
The Intricate Epigenetic and Transcriptional Alterations in Pediatric High-Grade Gliomas: Targeting the Crosstalk as the Oncogenic Achilles’ Heel

Abstract
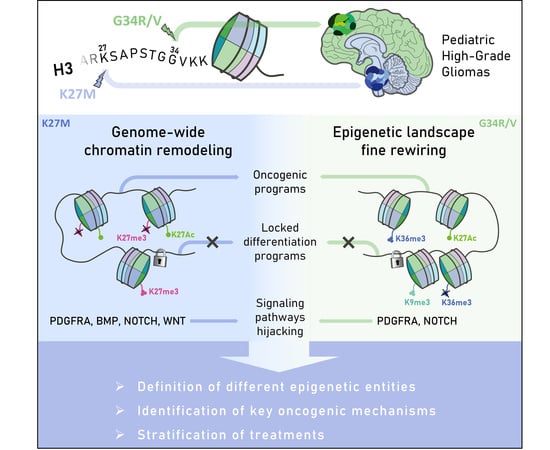
1. Introduction
2. Epigenetic Remodeling at the Root of pHGGs Etiology
2.1. Chromatin Reorganization Due to Genetic Events as Oncogenic Drivers in pHGGs
2.2. Establishment of a New pHGGs Classification: Epigenetic as a New Guide
3. Synergy between Transcriptional and Epigenetic Rewiring in pHGGs: A Matter of Oncogenic Window
3.1. Oncogenic Contribution of Non-Genetic Ontogenic Factors in Gliomagenesis
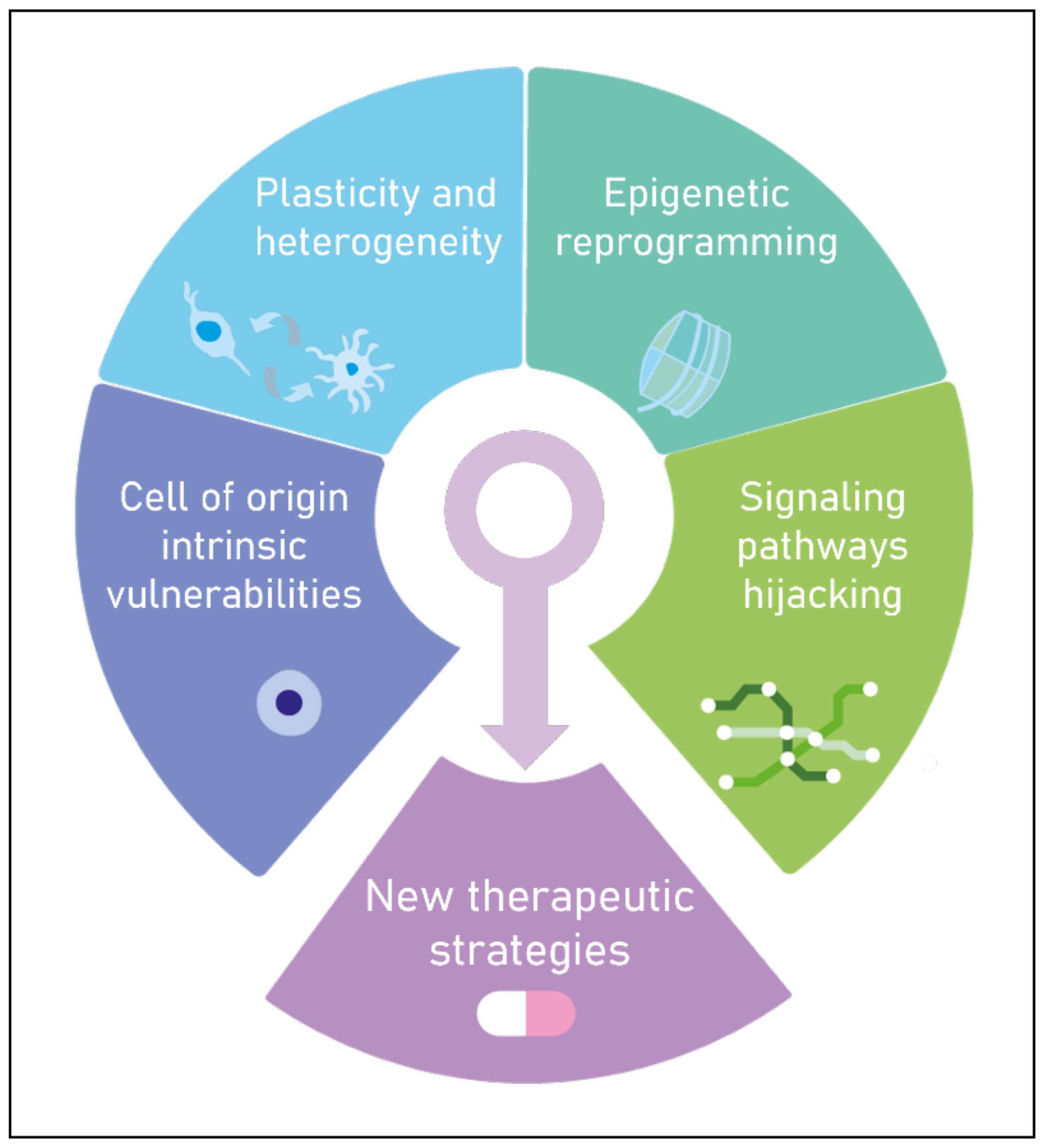
3.2. Importance of the Cell-of-Origin in the Activation of Oncogenic Transcriptional Networks
3.3. Hijacking of Transcriptional Developmental Pathways and Maintenance in an Immature Epigenetic State as the Core of pHGGs
4. Targeting the Synergistic Epigenetic/Transcriptional Oncogenic Node: A Path towards New Therapies
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-Oncology 2015, 16, x1–x36. [Google Scholar] [CrossRef] [PubMed]
- Broniscer, A.; Gajjar, A. Supratentorial High-Grade Astrocytoma and Diffuse Brainstem Glioma: Two Challenges for the Pediatric Oncologist. Oncologist 2004, 9, 197–206. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.J.; Aguilera, D.; Kramm, C.M. Treatment of High-Grade Glioma in Children and Adolescents. Neuro-Oncology 2011, 13, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Johung, T.B.; Monje, M. Diffuse Intrinsic Pontine Glioma: New Pathophysiological Insights and Emerging Therapeutic Targets. Curr. Neuropharmacol. 2017, 15, 88–97. [Google Scholar] [CrossRef]
- Jones, C.; Perryman, L.; Hargrave, D. Paediatric and Adult Malignant Glioma: Close Relatives or Distant Cousins? Nat. Rev. Clin. Oncol. 2012, 9, 400–413. [Google Scholar] [CrossRef]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated Molecular Genetic Profiling of Pediatric High-Grade Gliomas Reveals Key Differences With the Adult Disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The Landscape of Genomic Alterations across Childhood Cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef]
- Khuong-Quang, D.-A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.-Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M Mutation in Histone H3.3 Defines Clinically and Biologically Distinct Subgroups of Pediatric Diffuse Intrinsic Pontine Gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver Mutations in Histone H3.3 and Chromatin Remodelling Genes in Paediatric Glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic Histone H3 Alterations in Pediatric Diffuse Intrinsic Pontine Gliomas and Non-Brainstem Glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Guerreiro Stucklin, A.S.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E.; et al. Alterations in ALK/ROS1/NTRK/MET Drive a Group of Infantile Hemispheric Gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef]
- Clarke, M.; Mackay, A.; Ismer, B.; Pickles, J.C.; Tatevossian, R.G.; Newman, S.; Bale, T.A.; Stoler, I.; Izquierdo, E.; Temelso, S.; et al. Infant High-Grade Gliomas Comprise Multiple Subgroups Characterized by Novel Targetable Gene Fusions and Favorable Outcomes. Cancer Discov. 2020, 10, 942–963. [Google Scholar] [CrossRef]
- Bender, S.; Gronych, J.; Warnatz, H.-J.; Hutter, B.; Gröbner, S.; Ryzhova, M.; Pfaff, E.; Hovestadt, V.; Weinberg, F.; Halbach, S.; et al. Recurrent MET Fusion Genes Represent a Drug Target in Pediatric Glioblastoma. Nat. Med. 2016, 22, 1314–1320. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Grill, J.; Teuff, G.L.; Nysom, K.; Blomgren, K.; Hargrave, D.; MacCowage, G.; Bautista, F.; Van Vuurden, D.; Dangouloff-Ros, V.; Puget, S.; et al. DIPG-35. Biological medicine for diffuse intrinsic pontine glioma (DIPG) eradication: Results of the three arm biomarker-driven randomized BIOMEDE 1.0 trial. Neuro-Oncology 2020, 22, iii293–iii294. [Google Scholar] [CrossRef]
- Dufour, C.; Grill, J.; Lellouch-Tubiana, A.; Puget, S.; Chastagner, P.; Frappaz, D.; Doz, F.; Pichon, F.; Plantaz, D.; Gentet, J.C.; et al. High-Grade Glioma in Children under 5 Years of Age: A Chemotherapy Only Approach with the BBSFOP Protocol. Eur. J. Cancer 2006, 42, 2939–2945. [Google Scholar] [CrossRef]
- Frappaz, D.; Schell, M.; Thiesse, P.; Marec-Bérard, P.; Mottolese, C.; Perol, D.; Bergeron, C.; Philip, T.; Ricci, A.C.; Galand-Desme, S.; et al. Preradiation Chemotherapy May Improve Survival in Pediatric Diffuse Intrinsic Brainstem Gliomas: Final Results of BSG 98 Prospective Trial. Neuro-Oncology 2008, 10, 599–607. [Google Scholar] [CrossRef]
- Filbin, M.; Monje, M. Developmental Origins and Emerging Therapeutic Opportunities for Childhood Cancer. Nat. Med. 2019, 25, 367–376. [Google Scholar] [CrossRef]
- Campos, E.I.; Reinberg, D. Histones: Annotating Chromatin. Annu. Rev. Genet. 2009, 43, 559–599. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 Activity by a Gain-of-Function H3 Mutation Found in Pediatric Glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.W.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef]
- Chan, K.-M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The Histone H3.3K27M Mutation in Pediatric Glioma Reprograms H3K27 Methylation and Gene Expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.-H.; Woodfin, A.R.; et al. Therapeutic Targeting of Polycomb and BET Bromodomain Proteins in Diffuse Intrinsic Pontine Gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef]
- Stafford, J.M.; Lee, C.-H.; Voigt, P.; Descostes, N.; Saldaña-Meyer, R.; Yu, J.-R.; Leroy, G.; Oksuz, O.; Chapman, J.R.; Suarez, F.; et al. Multiple Modes of PRC2 Inhibition Elicit Global Chromatin Alterations in H3K27M Pediatric Glioma. Sci. Adv. 2018, 4, eaau5935. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 Is a Potential Therapeutic Target for H3K27M-Mutant Pediatric Gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M Induces Defective Chromatin Spread of PRC2-Mediated Repressive H3K27me2/Me3 and Is Essential for Glioma Tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Sarthy, J.F.; Meers, M.P.; Janssens, D.H.; Henikoff, J.G.; Feldman, H.; Paddison, P.J.; Lockwood, C.M.; Vitanza, N.A.; Olson, J.M.; Ahmad, K.; et al. Histone Deposition Pathways Determine the Chromatin Landscapes of H3.1 and H3.3 K27M Oncohistones. Elife 2020, 9, e61090. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, S.A.; Feldman, H.M.; Arora, S.; Hoellerbauer, P.; Toledo, C.M.; Corrin, P.; Carter, L.; Kufeld, M.; Bolouri, H.; Basom, R.; et al. Neural G0: A Quiescent-like State Found in Neuroepithelial-Derived Cells and Glioma. Mol. Syst. Biol. 2021, 17, e9522. [Google Scholar] [CrossRef] [PubMed]
- Galvao, R.P.; Kasina, A.; McNeill, R.S.; Harbin, J.E.; Foreman, O.; Verhaak, R.G.W.; Nishiyama, A.; Miller, C.R.; Zong, H. Transformation of Quiescent Adult Oligodendrocyte Precursor Cells into Malignant Glioma through a Multistep Reactivation Process. Proc. Natl. Acad. Sci. USA 2014, 111, E4214–E4223. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Osswald, M.; Ratliff, M.; Dogan, H.; Xie, R.; Weil, S.; Hoffmann, D.C.; Kurz, F.T.; Kessler, T.; Heiland, S.; et al. Tumor Cell Plasticity, Heterogeneity, and Resistance in Crucial Microenvironmental Niches in Glioma. Nat. Commun. 2021, 12, 1014. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Udugama, M.; Lin, W.; Hii, L.; Law, R.H.P.; Steer, D.L.; Das, P.P.; Mann, J.R.; Wong, L.H. Inhibition of a K9/K36 Demethylase by an H3.3 Point Mutation Found in Paediatric Glioblastoma. Nat. Commun. 2018, 9, 3142. [Google Scholar] [CrossRef]
- Jain, S.U.; Khazaei, S.; Marchione, D.M.; Lundgren, S.M.; Wang, X.; Weinberg, D.N.; Deshmukh, S.; Juretic, N.; Lu, C.; Allis, C.D.; et al. Histone H3.3 G34 Mutations Promote Aberrant PRC2 Activity and Drive Tumor Progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27354–27364. [Google Scholar] [CrossRef]
- Bressan, R.B.; Southgate, B.; Ferguson, K.M.; Blin, C.; Grant, V.; Alfazema, N.; Wills, J.C.; Marques-Torrejon, M.A.; Morrison, G.M.; Ashmore, J.; et al. Regional Identity of Human Neural Stem Cells Determines Oncogenic Responses to Histone H3.3 Mutants. Cell Stem Cell 2021, 28, 877–893.e9. [Google Scholar] [CrossRef]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3 Mutations Drive Pediatric Glioblastoma through Upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef]
- Schotta, G.; Lachner, M.; Sarma, K.; Ebert, A.; Sengupta, R.; Reuter, G.; Reinberg, D.; Jenuwein, T. A Silencing Pathway to Induce H3-K9 and H4-K20 Trimethylation at Constitutive Heterochromatin. Genes Dev. 2004, 18, 1251–1262. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial Patterns of Histone Acetylations and Methylations in the Human Genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic Histone H3 Methylation during Gene Induction: HYPB/Setd2 Mediates All H3K36 Trimethylation. EMBO J. 2008, 27, 406–420. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-Wide Maps of Chromatin State in Pluripotent and Lineage-Committed Cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of Chromatin Structure by Site-Specific Histone H3 Methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef]
- Bannister, A.J.; Schneider, R.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Spatial Distribution of Di- and Tri-Methyl Lysine 36 of Histone H3 at Active Genes. J. Biol. Chem. 2005, 280, 17732–17736. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B Driver Mutations Define Chondroblastoma and Giant Cell Tumor of Bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 Methylation Defines a Subset of Head and Neck Squamous Cell Carcinomas. Nat. Genet. 2017, 49, 180–185. [Google Scholar] [CrossRef]
- Fang, D.; Gan, H.; Lee, J.-H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J.; et al. The Histone H3.3K36M Mutation Reprograms the Epigenome of Chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef]
- Castel, D.; Kergrohen, T.; Tauziède-Espariat, A.; Mackay, A.; Ghermaoui, S.; Lechapt, E.; Pfister, S.M.; Kramm, C.M.; Boddaert, N.; Blauwblomme, T.; et al. Histone H3 Wild-Type DIPG/DMG Overexpressing EZHIP Extend the Spectrum Diffuse Midline Gliomas with PRC2 Inhibition beyond H3-K27M Mutation. Acta Neuropathol. 2020, 139, 1109–1113. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Hodgson, J.G.; VandenBerg, S.R.; Flaherty, P.; Polley, M.-Y.C.; Yu, M.; Fisher, P.G.; Rowitch, D.H.; Ford, J.M.; Berger, M.S.; et al. Oncogenic BRAF Mutation with CDKN2A Inactivation Is Characteristic of a Subset of Pediatric Malignant Astrocytomas. Cancer Res. 2010, 70, 512–519. [Google Scholar] [CrossRef]
- Nicolaides, T.P.; Li, H.; Solomon, D.A.; Hariono, S.; Hashizume, R.; Barkovich, K.; Baker, S.J.; Paugh, B.S.; Jones, C.; Forshew, T.; et al. Targeted Therapy for BRAFV600E Malignant Astrocytoma. Clin. Cancer Res. 2011, 17, 7595–7604. [Google Scholar] [CrossRef]
- Korshunov, A.; Schrimpf, D.; Ryzhova, M.; Sturm, D.; Chavez, L.; Hovestadt, V.; Sharma, T.; Habel, A.; Burford, A.; Jones, C.; et al. H3-/IDH-Wild Type Pediatric Glioblastoma Is Comprised of Molecularly and Prognostically Distinct Subtypes with Associated Oncogenic Drivers. Acta Neuropathol. 2017, 134, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Downing, J.R.; Wilson, R.K.; Zhang, J.; Mardis, E.R.; Pui, C.-H.; Ding, L.; Ley, T.J.; Evans, W.E. The Pediatric Cancer Genome Project. Nat. Genet. 2012, 44, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.C.; Lindquist, R.A.; Nguyen, T.; Sanai, N.; Barkovich, A.J.; Huang, E.J.; Rowitch, D.H.; Alvarez-Buylla, A. Postnatal Growth of the Human Pons: A Morphometric and Immunohistochemical Analysis. J. Comp. Neurol. 2015, 523, 449–462. [Google Scholar] [CrossRef]
- Thatcher, R.W.; Walker, R.A.; Giudice, S. Human Cerebral Hemispheres Develop at Different Rates and Ages. Science 1987, 236, 1110–1113. [Google Scholar] [CrossRef]
- Gibson, E.M.; Geraghty, A.C.; Monje, M. Bad Wrap: Myelin and Myelin Plasticity in Health and Disease. Dev. Neurobiol. 2018, 78, 123–135. [Google Scholar] [CrossRef]
- Lebel, C.; Gee, M.; Camicioli, R.; Wieler, M.; Martin, W.; Beaulieu, C. Diffusion Tensor Imaging of White Matter Tract Evolution over the Lifespan. Neuroimage 2012, 60, 340–352. [Google Scholar] [CrossRef]
- Proctor, D.T.; Stotz, S.C.; Scott, L.O.M.; de la Hoz, C.L.R.; Poon, K.W.C.; Stys, P.K.; Colicos, M.A. Axo-Glial Communication through Neurexin-Neuroligin Signaling Regulates Myelination and Oligodendrocyte Differentiation. Glia 2015, 63, 2023–2039. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting Neuronal Activity-Regulated Neuroligin-3 Dependency in High-Grade Glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef]
- Eze, U.C.; Bhaduri, A.; Haeussler, M.; Nowakowski, T.J.; Kriegstein, A.R. Single-Cell Atlas of Early Human Brain Development Highlights Heterogeneity of Human Neuroepithelial Cells and Early Radial Glia. Nat. Neurosci. 2021, 24, 584–594. [Google Scholar] [CrossRef]
- Carvalho, D.; Taylor, K.R.; Olaciregui, N.G.; Molinari, V.; Clarke, M.; Mackay, A.; Ruddle, R.; Henley, A.; Valenti, M.; Hayes, A.; et al. ALK2 Inhibitors Display Beneficial Effects in Preclinical Models of ACVR1 Mutant Diffuse Intrinsic Pontine Glioma. Commun. Biol. 2019, 2, 156. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Knoechel, B.; Roderick, J.E.; Williamson, K.E.; Zhu, J.; Lohr, J.G.; Cotton, M.J.; Gillespie, S.M.; Fernandez, D.; Ku, M.; Wang, H.; et al. An Epigenetic Mechanism of Resistance to Targeted Therapy in T Cell Acute Lymphoblastic Leukemia. Nat. Genet. 2014, 46, 364–370. [Google Scholar] [CrossRef]
- Van Gils, N.; Denkers, F.; Smit, L. Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 659253. [Google Scholar] [CrossRef]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and Oncogenic Programs in H3K27M Gliomas Dissected by Single-Cell RNA-Seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef]
- Monje, M.; Mitra, S.S.; Freret, M.E.; Raveh, T.B.; Kim, J.; Masek, M.; Attema, J.L.; Li, G.; Haddix, T.; Edwards, M.S.B.; et al. Hedgehog-Responsive Candidate Cell of Origin for Diffuse Intrinsic Pontine Glioma. Proc. Natl. Acad. Sci. USA 2011, 108, 4453–4458. [Google Scholar] [CrossRef]
- Ballester, L.Y.; Wang, Z.; Shandilya, S.; Miettinen, M.; Burger, P.C.; Eberhart, C.G.; Rodriguez, F.J.; Raabe, E.; Nazarian, J.; Warren, K.; et al. Morphologic Characteristics and Immunohistochemical Profile of Diffuse Intrinsic Pontine Gliomas. Am. J. Surg. Pathol. 2013, 37, 1357–1364. [Google Scholar] [CrossRef]
- Cordero, F.J.; Huang, Z.; Grenier, C.; He, X.; Hu, G.; McLendon, R.E.; Murphy, S.K.; Hashizume, R.; Becher, O.J. Histone H3.3K27M Represses P16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol. Cancer Res. 2017, 15, 1243–1254. [Google Scholar] [CrossRef]
- Misuraca, K.L.; Hu, G.; Barton, K.L.; Chung, A.; Becher, O.J. A Novel Mouse Model of Diffuse Intrinsic Pontine Glioma Initiated in Pax3-Expressing Cells. Neoplasia 2016, 18, 60–70. [Google Scholar] [CrossRef]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of Human Embryonic Stem Cells to Model Pediatric Gliomas with H3.3K27M Histone Mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.-H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155.e7. [Google Scholar] [CrossRef]
- Pathania, M.; De Jay, N.; Maestro, N.; Harutyunyan, A.S.; Nitarska, J.; Pahlavan, P.; Henderson, S.; Mikael, L.G.; Richard-Londt, A.; Zhang, Y.; et al. H3.3K27M Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 2017, 32, 684–700.e9. [Google Scholar] [CrossRef]
- Haag, D.; Mack, N.; Benites Goncalves da Silva, P.; Statz, B.; Clark, J.; Tanabe, K.; Sharma, T.; Jäger, N.; Jones, D.T.W.; Kawauchi, D.; et al. H3.3-K27M Drives Neural Stem Cell-Specific Gliomagenesis in a Human IPSC-Derived Model. Cancer Cell 2021, 39, 407–422.e13. [Google Scholar] [CrossRef]
- Palm, T.; Bolognin, S.; Meiser, J.; Nickels, S.; Träger, C.; Meilenbrock, R.-L.; Brockhaus, J.; Schreitmüller, M.; Missler, M.; Schwamborn, J.C. Rapid and Robust Generation of Long-Term Self-Renewing Human Neural Stem Cells with the Ability to Generate Mature Astroglia. Sci. Rep. 2015, 5, 16321. [Google Scholar] [CrossRef]
- Funato, K.; Smith, R.C.; Saito, Y.; Tabar, V. Dissecting the Impact of Regional Identity and the Oncogenic Role of Human-Specific NOTCH2NL in an HESC Model of H3.3G34R-Mutant Glioma. Cell Stem Cell 2021, 28, 894–905.e7. [Google Scholar] [CrossRef]
- Chen, C.C.L.; Deshmukh, S.; Jessa, S.; Hadjadj, D.; Lisi, V.; Andrade, A.F.; Faury, D.; Jawhar, W.; Dali, R.; Suzuki, H.; et al. Histone H3.3G34-Mutant Interneuron Progenitors Co-Opt PDGFRA for Gliomagenesis. Cell 2020, 183, 1617–1633.e22. [Google Scholar] [CrossRef]
- Hansen, D.V.; Lui, J.H.; Flandin, P.; Yoshikawa, K.; Rubenstein, J.L.; Alvarez-Buylla, A.; Kriegstein, A.R. Non-Epithelial Stem Cells and Cortical Interneuron Production in the Human Ganglionic Eminences. Nat. Neurosci. 2013, 16, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Wang, C.; Wang, L.; Zhou, X.; Tian, M.; Zhang, Q.; Zhang, Y.; Li, J.; Liu, Z.; Cai, Y.; et al. Subcortical Origins of Human and Monkey Neocortical Interneurons. Nat. Neurosci. 2013, 16, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Yanazawa, M.; Sugiyama, N.; Miura, H.; Iizuka-Kogo, A.; Kusaka, M.; Omichi, K.; Suzuki, R.; Kato-Fukui, Y.; Kamiirisa, K.; et al. Mutation of ARX Causes Abnormal Development of Forebrain and Testes in Mice and X-Linked Lissencephaly with Abnormal Genitalia in Humans. Nat. Genet. 2002, 32, 359–369. [Google Scholar] [CrossRef]
- Mi, D.; Carr, C.B.; Georgala, P.A.; Huang, Y.-T.; Manuel, M.N.; Jeanes, E.; Niisato, E.; Sansom, S.N.; Livesey, F.J.; Theil, T.; et al. Pax6 Exerts Regional Control of Cortical Progenitor Proliferation via Direct Repression of Cdk6 and Hypophosphorylation of PRb. Neuron 2013, 78, 269–284. [Google Scholar] [CrossRef]
- Castro, D.S.; Skowronska-Krawczyk, D.; Armant, O.; Donaldson, I.J.; Parras, C.; Hunt, C.; Critchley, J.A.; Nguyen, L.; Gossler, A.; Göttgens, B.; et al. Proneural BHLH and Brn Proteins Coregulate a Neurogenic Program through Cooperative Binding to a Conserved DNA Motif. Dev. Cell 2006, 11, 831–844. [Google Scholar] [CrossRef]
- Pevny, L.; Placzek, M. SOX Genes and Neural Progenitor Identity. Curr. Opin. Neurobiol. 2005, 15, 7–13. [Google Scholar] [CrossRef]
- Young, F.I.; Keruzore, M.; Nan, X.; Gennet, N.; Bellefroid, E.J.; Li, M. The Doublesex-Related Dmrta2 Safeguards Neural Progenitor Maintenance Involving Transcriptional Regulation of Hes1. Proc. Natl. Acad. Sci. USA 2017, 114, E5599–E5607. [Google Scholar] [CrossRef]
- Konno, D.; Iwashita, M.; Satoh, Y.; Momiyama, A.; Abe, T.; Kiyonari, H.; Matsuzaki, F. The Mammalian DM Domain Transcription Factor Dmrta2 Is Required for Early Embryonic Development of the Cerebral Cortex. PLoS ONE 2012, 7, e46577. [Google Scholar] [CrossRef]
- Bertacchi, M.; Romano, A.L.; Loubat, A.; Tran Mau-Them, F.; Willems, M.; Faivre, L.; Khau van Kien, P.; Perrin, L.; Devillard, F.; Sorlin, A.; et al. NR2F1 Regulates Regional Progenitor Dynamics in the Mouse Neocortex and Cortical Gyrification in BBSOAS Patients. EMBO J. 2020, 39, e104163. [Google Scholar] [CrossRef]
- Steinfeld, H.; Cho, M.T.; Retterer, K.; Person, R.; Schaefer, G.B.; Danylchuk, N.; Malik, S.; Wechsler, S.B.; Wheeler, P.G.; van Gassen, K.L.I.; et al. Mutations in HIVEP2 Are Associated with Developmental Delay, Intellectual Disability, and Dysmorphic Features. Neurogenetics 2016, 17, 159–164. [Google Scholar] [CrossRef]
- Yoshida, M.; Suda, Y.; Matsuo, I.; Miyamoto, N.; Takeda, N.; Kuratani, S.; Aizawa, S. Emx1 and Emx2 Functions in Development of Dorsal Telencephalon. Development 1997, 124, 101–111. [Google Scholar] [CrossRef]
- Ono, K.; Takebayashi, H.; Ikeda, K.; Furusho, M.; Nishizawa, T.; Watanabe, K.; Ikenaka, K. Regional- and Temporal-Dependent Changes in the Differentiation of Olig2 Progenitors in the Forebrain, and the Impact on Astrocyte Development in the Dorsal Pallium. Dev. Biol. 2008, 320, 456–468. [Google Scholar] [CrossRef]
- Vasconcelos, F.F.; Castro, D.S. Transcriptional Control of Vertebrate Neurogenesis by the Proneural Factor Ascl1. Front. Cell. Neurosci. 2014, 8, 412. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.; Zhang, R.; Taylor, V. Notch and Neurogenesis. Adv. Exp. Med. Biol. 2018, 1066, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Kaise, T.; Kageyama, R. Hes1 Oscillation Frequency Correlates with Activation of Neural Stem Cells. Gene Expr. Patterns 2021, 40, 119170. [Google Scholar] [CrossRef]
- Fiddes, I.T.; Lodewijk, G.A.; Mooring, M.; Bosworth, C.M.; Ewing, A.D.; Mantalas, G.L.; Novak, A.M.; van den Bout, A.; Bishara, A.; Rosenkrantz, J.L.; et al. Human-Specific NOTCH2NL Genes Affect Notch Signaling and Cortical Neurogenesis. Cell 2018, 173, 1356–1369.e22. [Google Scholar] [CrossRef]
- Hirano, K.; Namihira, M. LSD1 Mediates Neuronal Differentiation of Human Fetal Neural Stem Cells by Controlling the Expression of a Novel Target Gene, HEYL. Stem Cells 2016, 34, 1872–1882. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, H.; Ohtsuka, T.; Kageyama, R. Oscillations in Notch Signaling Regulate Maintenance of Neural Progenitors. Neuron 2008, 58, 52–64. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Bush, K.; Klein, R.H.; Cervantes, V.; Lewis, N.; Naqvi, A.; Carcaboso, A.M.; Lechpammer, M.; Knoepfler, P.S. Reciprocal H3.3 Gene Editing Identifies K27M and G34R Mechanisms in Pediatric Glioma Including NOTCH Signaling. Commun. Biol. 2020, 3, 363. [Google Scholar] [CrossRef]
- Brien, G.L.; Bressan, R.B.; Monger, C.; Gannon, D.; Lagan, E.; Doherty, A.M.; Healy, E.; Neikes, H.; Fitzpatrick, D.J.; Deevy, O.; et al. Simultaneous Disruption of PRC2 and Enhancer Function Underlies Histone H3.3-K27M Oncogenic Activity in Human Hindbrain Neural Stem Cells. Nat. Genet. 2021, 53, 1221–1232. [Google Scholar] [CrossRef]
- Angelini, C.; Trimouille, A.; Arveiler, B.; Espil-Taris, C.; Ichinose, N.; Lasseaux, E.; Tourdias, T.; Lacombe, D. CHN1 and Duane Retraction Syndrome: Expanding the Phenotype to Cranial Nerves Development Disease. Eur. J. Med. Genet. 2021, 64, 104188. [Google Scholar] [CrossRef]
- Maussion, G.; Diallo, A.B.; Gigek, C.O.; Chen, E.S.; Crapper, L.; Théroux, J.-F.; Chen, G.G.; Vasuta, C.; Ernst, C. Investigation of Genes Important in Neurodevelopment Disorders in Adult Human Brain. Hum. Genet. 2015, 134, 1037–1053. [Google Scholar] [CrossRef]
- Cosgaya, J.M.; Chan, J.R.; Shooter, E.M. The Neurotrophin Receptor P75NTR as a Positive Modulator of Myelination. Science 2002, 298, 1245–1248. [Google Scholar] [CrossRef]
- Anderson, S.A.; Eisenstat, D.D.; Shi, L.; Rubenstein, J.L. Interneuron Migration from Basal Forebrain to Neocortex: Dependence on Dlx Genes. Science 1997, 278, 474–476. [Google Scholar] [CrossRef]
- Anderson, S.A.; Qiu, M.; Bulfone, A.; Eisenstat, D.D.; Meneses, J.; Pedersen, R.; Rubenstein, J.L. Mutations of the Homeobox Genes Dlx-1 and Dlx-2 Disrupt the Striatal Subventricular Zone and Differentiation of Late Born Striatal Neurons. Neuron 1997, 19, 27–37. [Google Scholar] [CrossRef]
- Lin, H.-C.; He, Z.; Ebert, S.; Schörnig, M.; Santel, M.; Nikolova, M.T.; Weigert, A.; Hevers, W.; Kasri, N.N.; Taverna, E.; et al. NGN2 Induces Diverse Neuron Types from Human Pluripotency. Stem Cell Rep. 2021, 16, 2118–2127. [Google Scholar] [CrossRef]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M Mutations Define Two Subgroups of Diffuse Intrinsic Pontine Gliomas with Different Prognosis and Phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef]
- Nagaraja, S.; Quezada, M.A.; Gillespie, S.M.; Arzt, M.; Lennon, J.J.; Woo, P.J.; Hovestadt, V.; Kambhampati, M.; Filbin, M.G.; Suva, M.L.; et al. Histone Variant and Cell Context Determine H3K27M Reprogramming of the Enhancer Landscape and Oncogenic State. Mol. Cell 2019, 76, 965–980.e12. [Google Scholar] [CrossRef]
- Sedykh, I.; Keller, A.N.; Yoon, B.; Roberson, L.; Moskvin, O.V.; Grinblat, Y. Zebrafish Rfx4 Controls Dorsal and Ventral Midline Formation in the Neural Tube. Dev. Dyn. 2018, 247, 650–659. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain Tumour Cells Interconnect to a Functional and Resistant Network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hänggi, D.; Wick, W.; Winkler, F. Tumor Microtubes Convey Resistance to Surgical Lesions and Chemotherapy in Gliomas. Neuro-Oncology 2017, 19, 1316–1326. [Google Scholar] [CrossRef]
- Tanneberger, K.; Pfister, A.S.; Kriz, V.; Bryja, V.; Schambony, A.; Behrens, J. Structural and Functional Characterization of the Wnt Inhibitor APC Membrane Recruitment 1 (Amer1). J. Biol. Chem. 2011, 286, 19204–19214. [Google Scholar] [CrossRef]
- Stefanski, C.D.; Prosperi, J.R. Wnt-Independent and Wnt-Dependent Effects of APC Loss on the Chemotherapeutic Response. Int. J. Mol. Sci. 2020, 21, 7844. [Google Scholar] [CrossRef] [PubMed]
- Humphries, A.C.; Mlodzik, M. From Instruction to Output: Wnt/PCP Signaling in Development and Cancer. Curr. Opin. Cell Biol. 2018, 51, 110–116. [Google Scholar] [CrossRef]
- Brafman, D.; Willert, K. Wnt/β-Catenin Signaling during Early Vertebrate Neural Development. Dev. Neurobiol. 2017, 77, 1239–1259. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic Analysis of Diffuse Intrinsic Pontine Gliomas Identifies Three Molecular Subgroups and Recurrent Activating ACVR1 Mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent Activating ACVR1 Mutations in Diffuse Intrinsic Pontine Glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.-O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent Somatic Mutations in ACVR1 in Pediatric Midline High-Grade Astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The Genomic Landscape of Diffuse Intrinsic Pontine Glioma and Pediatric Non-Brainstem High-Grade Glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.-J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A Recurrent Mutation in the BMP Type I Receptor ACVR1 Causes Inherited and Sporadic Fibrodysplasia Ossificans Progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Haupt, J.; Xu, M.; Shore, E.M. Variable Signaling Activity by FOP ACVR1 Mutations. Bone 2018, 109, 232–240. [Google Scholar] [CrossRef]
- Pathogenic ACVR1R206H Activation by Activin A-Induced Receptor Clustering and Autophosphorylation. EMBO J. 2021, 40, e106317. [CrossRef]
- Hoeman, C.M.; Cordero, F.J.; Hu, G.; Misuraca, K.; Romero, M.M.; Cardona, H.J.; Nazarian, J.; Hashizume, R.; McLendon, R.; Yu, P.; et al. ACVR1 R206H Cooperates with H3.1K27M in Promoting Diffuse Intrinsic Pontine Glioma Pathogenesis. Nat. Commun. 2019, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.; Tian, R.; Zarrabi, I.; Hill, G.; Williams, E.; Sanchez-Duffhues, G.; Thorikay, M.; Ramachandran, P.; Siddaway, R.; Wong, J.F.; et al. Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell 2020, 37, 308–323.e12. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Burford, A.; Molinari, V.; Kessler, K.; Popov, S.; Clarke, M.; Taylor, K.R.; Pemberton, H.N.; Lord, C.J.; Gutteridge, A.; et al. Functional Diversity and Cooperativity between Subclonal Populations of Pediatric Glioblastoma and Diffuse Intrinsic Pontine Glioma Cells. Nat. Med. 2018, 24, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Nikbakht, H.; Panditharatna, E.; Mikael, L.G.; Li, R.; Gayden, T.; Osmond, M.; Ho, C.-Y.; Kambhampati, M.; Hwang, E.I.; Faury, D.; et al. Spatial and Temporal Homogeneity of Driver Mutations in Diffuse Intrinsic Pontine Glioma. Nat. Commun. 2016, 7, 11185. [Google Scholar] [CrossRef] [PubMed]
- Paugh, B.S.; Zhu, X.; Qu, C.; Endersby, R.; Diaz, A.K.; Zhang, J.; Bax, D.A.; Carvalho, D.; Reis, R.M.; Onar-Thomas, A.; et al. Novel Oncogenic PDGFRA Mutations in Pediatric High-Grade Gliomas. Cancer Res. 2013, 73, 6219–6229. [Google Scholar] [CrossRef]
- Zhang, G.; Lübke, L.; Chen, F.; Beil, T.; Takamiya, M.; Diotel, N.; Strähle, U.; Rastegar, S. Neuron-Radial Glial Cell Communication via BMP/Id1 Signaling Is Key to Long-Term Maintenance of the Regenerative Capacity of the Adult Zebrafish Telencephalon. Cells 2021, 10, 2794. [Google Scholar] [CrossRef]
- Rodriguez Viales, R.; Diotel, N.; Ferg, M.; Armant, O.; Eich, J.; Alunni, A.; März, M.; Bally-Cuif, L.; Rastegar, S.; Strähle, U. The Helix-Loop-Helix Protein Id1 Controls Stem Cell Proliferation during Regenerative Neurogenesis in the Adult Zebrafish Telencephalon. Stem Cells 2015, 33, 892–903. [Google Scholar] [CrossRef]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The Dynamic Role of Bone Morphogenetic Proteins in Neural Stem Cell Fate and Maturation. Dev. Neurobiol. 2012, 72, 1068–1084. [Google Scholar] [CrossRef]
- Marques, S.; Zeisel, A.; Codeluppi, S.; van Bruggen, D.; Mendanha Falcão, A.; Xiao, L.; Li, H.; Häring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte Heterogeneity in the Mouse Juvenile and Adult Central Nervous System. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef]
- Funa, K.; Sasahara, M. The Roles of PDGF in Development and During Neurogenesis in the Normal and Diseased Nervous System. J. Neuroimmune Pharm. 2014, 9, 168–181. [Google Scholar] [CrossRef]
- Santos, A.K.; Gomes, K.N.; Parreira, R.C.; Scalzo, S.; Pinto, M.C.X.; Santiago, H.C.; Birbrair, A.; Sack, U.; Ulrich, H.; Resende, R.R. Mouse Neural Stem Cell Differentiation and Human Adipose Mesenchymal Stem Cell Transdifferentiation Into Neuron- and Oligodendrocyte-like Cells With Myelination Potential. Stem Cell Rev. Rep. 2022, 18, 732–751. [Google Scholar] [CrossRef]
- Jessa, S.; Blanchet-Cohen, A.; Krug, B.; Vladoiu, M.; Coutelier, M.; Faury, D.; Poreau, B.; De Jay, N.; Hébert, S.; Monlong, J.; et al. Stalled Developmental Programs at the Root of Pediatric Brain Tumors. Nat. Genet. 2019, 51, 1702–1713. [Google Scholar] [CrossRef]
- Zhu, Q.; Fang, L.; Heuberger, J.; Kranz, A.; Schipper, J.; Scheckenbach, K.; Vidal, R.O.; Sunaga-Franze, D.Y.; Müller, M.; Wulf-Goldenberg, A.; et al. The Wnt-Driven Mll1 Epigenome Regulates Salivary Gland and Head and Neck Cancer. Cell Rep. 2019, 26, 415–428.e5. [Google Scholar] [CrossRef]
- Bray, S.; Musisi, H.; Bienz, M. Bre1 Is Required for Notch Signaling and Histone Modification. Dev. Cell 2005, 8, 279–286. [Google Scholar] [CrossRef]
- Park, W.-Y.; Hong, B.-J.; Lee, J.; Choi, C.; Kim, M.-Y. H3K27 Demethylase JMJD3 Employs the NF-ΚB and BMP Signaling Pathways to Modulate the Tumor Microenvironment and Promote Melanoma Progression and Metastasis. Cancer Res. 2016, 76, 161–170. [Google Scholar] [CrossRef]
- Akizu, N.; Estarás, C.; Guerrero, L.; Martí, E.; Martínez-Balbás, M.A. H3K27me3 Regulates BMP Activity in Developing Spinal Cord. Development 2010, 137, 2915–2925. [Google Scholar] [CrossRef]
- Sinha, S.; Biswas, M.; Chatterjee, S.S.; Kumar, S.; Sengupta, A. Pbrm1 Steers Mesenchymal Stromal Cell Osteolineage Differentiation by Integrating PBAF-Dependent Chromatin Remodeling and BMP/TGF-β Signaling. Cell Rep. 2020, 31, 107570. [Google Scholar] [CrossRef]
- Cohen, K.J.; Heideman, R.L.; Zhou, T.; Holmes, E.J.; Lavey, R.S.; Bouffet, E.; Pollack, I.F. Temozolomide in the Treatment of Children with Newly Diagnosed Diffuse Intrinsic Pontine Gliomas: A Report from the Children’s Oncology Group. Neuro. Oncol. 2011, 13, 410–416. [Google Scholar] [CrossRef]
- Jalali, R.; Raut, N.; Arora, B.; Gupta, T.; Dutta, D.; Munshi, A.; Sarin, R.; Kurkure, P. Prospective Evaluation of Radiotherapy with Concurrent and Adjuvant Temozolomide in Children with Newly Diagnosed Diffuse Intrinsic Pontine Glioma. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 113–118. [Google Scholar] [CrossRef]
- Lashford, L.S.; Thiesse, P.; Jouvet, A.; Jaspan, T.; Couanet, D.; Griffiths, P.D.; Doz, F.; Ironside, J.; Robson, K.; Hobson, R.; et al. Temozolomide in Malignant Gliomas of Childhood: A United Kingdom Children’s Cancer Study Group and French Society for Pediatric Oncology Intergroup Study. J. Clin. Oncol. 2002, 20, 4684–4691. [Google Scholar] [CrossRef]
- Findlay, I.J.; De Iuliis, G.N.; Duchatel, R.J.; Jackson, E.R.; Vitanza, N.A.; Cain, J.E.; Waszak, S.M.; Dun, M.D. Pharmaco-Proteogenomic Profiling of Pediatric Diffuse Midline Glioma to Inform Future Treatment Strategies. Oncogene 2022, 41, 461–475. [Google Scholar] [CrossRef]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 31, 635–652.e6. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Biery, M.C.; Myers, C.; Ferguson, E.; Zheng, Y.; Girard, E.J.; Przystal, J.M.; Park, G.; Noll, A.; Pakiam, F.; et al. Optimal Therapeutic Targeting by HDAC Inhibition in Biopsy-Derived Treatment-Naïve Diffuse Midline Glioma Models. Neuro-Oncology 2021, 23, 376–386. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.-A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally Defined Therapeutic Targets in Diffuse Intrinsic Pontine Glioma. Nat. Med. 2015, 21, 555–559. [Google Scholar] [CrossRef]
- Brown, Z.Z.; Müller, M.M.; Jain, S.U.; Allis, C.D.; Lewis, P.W.; Muir, T.W. Strategy for “Detoxification” of a Cancer-Derived Histone Mutant Based on Mapping Its Interaction with the Methyltransferase PRC2. J. Am. Chem. Soc. 2014, 136, 13498–13501. [Google Scholar] [CrossRef]
- Rakotomalala, A.; Bailleul, Q.; Savary, C.; Arcicasa, M.; Hamadou, M.; Huchedé, P.; Hochart, A.; Restouin, A.; Castellano, R.; Collette, Y.; et al. H3.3K27M Mutation Controls Cell Growth and Resistance to Therapies in Pediatric Glioma Cell Lines. Cancers 2021, 13, 5551. [Google Scholar] [CrossRef]
- Amani, V.; Prince, E.W.; Alimova, I.; Balakrishnan, I.; Birks, D.; Donson, A.M.; Harris, P.; Levy, J.M.M.; Handler, M.; Foreman, N.K.; et al. Polo-like Kinase 1 as a Potential Therapeutic Target in Diffuse Intrinsic Pontine Glioma. BMC Cancer 2016, 16, 647. [Google Scholar] [CrossRef]
- Dong, J.; Park, S.Y.; Nguyen, N.; Ezhilarasan, R.; Martinez-Ledesma, E.; Wu, S.; Henry, V.; Piao, Y.; Tiao, N.; Brunell, D.; et al. The Polo-like Kinase 1 Inhibitor Volasertib Synergistically Increases Radiation Efficacy in Glioma Stem Cells. Oncotarget 2018, 9, 10497–10509. [Google Scholar] [CrossRef]
- Truffaux, N.; Philippe, C.; Paulsson, J.; Andreiuolo, F.; Guerrini-Rousseau, L.; Cornilleau, G.; Le Dret, L.; Richon, C.; Lacroix, L.; Puget, S.; et al. Preclinical Evaluation of Dasatinib Alone and in Combination with Cabozantinib for the Treatment of Diffuse Intrinsic Pontine Glioma. Neuro-Oncology 2015, 17, 953–964. [Google Scholar] [CrossRef]
- Taylor, I.C.; Hütt-Cabezas, M.; Brandt, W.D.; Kambhampati, M.; Nazarian, J.; Chang, H.T.; Warren, K.E.; Eberhart, C.G.; Raabe, E.H. Disrupting NOTCH Slows Diffuse Intrinsic Pontine Glioma Growth, Enhances Radiation Sensitivity, and Shows Combinatorial Efficacy With Bromodomain Inhibition. J. Neuropathol. Exp. Neurol. 2015, 74, 778–790. [Google Scholar] [CrossRef]
- Peng, X.; Sun, Z.; Kuang, P.; Chen, J. Recent Progress on HDAC Inhibitors with Dual Targeting Capabilities for Cancer Treatment. Eur. J. Med. Chem. 2020, 208, 112831. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Zee, B.M.; Kalin, J.H.; Kim, M.; Guo, R.; Alexandrescu, S.; Blanco, M.A.; Giera, S.; Gillespie, S.M.; Das, J.; et al. Re-Programing Chromatin with a Bifunctional LSD1/HDAC Inhibitor Induces Therapeutic Differentiation in DIPG. Cancer Cell 2019, 36, 528–544.e10. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ran, D.; Jiang, J.; Pan, T.; Dan, Y.; Tang, Q.; Li, W.; Zhang, L.; Gan, L.; Gan, Z. Discovery of Novel 9H-Purin Derivatives as Dual Inhibitors of HDAC1 and CDK2. Bioorg. Med. Chem. Lett. 2019, 29, 2136–2140. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Tawa, G.J.; Henderson, M.J.; Danchik, C.; Liu, S.; Shah, P.; Wang, A.Q.; Dunn, G.; Kabir, M.; Padilha, E.C.; et al. Design, Synthesis, and Biological Evaluation of Quinazolin-4-One-Based Hydroxamic Acids as Dual PI3K/HDAC Inhibitors. J. Med. Chem. 2020, 63, 4256–4292. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Yin, H.; Zhao, C.; Cao, J.; Xu, W.; Zhang, Y. Design, Synthesis and Biological Evaluation of Novel Osimertinib-Based HDAC and EGFR Dual Inhibitors. Molecules 2019, 24, 2407. [Google Scholar] [CrossRef]
- Lu, D.; Yan, J.; Wang, L.; Liu, H.; Zeng, L.; Zhang, M.; Duan, W.; Ji, Y.; Cao, J.; Geng, M.; et al. Design, Synthesis, and Biological Evaluation of the First c-Met/HDAC Inhibitors Based on Pyridazinone Derivatives. ACS Med. Chem. Lett. 2017, 8, 830–834. [Google Scholar] [CrossRef]
- Liu, J.; Qian, C.; Zhu, Y.; Cai, J.; He, Y.; Li, J.; Wang, T.; Zhu, H.; Li, Z.; Li, W.; et al. Design, Synthesis and Evaluate of Novel Dual FGFR1 and HDAC Inhibitors Bearing an Indazole Scaffold. Bioorg. Med. Chem. 2018, 26, 747–757. [Google Scholar] [CrossRef]
- Duchatel, R.J.; Jackson, E.R.; Alvaro, F.; Nixon, B.; Hondermarck, H.; Dun, M.D. Signal Transduction in Diffuse Intrinsic Pontine Glioma. Proteomics 2019, 19, 1800479. [Google Scholar] [CrossRef]
- Madhukar, N.S.; Khade, P.K.; Huang, L.; Gayvert, K.; Galletti, G.; Stogniew, M.; Allen, J.E.; Giannakakou, P.; Elemento, O. A Bayesian Machine Learning Approach for Drug Target Identification Using Diverse Data Types. Nat. Commun. 2019, 10, 5221. [Google Scholar] [CrossRef]
- Graves, P.R.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial Protease ClpP Is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e9. [Google Scholar] [CrossRef]
- Allen, J.E.; Krigsfeld, G.; Mayes, P.A.; Patel, L.; Dicker, D.T.; Patel, A.S.; Dolloff, N.G.; Messaris, E.; Scata, K.A.; Wang, W.; et al. Dual Inactivation of Akt and ERK by TIC10 Signals Foxo3a Nuclear Translocation, TRAIL Gene Induction, and Potent Antitumor Effects. Sci. Transl. Med. 2013, 5, 171ra17. [Google Scholar] [CrossRef]
- Ishida, C.T.; Zhang, Y.; Bianchetti, E.; Shu, C.; Nguyen, T.T.T.; Kleiner, G.; Sanchez-Quintero, M.J.; Quinzii, C.M.; Westhoff, M.-A.; Karpel-Massler, G.; et al. Metabolic Reprogramming by Dual AKT/ERK Inhibition through Imipridones Elicits Unique Vulnerabilities in Glioblastoma. Clin. Cancer Res. 2018, 24, 5392–5406. [Google Scholar] [CrossRef]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 Induction through an Atypical Integrated Stress Response to ONC201 Triggers P53-Independent Apoptosis in Hematological Malignancies. Sci. Signal. 2016, 9, ra17. [Google Scholar] [CrossRef]
- Przystal, J.M.; Cianciolo Cosentino, C.; Yadavilli, S.; Zhang, J.; Laternser, S.; Bonner, E.R.; Prasad, R.; Dawood, A.A.; Lobeto, N.; Chin Chong, W.; et al. Imipridones Affect Tumor Bioenergetics and Promote Cell Lineage Differentiation in Diffuse Midline Gliomas. Neuro-Oncology 2022, noac041. [Google Scholar] [CrossRef]
- Duchatel, R.J.; Mannan, A.; Woldu, A.S.; Hawtrey, T.; Hindley, P.A.; Douglas, A.M.; Jackson, E.R.; Findlay, I.J.; Germon, Z.P.; Staudt, D.; et al. Preclinical and Clinical Evaluation of German-Sourced ONC201 for the Treatment of H3K27M-Mutant Diffuse Intrinsic Pontine Glioma. Neurooncol. Adv. 2021, 3, vdab169. [Google Scholar] [CrossRef]
- Arrillaga-Romany, I.; Chi, A.S.; Allen, J.E.; Oster, W.; Wen, P.Y.; Batchelor, T.T. A Phase 2 Study of the First Imipridone ONC201, a Selective DRD2 Antagonist for Oncology, Administered Every Three Weeks in Recurrent Glioblastoma. Oncotarget 2017, 8, 79298–79304. [Google Scholar] [CrossRef]
- Stein, M.N.; Bertino, J.R.; Kaufman, H.L.; Mayer, T.; Moss, R.; Silk, A.; Chan, N.; Malhotra, J.; Rodriguez, L.; Aisner, J.; et al. First-in-Human Clinical Trial of Oral ONC201 in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2017, 23, 4163–4169. [Google Scholar] [CrossRef]
- Chi, A.; Arrillaga-Romany, I.; Gardner, S.; Wen, P.; Batchelor, T.; Hall, M.; Odia, Y.; Khatua, S.; Zaky, W.; McGovern, S.; et al. ACTR-34. Integrated clinical experience with ONC201 in previously-treated H3 K27M-mutant glioma patients. Neuro-Oncology 2018, 20, vi19. [Google Scholar] [CrossRef][Green Version]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-Specific T Cells Engineered to Coexpress Tumor-Specific Receptors: Persistence and Antitumor Activity in Individuals with Neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor Activity and Long-Term Fate of Chimeric Antigen Receptor–Positive T Cells in Patients with Neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Thomas, S.; Straathof, K.; Himoudi, N.; Anderson, J.; Pule, M. An Optimized GD2-Targeting Retroviral Cassette for More Potent and Safer Cellular Therapy of Neuroblastoma and Other Cancers. PLoS ONE 2016, 11, e0152196. [Google Scholar] [CrossRef]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-Trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent Antitumor Efficacy of Anti-GD2 CAR T Cells in H3-K27M+ Diffuse Midline Gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T Cell Therapy for H3K27M-Mutated Diffuse Midline Gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Name of the Tumor Entity | Major Event | Associated Events | Age at Diagnosis [Years] | Median OS [Months] |
|---|---|---|---|---|
| Diffuse midline glioma, H3K27-altered | H3.3K27M | TP53, PDGFRA | 7 | 11 |
| H3.1K27M | ACVR1, PIK3CA | 5 | 15 | |
| EZHIP overexpression | ACVR1, PIK3CA | 10 | 16 | |
| Diffuse hemispheric glioma, H3G34- mutant | H3.3G34R | TP53, ATRX, PDGFRA | 15 | 18 |
| H3.3G34V | ||||
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype | MYCN | 14 | ||
| ø | PDGFRA | 10 | 21 | |
| EGFR, CDKN2A/B | 44 | |||
| Infant-type hemispheric glioma | ALK, ROS1, NTRK1/2/3 | ø | 0.23 | 23 |
| or MET fusions |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huchedé, P.; Leblond, P.; Castets, M. The Intricate Epigenetic and Transcriptional Alterations in Pediatric High-Grade Gliomas: Targeting the Crosstalk as the Oncogenic Achilles’ Heel. Biomedicines 2022, 10, 1311. https://doi.org/10.3390/biomedicines10061311
Huchedé P, Leblond P, Castets M. The Intricate Epigenetic and Transcriptional Alterations in Pediatric High-Grade Gliomas: Targeting the Crosstalk as the Oncogenic Achilles’ Heel. Biomedicines. 2022; 10(6):1311. https://doi.org/10.3390/biomedicines10061311
Chicago/Turabian StyleHuchedé, Paul, Pierre Leblond, and Marie Castets. 2022. "The Intricate Epigenetic and Transcriptional Alterations in Pediatric High-Grade Gliomas: Targeting the Crosstalk as the Oncogenic Achilles’ Heel" Biomedicines 10, no. 6: 1311. https://doi.org/10.3390/biomedicines10061311
APA StyleHuchedé, P., Leblond, P., & Castets, M. (2022). The Intricate Epigenetic and Transcriptional Alterations in Pediatric High-Grade Gliomas: Targeting the Crosstalk as the Oncogenic Achilles’ Heel. Biomedicines, 10(6), 1311. https://doi.org/10.3390/biomedicines10061311