
The Role of Vitamin D in Alzheimer’s Disease: A Transcriptional Regulator of Amyloidopathy and Gliopathy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Tissue collection
2.3. ELISA
2.4. qRT-PCR
2.5. Behavioral Test
2.6. Quantification and Statistical Analysis
3. Results
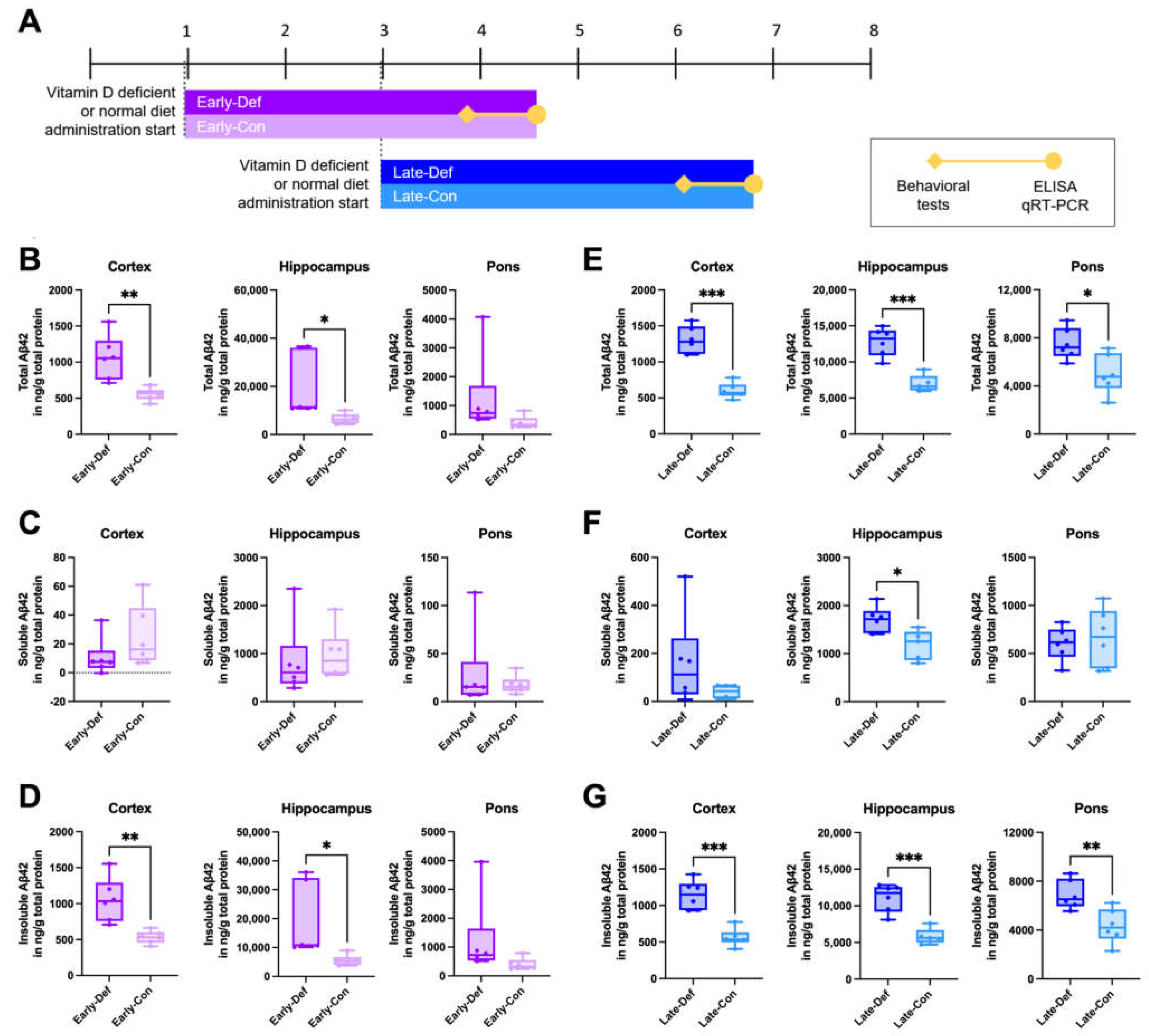
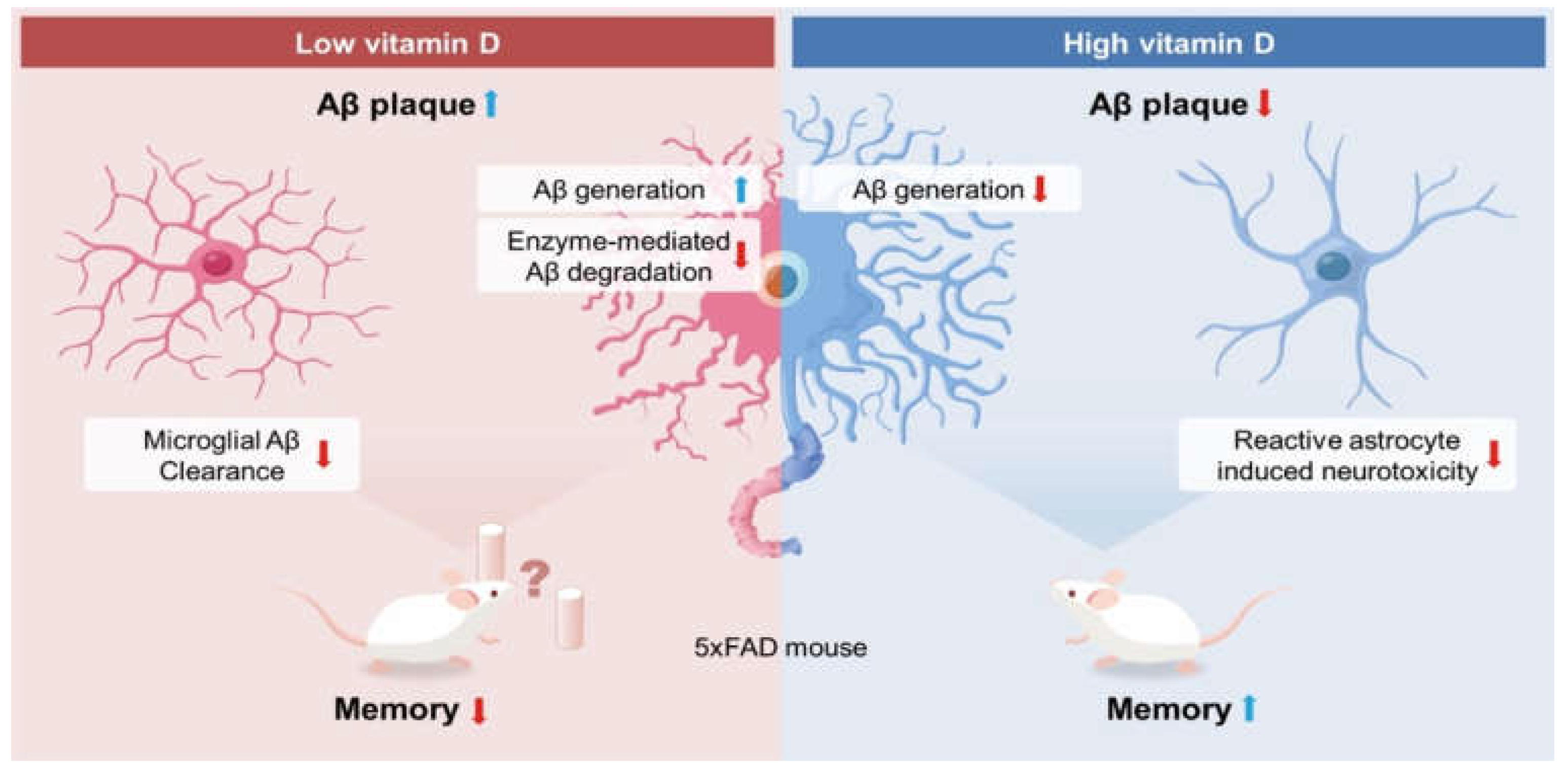
3.1. Vitamin D Deficiency Increased the Brain Aβ Load
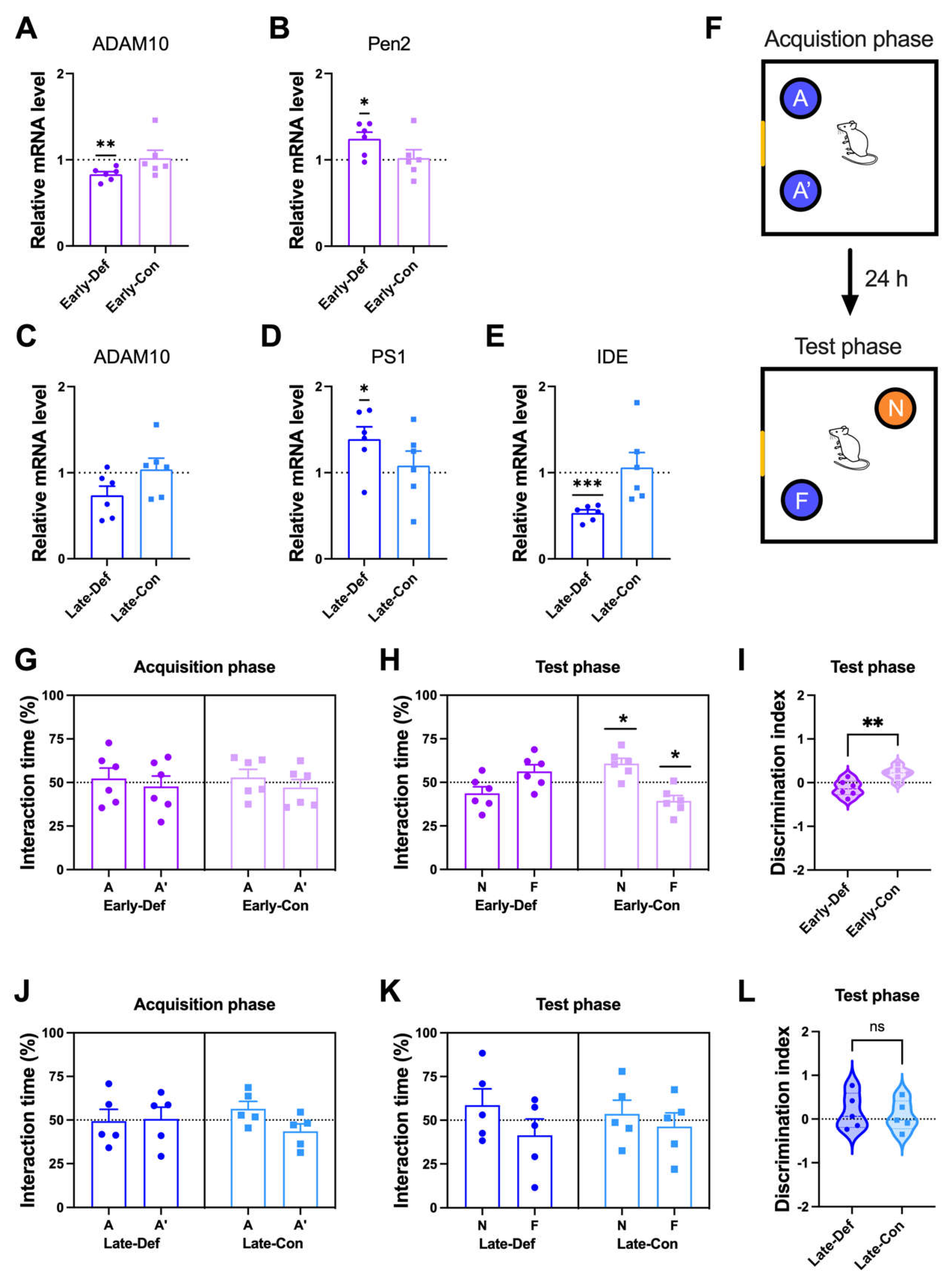
3.2. Vitamin D Deficiency Accelerated the Pathological Aβ Process and Reduced Aβ Degradation
3.3. Vitamin D Deficiency in the Early Stage Induced Memory Impairment
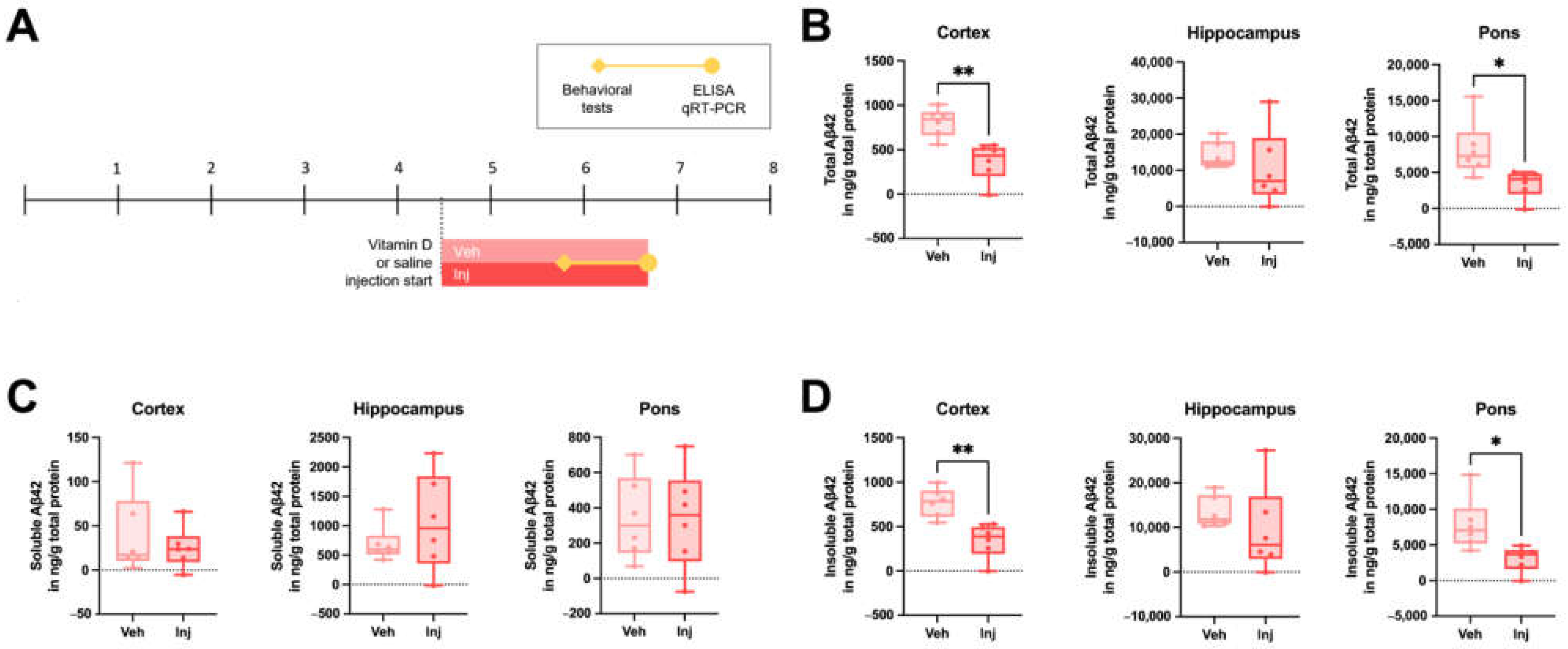
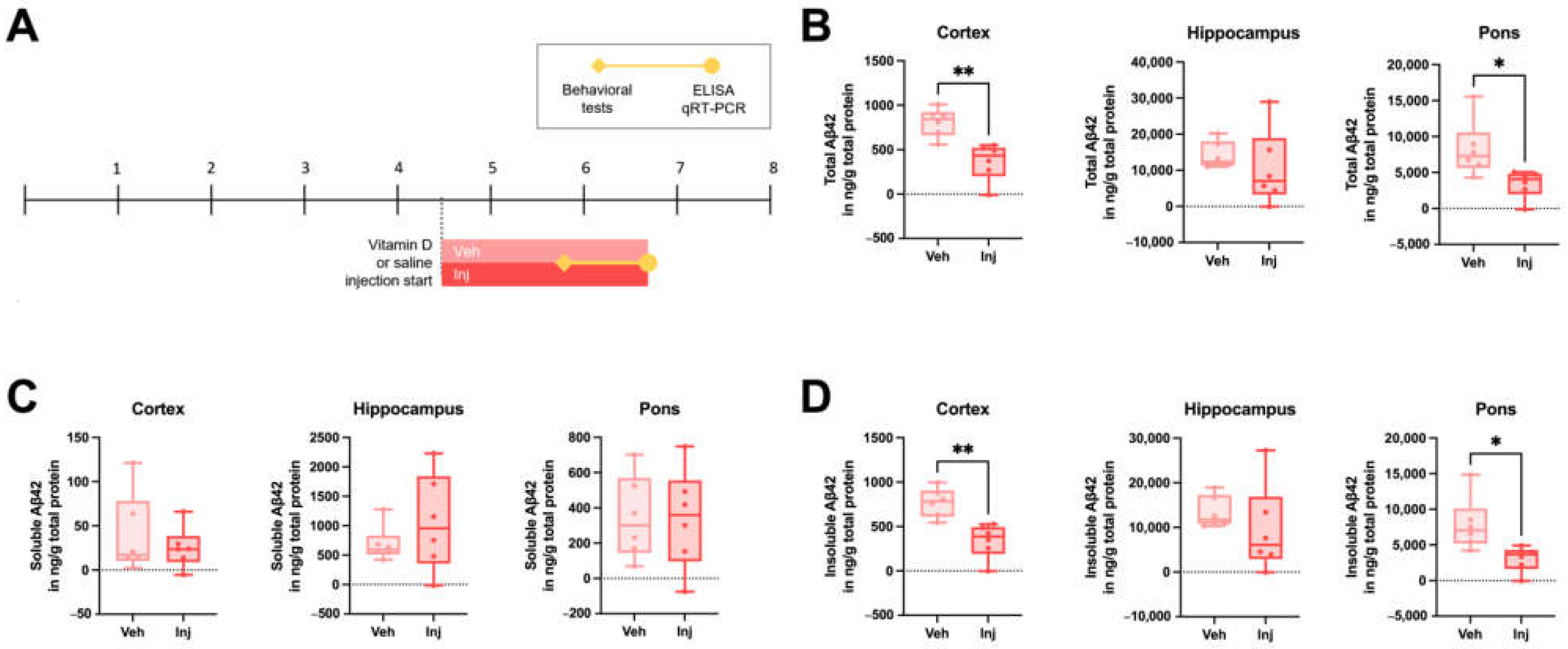
3.4. Vitamin D Injection in the Late-Stage Ameliorated the Brain Aβ Load
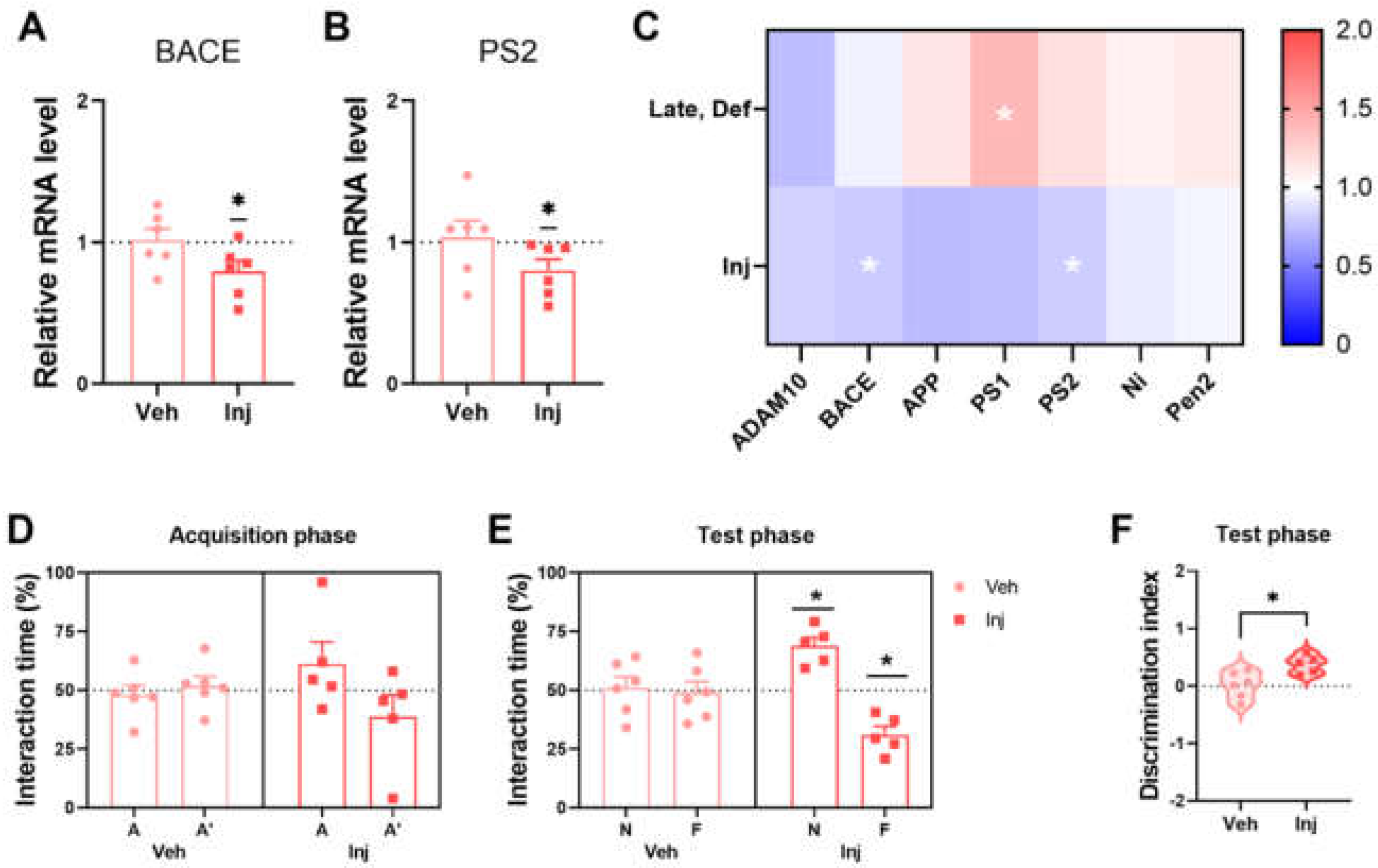
3.5. Vitamin D Injection Reduced the Pathological Aβ Process
3.6. Vitamin D Injection in the Late-Stage Restored Normal Memory Function
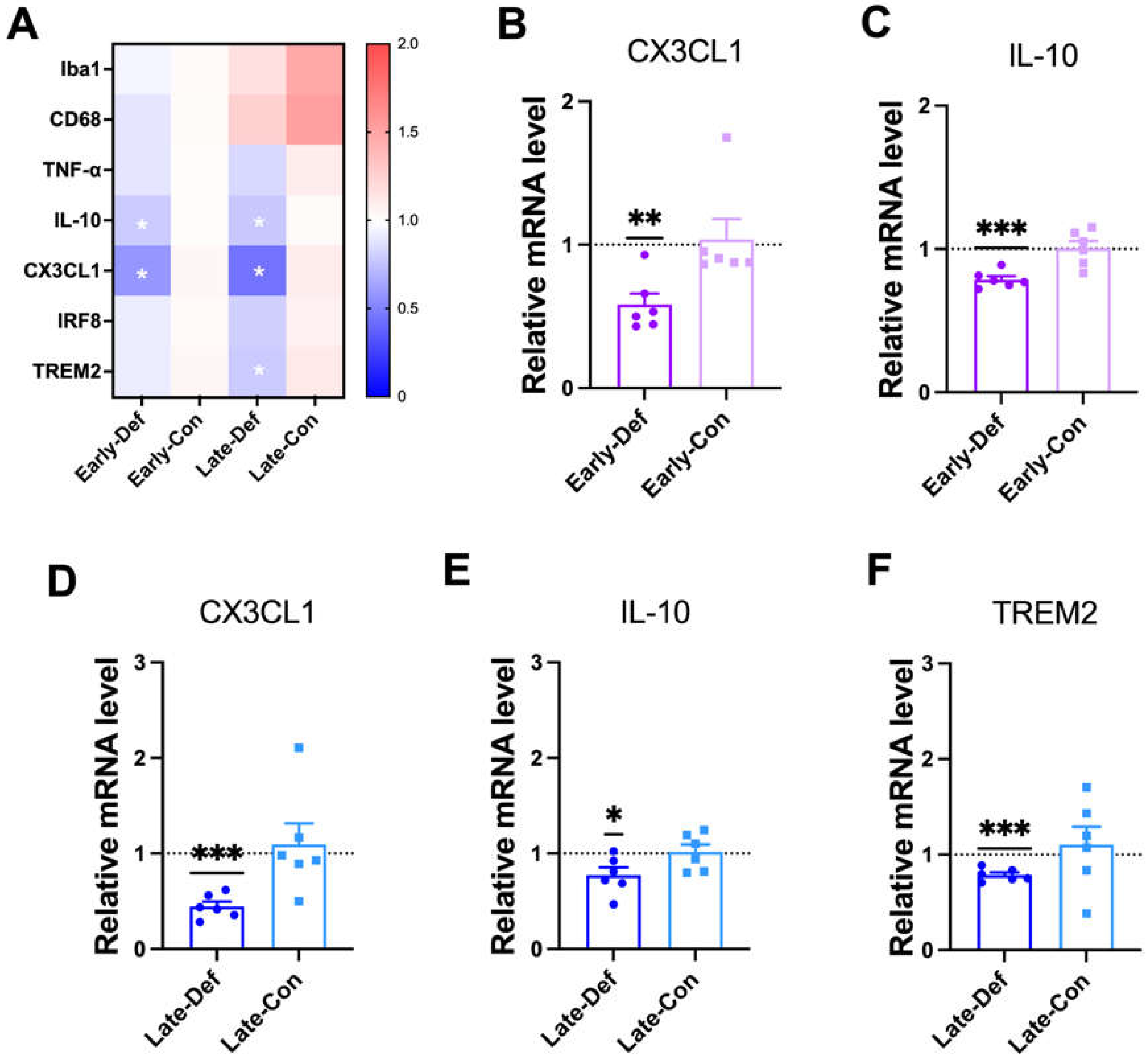
3.7. Vitamin D Deficiency Decreased Microglia-Related Aβ Clearance
3.8. Vitamin D Injection Attenuated Reactive Astrocytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afzal, S.; Bojesen, S.E.; Nordestgaard, B.G. Reduced 25-hydroxyvitamin D and risk of Alzheimer’s disease and vascular dementia. Alzheimer’s Dement. 2014, 10, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ji, H.F. Vitamin D deficiency is associated with increased risk of Alzheimer’s disease and dementia: Evidence from meta-analysis. Nutr. J. 2015, 14, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littlejohns, T.J.; Henley, W.E.; Lang, I.A.; Annweiler, C.; Beauchet, O.; Chaves, P.H.; Fried, L.; Kestenbaum, B.R.; Kuller, L.H.; Langa, K.M.; et al. Vitamin D and the risk of dementia and Alzheimer disease. Neurology 2014, 83, 920–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annweiler, C.; Llewellyn, D.J.; Beauchet, O. Low serum vitamin D concentrations in Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2013, 33, 659–674. [Google Scholar] [CrossRef] [Green Version]
- Chai, B.; Gao, F.; Wu, R.; Dong, T.; Gu, C.; Lin, Q.; Zhang, Y. Vitamin D deficiency as a risk factor for dementia and Alzheimer’s disease: An updated meta-analysis. BMC Neurol. 2019, 19, 284. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, Y.; Ji, H.F.; Shen, L. Vitamin D levels in Alzheimer’s and Parkinson’s diseases: A meta-analysis. Nutrition 2013, 29, 828–832. [Google Scholar] [CrossRef]
- Balion, C.; Griffith, L.E.; Strifler, L.; Henderson, M.; Patterson, C.; Heckman, G.; Llewellyn, D.J.; Raina, P. Vitamin D, cognition, and dementia: A systematic review and meta-analysis. Neurology 2012, 79, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Koduah, P.; Paul, F.; Dorr, J.M. Vitamin D in the prevention, prediction and treatment of neurodegenerative and neuroinflammatory diseases. EPMA J. 2017, 8, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Feige, J.; Moser, T.; Bieler, L.; Schwenker, K.; Hauer, L.; Sellner, J. Vitamin D Supplementation in Multiple Sclerosis: A Critical Analysis of Potentials and Threats. Nutrients 2020, 12, 783. [Google Scholar] [CrossRef] [Green Version]
- Pignolo, A.; Mastrilli, S.; Davi, C.; Arnao, V.; Aridon, P.; Dos Santos Mendes, F.A.; Gagliardo, C.; D’Amelio, M. Vitamin D and Parkinson’s Disease. Nutrients 2022, 14, 1220. [Google Scholar] [CrossRef]
- Bivona, G.; Gambino, C.M.; Iacolino, G.; Ciaccio, M. Vitamin D and the nervous system. Neurol. Res. 2019, 41, 827–835. [Google Scholar] [CrossRef]
- Garcion, E.; Wion-Barbot, N.; Montero-Menei, C.N.; Berger, F.; Wion, D. New clues about vitamin D functions in the nervous system. Trends Endocrinol. Metab. 2002, 13, 100–105. [Google Scholar] [CrossRef]
- Llewellyn, D.J.; Lang, I.A.; Langa, K.M.; Muniz-Terrera, G.; Phillips, C.L.; Cherubini, A.; Ferrucci, L.; Melzer, D. Vitamin D and risk of cognitive decline in elderly persons. Arch. Intern. Med. 2010, 170, 1135–1141. [Google Scholar] [CrossRef]
- Landel, V.; Annweiler, C.; Millet, P.; Morello, M.; Feron, F. Vitamin D, Cognition and Alzheimer’s Disease: The Therapeutic Benefit is in the D-Tails. J. Alzheimer’s Dis. 2016, 53, 419–444. [Google Scholar] [CrossRef] [Green Version]
- Latimer, C.S.; Brewer, L.D.; Searcy, J.L.; Chen, K.C.; Popovic, J.; Kraner, S.D.; Thibault, O.; Blalock, E.M.; Landfield, P.W.; Porter, N.M. Vitamin D prevents cognitive decline and enhances hippocampal synaptic function in aging rats. Proc. Natl. Acad. Sci. USA 2014, 111, E4359–E4366. [Google Scholar] [CrossRef] [Green Version]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D and brain health: The need for vitamin D supplementation and sensible sun exposure. J. Intern. Med. 2015, 277, 90–93. [Google Scholar] [CrossRef]
- Cui, X.; Gooch, H.; Petty, A.; McGrath, J.J.; Eyles, D. Vitamin D and the brain: Genomic and non-genomic actions. Mol. Cell Endocrinol. 2017, 453, 131–143. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell Sci. 2000, 113, 1857–1870. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimer’s Dis. 2001, 3, 75–80. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Thiel, A.; Lauer, A.A.; Winkler, J.; Lehmann, J.; Regner, L.; Nelke, C.; Janitschke, D.; Benoist, C.; Streidenberger, O.; et al. Vitamin D and Its Analogues Decrease Amyloid-beta (Abeta) Formation and Increase Abeta-Degradation. Int. J. Mol. Sci. 2017, 18, 2764. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.Z.; Sun, S.; Tan, C.C.; Yu, J.T.; Tan, L. The Role of ADAM10 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 303–322. [Google Scholar] [CrossRef]
- Grimm, M.O.; Lehmann, J.; Mett, J.; Zimmer, V.C.; Grosgen, S.; Stahlmann, C.P.; Hundsdorfer, B.; Haupenthal, V.J.; Rothhaar, T.L.; Herr, C.; et al. Impact of Vitamin D on amyloid precursor protein processing and amyloid-beta peptide degradation in Alzheimer’s disease. Neurodegener. Dis. 2014, 13, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Boontanrart, M.; Hall, S.D.; Spanier, J.A.; Hayes, C.E.; Olson, J.K. Vitamin D3 alters microglia immune activation by an IL-10 dependent SOCS3 mechanism. J. Neuroimmunol. 2016, 292, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Xu, P.; Li, G.; Qiao, Y.; Han, W.; Geng, C.; Liao, D.; Yang, M.; Chen, D.; Jiang, P. Vitamin D receptor activation regulates microglia polarization and oxidative stress in spontaneously hypertensive rats and angiotensin II-exposed microglial cells: Role of renin-angiotensin system. Redox Biol. 2019, 26, 101295. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, L.R.C.; Mimura, L.A.N.; Fraga-Silva, T.F.C.; Ishikawa, L.L.W.; Fernandes, A.A.H.; Zorzella-Pezavento, S.F.G.; Sartori, A. Calcitriol Prevents Neuroinflammation and Reduces Blood-Brain Barrier Disruption and Local Macrophage/Microglia Activation. Front. Pharmacol. 2020, 11, 161. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Lee, C.J. Reactive astrocytes in Alzheimer’s disease: A double-edged sword. Neurosci. Res. 2018, 126, 44–52. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Jiao, K.P.; Li, S.M.; Lv, W.Y.; Jv, M.L.; He, H.Y. Vitamin D3 repressed astrocyte activation following lipopolysaccharide stimulation in vitro and in neonatal rats. Neuroreport 2017, 28, 492–497. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Kearns, M.D.; Alvarez, J.A.; Tangpricha, V. Large, single-dose, oral vitamin D supplementation in adult populations: A systematic review. Endocr. Pract. 2014, 20, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Seibenhener, M.L.; Wooten, M.C. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J. Vis. Exp. 2015, 96, e52434. [Google Scholar] [CrossRef] [Green Version]
- Komada, M.; Takao, K.; Miyakawa, T. Elevated plus maze for mice. J. Vis. Exp. 2008, 22, e1088. [Google Scholar] [CrossRef] [Green Version]
- Denninger, J.K.; Smith, B.M.; Kirby, E.D. Novel Object Recognition and Object Location Behavioral Testing in Mice on a Budget. J. Vis. Exp. 2018, 141, e58593. [Google Scholar] [CrossRef]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A potential new target for inflammatory diseases. Mol. Interv. 2010, 10, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Horiuchi, M.; Wakayama, K.; Itoh, A.; Kawai, K.; Pleasure, D.; Ozato, K.; Itoh, T. Interferon regulatory factor 8/interferon consensus sequence binding protein is a critical transcription factor for the physiological phenotype of microglia. J. Neuroinflamm. 2012, 9, 227. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Coppola, K.; Schweig, J.E.; Crawford, F.; Mullan, M.; Paris, D. Distinct Signaling Pathways Regulate TREM2 Phagocytic and NFkappaB Antagonistic Activities. Front. Cell Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef] [Green Version]
- Calvello, R.; Cianciulli, A.; Nicolardi, G.; De Nuccio, F.; Giannotti, L.; Salvatore, R.; Porro, C.; Trotta, T.; Panaro, M.A.; Lofrumento, D.D. Vitamin D Treatment Attenuates Neuroinflammation and Dopaminergic Neurodegeneration in an Animal Model of Parkinson’s Disease, Shifting M1 to M2 Microglia Responses. J. Neuroimmune Pharmacol. 2017, 12, 327–339. [Google Scholar] [CrossRef]
- Ouma, S.; Suenaga, M.; Bolukbasi Hatip, F.F.; Hatip-Al-Khatib, I.; Tsuboi, Y.; Matsunaga, Y. Serum vitamin D in patients with mild cognitive impairment and Alzheimer’s disease. Brain Behav. 2018, 8, e00936. [Google Scholar] [CrossRef] [Green Version]
- Shih, E.J.; Lee, W.J.; Hsu, J.L.; Wang, S.J.; Fuh, J.L. Effect of vitamin D on cognitive function and white matter hyperintensity in patients with mild Alzheimer’s disease. Geriatr. Gerontol. Int. 2020, 20, 52–58. [Google Scholar] [CrossRef]
- Lee, Y.A.; Yoon, S.; Kim, S.; Youn, Y.C. Association of 25-hydroxyvitamin D status with brain volume changes. Food Sci. Nutr. 2021, 9, 4169–4175. [Google Scholar] [CrossRef]
- Perin, S.; Lai, J.; Pase, M.; Bransby, L.; Buckley, R.; Yassi, N.; Pietrzak, R.H.; Maruff, P.; Lim, Y.Y. Elucidating the association between depression, anxiety, and cognition in middle-aged adults: Application of dimensional and categorical approaches. J. Affect. Disord. 2022, 296, 559–566. [Google Scholar] [CrossRef]
- Gulpers, B.; Ramakers, I.; Hamel, R.; Kohler, S.; Oude Voshaar, R.; Verhey, F. Anxiety as a Predictor for Cognitive Decline and Dementia: A Systematic Review and Meta-Analysis. Am. J. Geriatr. Psychiatry 2016, 24, 823–842. [Google Scholar] [CrossRef]
- Mendez, M.F. The Relationship Between Anxiety and Alzheimer’s Disease. J. Alzheimer’s Dis. Rep. 2021, 5, 171–177. [Google Scholar] [CrossRef]
- Yang, T.; Wang, H.; Xiong, Y.; Chen, C.; Duan, K.; Jia, J.; Ma, F. Vitamin D Supplementation Improves Cognitive Function Through Reducing Oxidative Stress Regulated by Telomere Length in Older Adults with Mild Cognitive Impairment: A 12-Month Randomized Controlled Trial. J. Alzheimer’s Dis. 2020, 78, 1509–1518. [Google Scholar] [CrossRef]
- Santabarbara, J.; Lipnicki, D.M.; Bueno-Notivol, J.; Olaya-Guzman, B.; Villagrasa, B.; Lopez-Anton, R. Updating the evidence for an association between anxiety and risk of Alzheimer’s disease: A meta-analysis of prospective cohort studies. J. Affect. Disord. 2020, 262, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Sestier, M.V.; Doty, K.R.; Town, T. Innate Immunity Fights Alzheimer’s Disease. Trends Neurosci. 2015, 38, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landel, V.; Baranger, K.; Virard, I.; Loriod, B.; Khrestchatisky, M.; Rivera, S.; Benech, P.; Feron, F. Temporal gene profiling of the 5XFAD transgenic mouse model highlights the importance of microglial activation in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2016, 136, 10–17. [Google Scholar] [CrossRef]
- Lefebvre d’Hellencourt, C.; Montero-Menei, C.N.; Bernard, R.; Couez, D. Vitamin D3 inhibits proinflammatory cytokines and nitric oxide production by the EOC13 microglial cell line. J. Neurosci. Res. 2003, 71, 575–582. [Google Scholar] [CrossRef]
- Alessio, N.; Belardo, C.; Trotta, M.C.; Paino, S.; Boccella, S.; Gargano, F.; Pieretti, G.; Ricciardi, F.; Marabese, I.; Luongo, L.; et al. Vitamin D Deficiency Induces Chronic Pain and Microglial Phenotypic Changes in Mice. Int. J. Mol. Sci. 2021, 22, 3604. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef] [Green Version]
- d’Errico, P.; Ziegler-Waldkirch, S.; Aires, V.; Hoffmann, P.; Mezo, C.; Erny, D.; Monasor, L.S.; Liebscher, S.; Ravi, V.M.; Joseph, K.; et al. Microglia contribute to the propagation of Abeta into unaffected brain tissue. Nat. Neurosci. 2022, 25, 20–25. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Lian, H.; Zheng, H. Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J. Neurochem. 2016, 136, 475–491. [Google Scholar] [CrossRef]
- Zou, Y.; Mu, M.; Zhang, S.; Li, C.; Tian, K.; Li, Z.; Li, B.; Wang, W.; Cao, H.; Sun, Q.; et al. Vitamin D3 suppresses astrocyte activation and ameliorates coal dust-induced mood disorders in mice. J. Affect. Disord. 2022, 303, 138–147. [Google Scholar] [CrossRef]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef]
- Calton, E.K.; Keane, K.N.; Soares, M.J. The potential regulatory role of vitamin D in the bioenergetics of inflammation. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 367–373. [Google Scholar] [CrossRef]
- Fernandes de Abreu, D.A.; Eyles, D.; Feron, F. Vitamin D, a neuro-immunomodulator: Implications for neurodegenerative and autoimmune diseases. Psychoneuroendocrinology 2009, 34 (Suppl. S1), S265–S277. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Lagishetty, V.; Misharin, A.V.; Liu, N.Q.; Lisse, T.S.; Chun, R.F.; Ouyang, Y.; McLachlan, S.M.; Adams, J.S.; Hewison, M. Vitamin D deficiency in mice impairs colonic antibacterial activity and predisposes to colitis. Endocrinology 2010, 151, 2423–2432. [Google Scholar] [CrossRef] [Green Version]
- Zosky, G.R.; Berry, L.J.; Elliot, J.G.; James, A.L.; Gorman, S.; Hart, P.H. Vitamin D deficiency causes deficits in lung function and alters lung structure. Am. J. Respir. Crit. Care Med. 2011, 183, 1336–1343. [Google Scholar] [CrossRef]
- Weng, S.; Sprague, J.E.; Oh, J.; Riek, A.E.; Chin, K.; Garcia, M.; Bernal-Mizrachi, C. Vitamin D deficiency induces high blood pressure and accelerates atherosclerosis in mice. PLoS ONE 2013, 8, e54625. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhong, Y.; Shen, X.; Yang, K.; Cai, W. Maternal and early-life vitamin D deficiency enhances allergic reaction in an ovalbumin-sensitized BALB/c mouse model. Food Nutr. Res. 2018, 62, 1401. [Google Scholar] [CrossRef] [Green Version]
- Assa, A.; Vong, L.; Pinnell, L.J.; Avitzur, N.; Johnson-Henry, K.C.; Sherman, P.M. Vitamin D deficiency promotes epithelial barrier dysfunction and intestinal inflammation. J. Infect. Dis. 2014, 210, 1296–1305. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Orientation | Sequence |
|---|---|---|
| GAPDH | Forward | GGAGAAACCTGCCAAGTATG |
| Reverse | CATACCAGGAAATGAGCTTGAC | |
| BACE | Forward | AACGAATTGGCTTTGCTGTC |
| Reverse | AGCCACAGTCTTCCATGTCC | |
| ADAM10 | Forward | CTTCGCCGTTTCTCCTG |
| Reverse | CCAGGAGAGGAGCAGAA | |
| APP | Forward | GAACTACATCACCGCTGTGC |
| Reverse | CGCGGACATACTTCTTTAGC | |
| PS1 | Forward | GGTCGTGGCTACCATTAAGTC |
| Reverse | GCCCACAGTCTCGGTATCTT | |
| PS2 | Forward | CGCTGCTACAAGTTCATCCA |
| Reverse | TGAGCACTTCCCCAAGGTAG | |
| Ni | Forward | CACTATGTGCCATGCAGCTC |
| Reverse | GCTTGATGCTGAAGGTGCTT | |
| Pen2 | Forward | ATTGAACCTGTGCCGGAAGT |
| Reverse | GCCTCTCGGAAGAACCACAA | |
| Iba1 | Forward | GTCCTTGAAGCGAATGCTGG |
| Reverse | CATTCTCAAGATGGCAGATC | |
| CD68 | Forward | TTCACCTTGACCTGCTCTCTC |
| Reverse | GTAGGTTGATTGTCGTCTGCG | |
| TNF-α | Forward | TCTTCTGTCTACTGAACTTCGG |
| Reverse | AAGATGATCTGAGTGTGAGGG | |
| IL-10 | Forward | CGGGAAGACAATAACTGCACCC |
| Reverse | CGGTTAGCAGTATGTTGTCCAGC | |
| CX3CL1 | Forward | CAGCATCGACCGGTACCTT |
| Reverse | GCTGCACTGTCCGGTTGTT | |
| GFAP | Forward | TCCTGGAACAGCAAAACAAG |
| Reverse | CAGCCTCAGGTTGGTTTCAT | |
| MAO-B | Forward | TACTTGGGGACCGAGTGAAGCT |
| Reverse | CCAAAGCAGGTGGAATGGCACT | |
| IRF8 | Forward | GGATATGCCGCCTATGACACA |
| Reverse | CATCCGGCCCATACAACTTAG | |
| NEP | Forward | GATCAGCCTCTCGGTCCTTG |
| Reverse | TGTTTTGGATCAGTCGAGCAG | |
| IDE | Forward | CAAACCTCTCCTTCCAAGTCAGC |
| Reverse | TGTTCTCCGAGGTGCTCTGCAT | |
| TREM2 | Forward | CTACCAGTGTCAGAGTCTCCGA |
| Reverse | CCTCGAAACTCGATGACTCCTC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.; Park, M.; Lee, E.; Jung, J.; Kim, T. The Role of Vitamin D in Alzheimer’s Disease: A Transcriptional Regulator of Amyloidopathy and Gliopathy. Biomedicines 2022, 10, 1824. https://doi.org/10.3390/biomedicines10081824
Kang J, Park M, Lee E, Jung J, Kim T. The Role of Vitamin D in Alzheimer’s Disease: A Transcriptional Regulator of Amyloidopathy and Gliopathy. Biomedicines. 2022; 10(8):1824. https://doi.org/10.3390/biomedicines10081824
Chicago/Turabian StyleKang, Jiseung, Mincheol Park, Eunkyung Lee, Jieun Jung, and Tae Kim. 2022. "The Role of Vitamin D in Alzheimer’s Disease: A Transcriptional Regulator of Amyloidopathy and Gliopathy" Biomedicines 10, no. 8: 1824. https://doi.org/10.3390/biomedicines10081824
APA StyleKang, J., Park, M., Lee, E., Jung, J., & Kim, T. (2022). The Role of Vitamin D in Alzheimer’s Disease: A Transcriptional Regulator of Amyloidopathy and Gliopathy. Biomedicines, 10(8), 1824. https://doi.org/10.3390/biomedicines10081824