
Lipid Metabolism in Glioblastoma: From De Novo Synthesis to Storage


Abstract
:1. Introduction
2. Standard GBM Therapy
3. Lipid Metabolism Regulation in GBM
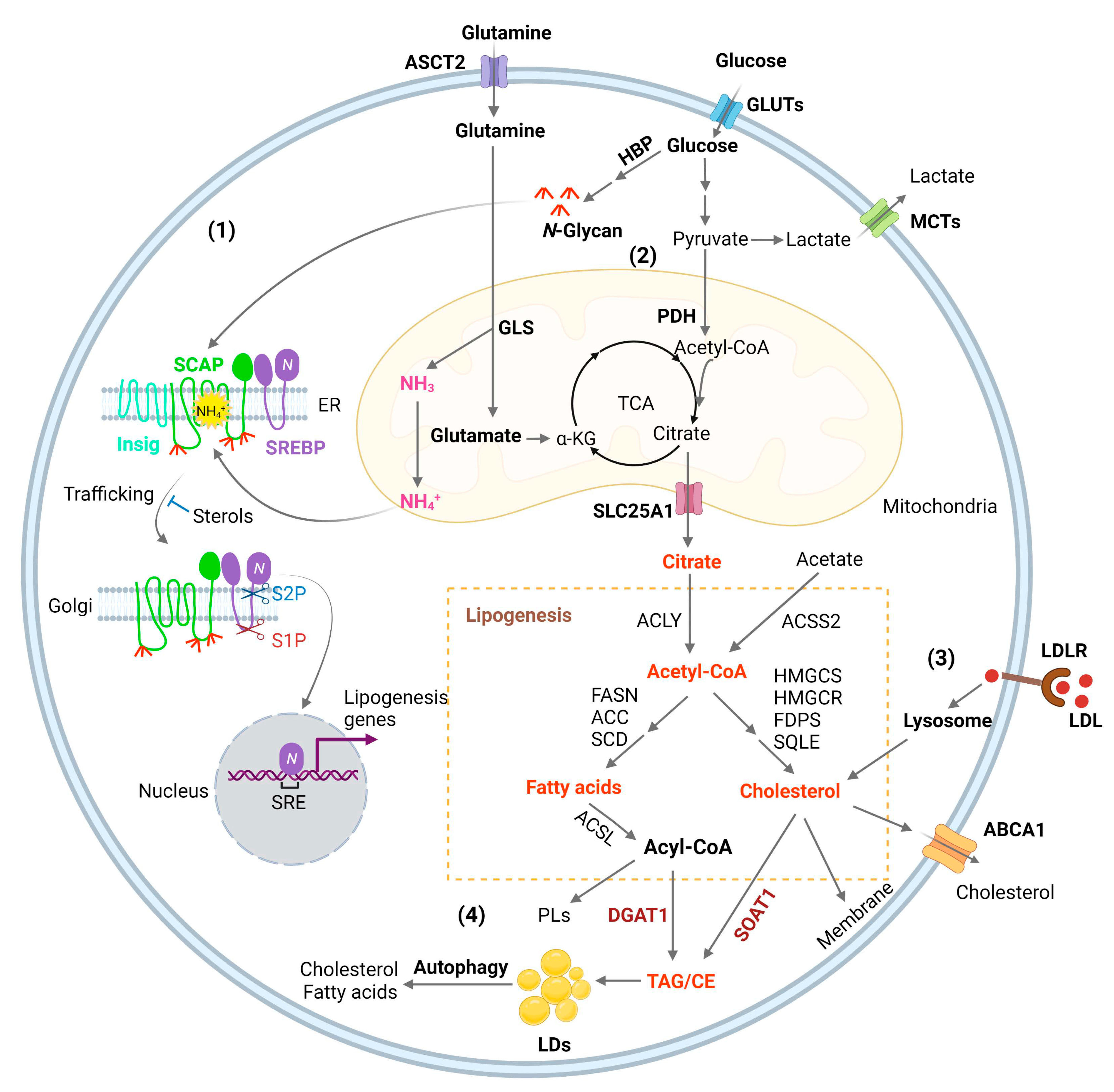
3.1. Lipogenesis
3.2. SREBP Activation, Connecting Glucose and Glutamine to Lipid Synthesis
3.3. Membrane Phospholipid Remodeling
3.4. Cholesterol Uptake
3.5. Lipid storage and Energy Homeostasis
4. The Basics of LDs
4.1. LD Formation
4.2. LD Size and Composition
4.3. LD Interaction and Proteomes
5. Inhibiting LD Formation for GBM Therapy
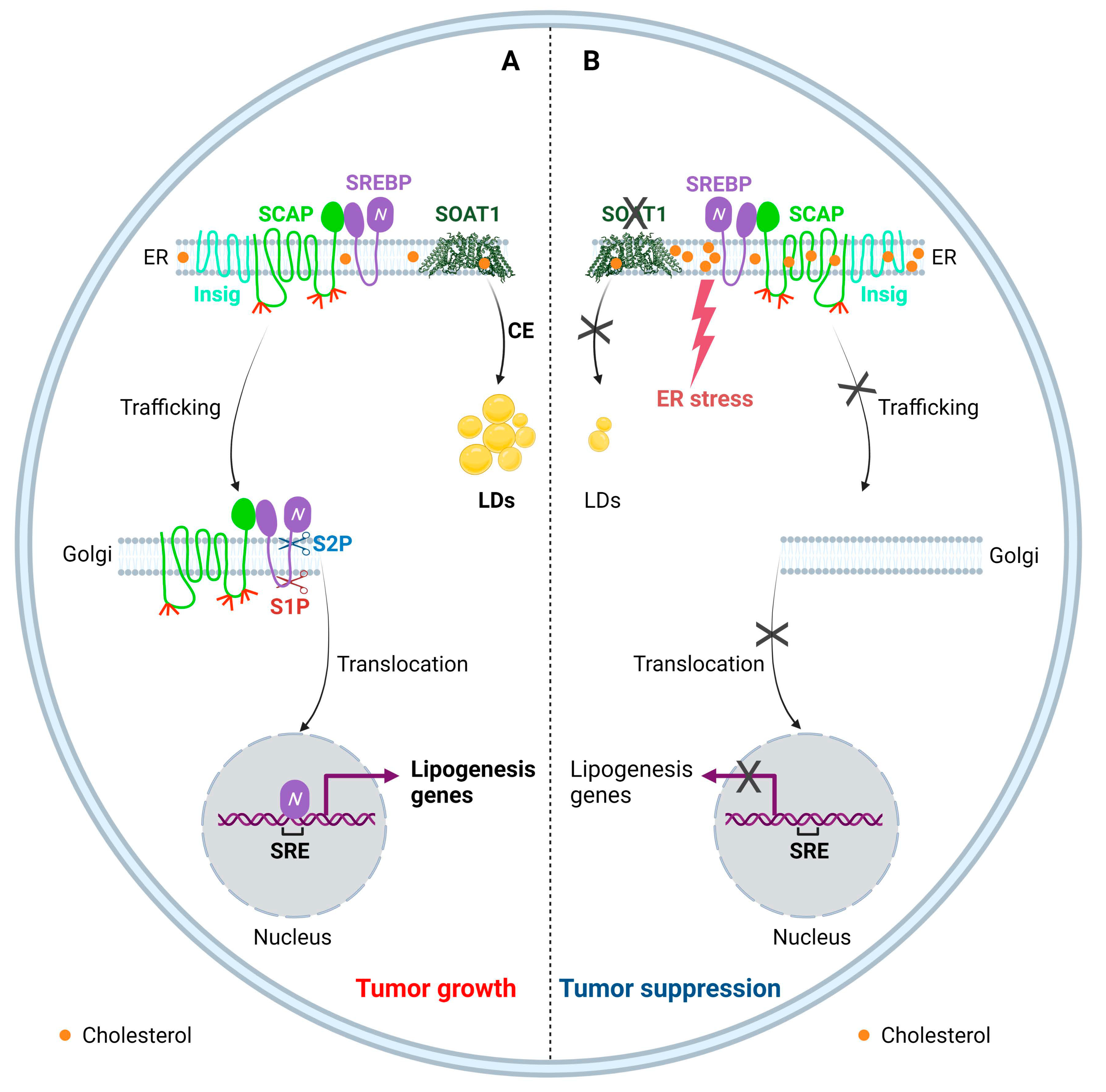
5.1. Targeting SOAT1 to Disrupt Cholesterol Homeostasis for GBM Therapy
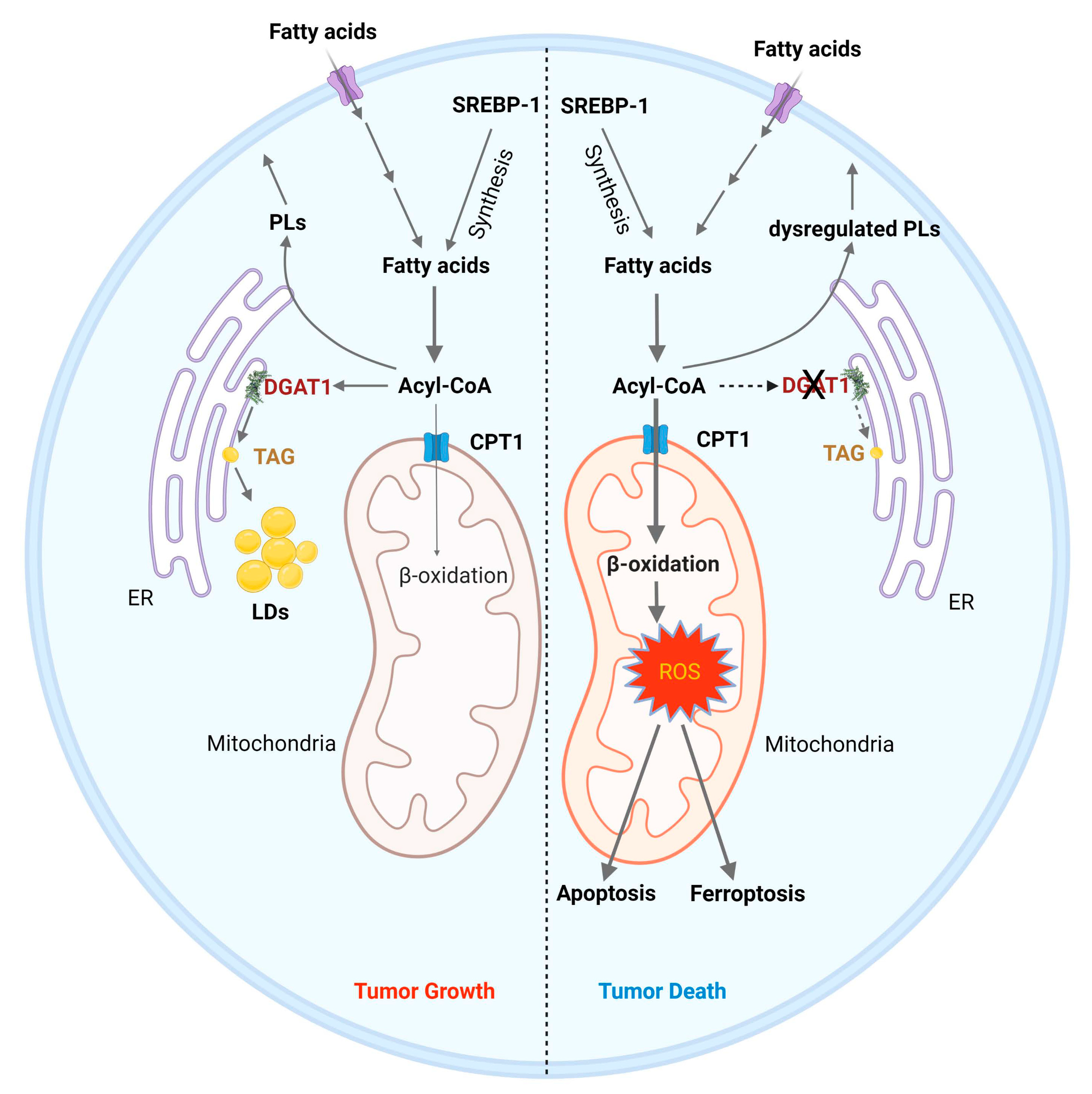
5.2. Targeting DGAT1 to Disrupt Fatty Acid Homeostasis for GBM Therapy
6. Other Targets in LDs
7. Conclusions and Future Directions

{kind=link}
{kind=link}
{kind=link}
| Target | Inhibitors | Types of Cancer | Preclinical Evidence | Clinical Trials | References |
|---|---|---|---|---|---|
| SREBPs | Fatostatin, Betulin, PF-429242, dipyridamole | GBM, breast, uterine, prostate, liver, lung, kidney, pancreatic and colon | Xenografts | [1,19,225,226,227,228,229,230,231,232,233,234,235] | |
| ACLY | SB-201076, Hydroxycitric acid, Bempedoic acid, BMS-303141, SB-204990, | Lung, prostate | Xenografts | [1,236,237] | |
| ACCs | ND-646, ND-654, soraphen A, TOFA | Lung, liver, breast | Xenografts, DEN-injured rats | [17,156,238,239,240,241,242,243,244] | |
| FASN | TVB-2640, C75, cerulenin, orlistat | Lung, colon, breast, GBM, astrocytoma, prostate, uterine | Xenografts | NCT03808558 NCT02223247 NCT02980029 NCT03179904 NCT05118776 NCT03032484 | [185,187,192,245,246,247,248,249] |
| SCD1 | A939572, MF-438, CAY10566 MK-8245 | Lung, pancreatic, kidney, GBM, ovarian | Xenograft, Pdx1Cre; LSL-KrasG12D mouse | NCT00972322 (type 2 diabetes) | [164,250,251,252,253,254,255,256] |
| ACSS2 | Tetrazoles, Pyridine Derivatives | [257,258] | |||
| SLC25A1 | CTPi2, BTA, CNASB | [259,260] | |||
| CPT1A | Etomoxir, Perhexiline, ST1326 | GBM, breast, leukemia, prostate, colon | Xenografts, Transgenic mice | [261,262,263,264,265,266,267,268] | |
| DGAT1 | A922500, AZD3988, PF-04620110 | GBM, liver, uterine, prostate | Xenografts | [17,156,242,243,244,269] | |
| SOAT1 | Avasimibe, ATR-101, K604, | Liver, colon, GBM, kidney | P53-deficient mice, AOM/DSS-treated and ApcMin/+ mice, xenografts | NCT01898715 | [185,187,192,246,247] |
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| 3-UTR | 3′-untranslational region |
| 25-HC | 25-hydroxycholesterol |
| α-KG | α-ketoglutaric acid |
| ABC | ATP-binding cassette |
| ABCA1 | ATP-binding cassette subfamily A member 1 |
| ACAT | acyl-CoA: cholesterol acyltransferase |
| ACC | acetyl-CoA carboxylases |
| ACLY | ATP-citrate lyase |
| ACSL | long-chain acyl-CoA synthetase |
| ACSS2 | acetyl-CoA synthetase 2 |
| AICAR | 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside |
| AKT | protein kinase B |
| AMPK | AMP-activated protein kinase |
| ASCT2 | alanine/serine/cysteine-preferring transporter 2 |
| ATGL | adipose triglyceride lipase |
| BBB | blood–brain barrier |
| COPII | coat protein complex II |
| CPT1 | carnitine palmitoyltransferase 1 |
| CE | cholesteryl esters |
| CESD | cholesterol storage disease |
| DGAT1 | diacylglycerol acyltransferase 1 |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial–mesenchymal transition |
| ER | endoplasmic reticulum |
| ESCC | esophageal squamous cell carcinoma |
| FAs | fatty acids |
| FAO | fatty acid oxidation |
| FASN | fatty acid synthase |
| FDPS | farnesyldiphosphate synthase |
| FIT | fat-storage-inducing transmembrane |
| GBM | glioblastoma |
| GLS | glutaminase |
| Gluts | glucose transporters |
| HBP | hexosamine biosynthesis pathway |
| HCC | hepatocellular carcinoma |
| HMGCS | hydroxymethylglutaryl-CoA synthase |
| HMGCR | hydroxymethylglutaryl-CoA reductase |
| HILPDA | hypoxia-inducible lipid droplet associated |
| Insig | insulin-inducible gene protein |
| IDH | isocitrate dehydrogenase |
| LDs | lipid droplets |
| LDL | low-density lipoprotein |
| LDLR | LDL receptor |
| IDOL | inducible degrader of LDLR |
| LPC | lysophosphatidylcholine |
| LPCATs | lysophosphatidylcholine acyltransferases |
| LXR | liver X receptor |
| MBOAT | membrane-bound O-acyltransferase |
| MCTs | monocarboxylate transporters |
| NASH | non-alcoholic steatohepatitis |
| NLSD | neutral lipid storage disease |
| OAA | oxaloacetate |
| PD1 | programmed cell death protein 1 |
| PDH | pyruvate dehydrogenase |
| PD-L1 | programmed death-ligand 1 |
| PI3K | phosphoinositide 3-kinases |
| PLs | phospholipids |
| PLA | phospholipase A |
| ROS | reactive oxygen species |
| S1P | site 1 protease |
| S2P | site 2 protease |
| SCAP | SREBP-cleavage activating protein |
| SCD1 | stearoyl-CoA desaturase 1 |
| SLC25A1 | tricarboxylate transport protein, mitochondrial |
| SOAT1 | sterol-O-acyltransferase 1 |
| SQLE | squalene monooxygenase |
| SRE | sterol regulatory element |
| SREBF1 | sterol regulatory element binding transcription factor 1 |
| SREBP | sterol regulatory element-binding protein |
| TAG | triacylglycerol |
| TCA cycle | tricarboxylic acid cycle |
| TCGA | the cancer genome atlas |
| TKI | tyrosine kinase inhibitors |
| TMZ | temozolomide |
| VEGF | vascular endothelial growth factor |
References
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Cockcroft, S. Mammalian lipids: Structure, synthesis and function. Essays Biochem. 2021, 65, 813–845. [Google Scholar] [CrossRef]
- Cisa-Wieczorek, S.; Hernandez-Alvarez, M.I. Deregulation of Lipid Homeostasis: A Fa(c)t in the Development of Metabolic Diseases. Cells 2020, 9, 2605. [Google Scholar] [CrossRef]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 128. [Google Scholar] [CrossRef] [Green Version]
- Estes, R.E.; Lin, B.; Khera, A.; Davis, M.Y. Lipid Metabolism Influence on Neurodegenerative Disease Progression: Is the Vehicle as Important as the Cargo? Front. Mol. Neurosci. 2021, 14, 788695. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.; Maciel, P.; Costa, V. Leading the way in the nervous system: Lipid Droplets as new players in health and disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158820. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. 2014, 9, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J.; Czernin, J.; Shyy, J.Y.; Watson, A.D.; Phelps, M.; et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Cloughesy, T.F.; Radu, C.G.; Mischel, P.S. AMPK: A metabolic checkpoint that regulates the growth of EGFR activated glioblastomas. Cell Cycle 2010, 9, 211–212. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242.e8. [Google Scholar] [CrossRef]
- Rudalska, R.; Harbig, J.; Snaebjornsson, M.T.; Klotz, S.; Zwirner, S.; Taranets, L.; Heinzmann, F.; Kronenberger, T.; Forster, M.; Cui, W.; et al. LXRalpha activation and Raf inhibition trigger lethal lipotoxicity in liver cancer. Nat. Cancer 2021, 2, 201–217. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, F.; Guo, D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern. Med. Rev. 2017, 3. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Geng, F.; Cheng, X.; Guo, Q.; Zhong, Y.; Cloughesy, T.F.; Yong, W.H.; Chakravarti, A.; Guo, D. Lipid Droplets Maintain Energy Homeostasis and Glioblastoma Growth via Autophagic Release of Stored Fatty Acids. iScience 2020, 23, 101569. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Geng, F.; Guo, D. DGAT1 protects tumor from lipotoxicity, emerging as a promising metabolic target for cancer therapy. Mol. Cell. Oncol. 2020, 7, 1805257. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Rock, K.; McArdle, O.; Forde, P.; Dunne, M.; Fitzpatrick, D.; O’Neill, B.; Faul, C. A clinical review of treatment outcomes in glioblastoma multiforme--the validation in a non-trial population of the results of a randomised Phase III clinical trial: Has a more radical approach improved survival? Br. J. Radiol. 2012, 85, e729–e733. [Google Scholar] [CrossRef]
- Janjua, T.I.; Rewatkar, P.; Ahmed-Cox, A.; Saeed, I.; Mansfeld, F.M.; Kulshreshtha, R.; Kumeria, T.; Ziegler, D.S.; Kavallaris, M.; Mazzieri, R.; et al. Frontiers in the treatment of glioblastoma: Past, present and emerging. Adv. Drug Deliv. Rev. 2021, 171, 108–138. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.H.; Guan, Y.L.; Liu, Q.; Wang, Y.; Cui, R.; Wang, Y.J. Astrocytoma progression scoring system based on the WHO 2016 criteria. Sci. Rep. 2019, 9, 96. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329. [Google Scholar] [CrossRef] [Green Version]
- Ringel, F.; Pape, H.; Sabel, M.; Krex, D.; Bock, H.C.; Misch, M.; Weyerbrock, A.; Westermaier, T.; Senft, C.; Schucht, P.; et al. Clinical benefit from resection of recurrent glioblastomas: Results of a multicenter study including 503 patients with recurrent glioblastomas undergoing surgical resection. Neuro-Oncology 2016, 18, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23 (Suppl. 2), iii1–iii105. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in re-current glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Schultz, N.; Mischel, P.S.; Cloughesy, T.F. Will kinase inhibitors make it as glioblastoma drugs? Curr. Top. Microbiol. Immunol. 2012, 355, 135–169. [Google Scholar]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology 2016, 18, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Kiesel, B.; Widhalm, G.; Rajky, O.; Ricken, G.; Wöhrer, A.; Dieckmann, K.; Filipits, M.; Brandstetter, A.; Weller, M.; et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology 2015, 17, 1064–1075. [Google Scholar] [CrossRef] [Green Version]
- Muir, M.; Gopakumar, S.; Traylor, J.; Lee, S.; Rao, G. Glioblastoma multiforme: Novel therapeutic targets. Expert Opin. Targets 2020, 24, 605–614. [Google Scholar] [CrossRef]
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626. [Google Scholar] [CrossRef] [Green Version]
- Bhagavan, N.V.; Ha, C.-E. Chapter 16—Lipids I: Fatty Acids and Eicosanoids. In Essentials of Medical Biochemistry, 2nd ed.; Bhagavan, N.V., Ha, C.-E., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 269–297. [Google Scholar]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa Kimmo, J.; Singh Dinesh, K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo Sara, G.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, N.; Wang, J.; Fu, S.; Wang, X.; Chen, D. Acetyl-CoA Synthetase 2 as a Therapeutic Target in Tumor Metabolism. Cancers 2022, 14, 2896. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shi, L.Z. Metabolic regulation of TH17 cells. Mol. Immunol. 2019, 109, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Ru, P.; Geng, F.; Liu, J.; Yoo, J.Y.; Wu, X.; Cheng, X.; Euthine, V.; Hu, P.; Guo, J.Y.; et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell 2015, 28, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30 Pt 6, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The carnitine system and cancer metabolic plasticity. Cell Death Dis. 2018, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.J.; Argus, J.P.; Zhu, Y.; Wilks, M.Q.; Marbois, B.N.; York, A.G.; Kidani, Y.; Pourzia, A.L.; Akhavan, D.; Lisiero, D.N.; et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 2013, 73, 2850–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ru, P.; Hu, P.; Geng, F.; Mo, X.; Cheng, C.; Yoo, J.Y.; Cheng, X.; Wu, X.; Guo, J.Y.; Nakano, I.; et al. Feedback Loop Regulation of SCAP/SREBP-1 by miR-29 Modulates EGFR Signaling-Driven Glioblastoma Growth. Cell Rep. 2016, 16, 1527–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ru, P.; Guo, D. microRNA-29 mediates a novel negative feedback loop to regulate SCAP/SREBP-1 and lipid metabolism. RNA Dis. 2017, 4, e1525. [Google Scholar] [PubMed] [Green Version]
- Jin, X.; Demere, Z.; Nair, K.; Ali, A.; Ferraro, G.B.; Natoli, T.; Deik, A.; Petronio, L.; Tang, A.A.; Zhu, C.; et al. A metastasis map of human cancer cell lines. Nature 2020, 588, 331–336. [Google Scholar] [CrossRef]
- Venneti, S.; Dunphy, M.P.; Zhang, H.; Pitter, K.L.; Zanzonico, P.; Campos, C.; Carlin, S.D.; La Rocca, G.; Lyashchenko, S.; Ploessl, K.; et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 2015, 7, 274ra217. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Geng, F.; Li, Z.; Zhong, Y.; Wang, H.; Cheng, X.; Zhao, Y.; Mo, X.; Horbinski, C.; Duan, W.; et al. Ammonia stimulates SCAP/Insig dissociation and SREBP-1 activation to promote lipogenesis and tumour growth. Nat. Metab. 2022, 4, 575–588. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Jeon, T.I.; Osborne, T.F. SREBPs: Metabolic integrators in physiology and metabolism. Trends Endocrinol. Metab. 2012, 23, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 2018, 87, 783–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.K.; Lopez, J.M.; Sanchez, H.B.; Osborne, T.F. Sterol regulation of fatty acid synthase promoter. Coordinate feedback regulation of two major lipid pathways. J. Biol. Chem. 1995, 270, 25578–25583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.P.; Seemann, J.; Goldstein, J.L.; Brown, M.S. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc. Natl Acad. Sci. USA 2007, 104, 6519–6526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espenshade, P.J.; Cheng, D.; Goldstein, J.L.; Brown, M.S. Autocatalytic processing of site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 1999, 274, 22795–22804. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Espenshade, P.J.; Slaughter, C.A.; Jaen, J.C.; Brown, M.S.; Goldstein, J.L. Secreted site-1 protease cleaves peptides corresponding to luminal loop of sterol regulatory element-binding proteins. J. Biol. Chem. 1999, 274, 22805–22812. [Google Scholar] [CrossRef] [Green Version]
- Rawson, R.B.; Zelenski, N.G.; Nijhawan, D.; Ye, J.; Sakai, J.; Hasan, M.T.; Chang, T.Y.; Brown, M.S.; Goldstein, J.L. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell 1997, 1, 47–57. [Google Scholar] [CrossRef]
- Duncan, E.A.; Dave, U.P.; Sakai, J.; Goldstein, J.L.; Brown, M.S. Second-site cleavage in sterol regulatory element-binding protein occurs at transmembrane junction as determined by cysteine panning. J. Biol. Chem. 1998, 273, 17801–17809. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Sakai, J.; Brown, M.S.; Goldstein, J.L. Regulated cleavage of sterol regulatory element binding proteins requires sequences on both sides of the endoplasmic reticulum membrane. J. Biol. Chem. 1996, 271, 10379–10384. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Yabe, D.; Brown, M.S.; Goldstein, J.L. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 12753–12758. [Google Scholar] [CrossRef] [Green Version]
- Espenshade, P.J.; Li, W.P.; Yabe, D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc. Natl. Acad. Sci. USA 2002, 99, 11694–11699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, C.M.; Goldstein, J.L.; Brown, M.S. Cholesterol-induced conformational change in SCAP enhanced by Insig proteins and mimicked by cationic amphiphiles. Proc. Natl. Acad. Sci. USA 2003, 100, 10647–10652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D. SCAP links glucose to lipid metabolism in cancer cells. Mol. Cell. Oncol. 2016, 3, e1132120. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Espenshade, P.J. Sugar Makes Fat by Talking to SCAP. Cancer Cell 2015, 28, 548–549. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Guo, J.Y.; Geng, F.; Wu, X.; Cheng, X.; Li, Q.; Guo, D. Analysis of SCAP N-glycosylation and Trafficking in Human Cells. J. Vis. Exp. 2016, 117, e54709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Hu, B. 21-Bio-based chemicals from biorefining: Lipid and wax conversion and utilization. In Advances in Biorefineries; Waldron, K., Ed.; Woodhead Publishing: Thorston, UK, 2014; pp. 693–720. [Google Scholar]
- Currie, E.; Schulze, A.; Zechner, R.; Walther Tobias, C.; Farese Robert, V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat. Rev. Cancer 2021, 21, 753–766. [Google Scholar] [CrossRef]
- Pérez-Chacón, G.; Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Control of free arachidonic acid levels by phospholipases A2 and lysophospholipid acyltransferases. Biochim. Bi-Ophysica Acta (BBA)-Mol. Cell Biol. Lipids 2009, 1791, 1103–1113. [Google Scholar] [CrossRef] [Green Version]
- Cifarelli, V.; Abumrad, N.A. Chapter 48—Enterocyte Fatty Acid Handling Proteins and Chylomicron Formation. In Physiology of the Gastrointestinal Tract, 6th ed.; Said, H.M., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 1087–1107. [Google Scholar]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef]
- Saito, R.F.; Rangel, M.C.; Halman, J.R.; Chandler, M.; de Sousa Andrade, L.N.; Odete-Bustos, S.; Furuya, T.K.; Carrasco, A.G.M.; Chaves-Filho, A.B.; Yoshinaga, M.Y.; et al. Simultaneous silencing of lyso-phosphatidylcholine acyltransferases 1–4 by nucleic acid nanoparticles (NANPs) improves radiation response of melanoma cells. Nanomed. Nanotechnol. Biol. Med. 2021, 36, 102418. [Google Scholar] [CrossRef]
- Shi, C.; Qiao, S.; Wang, S.; Wu, T.; Ji, G.J.I.J.C.E.M. Recent progress of lysophosphatidylcholine acyltransferases in metabolic disease and cancer. Int. J. Clin. Exp. Med. 2018, 11, 8941–8953. [Google Scholar]
- Bi, J.; Ichu, T.-A.; Zanca, C.; Yang, H.; Zhang, W.; Gu, Y.; Chowdhry, S.; Reed, A.; Ikegami, S.; Turner, K.M.; et al. Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers De-pendent on Membrane Lipid Remodeling. Cell Metab. 2019, 30, 525–538.e8. [Google Scholar] [CrossRef] [PubMed]
- He, R.-Q.; Li, J.-D.; Du, X.-F.; Dang, Y.-W.; Yang, L.-J.; Huang, Z.-G.; Liu, L.-M.; Liao, L.-F.; Yang, H.; Chen, G. LPCAT1 overexpression promotes the progression of hepatocellular carcinoma. Cancer Cell Int. 2021, 21, 442. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wang, Q.; Zhang, X.; Wang, X.; Qin, C.; Sheng, Z.; Yin, H.; Jiang, C.; Li, J.; Xu, T. Lysophosphatidylcholine acyltransferase 1 upregulation and concomitant phospholipid alterations in clear cell renal cell carcinoma. J. Exp. Clin. Cancer Res. CR 2017, 36, 66. [Google Scholar] [CrossRef] [Green Version]
- Tao, M.; Luo, J.; Gu, T.; Yu, X.; Song, Z.; Jun, Y.; Gu, H.; Han, K.; Huang, X.; Yu, W.; et al. LPCAT1 reprogramming cholesterol metabolism promotes the progression of esophageal squamous cell car-cinoma. Cell Death Dis. 2021, 12, 845. [Google Scholar] [CrossRef]
- Uehara, T.; Kikuchi, H.; Miyazaki, S.; Iino, I.; Setoguchi, T.; Hiramatsu, Y.; Ohta, M.; Kamiya, K.; Morita, Y.; Tanaka, H.; et al. Overexpression of Lysophosphatidylcholine Acyltransferase 1 and Con-comitant Lipid Alterations in Gastric Cancer. Ann. Surg. Oncol. 2016, 23 (Suppl. 2), S206–S213. [Google Scholar] [CrossRef] [Green Version]
- Lebok, P.; von Hassel, A.; Meiners, J.; Hube-Magg, C.; Simon, R.; Höflmayer, D.; Hinsch, A.; Dum, D.; Fraune, C.; Göbel, C.; et al. Up-regulation of lysophosphatidylcholine acyltransferase 1 (LPCAT1) is linked to poor prognosis in breast cancer. Aging 2019, 11, 7796–7804. [Google Scholar] [CrossRef]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 2020, 5, 108. [Google Scholar] [CrossRef]
- Mbah, N.E.; Lyssiotis, C.A. Metabolic regulation of ferroptosis in the tumor microenvironment. J. Biol. Chem. 2022, 298, 101617. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeagle, P. L: Modulation of membrane function by cholesterol. Biochimie 1991, 73, 1303–1310. [Google Scholar] [CrossRef]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Holthuis, J.C.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Iaea, D.B.; Maxfield, F.R. Cholesterol trafficking and distribution. Essays Biochem. 2015, 57, 43–55. [Google Scholar]
- Holmes, M.V.; Ala-Korpela, M. What is ‘LDL cholesterol’? Nat. Rev. Cardiol. 2019, 16, 197–198. [Google Scholar] [CrossRef]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Segrest, J.P.; Jones, M.K.; De Loof, H.; Dashti, N. Structure of apolipoprotein B-100 in low density lipoproteins. J. Lipid Res. 2001, 42, 1346–1367. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.L.; Anderson, R.G.; Brown, M.S. Receptor-mediated endocytosis and the cellular uptake of low density lipoprotein. Ciba Found. Symp. 1982, 92, 77–95. [Google Scholar]
- Goldstein, J.L.; Basu, S.K.; Brown, M.S. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983, 98, 241–260. [Google Scholar] [PubMed]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [Green Version]
- Lipke, K.; Kubis-Kubiak, A.; Piwowar, A. Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge. Cells 2022, 11, 844. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Cohen, S. Lipid Droplets as Organelles. Int. Rev. Cell Mol. Biol. 2018, 337, 83–110. [Google Scholar]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.A.; Olzmann, J.A. Protein Quality Control and Lipid Droplet Metabolism. Annu. Rev. Cell Dev. Biol. 2020, 36, 115–139. [Google Scholar] [CrossRef]
- Lundquist, P.K.; Shivaiah, K.K.; Espinoza-Corral, R. Lipid droplets throughout the evolutionary tree. Prog. Lipid Res. 2020, 78, 101029. [Google Scholar] [CrossRef]
- Coleman, R.A. The “discovery” of lipid droplets: A brief history of organelles hidden in plain sight. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158762. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Qiao, J.; Guo, H. The dynamic behavior of lipid droplets in the pre-metastatic niche. Cell Death Dis. 2020, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Thiam, A.R.; Forêt, L. The physics of lipid droplet nucleation, growth and budding. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861 Pt A, 715–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben M’barek, K.; Ajjaji, D.; Chorlay, A.; Vanni, S.; Forêt, L.; Thiam, A.R. ER Membrane Phospholipids and Surface Tension Control Cellular Lipid Droplet Formation. Dev. Cell 2017, 41, 591–604.e7. [Google Scholar] [CrossRef] [Green Version]
- Chorlay, A.; Thiam, A.R. An Asymmetry in Monolayer Tension Regulates Lipid Droplet Budding Direction. Biophys. J. 2018, 114, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, V.; Ojha, N.; Golden, A.; Prinz, W.A. A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J. Cell Biol. 2015, 211, 261–271. [Google Scholar] [CrossRef]
- Gross, D.A.; Zhan, C.; Silver, D.L. Direct binding of triglyceride to fat storage-inducing transmembrane proteins 1 and 2 is important for lipid droplet formation. Proc. Natl. Acad. Sci. USA 2011, 108, 19581–19586. [Google Scholar] [CrossRef] [Green Version]
- Pagac, M.; Cooper, D.E.; Qi, Y.; Lukmantara, I.E.; Mak, H.Y.; Wu, Z.; Tian, Y.; Liu, Z.; Lei, M.; Du, X.; et al. SEIPIN Regulates Lipid Droplet Expansion and Adipocyte Development by Modulating the Activity of Glycerol-3-phosphate Acyltransferase. Cell Rep. 2016, 17, 1546–1559. [Google Scholar] [CrossRef] [Green Version]
- Fei, W.; Shui, G.; Gaeta, B.; Du, X.; Kuerschner, L.; Li, P.; Brown, A.J.; Wenk, M.R.; Parton, R.G.; Yang, H. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J. Cell Biol. 2008, 180, 473–482. [Google Scholar] [CrossRef]
- Szymanski, K.M.; Binns, D.; Bartz, R.; Grishin, N.V.; Li, W.P.; Agarwal, A.K.; Garg, A.; Anderson, R.G.; Goodman, J.M. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl. Acad. Sci. USA 2007, 104, 20890–20895. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Becuwe, M.; Housden, B.E.; Chitraju, C.; Porras, A.J.; Graham, M.M.; Liu, X.N.; Thiam, A.R.; Savage, D.B.; Agarwal, A.K.; et al. Seipin is required for converting nascent to mature lipid droplets. eLife 2016, 5, e16582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Li, Y.; Wu, L.; Li, Y.; Zhao, D.; Yu, J.; Huang, T.; Ferguson, C.; Parton, R.G.; Yang, H.; et al. Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions. J. Cell Biol. 2018, 217, 975–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Xu, D.; Zhou, L.; Xie, B.; Yu, L.; Yang, H.; Huang, L.; Ye, J.; Deng, H.; Yuan, Y.A.; et al. Rab8a-AS160-MSS4 regulatory circuit controls lipid droplet fusion and growth. Dev. Cell 2014, 30, 378–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Sun, Z.; Wu, L.; Xu, W.; Schieber, N.; Xu, D.; Shui, G.; Yang, H.; Parton, R.G.; Li, P. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J. Cell Biol. 2011, 195, 953–963. [Google Scholar] [CrossRef]
- Wilfling, F.; Wang, H.; Haas, J.T.; Krahmer, N.; Gould, T.J.; Uchida, A.; Cheng, J.X.; Graham, M.; Christiano, R.; Fröhlich, F.; et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 2013, 24, 384–399. [Google Scholar] [CrossRef] [Green Version]
- Bermúdez, M.A.; Balboa, M.A.; Balsinde, J. Lipid Droplets, Phospholipase A2, Arachidonic Acid, and Atherosclerosis. Biomedicines 2021, 9, 1891. [Google Scholar] [CrossRef]
- Rinia, H.A.; Burger, K.N.J.; Bonn, M.; Müller, M. Quantitative label-free imaging of lipid composition and packing of individual cellular lipid droplets using multiplex CARS microscopy. Biophys. J. 2008, 95, 4908–4914. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xu, L.; Zhu, L.; Liu, Y.; Yang, S.; Zhao, M. Lipid Droplets, the Central Hub Integrating Cell Metabolism and the Immune System. Front. Physiol. 2021, 12, 746749. [Google Scholar] [CrossRef]
- Suzuki, M.; Shinohara, Y.; Ohsaki, Y.; Fujimoto, T. Lipid droplets: Size matters. J. Electron. Microsc. 2011, 60 (Suppl. 1), S101–S116. [Google Scholar] [CrossRef]
- Yang, H.; Galea, A.; Sytnyk, V.; Crossley, M. Controlling the size of lipid droplets: Lipid and protein factors. Curr. Opin. Cell Biol. 2012, 24, 509–516. [Google Scholar] [CrossRef]
- Scorletti, E.; Carr, R.M. A new perspective on NAFLD: Focusing on lipid droplets. J. Hepatol. 2022, 76, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, K.; Lee, Y.K.; Londos, C.; Raaka, B.M.; Dalen, K.T.; Kimmel, A.R. Perilipin family members preferentially sequester to either triacylglycerol-specific or cholesteryl-ester-specific intracellular lipid storage droplets. J. Cell Sci. 2012, 125 Pt 17, 4067–4076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Ohsaki, Y.; Tatematsu, T.; Shinohara, Y.; Maeda, T.; Cheng, J.; Fujimoto, T. Translation inhibitors induce formation of cholesterol ester-rich lipid droplets. PLoS ONE 2012, 7, e42379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herker, E.; Vieyres, G.; Beller, M.; Krahmer, N.; Bohnert, M. Lipid Droplet Contact Sites in Health and Disease. Trends Cell Biol. 2021, 31, 345–358. [Google Scholar] [CrossRef]
- Prinz, W.A. Bridging the gap: Membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 2014, 205, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef]
- Valm, A.M.; Cohen, S.; Legant, W.R.; Melunis, J.; Hershberg, U.; Wait, E.; Cohen, A.R.; Davidson, M.W.; Betzig, E.; Lippincott-Schwartz, J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 2017, 546, 162–167. [Google Scholar] [CrossRef]
- Shai, N.; Yifrach, E.; van Roermund, C.W.T.; Cohen, N.; Bibi, C.; Ijlst, L.; Cavellini, L.; Meurisse, J.; Schuster, R.; Zada, L.; et al. Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 2018, 9, 1761. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Liang, Y.; Li, J.; Liu, Y.; Zhang, J.; Zhang, A.; Fu, J.; Jiang, G. Specific accumulation of lipid droplets in hepatocyte nuclei of PFOA-exposed BALB/c mice. Sci. Rep. 2013, 3, 2174. [Google Scholar] [CrossRef] [Green Version]
- Uzbekov, R.; Roingeard, P. Nuclear lipid droplets identified by electron microscopy of serial sections. BMC Res. Notes 2013, 6, 386. [Google Scholar] [CrossRef] [Green Version]
- Layerenza, J.P.; Gonzalez, P.; Garcia de Bravo, M.M.; Polo, M.P.; Sisti, M.S.; Ves-Losada, A. Nuclear lipid droplets: A novel nuclear domain. Biochim. Biophys. Acta 2013, 1831, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.D.; Siniossoglou, S. New kid on the block: Lipid droplets in the nucleus. FEBS J. 2020, 287, 4838–4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanauska, A.; Kohler, A. The Inner Nuclear Membrane Is a Metabolically Active Territory that Generates Nuclear Lipid Droplets. Cell 2018, 174, 700–715.e8. [Google Scholar] [CrossRef] [Green Version]
- Ohsaki, Y.; Kawai, T.; Yoshikawa, Y.; Cheng, J.; Jokitalo, E.; Fujimoto, T. PML isoform II plays a critical role in nuclear lipid droplet formation. J. Cell Biol. 2016, 212, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltysik, K.; Ohsaki, Y.; Tatematsu, T.; Cheng, J.; Fujimoto, T. Nuclear lipid droplets derive from a lipoprotein precursor and regulate phosphatidylcholine synthesis. Nat. Commun. 2019, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Y.; Cui, L.; Deng, Y.; Xu, S.; Yu, J.; Cichello, S.; Serrero, G.; Ying, Y.; Liu, P. Morphologically and Functionally Distinct Lipid Droplet Subpopulations. Sci. Rep. 2016, 6, 29539. [Google Scholar] [CrossRef] [Green Version]
- Bersuker, K.; Peterson, C.W.H.; To, M.; Sahl, S.J.; Savikhin, V.; Grossman, E.A.; Nomura, D.K.; Olzmann, J.A. A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Dev. Cell 2018, 44, 97–112.e7. [Google Scholar] [CrossRef]
- Aboumrad, M.H.; Horn, R.C., Jr.; Fine, G. Lipid-secreting mammary carcinoma. Report of a case associated with Paget’s disease of the nipple. Cancer 1963, 16, 521–525. [Google Scholar] [CrossRef]
- Wright, D.H. Lipid content of malignant lymphomas. J. Clin. Pathol. 1968, 21, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Fitchev, P.S.; Cornwell, M.L.; Greenberg, J.; Cabe, M.; Weber, C.R.; Roy, H.K.; Crawford, S.E.; Savkovic, S.D. FOXO3 growth inhibition of colonic cells is dependent on intraepithelial lipid droplet density. J. Biol. Chem. 2013, 288, 16274–16281. [Google Scholar] [CrossRef] [Green Version]
- Fazolini, N.P.; Cruz, A.L.; Werneck, M.B.; Viola, J.P.; Maya-Monteiro, C.M.; Bozza, P.T. Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation, cytokine production and increased cell proliferation. Cell Cycle 2015, 14, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Penrose, H.; Heller, S.; Cable, C.; Makboul, R.; Chadalawada, G.; Chen, Y.; Crawford, S.E.; Savkovic, S.D. Epidermal growth factor receptor mediated proliferation depends on increased lipid droplet density regulated via a negative regulatory loop with FOXO3/Sirtuin6. Biochem. Biophys. Res. Commun. 2016, 469, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Corbet, C.; Bastien, E.; Santiago de Jesus, J.P.; Dierge, E.; Martherus, R.; Vander Linden, C.; Doix, B.; Degavre, C.; Guilbaud, C.; Petit, L.; et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat. Commun. 2020, 11, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudetti, A.M.; De Domenico, S.; Ragusa, A.; Lunetti, P.; Gaballo, A.; Franck, J.; Simeone, P.; Nicolardi, G.; De Nuccio, F.; Santino, A.; et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Marafioti, M.G.; Pagliari, F.; Jansen, J.; Aversa, I.; Hanley, R.; Nistico, C.; Garcia-Calderon, D.; Genard, G.; Guerreiro, J.F.; et al. Lipid droplets and ferritin heavy chain: A devilish liaison in human cancer cell radioresistance. eLife 2021, 10, e72943. [Google Scholar] [CrossRef]
- Nistico, C.; Pagliari, F.; Chiarella, E.; Fernandes Guerreiro, J.; Marafioti, M.G.; Aversa, I.; Genard, G.; Hanley, R.; Garcia-Calderon, D.; Bond, H.M.; et al. Lipid Droplet Biosynthesis Impairment through DGAT2 Inhibition Sensitizes MCF7 Breast Cancer Cells to Radiation. Int. J. Mol. Sci. 2021, 22, 10102. [Google Scholar] [CrossRef]
- Lettiero, B.; Inasu, M.; Kimbung, S.; Borgquist, S. Insensitivity to atorvastatin is associated with increased accumulation of intracellular lipid droplets and fatty acid metabolism in breast cancer cells. Sci. Rep. 2018, 8, 5462. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Hitz, C.A.; Gijon, M.A.; Bergman, B.C.; Eckel, R.H.; Jacobsen, B.M. Progestin modulates the lipid profile and sensitivity of breast cancer cells to docetaxel. Mol. Cell Endocrinol. 2012, 363, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Rak, S.; De Zan, T.; Stefulj, J.; Kosovic, M.; Gamulin, O.; Osmak, M. FTIR spectroscopy reveals lipid droplets in drug resistant laryngeal carcinoma cells through detection of increased ester vibra-tional bands intensity. Analyst 2014, 139, 3407–3415. [Google Scholar] [CrossRef]
- Verbrugge, S.E.; Al, M.; Assaraf, Y.G.; Kammerer, S.; Chandrupatla, D.M.; Honeywell, R.; Musters, R.P.; Giovannetti, E.; O’Toole, T.; Scheffer, G.L.; et al. Multifactorial resistance to aminopeptidase inhibitor prodrug CHR2863 in myeloid leukemia cells: Down-regulation of carboxylesterase 1, drug sequestration in lipid droplets and pro-survival activation ERK/Akt/mTOR. Oncotarget 2016, 7, 5240–5257. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.R.; Zeng, X.; Zhao, J.; Liu, Y.; Hou, G.; Liu, H.; Hou, S.X. The lipolysis pathway sustains normal and transformed stem cells in adult Drosophila. Nature 2016, 538, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Hershey, B.J.; Vazzana, R.; Joppi, D.L.; Havas, K.M. Lipid Droplets Define a Sub-Population of Breast Cancer Stem Cells. J. Clin. Med. 2019, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Han, Y.; Rodriguez Sillke, Y.; Deng, H.; Siddiqui, S.; Treese, C.; Schmidt, F.; Friedrich, M.; Keye, J.; Wan, J.; et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol. Med. 2019, 11, e10698. [Google Scholar] [CrossRef]
- Siddiqui, S.; Glauben, R. Fatty Acid Metabolism in Myeloid-Derived Suppressor Cells and Tumor-Associated Macrophages: Key Factor in Cancer Immune Evasion. Cancers 2022, 14, 250. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef] [PubMed]
- Schade, D.S.; Shey, L.; Eaton, R.P. Cholesterol Review: A Metabolically Important Molecule. Endocr. Pract. 2020, 26, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, A.; Goldstein, J.L.; McDonald, J.G.; Brown, M.S. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 2008, 8, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Zelcer, N.; Tontonoz, P. Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Investig. 2006, 116, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Calkin, A.C.; Tontonoz, P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1513–1518. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.Y.; Chang, C.C.; Ohgami, N.; Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Hai, Q.; Smith, J.D. Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era. Metabolites 2021, 11, 543. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K. A superfamily of membrane-bound O-acyltransferases with implications for wnt signaling. Trends Biochem. Sci. 2000, 25, 111–112. [Google Scholar] [CrossRef]
- Oelkers, P.; Behari, A.; Cromley, D.; Billheimer, J.T.; Sturley, S.L. Characterization of two human genes encoding acyl coenzyme A:cholesterol acyltransferase-related enzymes. J. Biol. Chem. 1998, 273, 26765–26771. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Sakashita, N.; Ornvold, K.; Lee, O.; Chang, E.T.; Dong, R.; Lin, S.; Lee, C.Y.; Strom, S.C.; Kashyap, R.; et al. Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. J. Biol. Chem. 2000, 275, 28083–28092. [Google Scholar] [CrossRef]
- Chang, T.Y.; Li, B.L.; Chang, C.C.; Urano, Y. Acyl-coenzyme A: Cholesterol acyltransferases. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1–E9. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, Y.; Chang, C.C.; Chang, T.Y. ACAT1/SOAT1 as a therapeutic target for Alzheimer’s disease. Future Med. Chem. 2015, 7, 2451–2467. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Y.; Hao, S.; Qin, Y.; Wu, Y. Knockdown of sterol O-acyltransferase 1 (SOAT1) suppresses SCD1-mediated lipogenesis and cancer procession in prostate cancer. Prostaglandins Other Lipid Mediat. 2021, 153, 106537. [Google Scholar] [CrossRef]
- Eckhardt, C.; Sbiera, I.; Krebs, M.; Sbiera, S.; Spahn, M.; Kneitz, B.; Joniau, S.; Fassnacht, M.; Kubler, H.; Weigand, I.; et al. High expression of Sterol-O-Acyl transferase 1 (SOAT1), an enzyme involved in cholesterol metabolism, is associated with earlier biochemical recurrence in high risk prostate cancer. Prostate Cancer Prostatic Dis. 2021. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B.; et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Xu, H.; Xia, H.; Tang, Q.; Bi, F. Simultaneously targeting SOAT1 and CPT1A ameliorates hepatocellular carcinoma by disrupting lipid homeostasis. Cell Death Discov. 2021, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, X.; Chen, Y.; Chen, G.; Winkler, C.A.; An, P.; Lyu, J. Impacts of the SOAT1 genetic variants and protein expression on HBV-related hepatocellular carcinoma. BMC Cancer 2021, 21, 615. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gu, L.; Lin, X.; Zhou, X.; Lu, B.; Liu, C.; Li, Y.; Prochownik, E.V.; Karin, M.; Wang, F.; et al. P53 deficiency affects cholesterol esterification to exacerbate hepatocarcinogenesis. Hepatology 2022. [Google Scholar] [CrossRef] [PubMed]
- Oni, T.E.; Biffi, G.; Baker, L.A.; Hao, Y.; Tonelli, C.; Somerville, T.D.D.; Deschenes, A.; Belleau, P.; Hwang, C.I.; Sanchez-Rivera, F.J.; et al. SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J. Exp. Med. 2020, 217, e20192389. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Luo, L.M.; Huang, T.S.; Feng, L.F. SOAT1 is a new prognostic factor of colorectal cancer. Ir. J. Med. Sci. 2021, 191, 1549–1554. [Google Scholar] [CrossRef]
- Zhu, T.; Wang, Z.; Zou, T.; Xu, L.; Zhang, S.; Chen, Y.; Chen, C.; Zhang, W.; Wang, S.; Ding, Q.; et al. SOAT1 Promotes Gastric Cancer Lymph Node Metastasis Through Lipid Synthesis. Front. Pharm. 2021, 12, 769647. [Google Scholar] [CrossRef]
- Mo, Y.; Lin, L.; Zhang, J.; Yu, C. SOAT1 enhances lung cancer invasiveness by stimulating AKT-mediated mitochondrial fragmentation. Biochem. Cell Biol. 2022, 100, 68–74. [Google Scholar] [CrossRef]
- Smith, D.C.; Kroiss, M.; Kebebew, E.; Habra, M.A.; Chugh, R.; Schneider, B.J.; Fassnacht, M.; Jafarinasabian, P.; Ijzerman, M.M.; Lin, V.H.; et al. A phase 1 study of nevanimibe HCl, a novel adrenal-specific sterol O-acyltransferase 1 (SOAT1) inhibitor, in adrenocortical carcinoma. Investig. New Drugs 2020, 38, 1421–1429. [Google Scholar] [CrossRef]
- Cheng, X.; Li, J.; Guo, D. SCAP/SREBPs are Central Players in Lipid Metabolism and Novel Metabolic Targets in Cancer Therapy. Curr. Top. Med. Chem. 2018, 18, 484–493. [Google Scholar] [CrossRef]
- Lohr, M.; Hartig, W.; Schulze, A.; Kroiss, M.; Sbiera, S.; Lapa, C.; Mages, B.; Strobel, S.; Hundt, J.E.; Bohnert, S.; et al. SOAT1: A Suitable Target for Therapy in High-Grade Astrocytic Glioma? Int. J. Mol. Sci. 2022, 23, 3726. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Shaw, J.L.; Haigis, M.C.; Greka, A. Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity. Mol. Cell 2021, 81, 3708–3730. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Hardy, S.; El-Assaad, W.; Przybytkowski, E.; Joly, E.; Prentki, M.; Langelier, Y. Saturated fatty acid-induced apoptosis in MDA-MB-231 breast cancer cells. A role for cardiolipin. J. Biol. Chem. 2003, 278, 31861–31870. [Google Scholar] [CrossRef] [Green Version]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef]
- Bhatt-Wessel, B.; Jordan, T.W.; Miller, J.H.; Peng, L. Role of DGAT enzymes in triacylglycerol metabolism. Arch. Biochem. Biophys. 2018, 655, 1–11. [Google Scholar] [CrossRef]
- Hernandez-Corbacho, M.J.; Obeid, L.M. A novel role for DGATs in cancer. Adv. Biol. Regul. 2019, 72, 89–101. [Google Scholar] [CrossRef]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [Green Version]
- Cases, S.; Stone, S.J.; Zhou, P.; Yen, E.; Tow, B.; Lardizabal, K.D.; Voelker, T.; Farese, R.V., Jr. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 2001, 276, 38870–38876. [Google Scholar] [CrossRef] [Green Version]
- van Rijn, J.M.; Ardy, R.C.; Kuloglu, Z.; Harter, B.; van Haaften-Visser, D.Y.; van der Doef, H.P.J.; van Hoesel, M.; Kansu, A.; van Vugt, A.H.M.; Thian, M.; et al. Intestinal Failure and Aberrant Lipid Metabolism in Patients With DGAT1 Deficiency. Gastroenterology 2018, 155, 130–143.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephen, J.; Vilboux, T.; Haberman, Y.; Pri-Chen, H.; Pode-Shakked, B.; Mazaheri, S.; Marek-Yagel, D.; Barel, O.; Di Segni, A.; Eyal, E.; et al. Congenital protein losing enteropathy: An inborn error of lipid metabolism due to DGAT1 mutations. Eur. J. Hum. Genet. 2016, 24, 1268–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, S.J.; Myers, H.M.; Watkins, S.M.; Brown, B.E.; Feingold, K.R.; Elias, P.M.; Farese, R.V., Jr. Lipopenia and skin barrier abnormalities in DGAT2-deficient mice. J. Biol. Chem. 2004, 279, 11767–11776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.J.; Cases, S.; Jensen, D.R.; Chen, H.C.; Sande, E.; Tow, B.; Sanan, D.A.; Raber, J.; Eckel, R.H.; Farese, R.V., Jr. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat. Nat. Genet 2000, 25, 87–90. [Google Scholar] [CrossRef]
- Xia, L.; Wang, Y.; Cai, S.; Xu, M. DGAT1 Expression Promotes Ovarian Cancer Progression and Is Associated with Poor Prognosis. J. Immunol. Res. 2021, 2021, 6636791. [Google Scholar] [CrossRef]
- Nardi, F.; Franco, O.E.; Fitchev, P.; Morales, A.; Vickman, R.E.; Hayward, S.W.; Crawford, S.E. DGAT1 Inhibitor Suppresses Prostate Tumor Growth and Migration by Regulating Intracellular Lipids and Non-Centrosomal MTOC Protein GM130. Sci. Rep. 2019, 9, 3035. [Google Scholar] [CrossRef]
- de la Rosa Rodriguez, M.A.; Deng, L.; Gemmink, A.; van Weeghel, M.; Aoun, M.L.; Warnecke, C.; Singh, R.; Borst, J.W.; Kersten, S. Hypoxia-inducible lipid droplet-associated induces DGAT1 and promotes lipid storage in hepatocytes. Mol. Metab. 2021, 47, 101168. [Google Scholar] [CrossRef]
- Morales, A.; Greenberg, M.; Nardi, F.; Gil, V.; Hayward, S.W.; Crawford, S.E.; Franco, O.E. Loss of ephrin B2 receptor (EPHB2) sets lipid rheostat by regulating proteins DGAT1 and ATGL inducing lipid droplet storage in prostate cancer cells. Lab. Investig. 2021, 101, 921–934. [Google Scholar] [CrossRef]
- Mitra, R.; Le, T.T.; Gorjala, P.; Goodman, O.B., Jr. Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer 2017, 17, 631. [Google Scholar] [CrossRef]
- He, P.; Cheng, S.; Hu, F.; Ma, Z.; Xia, Y. Up-regulation of DGAT1 in cancer tissues and tumor-infiltrating macrophages influenced survival of patients with gastric cancer. BMC Cancer 2021, 21, 252. [Google Scholar] [CrossRef]
- de la Rosa Rodriguez, M.A.; Kersten, S. Regulation of lipid droplet homeostasis by hypoxia inducible lipid droplet associated HILPDA. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158738. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Johnson, S.M.; Liu, J. Hypoxia, hypoxia-inducible gene 2 (HIG2)/HILPDA, and intracellular lipolysis in cancer. Cancer Lett. 2020, 493, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.G.; Wang, C.; Liu, D.Y.; Zhang, X.; Wang, L.; Yan, M.; Zhang, W.; Zhu, J.; Li, Z.C.; Mi, C.; et al. Hypoxia upregulates HIG2 expression and contributes to bevacizumab resistance in glioblastoma. Oncotarget 2016, 7, 47808–47820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Li, Y.; Grisé, A.; Wang, H. CGI-58: Versatile Regulator of Intracellular Lipid Droplet Homeostasis. Adv. Exp. Med. Biol. 2020, 1276, 197–222. [Google Scholar] [PubMed]
- Shi, Z.; Luo, X.; Zhao, H.; Huang, B.; Wang, Y.; Chen, X.; Yu, J. Clinicalpathologic and Prognostic Significance of CGI-58 in Endometrial Cancer. J. Cancer 2021, 12, 7374–7379. [Google Scholar] [CrossRef]
- Chen, G.; Zhou, G.; Lotvola, A.; Granneman, J.G.; Wang, J. ABHD5 suppresses cancer cell anabolism through lipolysis-dependent activation of the AMPK/mTORC1 pathway. J. Biol. Chem. 2021, 296, 100104. [Google Scholar] [CrossRef]
- Long, T.; Sun, Y.; Hassan, A.; Qi, X.; Li, X. Structure of nevanimibe-bound tetrameric human ACAT1. Nature 2020, 581, 339–343. [Google Scholar] [CrossRef]
- Qian, H.; Zhao, X.; Yan, R.; Yao, X.; Gao, S.; Sun, X.; Du, X.; Yang, H.; Wong, C.C.L.; Yan, N. Structural basis for catalysis and substrate specificity of human ACAT1. Nature 2020, 581, 333–338. [Google Scholar] [CrossRef]
- Guan, C.; Niu, Y.; Chen, S.C.; Kang, Y.; Wu, J.X.; Nishi, K.; Chang, C.C.Y.; Chang, T.Y.; Luo, T.; Chen, L. Structural insights into the inhibition mechanism of human sterol O-acyltransferase 1 by a competitive inhibitor. Nat. Commun. 2020, 11, 2478. [Google Scholar] [CrossRef]
- Sui, X.; Wang, K.; Gluchowski, N.L.; Elliott, S.D.; Liao, M.; Walther, T.C.; Farese, R.V., Jr. Structure and catalytic mechanism of a human triacylglycerol-synthesis enzyme. Nature 2020, 581, 323–328. [Google Scholar] [CrossRef]
- Wang, L.; Qian, H.; Nian, Y.; Han, Y.; Ren, Z.; Zhang, H.; Hu, L.; Prasad, B.V.V.; Laganowsky, A.; Yan, N.; et al. Structure and mechanism of human diacylglycerol O-acyltransferase 1. Nature 2020, 581, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-A.; Xiong, X.; Zaytseva, Y.Y.; Napier, D.L.; Vallee, E.; Li, A.T.; Wang, C.; Weiss, H.L.; Evers, B.M.; Gao, T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018, 9, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, Z.; Xie, Z.; Xu, W.; Zhao, X.; Jin, G.; Sun, X.; Huang, B.; Tang, P.; Wang, G.; Shen, S.; et al. SREBP-2 aggravates breast cancer associated osteolysis by promoting osteoclastogenesis and breast cancer metastasis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Chen, S.; Li, W. Fatostatin inhibits the development of endometrial carcinoma in endometrial carcinoma cells and a xenograft model by targeting lipid metabolism. Arch. Bio-Chem. Biophys. 2020, 684, 108327. [Google Scholar] [CrossRef] [PubMed]
- Brovkovych, V.; Izhar, Y.; Danes, J.M.; Dubrovskyi, O.; Sakallioglu, I.T.; Morrow, L.M.; Atilla-Gokcumen, G.E.; Frasor, J. Fatostatin induces pro- and anti-apoptotic lipid accumulation in breast cancer. Oncogenesis 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, Y.T.; Hu, P.; Huang, W.C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 2014, 13, 855–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, F.; Feng, F.; Wang, L.; Wang, X.; Li, Z.; Cao, Y. SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis. 2019, 10, 672. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yan, H.; Zhao, L.; Jia, W.; Yang, H.; Liu, L.; Zhou, X.; Miao, P.; Sun, X.; Song, S.; et al. Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells. Oncotarget 2016, 7, 52392–52403. [Google Scholar] [CrossRef] [Green Version]
- Caruana, B.T.; Skoric, A.; Brown, A.J.; Lutze-Mann, L.H. Site-1 protease, a novel metabolic target for glioblastoma. Biochem. Biophys. Res. Commun. 2017, 490, 760–766. [Google Scholar] [CrossRef]
- Wang, T.-B.; Geng, M.; Jin, H.; Tang, A.-G.; Sun, H.; Zhou, L.-Z.; Chen, B.-H.; Shen, G.; Sun, Q. SREBP1 site 1 protease inhibitor PF-429242 suppresses renal cell carcinoma cell growth. Cell Death Dis. 2021, 12, 717. [Google Scholar] [CrossRef]
- Siqingaowa Sekar, S.; Gopalakrishnan, V.; Taghibiglou, C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem. Biophys. Res. Commun. 2017, 488, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Esquejo, R.M.; Roqueta-Rivera, M.; Shao, W.; Phelan, P.E.; Seneviratne, U.; Am Ende, C.W.; Hershberger, P.M.; Machamer, C.E.; Espenshade, P.J.; Osborne, T.F. Dipyridamole Inhibits Lipogenic Gene Expression by Retaining SCAP-SREBP in the Endoplasmic Reticulum. Cell Chem. Biol. 2021, 28, 169–179.e7. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C. ATP-citrate lyase (ACLY) inhibitors as therapeutic agents: A patenting perspective. Expert Opin. Ther. Pat. 2022, 32, 731–742. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e175. [Google Scholar] [CrossRef] [Green Version]
- Tan, K. Investigating the Role of Acetyl-CoA Carboxylases in Breast Cancer Progression. Doctoral Dissertation, The University of Sydney, Camperdown, Australia, 2021. [Google Scholar]
- Pizer, E.S.; Thupari, J.; Han, W.F.; Pinn, M.L.; Chrest, F.J.; Frehywot, G.L.; Townsend, C.A.; Kuhajda, F.P. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000, 60, 213–218. [Google Scholar]
- Qi, J.; Lang, W.; Geisler, J.G.; Wang, P.; Petrounia, I.; Mai, S.; Smith, C.; Askari, H.; Struble, G.T.; Williams, R.; et al. The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and-2. J. Lipid Res. 2012, 53, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Balaban, S.; Nassar, Z.D.; Zhang, A.Y.; Hosseini-Beheshti, E.; Centenera, M.M.; Schreuder, M.; Lin, H.-M.; Aishah, A.; Varney, B.; Liu-Fu, F.; et al. Extracellular fatty acids are the major contributor to lipid synthesis in prostate cancer. Mol. Cancer Res. 2019, 17, 949–962. [Google Scholar] [CrossRef]
- Dow, R.L.; Li, J.C.; Pence, M.P.; Gibbs, E.M.; LaPerle, J.L.; Litchfield, J.; Piotrowski, D.W.; Munchhof, M.J.; Manion, T.B.; Zavadoski, W.J.; et al. Discovery of PF-04620110, a Potent, Selective, and Orally Bioavailable Inhibitor of DGAT-1. ACS Med. Chem. Lett. 2011, 2, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.; Infante, J.; Arkenau, H.T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gu, L.; Lin, X.; Zhang, J.; Tang, Y.; Zhou, X.; Lu, B.; Lin, X.; Liu, C.; Prochownik, E.V.; et al. Ceramide-mediated gut dysbiosis enhances cholesterol esterification and promotes colorectal tu-morigenesis in mice. JCI Insight 2022, 7, e150607. [Google Scholar] [CrossRef] [PubMed]
- Ohmoto, T.; Nishitsuji, K.; Yoshitani, N.; Mizuguchi, M.; Yanagisawa, Y.; Saito, H.; Sakashita, N. K604, a specific acyl-CoA:cholesterol acyltransferase 1 inhibitor, suppresses proliferation of U251-MG glioblastoma cells. Mol. Med. Rep. 2015, 12, 6037–6042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rae, C.; Fragkoulis, G.I.; Chalmers, A.J. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv. Radiat. Oncol. 2020, 5, 994–1005. [Google Scholar] [CrossRef]
- Grube, S.; Dünisch, P.; Freitag, D.; Klausnitzer, M.; Sakr, Y.; Walter, J.; Kalff, R.; Ewald, C. Overexpression of fatty acid synthase in human gliomas correlates with the WHO tumor grade and inhibition with Orlistat reduces cell viability and triggers apoptosis. J. Neuro-Oncol. 2014, 118, 277–287. [Google Scholar] [CrossRef]
- Hu, X.; Xiang, J.; Li, Y.; Xia, Y.; Xu, S.; Gao, X.; Qiao, S. Inhibition of Stearoyl-CoA Desaturase 1 Potentiates Anti-tumor Activity of Amodiaquine in Non-small Cell Lung Cancer. Biol. Pharm. Bull. 2022, 45, 438–445. [Google Scholar] [CrossRef]
- Skrypek, K.; Balog, S.; Eriguchi, Y.; Asahina, K. Inhibition of Stearoyl-CoA Desaturase Induces the Unfolded Protein Response in Pancreatic Tumors and Suppresses Their Growth. Pancreas 2021, 50, 219–226. [Google Scholar] [CrossRef]
- She, K.; Fang, S.; Du, W.; Fan, X.; He, J.; Pan, H.; Huang, L.; He, P.; Huang, J. SCD1 is required for EGFR-targeting cancer therapy of lung cancer via re-activation of EGFR/PI3K/AKT signals. Cancer Cell Int. 2019, 19, 103. [Google Scholar] [CrossRef]
- von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef] [Green Version]
- Pisanu, M.E.; Noto, A.; De Vitis, C.; Morrone, S.; Scognamiglio, G.; Botti, G.; Venuta, F.; Diso, D.; Jakopin, Z.; Padula, F.; et al. Blockade of Stearoyl-CoA-desaturase 1 activity reverts resistance to cisplatin in lung cancer stem cells. Cancer Lett. 2017, 406, 93–104. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Maugeri-Saccà, M.; Fattore, L.; Bruschini, S.; De Vitis, C.; Tabbì, E.; Bellei, B.; Migliano, E.; Kovacs, D.; Camera, E.; et al. Inhibition of Stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-mutated melanoma. J. Exp. Clin. Cancer Res. CR 2018, 37, 318. [Google Scholar] [CrossRef] [PubMed]
- Oballa, R.M.; Belair, L.; Black, W.C.; Bleasby, K.; Chan, C.C.; Desroches, C.; Du, X.; Gordon, R.; Guay, J.; Guiral, S.; et al. Development of a liver-targeted stearoyl-CoA desaturase (SCD) inhibitor (MK-8245) to establish a therapeutic window for the treatment of diabetes and dyslipidemia. J. Med. Chem. 2011, 54, 5082–5096. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, R.W. Novel Substituted Tetrazoles as ACSS2 Inhibitors for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 1894–1895. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, R.W. Amide-Substituted Condensed Pyridine Derivatives as ACSS2 Inhibitors for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 1870–1871. [Google Scholar] [CrossRef] [PubMed]
- Kalthoff, C.; Xiang, K.; Muench, C.; Hlouschek, J.; Jendrossek, V.; Matschke, J. Abstract PO-022: Inhibition of the citrate carrier SLC25A1 affects repair of radiation-induced DNA damage, increases radiosensitivity, and enhances lethality of IR in combination with DNA repair defects or DSB inhibitors. Clin. Cancer Res. 2021, 27 (Suppl. 8), PO-022. [Google Scholar] [CrossRef]
- Hlouschek, J.; Hansel, C.; Jendrossek, V.; Matschke, J. The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells With Tolerance to Cycling Severe Hypoxia. Front. Oncol. 2018, 8, 170. [Google Scholar] [CrossRef]
- Reis, L.M.D.; Adamoski, D.; Ornitz Oliveira Souza, R.; Rodrigues Ascenção, C.F.; Sousa de Oliveira, K.R.; Corrêa-da-Silva, F.; Malta de Sá Patroni, F.; Meira Dias, M.; Consonni, S.R.; Mendes de Moraes-Vieira, P.M.; et al. Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. J. Biol. Chem. 2019, 294, 9342–9357. [Google Scholar] [CrossRef]
- Jiang, N.; Xie, B.; Xiao, W.; Fan, M.; Xu, S.; Duan, Y.; Hamsafar, Y.; Evans, A.C.; Huang, J.; Zhou, W.; et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat. Commun. 2022, 13, 1511. [Google Scholar] [CrossRef]
- Galicia-Vázquez, G.; Aloyz, R. Ibrutinib Resistance Is Reduced by an Inhibitor of Fatty Acid Oxidation in Primary CLL Lymphocytes. Front. Oncol. 2018, 8, 411. [Google Scholar] [CrossRef]
- Flaig, T.W.; Salzmann-Sullivan, M.; Su, L.J.; Zhang, Z.; Joshi, M.; Gijón, M.A.; Kim, J.; Arcaroli, J.J.; Van Bokhoven, A.; Lucia, M.S.; et al. Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 2017, 8, 56051–56065. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakal, B.; Li, C.M.Y.; Li, R.; Yeo, K.; Wright, J.A.; Gieniec, K.A.; Vrbanac, L.; Sammour, T.; Lawrence, M.; Thomas, M.; et al. The Antianginal Drug Perhexiline Displays Cytotoxicity against Colorectal Cancer Cells In Vitro: A Potential for Drug Repurposing. Cancers 2022, 14, 1043. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Kesarwani, P.; Guastella, A.R.; Kumar, P.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Perhexiline Demonstrates FYN-mediated Antitumor Activity in Glioblastoma. Mol. Cancer Ther. 2020, 19, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Simon, J.M.; Liao, C.; Zhang, C.; Hu, L.; Zurlo, G.; Liu, X.; Fan, C.; Hepperla, A.; Jia, L.; et al. An oncogenic JMJD6-DGAT1 axis tunes the epigenetic regulation of lipid droplet formation in clear cell renal cell carcinoma. Mol. Cell, 2022; in press. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kou, Y.; Geng, F.; Guo, D. Lipid Metabolism in Glioblastoma: From De Novo Synthesis to Storage. Biomedicines 2022, 10, 1943. https://doi.org/10.3390/biomedicines10081943
Kou Y, Geng F, Guo D. Lipid Metabolism in Glioblastoma: From De Novo Synthesis to Storage. Biomedicines. 2022; 10(8):1943. https://doi.org/10.3390/biomedicines10081943
Chicago/Turabian StyleKou, Yongjun, Feng Geng, and Deliang Guo. 2022. "Lipid Metabolism in Glioblastoma: From De Novo Synthesis to Storage" Biomedicines 10, no. 8: 1943. https://doi.org/10.3390/biomedicines10081943
APA StyleKou, Y., Geng, F., & Guo, D. (2022). Lipid Metabolism in Glioblastoma: From De Novo Synthesis to Storage. Biomedicines, 10(8), 1943. https://doi.org/10.3390/biomedicines10081943