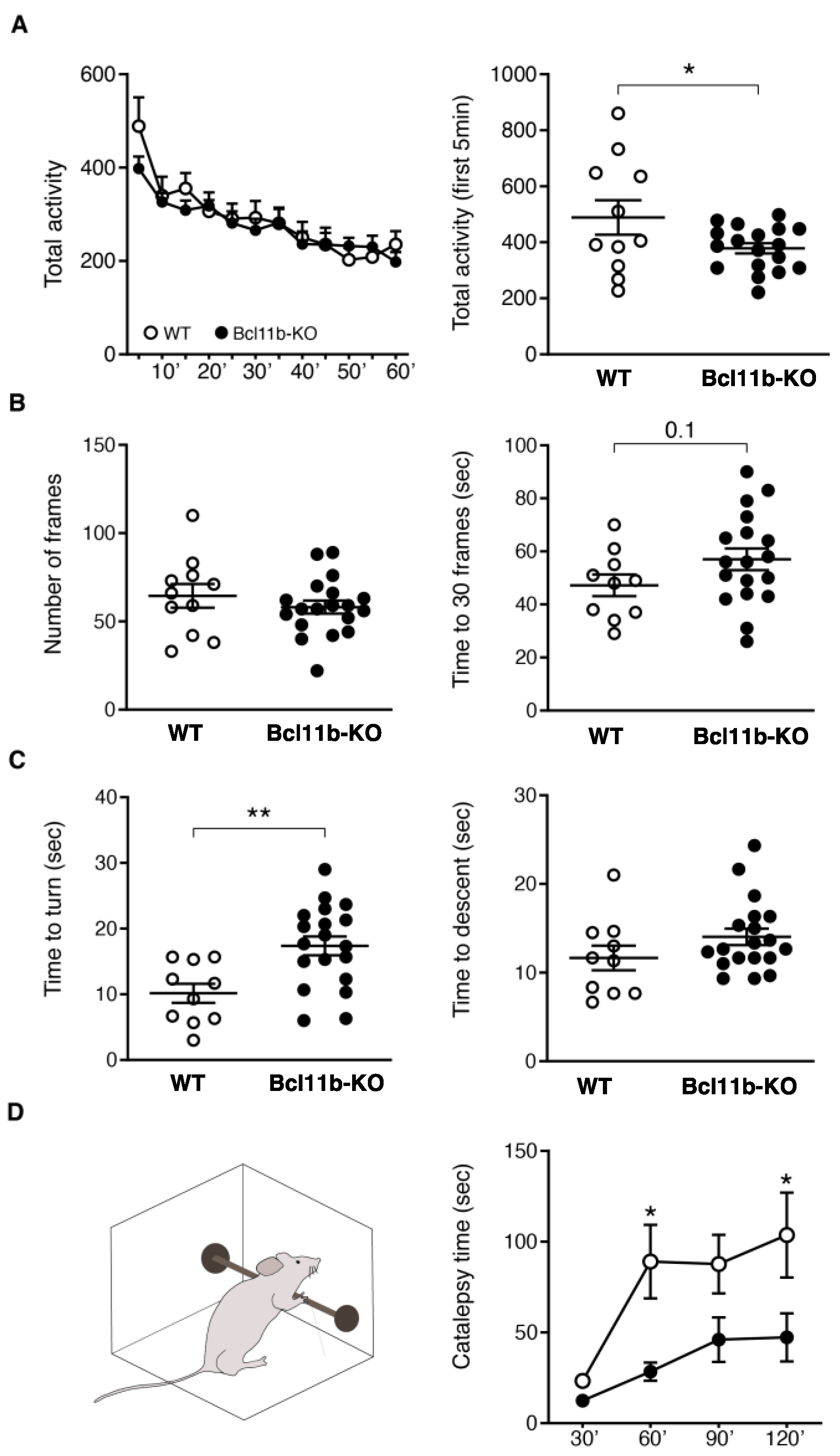
Behavioral Testing of Bcl11b-Deletion Mice
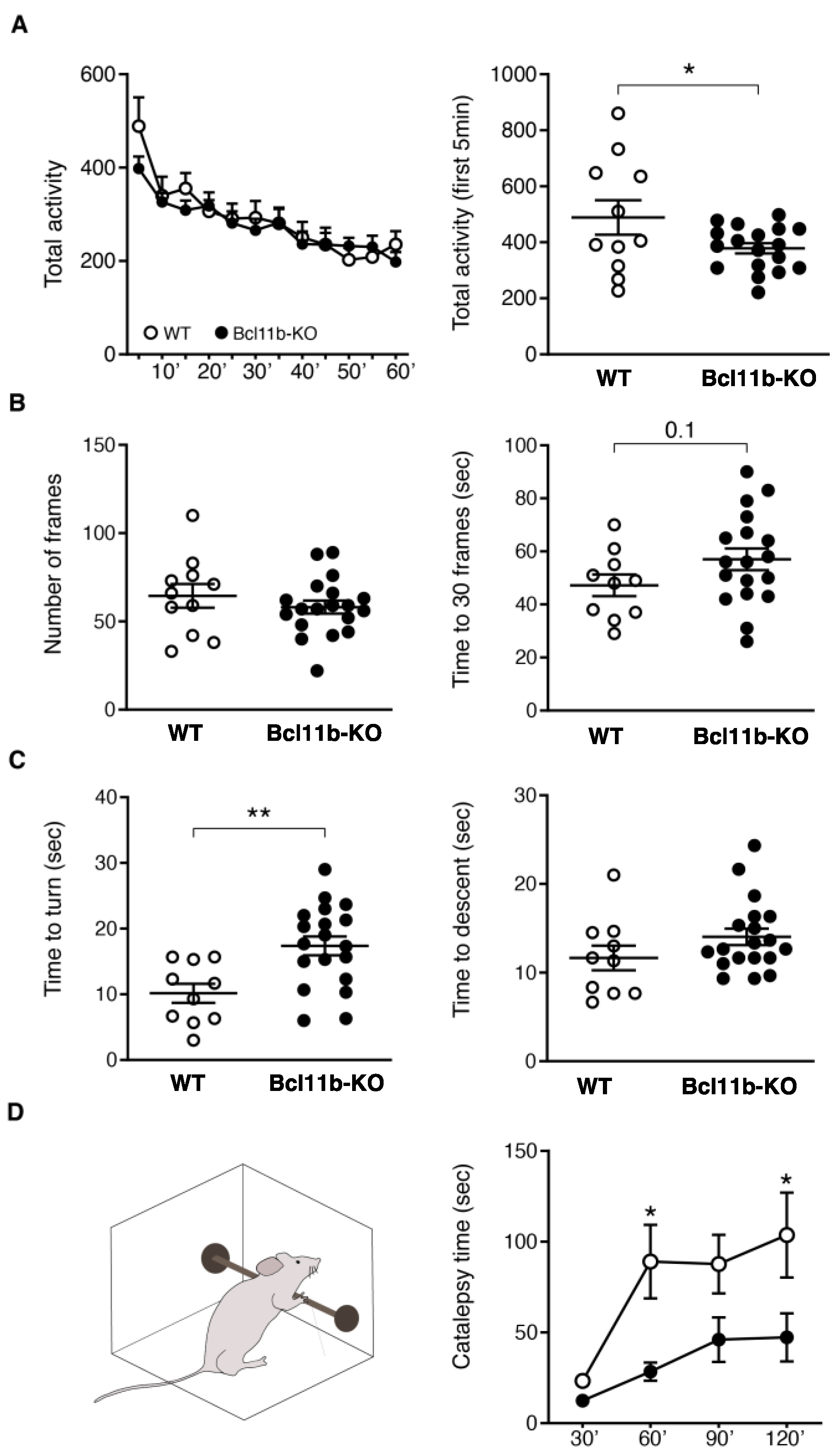
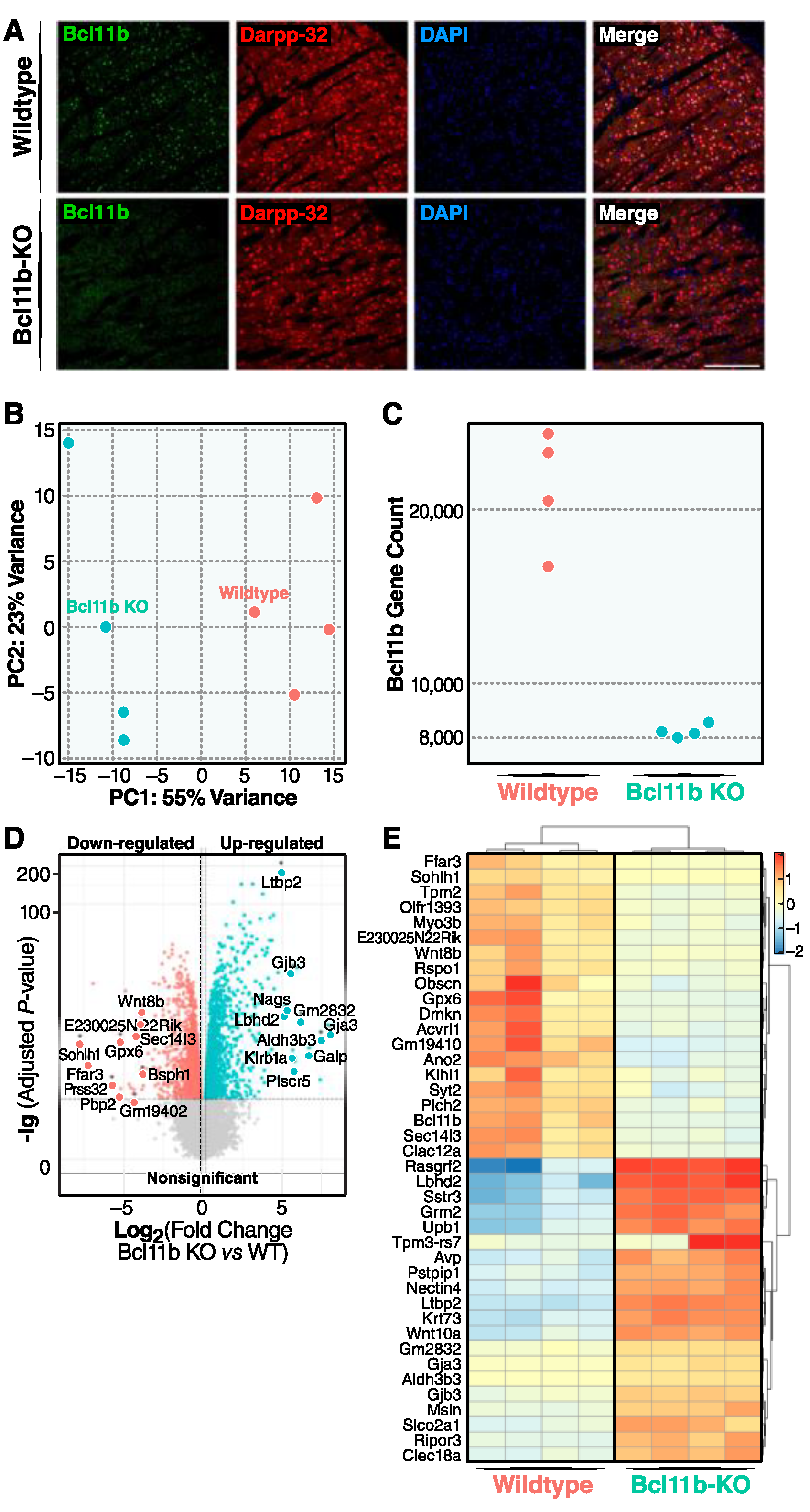
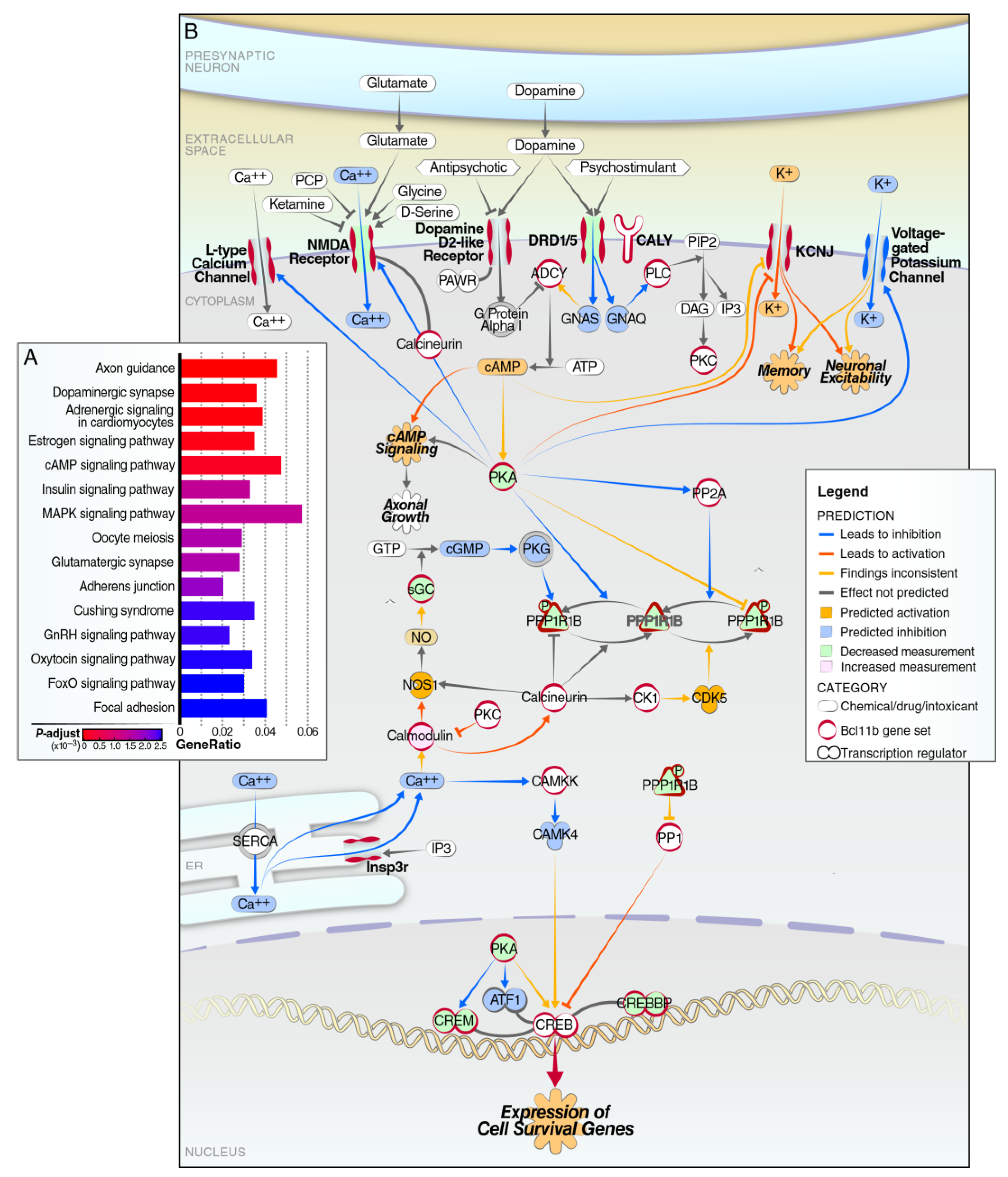
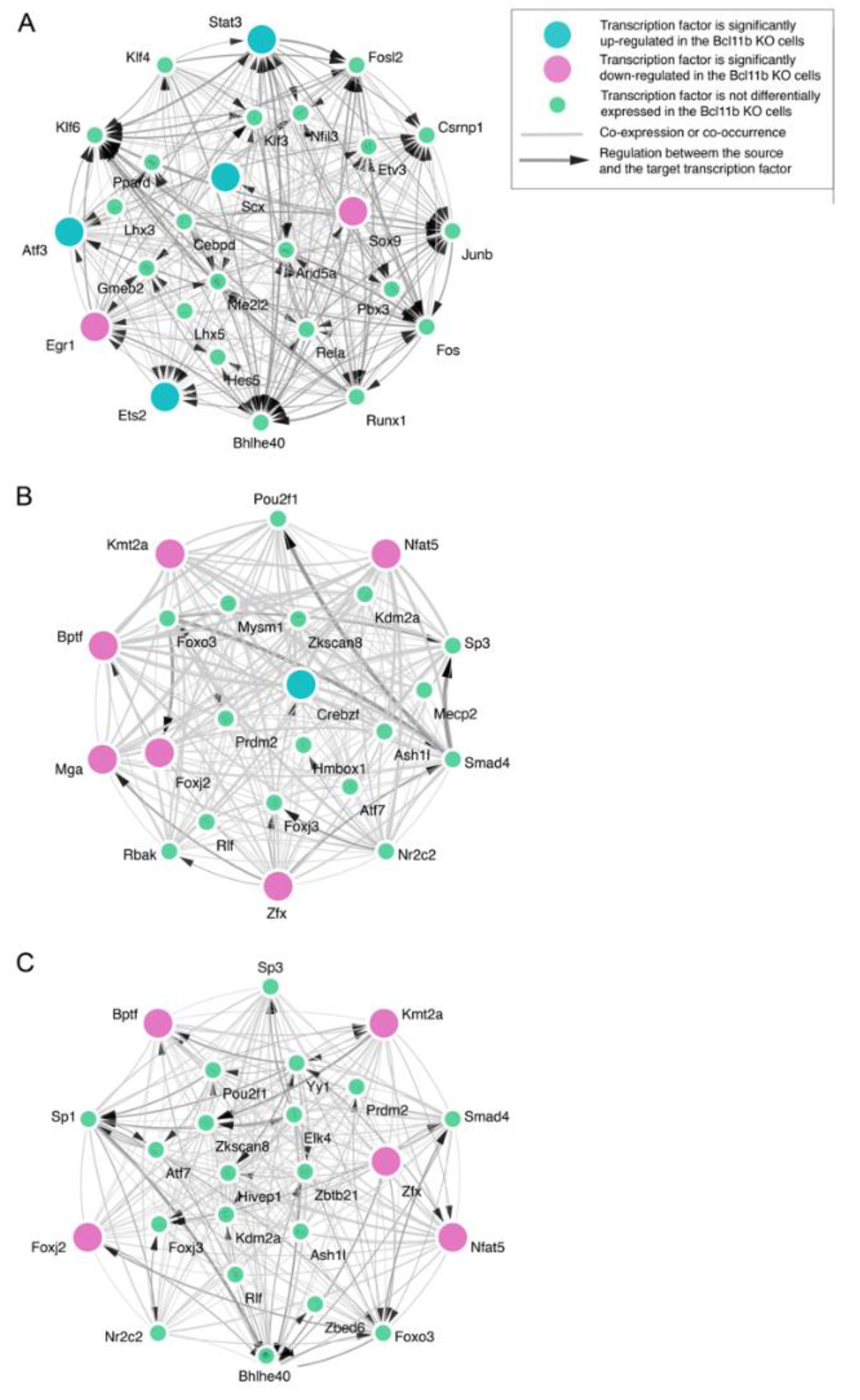
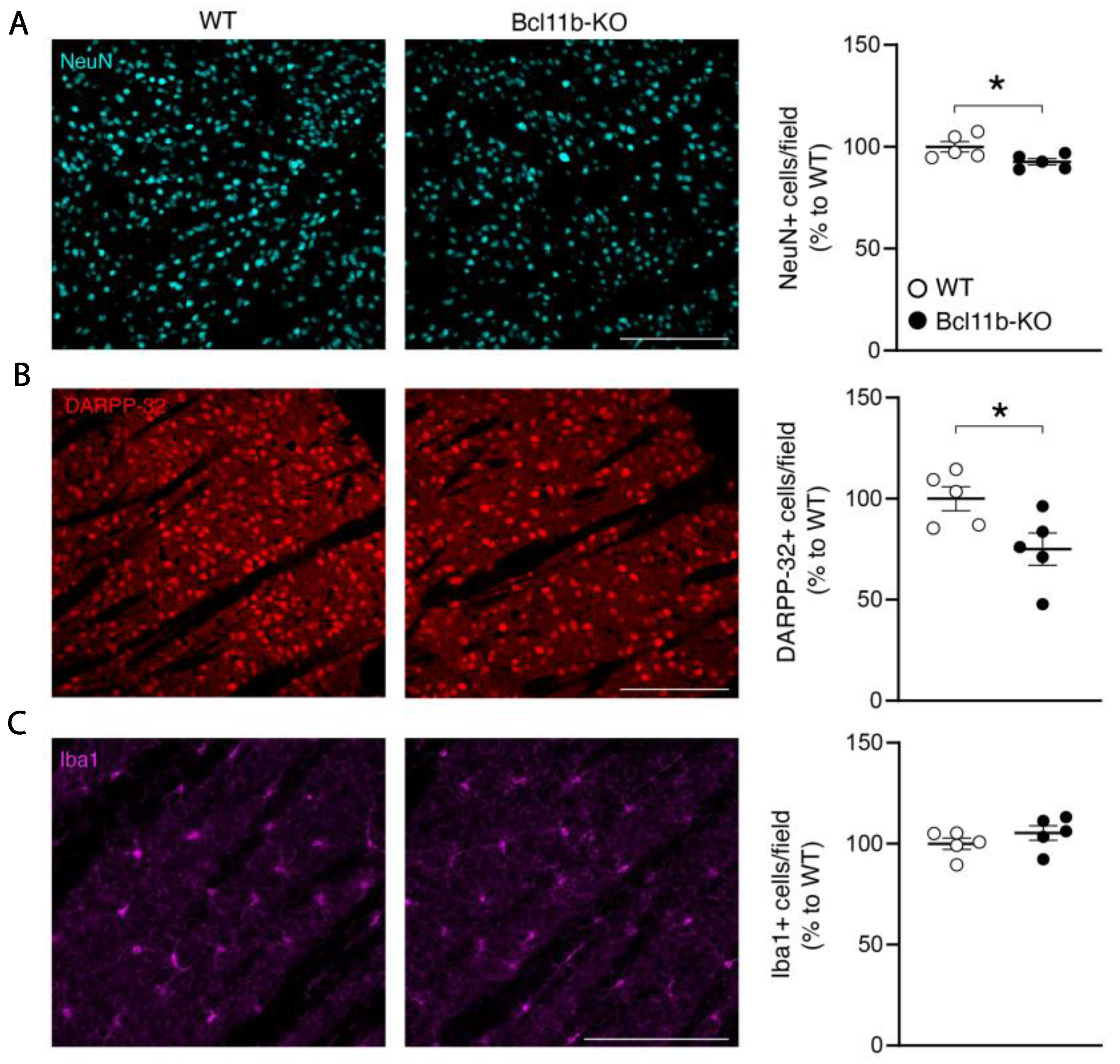
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ring, K.L.; An, M.C.; Zhang, N.; O’Brien, R.N.; Ramos, E.M.; Gao, F.; Atwood, R.; Bailus, B.J.; Melov, S.; Mooney, S.D.; et al. Genomic Analysis Reveals Disruption of Striatal Neuronal Development and Therapeutic Targets in Human Huntington’s Disease Neural Stem Cells. Stem Cell Rep. 2015, 5, 1023–1038. [Google Scholar] [CrossRef] [PubMed]
- Domingo, A.; Yadav, R.; Shah, S.; Hendriks, W.T.; Erdin, S.; Gao, D.; O’Keefe, K.; Currall, B.; Gusella, J.F.; Sharma, N.; et al. Dystonia-specific mutations in THAP1 alter transcription of genes associated with neurodevelopment and myelin. Am. J. Hum. Genet. 2021, 108, 2145–2158. [Google Scholar] [CrossRef] [PubMed]
- Cirnaru, M.D.; Creus-Muncunill, J.; Nelson, S.; Lewis, T.B.; Watson, J.; Ellerby, L.M.; Gonzalez-Alegre, P.; Ehrlich, M.E. Striatal Cholinergic Dysregulation after Neonatal Decrease in X-Linked Dystonia Parkinsonism-Related TAF1 Isoforms. Mov. Disord. 2021, 36, 2780–2794. [Google Scholar] [CrossRef]
- Aneichyk, T.; Hendriks, W.T.; Yadav, R.; Shin, D.; Gao, D.; Vaine, C.A.; Collins, R.L.; Domingo, A.; Currall, B.; Stortchevoi, A.; et al. Dissecting the Causal Mechanism of X-Linked Dystonia-Parkinsonism by Integrating Genome and Transcriptome Assembly. Cell 2018, 172, 897–909.e21. [Google Scholar] [CrossRef] [PubMed]
- Egervari, G.; Siciliano, C.A.; Whiteley, E.L.; Ron, D. Alcohol and the brain: From genes to circuits. Trends Neurosci. 2021, 44, 1004–1015. [Google Scholar] [CrossRef]
- Van Hees, L.; Didone, V.; Charlet-Briart, M.; Van Ingelgom, T.; Alexandre, A.; Quertemont, E.; Nguyen, L.; Laguesse, S. Voluntary alcohol binge-drinking in adolescent C57Bl6 mice induces delayed appearance of behavioural defects in both males and females. Addict. Biol. 2021, 27, e13102. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Lindenberg, A. From maps to mechanisms through neuroimaging of schizophrenia. Nature 2010, 468, 194–202. [Google Scholar] [CrossRef]
- Lennon, M.J.; Jones, S.P.; Lovelace, M.D.; Guillemin, G.J.; Brew, B.J. Bcl11b-A Critical Neurodevelopmental Transcription Factor-Roles in Health and Disease. Front. Cell Neurosci. 2017, 11, 89. [Google Scholar] [CrossRef]
- Volpicelli, F.; Perrone-Capano, C.; Bellenchi, G.C.; Colucci-D’Amato, L.; di Porzio, U. Molecular Regulation in Dopaminergic Neuron Development. Cues to Unveil Molecular Pathogenesis and Pharmacological Targets of Neurodegeneration. Int. J. Mol. Sci. 2020, 21, e13102. [Google Scholar] [CrossRef]
- Avram, D.; Fields, A.; Pretty On Top, K.; Nevrivy, D.J.; Ishmael, J.E.; Leid, M. Isolation of a novel family of C(2)H(2) zinc finger proteins implicated in transcriptional repression mediated by chicken ovalbumin upstream promoter transcription factor (COUP-TF) orphan nuclear receptors. J. Biol. Chem. 2000, 275, 10315–10322. [Google Scholar] [CrossRef] [Green Version]
- Canovas, J.; Berndt, F.A.; Sepulveda, H.; Aguilar, R.; Veloso, F.A.; Montecino, M.; Oliva, C.; Maass, J.C.; Sierralta, J.; Kukuljan, M. The Specification of Cortical Subcerebral Projection Neurons Depends on the Direct Repression of TBR1 by CTIP1/BCL11a. J. Neurosci. 2015, 35, 7552–7564. [Google Scholar] [CrossRef] [PubMed]
- Leid, M.; Ishmael, J.E.; Avram, D.; Shepherd, D.; Fraulob, V.; Dolle, P. CTIP1 and CTIP2 are differentially expressed during mouse embryogenesis. Gene Expr. Patterns 2004, 4, 733–739. [Google Scholar] [CrossRef]
- Wiegreffe, C.; Simon, R.; Peschkes, K.; Kling, C.; Strehle, M.; Cheng, J.; Srivatsa, S.; Liu, P.; Jenkins, N.A.; Copeland, N.G.; et al. Bcl11a (Ctip1) Controls Migration of Cortical Projection Neurons through Regulation of Sema3c. Neuron 2015, 87, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Satterwhite, E.; Sonoki, T.; Willis, T.G.; Harder, L.; Nowak, R.; Arriola, E.L.; Liu, H.; Price, H.P.; Gesk, S.; Steinemann, D.; et al. The BCL11 gene family: Involvement of BCL11A in lymphoid malignancies. Blood 2001, 98, 3413–3420. [Google Scholar] [CrossRef]
- Arlotta, P.; Molyneaux, B.J.; Jabaudon, D.; Yoshida, Y.; Macklis, J.D. Ctip2 controls the differentiation of medium spiny neurons and the establishment of the cellular architecture of the striatum. J. Neurosci. 2008, 28, 622–632. [Google Scholar] [CrossRef]
- Chandwani, S.; Keilani, S.; Ortiz-Virumbrales, M.; Morant, A.; Bezdecny, S.; Ehrlich, M.E. Induction of DARPP-32 by brain-derived neurotrophic factor in striatal neurons in vitro is modified by histone deacetylase inhibitors and Nab2. PLoS ONE 2013, 8, e76842. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.A.; Kass, K.E.; Gilmartin, T.; Stanwood, G.D.; Woodward, E.L.; Head, S.R.; Sutcliffe, J.G.; Thomas, E.A. Selective deficits in the expression of striatal-enriched mRNAs in Huntington’s disease. J. Neurochem. 2006, 96, 743–757. [Google Scholar] [CrossRef]
- Golonzhka, O.; Liang, X.; Messaddeq, N.; Bornert, J.M.; Campbell, A.L.; Metzger, D.; Chambon, P.; Ganguli-Indra, G.; Leid, M.; Indra, A.K. Dual role of COUP-TF-interacting protein 2 in epidermal homeostasis and permeability barrier formation. J. Investig. Dermatol. 2009, 129, 1459–1470. [Google Scholar] [CrossRef]
- Desplats, P.A.; Lambert, J.R.; Thomas, E.A. Functional roles for the striatal-enriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington’s disease. Neurobiol. Dis. 2008, 31, 298–308. [Google Scholar] [CrossRef]
- Fjodorova, M.; Louessard, M.; Li, Z.; De La Fuente, D.C.; Dyke, E.; Brooks, S.P.; Perrier, A.L.; Li, M. CTIP2-Regulated Reduction in PKA-Dependent DARPP32 Phosphorylation in Human Medium Spiny Neurons: Implications for Huntington Disease. Stem Cell Rep. 2019, 13, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Cantle, J.P.; Chatzopoulou, D.; Wang, N.; Gao, F.; Al-Ramahi, I.; Lu, X.H.; Ramos, E.M.; El-Zein, K.; Zhao, Y.; et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 2016, 19, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria-Rekalde, E.; Alzola-Aldamizetxebarria, S.; Flunkert, S.; Hable, I.; Daurer, M.; Neddens, J.; Hutter-Paier, B. Quantification of Huntington’s Disease Related Markers in the R6/2 Mouse Model. Front. Mol. Neurosci. 2020, 13, 617229. [Google Scholar] [CrossRef] [PubMed]
- Arlotta, P.; Molyneaux, B.J.; Chen, J.; Inoue, J.; Kominami, R.; Macklis, J.D. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron 2005, 45, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Onorati, M.; Castiglioni, V.; Biasci, D.; Cesana, E.; Menon, R.; Vuono, R.; Talpo, F.; Laguna Goya, R.; Lyons, P.A.; Bulfamante, G.P.; et al. Molecular and functional definition of the developing human striatum. Nat. Neurosci. 2014, 17, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Precious, S.V.; Cambray, S.; Risner-Janiczek, J.R.; Kelly, C.; Noakes, Z.; Fjodorova, M.; Heuer, A.; Ungless, M.A.; Rodriguez, T.A.; et al. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development 2015, 142, 1375–1386. [Google Scholar] [CrossRef]
- Fullard, J.F.; Hauberg, M.E.; Bendl, J.; Egervari, G.; Cirnaru, M.D.; Reach, S.M.; Motl, J.; Ehrlich, M.E.; Hurd, Y.L.; Roussos, P. An atlas of chromatin accessibility in the adult human brain. Genome Res. 2018, 28, 1243–1252. [Google Scholar] [CrossRef]
- Ivkovic, S.; Ehrlich, M.E. Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J. Neurosci. 1999, 19, 5409–5419. [Google Scholar] [CrossRef]
- Tang, B.; Di Lena, P.; Schaffer, L.; Head, S.R.; Baldi, P.; Thomas, E.A. Genome-wide identification of Bcl11b gene targets reveals role in brain-derived neurotrophic factor signaling. PLoS ONE. 2011, 6, e23691. [Google Scholar] [CrossRef]
- Hobert, O. Terminal Selectors of Neuronal Identity. Curr. Top. Dev. Biol. 2016, 116, 455–475. [Google Scholar] [CrossRef]
- Bogush, A.I.; McCarthy, L.E.; Tian, C.; Olm, V.; Gieringer, T.; Ivkovic, S.; Ehrlich, M.E. DARPP-32 genomic fragments drive Cre expression in postnatal striatum. Genesis 2005, 42, 37–46. [Google Scholar] [CrossRef]
- Cirnaru, M.D.; Song, S.; Tshilenge, K.T.; Corwin, C.; Mleczko, J.; Galicia Aguirre, C.; Benlhabib, H.; Bendl, J.; Apontes, P.; Fullard, J.; et al. Unbiased identification of novel transcription factors in striatal compartmentation and striosome maturation. Elife 2021, 10, e65979. [Google Scholar] [CrossRef] [PubMed]
- Merienne, N.; Meunier, C.; Schneider, A.; Seguin, J.; Nair, S.S.; Rocher, A.B.; Le Gras, S.; Keime, C.; Faull, R.; Pellerin, L.; et al. Cell-Type-Specific Gene Expression Profiling in Adult Mouse Brain Reveals Normal and Disease-State Signatures. Cell Rep. 2019, 26, 2477–2493.e9. [Google Scholar] [CrossRef] [PubMed]
- Creus-Muncunill, J.; Badillos-Rodriguez, R.; Garcia-Forn, M.; Masana, M.; Garcia-Diaz Barriga, G.; Guisado-Corcoll, A.; Alberch, J.; Malagelada, C.; Delgado-Garcia, J.M.; Gruart, A.; et al. Increased translation as a novel pathogenic mechanism in Huntington’s disease. Brain 2019, 142, 3158–3175. [Google Scholar] [CrossRef] [PubMed]
- Creus-Muncunill, J.; Guisado-Corcoll, A.; Venturi, V.; Pantano, L.; Escaramis, G.; Garcia de Herreros, M.; Solaguren-Beascoa, M.; Gamez-Valero, A.; Navarrete, C.; Masana, M.; et al. Huntington’s disease brain-derived small RNAs recapitulate associated neuropathology in mice. Acta Neuropathol. 2021, 141, 565–584. [Google Scholar] [CrossRef] [PubMed]
- Walf, A.A.; Frye, C.A. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat. Protoc. 2007, 2, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.C.; Wernig, M.; Duncan, I.D.; Brustle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129–1133. [Google Scholar] [CrossRef]
- Naphade, S.; Embusch, A.; Madushani, K.L.; Ring, K.L.; Ellerby, L.M. Altered Expression of Matrix Metalloproteinases and Their Endogenous Inhibitors in a Human Isogenic Stem Cell Model of Huntington’s Disease. Front. Neurosci. 2017, 11, 736. [Google Scholar] [CrossRef]
- Kemp, P.J.; Rushton, D.J.; Yarova, P.L.; Schnell, C.; Geater, C.; Hancock, J.M.; Wieland, A.; Hughes, A.; Badder, L.; Cope, E.; et al. Improving and accelerating the differentiation and functional maturation of human stem cell-derived neurons: Role of extracellular calcium and GABA. J. Physiol. 2016, 594, 6583–6594. [Google Scholar] [CrossRef]
- Feijen, A.; Goumans, M.J.; van den Eijnden-van Raaij, A.J. Expression of activin subunits, activin receptors and follistatin in postimplantation mouse embryos suggests specific developmental functions for different activins. Development 1994, 120, 3621–3637. [Google Scholar] [CrossRef]
- Maira, M.; Long, J.E.; Lee, A.Y.; Rubenstein, J.L.; Stifani, S. Role for TGF-beta superfamily signaling in telencephalic GABAergic neuron development. J. Neurodev. Disord. 2010, 2, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Liu, X.; Wen, J.; Tang, F.; Zhou, Y.; Jing, N.; Jin, Y. SOX21 Ensures Rostral Forebrain Identity by Suppression of WNT8B during Neural Regionalization of Human Embryonic Stem Cells. Stem Cell Rep. 2019, 13, 1038–1052. [Google Scholar] [CrossRef]
- Easton, A.C.; Rotter, A.; Lourdusamy, A.; Desrivieres, S.; Fernandez-Medarde, A.; Biermann, T.; Fernandes, C.; Santos, E.; Kornhuber, J.; Schumann, G.; et al. Rasgrf2 controls dopaminergic adaptations to alcohol in mice. Brain Res. Bull. 2014, 109, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.E.; Olevska, A.; Morella, I.; Fasano, S.; Santos, E.; Brambilla, R.; Spanagel, R. The Inhibition of RasGRF2, But Not RasGRF1, Alters Cocaine Reward in Mice. J. Neurosci. 2019, 39, 6325–6338. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Neumann, K.; Duhan, V.; Namineni, S.; Hansen, A.L.; Wartewig, T.; Kurgyis, Z.; Holm, C.K.; Heikenwalder, M.; Lang, K.S.; et al. The uric acid crystal receptor Clec12A potentiates type I interferon responses. Proc. Natl. Acad. Sci. USA 2019, 116, 18544–18549. [Google Scholar] [CrossRef]
- Barbeito, P.; Garcia-Gonzalo, F.R. HTR6 and SSTR3 targeting to primary cilia. Biochem. Soc. Trans. 2021, 49, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Liu, Q.; Xu, H.; Wu, Z.; Zhou, L.; Gu, Z.; Gong, P.; Shen, J. Upregulated Expression of SSTR3 is Involved in Neuronal Apoptosis After Intracerebral Hemorrhage in Adult Rats. Cell Mol. Neurobiol. 2017, 37, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.; Hueske, E.; Drammis, S.M.; Toro Arana, S.E.; Nelson, E.D.; Carter, C.W.; Delcasso, S.; Rodriguez, R.X.; Lutwak, H.; DiMarco, K.S.; et al. Striosomes Mediate Value-Based Learning Vulnerable in Age and a Huntington’s Disease Model. Cell 2020, 183, 918–934.e49. [Google Scholar] [CrossRef]
- Hedreen, J.C.; Folstein, S.E. Early loss of neostriatal striosome neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1995, 54, 105–120. [Google Scholar] [CrossRef]
- Morigaki, R.; Lee, J.H.; Yoshida, T.; Wuthrich, C.; Hu, D.; Crittenden, J.R.; Friedman, A.; Kubota, Y.; Graybiel, A.M. Spatiotemporal Up-Regulation of Mu Opioid Receptor 1 in Striatum of Mouse Model of Huntington’s Disease Differentially Affecting Caudal and Striosomal Regions. Front. Neuroanat. 2020, 14, 608060. [Google Scholar] [CrossRef]
- Schmeier, S.; Alam, T.; Essack, M.; Bajic, V.B. TcoF-DB v2: Update of the database of human and mouse transcription co-factors and transcription factor interactions. Nucleic Acids Res. 2017, 45, D145–D150. [Google Scholar] [CrossRef]
- Keilani, S.; Chandwani, S.; Dolios, G.; Bogush, A.; Beck, H.; Hatzopoulos, A.K.; Rao, G.N.; Thomas, E.A.; Wang, R.; Ehrlich, M.E. Egr-1 induces DARPP-32 expression in striatal medium spiny neurons via a conserved intragenic element. J. Neurosci. 2012, 32, 6808–6818. [Google Scholar] [CrossRef]
- Genin, E.C.; Caron, N.; Vandenbosch, R.; Nguyen, L.; Malgrange, B. Concise review: Forkhead pathway in the control of adult neurogenesis. Stem Cells 2014, 32, 1398–1407. [Google Scholar] [CrossRef]
- Schmidt, M.; Fernandez de Mattos, S.; van der Horst, A.; Klompmaker, R.; Kops, G.J.; Lam, E.W.; Burgering, B.M.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef]
- Bochynska, A.; Luscher-Firzlaff, J.; Luscher, B. Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin. Cells 2018, 7, 17. [Google Scholar] [CrossRef]
- Zhu, L.; Li, Q.; Wong, S.H.; Huang, M.; Klein, B.J.; Shen, J.; Ikenouye, L.; Onishi, M.; Schneidawind, D.; Buechele, C.; et al. ASH1L Links Histone H3 Lysine 36 Dimethylation to MLL Leukemia. Cancer Discov. 2016, 6, 770–783. [Google Scholar] [CrossRef]
- Dhar, S.S.; Alam, H.; Li, N.; Wagner, K.W.; Chung, J.; Ahn, Y.W.; Lee, M.G. Transcriptional repression of histone deacetylase 3 by the histone demethylase KDM2A is coupled to tumorigenicity of lung cancer cells. J. Biol. Chem. 2014, 289, 7483–7496. [Google Scholar] [CrossRef]
- Gao, R.; Dong, R.; Du, J.; Ma, P.; Wang, S.; Fan, Z. Depletion of histone demethylase KDM2A inhibited cell proliferation of stem cells from apical papilla by de-repression of p15INK4B and p27Kip1. Mol. Cell. Biochem. 2013, 379, 115–122. [Google Scholar] [CrossRef]
- Vashishtha, M.; Ng, C.W.; Yildirim, F.; Gipson, T.A.; Kratter, I.H.; Bodai, L.; Song, W.; Lau, A.; Labadorf, A.; Vogel-Ciernia, A.; et al. Targeting H3K4 trimethylation in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, E3027–E3036. [Google Scholar] [CrossRef]
- Kunii, Y.; Hino, M.; Matsumoto, J.; Nagaoka, A.; Nawa, H.; Kakita, A.; Akatsu, H.; Hashizume, Y.; Yabe, H. Differential protein expression of DARPP-32 versus Calcineurin in the prefrontal cortex and nucleus accumbens in schizophrenia and bipolar disorder. Sci. Rep. 2019, 9, 14877. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Soto, C. Role of calcineurin in neurodegeneration produced by misfolded proteins and endoplasmic reticulum stress. Curr. Opin. Cell Biol. 2011, 23, 223–230. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, L.; Zheng, X.; Liu, S.; Che, F. Role of Ash1l in Tourette syndrome and other neurodevelopmental disorders. Dev Neurobiol. 2021, 81, 79–91. [Google Scholar] [CrossRef]
- Francelle, L.; Galvan, L.; Brouillet, E. Possible involvement of self-defense mechanisms in the preferential vulnerability of the striatum in Huntington’s disease. Front. Cell Neurosci. 2014, 8, 295. [Google Scholar] [CrossRef]
- Sanberg, P.R. Haloperidol-induced catalepsy is mediated by postsynaptic dopamine receptors. Nature 1980, 284, 472–473. [Google Scholar] [CrossRef]
- Becanovic, K.; Pouladi, M.A.; Lim, R.S.; Kuhn, A.; Pavlidis, P.; Luthi-Carter, R.; Hayden, M.R.; Leavitt, B.R. Transcriptional changes in Huntington disease identified using genome-wide expression profiling and cross-platform analysis. Hum. Mol. Genet. 2010, 19, 1438–1452. [Google Scholar] [CrossRef]
- Brochier, C.; Gaillard, M.C.; Diguet, E.; Caudy, N.; Dossat, C.; Segurens, B.; Wincker, P.; Roze, E.; Caboche, J.; Hantraye, P.; et al. Quantitative gene expression profiling of mouse brain regions reveals differential transcripts conserved in human and affected in disease models. Physiol. Genom. 2008, 33, 170–179. [Google Scholar] [CrossRef]
- Hervas-Corpion, I.; Guiretti, D.; Alcaraz-Iborra, M.; Olivares, R.; Campos-Caro, A.; Barco, A.; Valor, L.M. Early alteration of epigenetic-related transcription in Huntington’s disease mouse models. Sci. Rep. 2018, 8, 9925. [Google Scholar] [CrossRef]
- Labadorf, A.; Hoss, A.G.; Lagomarsino, V.; Latourelle, J.C.; Hadzi, T.C.; Bregu, J.; MacDonald, M.E.; Gusella, J.F.; Chen, J.F.; Akbarian, S.; et al. RNA Sequence Analysis of Human Huntington Disease Brain Reveals an Extensive Increase in Inflammatory and Developmental Gene Expression. PLoS ONE 2015, 10, e0143563. [Google Scholar] [CrossRef]
- Le Gras, S.; Keime, C.; Anthony, A.; Lotz, C.; De Longprez, L.; Brouillet, E.; Cassel, J.C.; Boutillier, A.L.; Merienne, K. Altered enhancer transcription underlies Huntington’s disease striatal transcriptional signature. Sci. Rep. 2017, 7, 42875. [Google Scholar] [CrossRef]
- Novati, A.; Hentrich, T.; Wassouf, Z.; Weber, J.J.; Yu-Taeger, L.; Deglon, N.; Nguyen, H.P.; Schulze-Hentrich, J.M. Environment-dependent striatal gene expression in the BACHD rat model for Huntington disease. Sci. Rep. 2018, 8, 5803. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.; Creus Muncunill, J.; Galicia Aguirre, C.; Tshilenge, K.-T.; Hamilton, B.W.; Gerencser, A.A.; Benlhabib, H.; Cirnaru, M.-D.; Leid, M.; Mooney, S.D.; et al. Postnatal Conditional Deletion of Bcl11b in Striatal Projection Neurons Mimics the Transcriptional Signature of Huntington’s Disease. Biomedicines 2022, 10, 2377. https://doi.org/10.3390/biomedicines10102377
Song S, Creus Muncunill J, Galicia Aguirre C, Tshilenge K-T, Hamilton BW, Gerencser AA, Benlhabib H, Cirnaru M-D, Leid M, Mooney SD, et al. Postnatal Conditional Deletion of Bcl11b in Striatal Projection Neurons Mimics the Transcriptional Signature of Huntington’s Disease. Biomedicines. 2022; 10(10):2377. https://doi.org/10.3390/biomedicines10102377
Chicago/Turabian StyleSong, Sicheng, Jordi Creus Muncunill, Carlos Galicia Aguirre, Kizito-Tshitoko Tshilenge, B. Wade Hamilton, Akos A. Gerencser, Houda Benlhabib, Maria-Daniela Cirnaru, Mark Leid, Sean D. Mooney, and et al. 2022. "Postnatal Conditional Deletion of Bcl11b in Striatal Projection Neurons Mimics the Transcriptional Signature of Huntington’s Disease" Biomedicines 10, no. 10: 2377. https://doi.org/10.3390/biomedicines10102377
APA StyleSong, S., Creus Muncunill, J., Galicia Aguirre, C., Tshilenge, K.-T., Hamilton, B. W., Gerencser, A. A., Benlhabib, H., Cirnaru, M.-D., Leid, M., Mooney, S. D., Ellerby, L. M., & Ehrlich, M. E. (2022). Postnatal Conditional Deletion of Bcl11b in Striatal Projection Neurons Mimics the Transcriptional Signature of Huntington’s Disease. Biomedicines, 10(10), 2377. https://doi.org/10.3390/biomedicines10102377