
Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke


Abstract
1. Introduction
2. Oxidative Stress in the Pathophysiology of Ischemia/Reperfusion Injuries after Acute Ischemic Stroke
- ‑
- It has the highest metabolic activity per unit weight compared to other organs;
- ‑
- It has low levels of antioxidant enzymes, such as superoxide dismutase, catalase, glutathione peroxidase, heme oxygenase-1;
- ‑
- Upon release, neurotransmitters contribute to cellular calcium overload and, through their metabolism, generate ROS;
- ‑
- Brain cells have a higher membrane surface/cytoplasmic volume ratio, and the plasmalemma is rich in cholesterol, is arranged in lipid rafts, has polyunsaturated fatty acids, and is very susceptible to oxidative damage;
- ‑
- The brain has lower levels of cytochrome c oxidase, leading to increased superoxide generation during adenosine triphosphate (ATP) generation;
- ‑
- Iron, released from damaged cerebral tissue, can catalyze the generation of free radicals.
- ‑
- ROS oxidize, degrade, or cleave proteins, leading to protein aggregation, modifications in ion channel activities, and enzyme inactivation [24].
- ‑
- By attacking the carbon–carbon bonds of polyunsaturated fatty acids, ROS initiate lipid peroxidation, a self-propagating chain of events leading to the generation of unstable lipid radicals which further react with oxygen to form lipid peroxyl radicals [25]. Peroxidation of membrane lipids alters the bi-layer thickness, membrane fluidity, and membrane permeability.
- ‑
- ROS can directly damage deoxyribonucleic acids (DNA) by causing double strand breaks, structural changes, DNA mutations, or protein-DNA cross-links [26].
- ‑
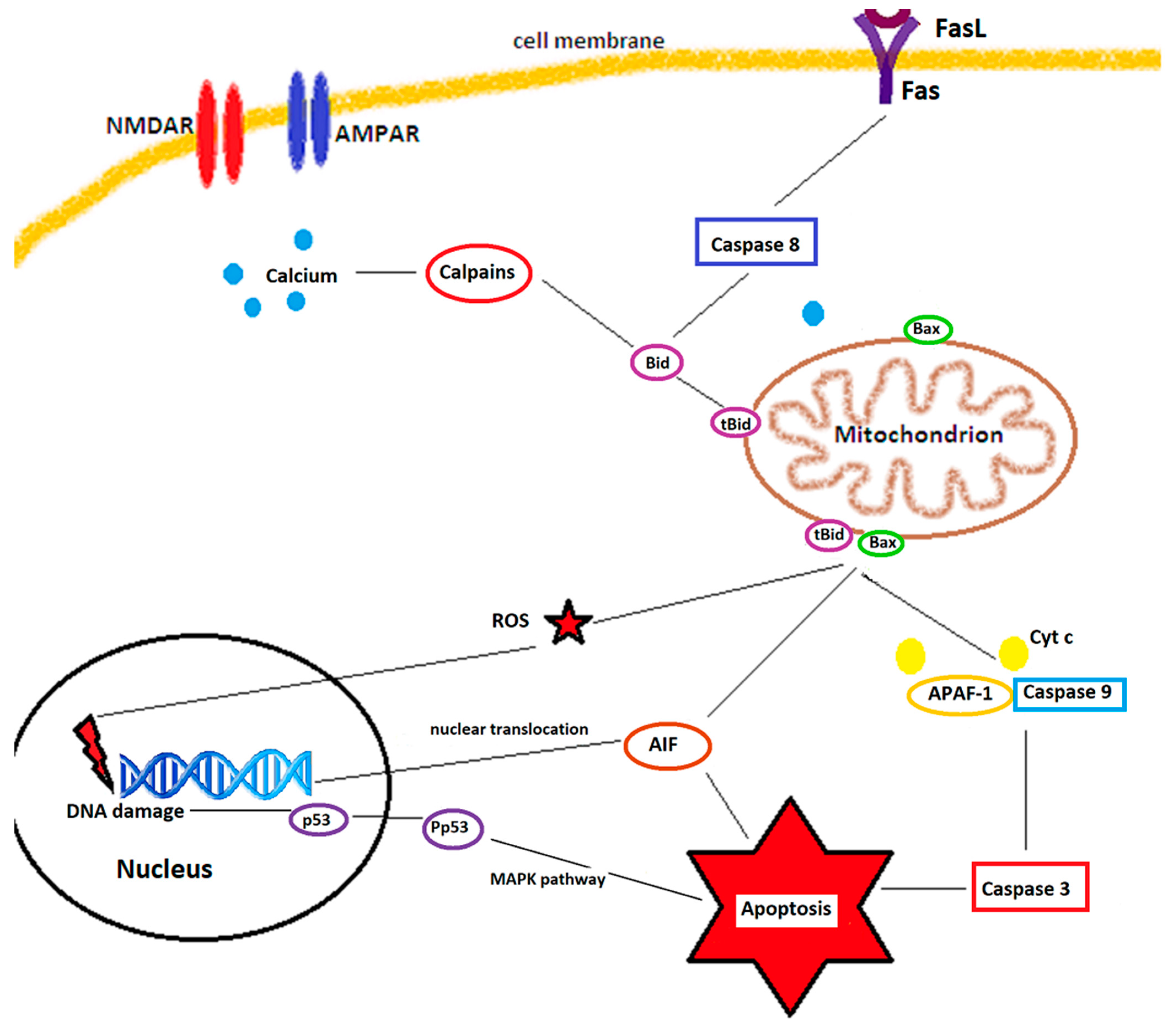
- They also regulate several apoptosis and necrosis signaling cascades. ROS can activate p53, a key molecule in ROS-induced cell death [27], which, in turn, upregulates PUMA (p53 upregulated modulator of apoptosis). ROS can open the mitochondrial permeability transition pore (MPTP), leading to mitochondrial swelling and cytochrome c release, thereby initiating apoptosis [28]. The MAPK (mitogen activated protein kinase) pathway, also triggered by ROS, has 3 main members: c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated kinase 1/2 (ERK 1/2), and p38 MAPK. While ERK 1/2 has a controversial role in cell death and appears to be rather neuroprotective against ischemia/reperfusion injuries [15], JNK and p38 MAPK, activated by ROS through ASK1 (apoptosis signal-regulating kinase 1), significantly contribute to apoptosis during reperfusion after an ischemic insult [29,30].
2.1. Mitochondria as a Source of ROS and Their Implication in Cerebral Ischemia/Reperfusion Injuries
2.2. Nitric Oxide Synthases as Sources of Reactive Oxygen Species
2.3. NADPH Oxidase as a Source of ROS
2.4. Xanthine Oxidase as Source of ROS
3. Antioxidative Signaling Pathways
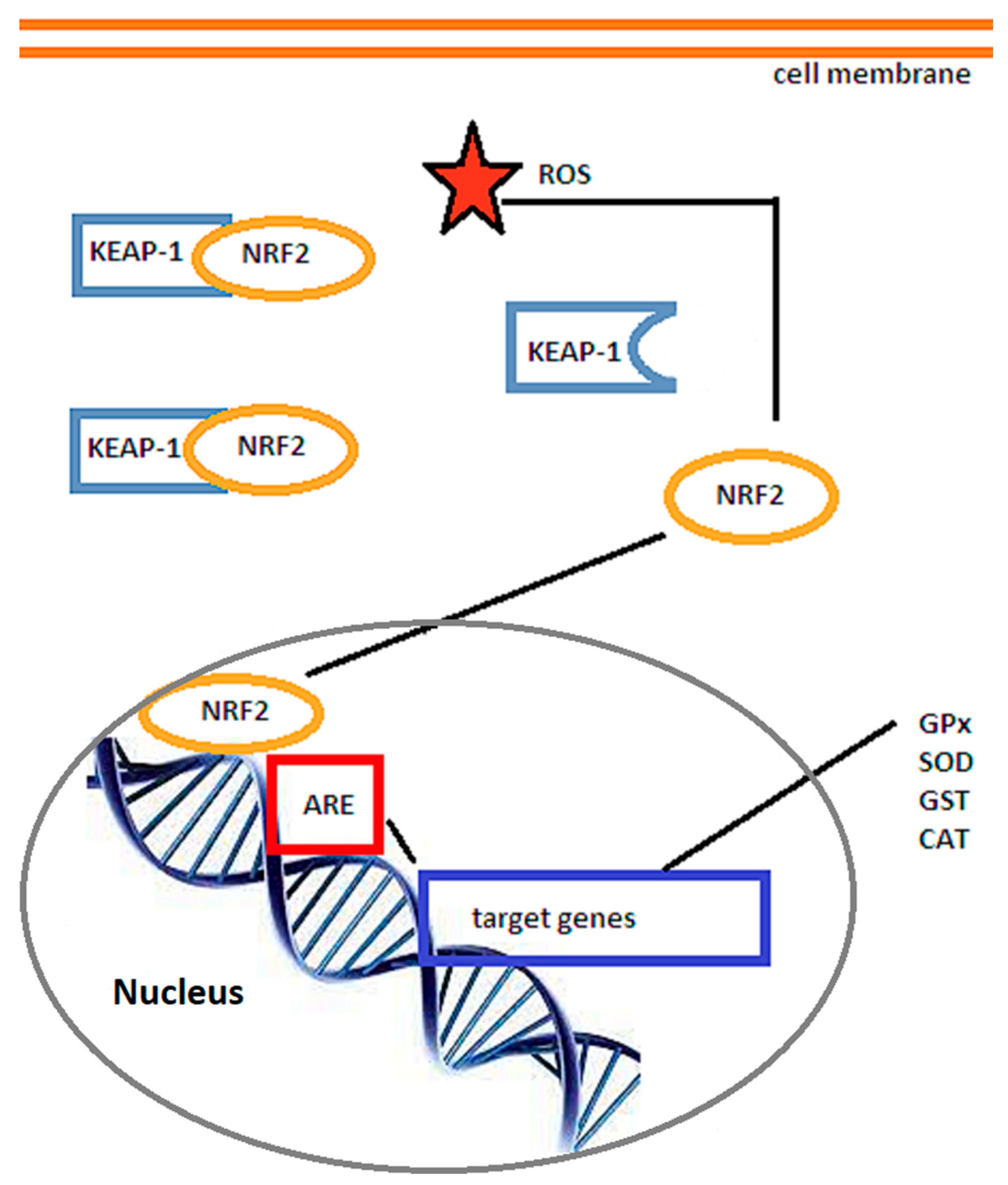
3.1. The Nrf2/ARE Signaling Pathway

3.2. The SIRT/FOXO Signaling Pathway
4. From Bench to Bedside: Translating Knowledge on Oxidative Stress in Ischemic Stroke into Therapeutic Approaches
4.1. Upregulation of Endogenous Antioxidant Defenses
4.2. Preventing the Generation of ROS during Cerebral Ischemia
- NOX inhibition would be one possibility, although it is not yet clear which NOX isoform and which cell types play key roles in ischemia/reperfusion-induced ROS production [95]. NOX 2 promotes the production of superoxide, while NOX4 facilitates hydrogen peroxide production [128,129]. NOX2- and NOX4-deficient mice exhibited decreased infarct size compared to wild-type animals [130].
- 2.
- Xanthine oxidase inhibitor allopurinol has long been used in humans to treat gout. It can also reduce superoxide formation [129]. Despite encouraging results in experimental studies [135], it did not show any benefit in clinical setting [136]. Other small molecules acting as XO inhibitors have been developed and are being tested, such as TEI-6720 [137], BOF-4272 [138], or febuxostat [139].
- 3.
- The generation of ROS by cyclooxygenases (COXs) and lipoxygenases (LOXs) have also been targeted in animal models of stroke. COX-2 knockout in mice and COX-2 inhibition with NS-398 resulted in reduced infarct size after middle cerebral artery occlusion [140,141]. Similarly, 12/15-LOX knockout or inhibition with baicalein or LOXBlock-1 led to reduced infarct size after transient middle cerebral artery occlusion in animal experiments [142,143].
4.3. Free Radical Scavengers
- Lipoic acid (ALA, 1,2-dithiolane-3-pentanoic acid) recycles vitamin E and C [144] and is a free radical scavenger and a co-factor in the mitochondrial dehydrogenase complexes [145]. Pre-treatment with lipoic acid reduced infarct size [146] and improved functional recovery in animal models of stroke [147]. In a retrospective study of 172 thrombolysed acute ischemic stroke patients, 47 of which received alpha lipoic acid 600 mg/day, improved outcome was demonstrated both at 3 months and at one year [148]. Currently, another clinical trial, IMPORTANT, NCT 04041167, is recruiting diabetic acute ischemic stroke patients undergoing reperfusion therapies to assess the effect of the drug on outcome and complication rate [149].
- NXY-059 (disodium 2,4-sulphophenyl-N-tert-butylnitrone, Cerovive), acts by adding a free radical to a nitrone spin trap, resulting in the generation of a spin adduct without the formation of free radicals, thereby terminating the radical chain reaction [129]. Despite reducing infarct size in a rat permanent middle cerebral artery occlusion model [150], improving functional outcome in monkeys [151], and reducing disability at 90 days when given within 6 h after stroke onset to 1722 patients in the SAINT I clinical trial [152], a subsequently conducted phase III clinical trial (SAINT II), which enrolled 3306 patients, failed to show clinical efficacy [153]. The discrepancy in the results was attributed to the poor BBB availability of NXY-059 [154] as well as to statistical weakness of the SAINT I trial [155].
- Tirilazad (U-74006F) is a potent inhibitor of oxygen free radical-induced lipid peroxidation in microvascular and nervous tissue [156]. A meta-analysis of several experimental studies found that the molecule reduced infarct volume by almost a third and improved neurobehavioral score by almost 50% [157]. As such, the RANTTAS trial began enrolling 660 patients who were administered tirilazad 6 mg/kg/day for 3 days, started within 12 h from stroke onset, with disability measured at 90 days by the Glasgow Outcome Scale and Barthel Index set as primary outcome. The trial was prematurely terminated due to lack of efficacy [158]. A second trial, using higher doses (12–15 mg/kg/day) and given within 4 h from symptom onset [159], was stopped prematurely due to safety concerns raised by TESS II, a parallel study running in Europe. A meta-analysis of trials with tirilazad concluded that the drug actually increases death and disability after ischemic stroke [160].
- N-acetyl cysteine is a glutathione precursor with a free thiol group, through which it can react with ROS [161]. In rat stroke models, it reduced infarct size and improved neurological score [162]. After reports of the beneficial effects of oral N-acetylcysteine in ischemic stroke [163], two phase 2 clinical trials (NCT 04918719 and NCT 04920448), with the drug administered intravenously, are planned but are not yet recruiting [149].
- Citicoline is a natural compound which, by stabilizing cell membranes and preventing lipid peroxidation, acts as an antioxidant [164]. As with other antioxidants, it showed efficacy in animal models by reducing lesion volume [165]. However, the ICTUS phase 3 trial (NCT 00331890), the only citicoline trial listed with published results [149], failed to show efficacy in moderate-to-severe acute ischemic stroke [166].
- Edaravone (5-methyl-2-phenyl-4H-pyrazol-3-one, or MCI-186) is a lipophilic free radical scavenger able to scavenge superoxide, hydroxyl, and peroxide radicals [167] and has been approved in Asia for the treatment of acute ischemic stroke since 2002 [23]. In experimental settings, it reduces MMP-9 activation and recombinant tissue plasminogen activator-induced blood–brain barrier damage in rodents [168], raising the possibility of extending the time window for thrombolysis. Indeed, in a clinical trial, edaravone improved the outcome of acute ischemic stroke patients if given simultaneously with reperfusion therapy, with 80% of patients showing “remarkable” or “good” recovery as compared to patients who received edaravone after alteplase [169]. A recent study which enrolled over 10,000 patients with acute ischemic stroke showed that edaravone given within 48 h after endovascular revascularization was associated with greater functional independence at hospital discharge, lower in-hospital mortality, and reduced intracranial bleeding after admission [170]. A clinical trial including edaravone administered in a cocktail, together with dexamethasone and argatroban, 30 to 60 min after thrombectomy (NCT 04202549) is ongoing, while another trial, NCT04817527, or EXISTENT, will explore the safety and efficacy of edaravone dexborneol for patients with acute ischemic stroke receiving endovascular therapy in extended time windows, but is currently not recruiting patients [149].
- Melatonin is an endogenous molecule synthesized in the pineal gland. It has been shown to be able to efficiently scavenge oxygen-centered free radicals, inhibit oxidative damage of biological molecules [171], and upregulate antioxidant defenses such as glutathione reductase, glutathione peroxidase, catalase, and SOD [172]. The efficacy of 14 mg melatonin daily supplementation in acute ischemic stroke was planned to be evaluated in a phase 4 clinical trial (NCT 01863277), but the current status of the study is unknown [149].
4.4. Degradation of Free Radicals
- Ebselen reacts with peroxynitrite radicals and inhibits glutathione peroxidase-like activity [129]. In rodent stroke models, Ebselen reduced lesion size and improved recovery [173], although if administered after onset of ischemia, the protective effect was more modest [174]. In a clinical setting, 302 patients who received Ebselen within 48 h from stroke onset and continued for 2 weeks showed a slightly better outcome at 1 month, but the difference between the active and placebo arm at 3 months failed to reach statistical significance [175].
- Lubeluzole inhibits the glutamate-mediated nitric oxide synthase pathway, thereby reducing NO levels and peroxynitrite production [176]. In animal models, lubeluzole was able to reduce infarct size by 50% when given 15 min, and by one third when given 30 min after ischemia onset [177]. However, in clinical trials Lubeluzole failed to improve outcome and caused heart conduction disorders and Q-T prolongation [178,179].
4.5. Mitochondria-Targeted Antioxidants
- Another mitochondria-targeted antioxidant is Mito-Q10, which is able to mildly uncouple mitochondrial respiration and phosphorylation and, thereby, reduce the mitochondrial generation of ROS as well as protect mitochondria from oxidative damage caused by hydrogen peroxide [183]. Although intensely investigated in cardiovascular disease and neurodegenerative diseases, it has not been evaluated in ischemic stroke.
- Preventing MPTP opening with cyclophilin D was the subject of intense research in the 1990s for attenuating ischemia/reperfusion injuries in the liver, heart, and brain, but the risks often outweigh the benefits [184]. However, more recently, cyclosporin A analogues, such as alisporivir/debio-025, sanglifehrin A, or NIM811, are being tested in animal models for attenuated ischemia/reperfusion injuries in myocardium, liver, or brain [185].
4.6. Novel Experimental Approaches
- 1.
- The inhalation of gases is appealing because gases are able to rapidly penetrate biological membranes and diffuse into the cytosol, mitochondria, and even the nucleus [186]. Beneficial effects in terms of infarct size were obtained with hydrogen gas in transient middle cerebral artery occlusion [186], normobaric oxygen [187], or inhalation of nitric oxide [188].
- 2.
- 3.
- Nrf2-activating drugs, such as omaveloxolone or auranofin, are currently being evaluated. Nrf2 is one of the most important transcription factors, with pleiotropic actions [192] and would thereby induce endogenous cytoprotective pathways. In humans, omaveloxolone increased its plasma concentration 1 h after administration without significant side effects except for upper respiratory tract infections, nasopharyngitis, diarrhoea, nausea, and fatigue [193]. Auranofin is an oral anti-arthritic drug with Nrf2-dependent antioxidative action [194]. Sulforaphane, present in cruciferous vegetables such as broccoli, is also a potent Nrf2 stimulator, although these multi-targeted dietary antioxidants have a diminished bioavailability [195]. Research has revealed that Nrf2 activity peaked at 8 h after transient middle cerebral artery occlusion in the peri-infarct zone [196], followed by expression of down-stream antioxidants such as glutathione, thioredoxin, or heme oxygenase 1, which increased 24–72 h after reperfusion [196]. Brain edema and neuronal death were significantly decreased by Nrf2-inducing drugs in mouse and rat stroke models [100].
- 4.
- Upregulation of SIRT1 has also been shown to be neuroprotective against cerebral ischemia/reperfusion injuries [197]. Resveratrol was shown to upregulate the Sirt1-PGC-1α signaling pathways [198] as well as the expression of brain-derived neurotrophic factor (BDNF) [199]. However, it also has a limited ability to cross the BBB [195]. Similarly, SIRT6 overexpression in the brain of mice through gene transfer was able to reduce ischemia/reperfusion tissue damage, oxidative stress, and neurological deficits through increases of total and nuclear Nrf2 levels [108].
- 5.
- Leptin is a hormone previously studied in relation to energy expenditure and satiety. However, more recently, complex signaling cascades ignited by leptin binding to its receptors have been documented, including the Janus kinase (JAK)/signal transducer and activator of transcription (STAT), Ras/extracellular signal-regulated kinase (ERK)1/2, phosphoinositide-3 kinase (PI3K)/Akt/forkhead box O1, adenosine monophosphate kinase (AMPK) and mTOR/ribosomal protein 6 kinase pathways, through which leptin inhibits the release of presynaptic glutamate (JAK2/PI3K pathway) [200], promotes mitochondrial biogenesis (JAK/STAT pathway) [201], increases SOD levels [202] and has also an anti-inflammatory action (JAK2/STAT3 and p38MAPK/ERK1/2 pathways) [203]. Barriers imposed by drug pharmacokinetics [204] could be overcome by special formulations or synthetic polymers, delivered mainly to the damaged areas.
- 6.
- MicroRNAs (miRNAs) are non-coding RNAs which modulate gene expression at the post-transcriptional level [205]. Research has shown that in experimental middle cerebral artery occlusion in mice, miR-98-5p was downregulated in brain tissue samples, and that upregulation of miR-98-5p enhanced SOD activity and protected against oxidative stress through upregulation of the NAD(P)H quinone oxidoreductase 1 and heme oxygenase 1 levels [206]. In addition, miR-98-5p inhibited apoptosis by increasing Bcl-2 levels and by reducing Bax and cleaved caspase-3 levels [206]. Other microRNAs, such as miR133b [207] or miR-124-3p [208], can modulate brain plasticity and neuroinflammation following ischemic stroke [209], contributing to angiogenesis, neurogenesis, oligodendrogenesis, and astrogenesis, and improve functional recovery if administered in a prolonged time window after the ischemic insult (days to months after stroke) [210]. Technical issues relating to the appropriate timing of mesenchymal stem cell transfer, the route of administration, and the characterization of the stem cells need to be solved [211], but clinical trials have already been carried out on a small scale and show the therapy to be safe and to lead to significant functional improvement [212]. Similar effects were obtained with mesenchymal stem cell-derived extracellular vesicles, which are easier to deliver and contain proteins, lipids, nucleic acids with various forms of RNA, and membrane receptors. They are less immunogenic than stem cells and have lower risk of microvascular thrombosis, are able to cross the blood–brain barrier, and the miRNAs contained can be easily genetically modified [209].
- 7.
- Novel drug design, by dispersing or dissolving the drug within a polymer matrix, entrapping the drug inside lipid vesicles, encapsulating or adsorbing the active molecules on the surface of nanoparticles, are able to improve drug pharmacokinetics, pharmacodynamics and safety, and prevent off-target interactions [213]. Their small size, stability, long serum half-life, and ability to cross the BBB makes them a promising approach to deliver antioxidants in acute ischemic stroke [214], especially since in this setting activation of immune cells and the compromised BBB integrity potentiates the leakage of molecules [215]. The biological activities of nanomaterials, 1–500 nm in size, are related to their size, shape, and surface modifications [216]. They can be subdivided into [215]:
- ‑
- Inorganic nanoparticles, such as cerium oxide nanoparticles, platinum nanoparticles, gold nanoparticles, or superparamagnetic iron oxide nanoparticles;
- ‑
- Carbon allotropes, either spherical (fullerenes) or carbon nanotubes;
- ‑
- Lipid-based nanoparticles, such as liposomes, non-liposomal lipid nanoparticles (polyethylene glycol, i.e., PEGylated lipid nanoparticles, nanostructured lipid carriers, and nanoemulsions);
- ‑
- Polymer-based carriers, including micelles, dendrimers (branched polymers surrounding in repeated layers a central molecule), and nanogels.
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Feigin, V.L.; Lawes, C.M.; Bennett, D.A.; Barker-Collo, S.L.; Parag, V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: A systematic review. Lancet Neurol. 2009, 8, 355–369. [Google Scholar] [CrossRef]
- Danaei, G.; Finucane, M.M.; Lu, Y.; Singh, G.M.; Cowan, M.J.; Paciorek, C.J.; Lin, J.K.; Farzadfar, F.; Khang, Y.H.; Stevens, G.A.; et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980s: Systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet 2011, 378, 31–40. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Li, L.; Scott, C.A.; Rothwell, P.M.; on behalf of the Oxford Vascular Study. Trends in stroke incidence in high-income countries in the 21st century. Population-based study and systematic review. Stroke 2020, 51, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- The National Institute of Neurological Disorders and Stroke rt-PA Study Group. Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 1995, 333, 1581–1588. [Google Scholar] [CrossRef]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Dávalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef]
- Furlan, A.; Higashida, R.; Wechsler, L.; Gent, M.; Rowley, H.; Kase, C.; Pessin, M.; Ahuja, A.; Callahan, F.; Clark, W.M.; et al. Intra-arterial prourokinase for acute ischemic stroke. The PROACT II study: A randomized controlled trial. Prolyse in Acute Cerebral Thromboembolism. JAMA 1999, 282, 2003–2011. [Google Scholar] [CrossRef]
- Alexandrov, A.V.; Köhrmann, M.; Soinne, L.; Tsivgoulis, G.; Barreto, A.D.; Demchuk, A.M.; Sharma, V.K.; Mikulik, R.; Muir, K.W.; Brandt, G.; et al. Safety and efficacy of sonothrombolysis for acute ischaemic stroke: A multicentre, double-blind, phase 3, randomised controlled trial. Lancet Neurol. 2019, 18, 338–347. [Google Scholar] [CrossRef]
- Smith, W.S.; Sung, G.; Starkman, S.; Saver, J.L.; Kidwell, C.S.; Gobin, Y.P.; Lutsep, H.L.; Nesbit, G.M.; Grobelny, T.; Rymer, M.M.; et al. Safety and efficacy of mechanical embolectomy in acute ischemic stroke: Results of the MERCI trial. Stroke 2005, 36, 1432–1438. [Google Scholar] [CrossRef]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, M.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, D.M.; Hoh, B.; et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar]
- De Sousa, D.A.; von Martial, R.; Abilleira, S.; Gattringer, T.; Kobayashi, A.; Gallofre, M.; Fazekas, F.; Szikora, I.; Feigin, V.; Caso, V.; et al. Access to and delivery of acute ischaemic stroke treatments: A survey of national scientific societies and stroke experts in 44 European countries. Eur. Stroke J. 2019, 4, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Rha, J.-H.; Saver, J.L. The impact of recanalization on ischemic stroke outcome: A meta-analysis. Stroke 2007, 38, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A.; Ardelean, I.A. Molecular pathophysiological mechanisms of ischemia/reperfusion injuries after recanalization therapy for acute ischemic stroke. J. Integr. Neurosci. 2021, 20, 727–744. [Google Scholar]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxidative Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell. Mol. Biol. 2012, 298, 229–317. [Google Scholar]
- Simion, A.; Jurcau, A. The role of antioxidant treatment in acute ischemic stroke: Past, present and future. Neurol. Res. Surg. 2019, 2, 1–7. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Cognition, Statins, and Cholesterol in Elderly Ischemic Stroke Patients: A Neurologist’s Perspective. Medicina 2021, 57, 616. [Google Scholar] [CrossRef]
- Polidori, C.M.; Cherubini, A.; Stahl, W.; Senin, U.; Sies, H.; Mecocci, P. Plasma carotenoid and malondialdehyde levels in ischemic stroke patients: Relationship to early outcome. Free Radic. Res. 2002, 36, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Jurcau, A. The role of antioxidant treatment in acute ischemic stroke: A clinical study. Rom. J. Neurol. 2007, 6, 181–188. [Google Scholar]
- Menon, B.; Ramalingam, K.; Kumar, R. Evaluating the Role of Oxidative Stress in Acute Ischemic Stroke. J. Neurosci. Rural Pract. 2020, 11, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.-L.; Guo, Z.-N.; Yang, Y. Free radical damage in ischemia-reperfusion injury: An obstacle in acute ischemic stroke after revascularization therapy. Oxidative Med. Cell Longev. 2018, 2018, 3804979. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, S. Targeting oxidative stress for the treatment of ischemic stroke: Upstream and downstream therapeutic strategies. Brain Circ. 2016, 2, 153–163. [Google Scholar] [PubMed]
- Fucci, L.; Oliver, C.N.; Coon, M.J.; Stadtman, E.R. Inactivation of key metabolic enzymes by mixed-function oxidation reactions: Possible implication in protein turnover and ageing. Proc. Natl. Acad. Sci. USA 1983, 80, 1521–1525. [Google Scholar] [CrossRef]
- Hall, E.D.; Braughler, J.M. Central nervous system trauma and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals in lipid peroxidation. Free Radic. Biol. Med. 1989, 6, 303–313. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Saito, A.; Hayashi, T.; Okuno, S.; Nishi, T.; Chan, P.H. Modulation of p53 degradation via MDM2-mediated ubiquitylation and the ubiquitin–proteasome system during reperfusion after stroke: Role of oxidative stress. J. Cereb. Blood Flow Metab. 2005, 25, 267–280. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Song, J.; Cho, K.J.; Cheon, S.Y.; Kim, H.S.D.; Park, K.A.; Lee, W.T.; Lee, J.E. Apoptosis signal-regulating kinase 1 (ASK1) is linked to neural stem cell differentiation after ischemic brain injury. Exp. Mol. Med. 2013, 45, e69. [Google Scholar] [CrossRef][Green Version]
- Russo, E.; Nguyen, H.; Lippert, T.; Tuazon, J.; Borlongan, C.V.; Napoli, E. Mitochondrial targeting as novel therapy for stroke. Brain Circ. 2018, 4, 84–94. [Google Scholar] [PubMed]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol.-Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Efremov, R.G.; Baradaran, R.; Sazanov, L.A. The architecture of respiratory complex I. Nature 2010, 465, 441–445. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhai, Y.; Lou, J. Thiabendazole inhibits ubiquinone reduction activity of mitochondrial respiratory complex II via a water molecule mediated binding feature. Protein Cell 2011, 2, 531–542. [Google Scholar] [CrossRef]
- Crofts, A.R. The cytochrome bc1 complex: Function in the context of structure. Annu. Rev. Physiol. 2004, 66, 689–733. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qin, L.; Ferguson-Miller, S. Crystallographic and online spectral evidence for role of conformational change and conserved water in cytochrome oxidase proton pump. Proc. Natl. Acad. Sci. USA 2011, 108, 1284–1289. [Google Scholar] [CrossRef]
- Nakamoto, R.K.; Baylis Scanlon, J.A.; Al-Shawi, M.K. The rotary mechanism of the ATP synthase. Arch. Biochem. Biophys. 2008, 476, 43–50. [Google Scholar] [CrossRef]
- Gunter, T.E.; Pfeiffer, D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. Cell Physiol. 1990, 258, C755–C786. [Google Scholar] [CrossRef]
- McKormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hutteman, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef]
- Zaidan, E.; Sims, N.R. The calcium content of mitochondria from brain subregions following short-term forebrain ischemia and recirculation in the rat. J. Neurochem. 1994, 63, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Bender, E.; Kadenbach, B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000, 466, 130–134. [Google Scholar] [CrossRef]
- Liu, R.R.; Murphy, T.H. Reversible cyclosporin A-sensitive mitochondrial depolarization occurs within minutes of stroke onset in mouse somatosensory cortex in vivo: A two-photon imaging study. J. Biol. Chem. 2009, 184, 36109–36117. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S. Cooperation of a “reactive oxygen cycle” with the Q cycle and the proton cycle in the respiratory chain-superoxide generating and cycling mechanisms in mitochondria. J. Bioenerg. Biomembr. 1999, 31, 367–376. [Google Scholar] [CrossRef]
- Yuan, J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis 2009, 14, 469–477. [Google Scholar] [CrossRef]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 15. [Google Scholar] [CrossRef]
- Kohr, G. NMDA receptor function: Subunit composition versus spatial distribution. Cell Tissue Res. 2006, 326, 439–446. [Google Scholar] [CrossRef]
- Chen, M.; Lu, T.J.; Chen, X.-J.; Zhou, Y.; Chen, Q.; Feng, X.-Y.; Xu, L.; Duan, W.-H.; Xiong, Z.-Q. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 2008, 39, 3042–3048. [Google Scholar] [CrossRef]
- Downward, J. How BAD phosphorylation is good for survival. Nat. Cell. Biol. 1999, 1, E33–E35. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Tamatani, M.; Matsuzaki, H.; Namikawa, K.; Kiyama, H.; Vitek, M.P.; Mitsuda, N.; Tohyama, N. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J. Biol. Chem. 2001, 276, 5256–5264. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pellegrino, C.; Rama, S.; Dumalska, I.; Salyha, Y.; Ben-Ari, Y.; Medina, I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J. Physiol. 2006, 572, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Connelly, J.; Levitan, E.S.; Sun, D.; Wang, J.Q. Calcium/calmodulin-dependent protein kinase II in cerebrovascular diseases. Transl. Stroke Res. 2021, 12, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.; Finkelstein, J.P.; Gálvez, J.; Sánchez, G.; Donoso, P.; Behrens, M.I.; Hidalgo, C. Ischemia enhances activation by Ca2+ and redox modification of ryanodine receptor channels from rat brain cortex. J. Neurosci. 2008, 28, 9463–9472. [Google Scholar] [CrossRef]
- Szydlowska, K.; Tymianski, M. Calcium ischemia and excitotoxicity. Cell Calcium 2010, 47, 122–129. [Google Scholar] [CrossRef]
- Ren, M.; Phoon, C.K.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 2014, 55, 1–16. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: Implications for pharmacological cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1341–H1352. [Google Scholar] [CrossRef]
- Kagan, V.E.; Bayir, H.A.; Belikova, N.A.; Kapralov, O.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Stoyanovsky, D.A.; Wipf, P.; Kochanek, P.M.; et al. Cytochrome c/cardiolipin relations in mitochondria: A kiss of death. Free Radic. Biol. Med. 2009, 46, 1439–1453. [Google Scholar] [CrossRef]
- Garcia Fernandez, M.; Troiano, L.; Moretti, L.; Nasi, M.; Pinti, M.; Salvioli, S.; Dobrucki, J.; Cossarizza, A. Early changes in intramitochondrial cardiolipin distribution during apoptosis. Cell Growth Differ. 2002, 13, 449–455. [Google Scholar]
- Robinson, N.C. Functional binding of cardiolipin to cytochrome c oxidase. J. Bioenerg. Biomembr. 1993, 25, 153–163. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Inta, I.; Paxian, I.; Maegele, W.; Zhang, M.; Pizzi, P.; Spano, P.; Sarnico, I.; Muhammad, S.; Herrmann, O.; Inta, D.; et al. Bim and Noxa are candidates to mediate the deleterious effect of the NF-kappa B subunit RelA in cerebral ischemia. J. Neurosci. 2006, 26, 12896–12903. [Google Scholar] [CrossRef] [PubMed]
- Carinci, M.; Vezzani, B.; Patergnani, S.; Ludewig, P.; Lessman, K.; Magnus, T.; Casetta, I.; Pugliatti, M.; Pinton, P.; Giorgi, C. Different roles of mitochondria in cell death and inflammation: Focusing on mitochondrial quality control in ischemic stroke and reperfusion. Biomedicines 2021, 9, 169. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Kroemer, G. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.M.; Bratton, S.B.; Butterworth, M.; MacFarlane, M.; Cohen, G.M. Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J. Biol. Chem. 2002, 277, 11345–11351. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yenari, M.A.; Cheng, D.; Barreto-Chang, O.L.; Sapolsky, R.M.; Steinberg, G.K. Bcl-2 Transfection via Herpes Simplex Virus Blocks Apoptosis-Inducing Factor Translocation after Focal Ischemia in the Rat. Br. J. Pharmacol. 2004, 24, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.A.; Graham, R.M.; Thompson, J.W.; Spiga, M.-G.; Frazier, D.P.; Wilson, A.; Bishopric, N.H. Redox stress and the contributions of BH3-only proteins to infarction. Antioxid. Redox Signal. 2006, 8, 1667–1676. [Google Scholar] [CrossRef]
- Culmsee, C.; Krieglstein, J. Ischaemic brain damage after stroke: New insights into efficient therapeutic strategies. International Symposium on neurodegeneration and neuroprotection. EMBO Rep. 2007, 8, 129–133. [Google Scholar] [CrossRef]
- Chen, S.D.; Lin, T.K.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.V. Roles of PTEN-induced putative kinase 1 and dynamin-related protein 1 in transient global ischemia-induced hippocampal neuronal injury. Biochem. Biophys. Res. Commun. 2015, 460, 397–403. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Grohm, J.; Kim, S.W.; Mamrak, U.; Tobaben, S.; Cassidy-Stone, A.; Nunnari, J.; Plesnila, N.; Culmsee, C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012, 19, 1446–1458. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-L.; Mukda, S.; Chen, S.-D. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Karbowski, M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin. Cell Dev. Biol. 2009, 20, 365–374. [Google Scholar] [CrossRef]
- Li, S.; Sun, X.; Xu, L.; Sun, R.; Ma, Z.; Deng, X.; Liu, B.; Fu, Q.; Qu, R.; Ma, S. Baicalin attenuates in vivo and in vitro hyperglycemia-exacerbated ischemia/reperfusion injury by regulating mitochondrial function in a manner dependent on AMPK. Eur. J. Pharmacol. 2017, 815, 118–126. [Google Scholar] [CrossRef]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-e-Silva, O. Ischemia/reperfusion injury revisited: An overview of the latest pharmacological strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef]
- Ma, S.; Wang, Y.; Chen, Y.; Cao, F. The role of autophagy in myocardial ischemia/reperfusion injury. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 271–276. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.-R.; Wang, X.; Hu, W.-W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Fels, J.A.; Manfredi, G. Sex differences in ischemia/reperfusion injury: The role of mitochondrial permeability transition. Neurochem. Res. 2019, 44, 2336–2345. [Google Scholar] [CrossRef]
- Chen, X.-M.; Chen, H.-S.; Xu, M.-J.; Shen, J.-G. Targeting reactive nitrogen species: A promising therapeutic strategy for cerebral ischemia-reperfusion injury. Acta Pharmacol. Sin. 2013, 34, 67–77. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Oxidative stress in the pathogenesis of Alzheimer’s disease and cerebrovascular disease with therapeutic implications. CNS Neurol. Disord. Drug Targets 2020, 19, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, H.; Diya, J.B.; Shashikumar, S.; Rajanikat, G.K. Oxidative stress-assassin behind the ischemic stroke. Folia Neuropathol. 2012, 50, 219–230. [Google Scholar] [CrossRef]
- Iadecola, C.; Xu, X.; Zhang, F.; El-Fakahany, E.E.; Ross, M.E. Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1995, 15, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, P.L.; Ma, J.; Meng, W.; Ayata, C.; Fishman, M.C.; Moskowitz, M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood Flow Metab. 1996, 16, 981–987. [Google Scholar] [CrossRef]
- Ferriero, D.M.; Holtzman, D.M.; Black, S.M.; Sheldon, R.A. Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury. Neurobiol. Dis. 1996, 3, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Zhang, F.; Casey, R.; Nagayama, M.; Ross, M.E. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J. Neurosci. 1997, 17, 9157–9164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Reif, D.; MacDonald, J.; Tang, W.X.; Kamp, D.K.; Gentile, R.J.; Shakespeare, W.C.; Murray, R.J.; Chopp, M. ARL 17477, a potent and selective neuronal NOS inhibitor decreases infarct volume after transient middle cerebral artery occlusion in rats. J. Cereb. Blood Flow Metab. 1996, 16, 599–604. [Google Scholar] [CrossRef]
- Parmentier, S.; Böhme, G.A.; Lerouet, D.; Damour, D.; Stutzmann, J.M.; Margaill, I.; Plotkine, M. Selective inhibition of inducible nitric oxide synthase prevents ischaemic brain injury. Br. J. Pharmacol. 1999, 127, 546–552. [Google Scholar] [CrossRef]
- Chang, D.I.; Hosomi, N.; Lucero, J.; Heo, J.H.; Abumiya, T.; Mazar, A.; del Zoppo, G.J. Activation systems for latent matrix metalloproteinase-2 are upregulated immediately after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 1408–1419. [Google Scholar] [CrossRef]
- Rosenberg, G.A.; Yang, Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg. Focus 2007, 22, E4. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Wang, D.L. Nitric oxide inhibits matrix metalloproteinase-2 expression via the induction of activating transcription factor 3 in endothelial cells. Mol. Pharmacol. 2004, 65, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Moro, M.A.; Almeida, A.; Bolanos, J.P.; Lizasoain, I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic. Biol. Med. 2005, 39, 1291–1304. [Google Scholar] [CrossRef]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T.S. The role of nitric oxide pathway in brain injury and its treatment—From bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.N.; Cairns, B.; Kim, J.Y.; Yenari, M.A. NADPH oxidase in stroke and cerebrovascular disease. Neurol. Res. 2012, 34, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef]
- Suzuki, G.; Okamoto, K.; Kusano, T.; Matsuda, Y.; Fuse, A.; Yokota, H. Evaluation of neuronal protective effects of xanthine oxidoreductase inhibitors on severe whole-brain ischemia in mouse model and analysis of xanthine oxidoreductase activity in the mouse brain. Neurol. Med.-Chir. 2015, 55, 77–85. [Google Scholar] [CrossRef]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef]
- Wu, L.; Xiong, X.; Wu, X.; Ye, Y.; Jian, Z.; Zhi, Z.; Gu, L. Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front. Mol. Sci. 2020, 13, 28. [Google Scholar] [CrossRef]
- Liu, L.; Locascio, L.M.; Doré, S. Critical role of Nrf2 in experimental ischemic stroke. Front. Pharmacol. 2019, 10, 153. [Google Scholar] [CrossRef]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Mazur, A.; Fangman, M.; Ashouri, R.; Arcenas, A.; Doré, S. Nrf2 as a therapeutic target in ischemic stroke. Exp. Opin. Therap. Targets 2021, 25, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Rojo, A.I.; Velasco, D.; De Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Kim, H.M.; Shin, H.Y.; Jeong, H.J.; An, H.J.; Kim, N.S.; Chae, H.J.; Kim, H.R.; Song, H.J.; Kim, K.Y.; Baek, S.H.; et al. Reduced IL-2 but elevated IL-4, IL-6, and IgE serum levels in patients with cerebral infarction during the acute stage. J. Mol. Neurosci. 2000, 14, 191–196. [Google Scholar] [CrossRef]
- McBean, G.J.; López, M.G.; Wallner, F.K. Redox-based therapeutics in neurodegenerative disease. Br. J. Pharmacol. 2017, 174, 1750–1770. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; Ye, X.; Hao, Q.; Zhang, T.; Cui, G.; Yu, M. Nrf2/ARE pathway inhibits ROS-induced NLRP3 inflammasome activation in BV2 cells after cerebral ischemia reperfusion. Inflamm. Res. 2018, 67, 57–65. [Google Scholar] [CrossRef]
- Zhang, W.; Wei, R.; Zhang, L.; Tan, Y.; Qian, C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through Nrf2 activation. Neuroscience 2017, 366, 95–104. [Google Scholar] [CrossRef]
- Vasconcelos, A.R.; dos Santos, N.B.; Scavone, C.; Munhoz, C.D. Nrf2/ARE pathway modulation by dietary energy regulation in neurological disorders. Front. Pharamacol. 2019, 10, 33. [Google Scholar] [CrossRef]
- She, D.T.; Wong, L.J.; Baik, S.H.; Arumugam, T.V. SIRT 2 inhibition confers neuroprotection by downregulation of FOXO3a and MAPK signaling pathways in ischemic stroke. Mol. Neurobiol. 2018, 55, 9188–9203. [Google Scholar] [CrossRef]
- Khoury, N.; Koronowski, K.B.; Young, J.I.; Perez-Pinzon, M.A. The NAD+-Dependent Family of Sirtuins in Cerebral Ischemia and Preconditioning. Antioxid. Redox Signal. 2018, 28, 691–710. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT 1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef]
- Kalaivani, P.; Ganesh, M.; Sathiya, S.; Ranju, V.; Gayathiri, V.; Saravana Babu, C. Alteration in bioenergetic regulators, SirT1 and Parp1 expression precedes oxidative stress in rats subjected to transient cerebral focal ischemia: Molecular and histopathologic evidences. J. Stroke Cerebrovasc. Dis. 2014, 23, 2753–2766. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. SIRT 3 blocks the cardiac hypertrophic response by augmenting FOXO3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [PubMed]
- Shi, L.; Rocha, M.; Leak, R.K.; Zhao, J.; Bhatia, T.N.; Mu, H.; Wei, Z.; Yu, F.; Weiner, S.L.; Ma, F.; et al. A new era for stroke therapy: Integrating neurovascular protection with optimal reperfusion. J. Cereb. Blood Flow Metab. 2018, 38, 2073–2091. [Google Scholar] [CrossRef]
- Tymianski, M. Combining neuroprotection with endovascular treatment of acute stroke: Is there hope? Stroke 2017, 48, 1700–1705. [Google Scholar] [CrossRef]
- Campbell, B.C.V.; Christensen, S.; Tress, B.M.; Churilov, L.; Desmond, P.M.; Parsons, M.W.; Barber, P.A.; Levi, C.R.; Bladin, C.; Donnan, C.A.; et al. Failure of collateral blood flow is associated with infarct growth in ischemic stroke. J. Cereb. Blood Flow Metab. 2013, 33, 1168–1172. [Google Scholar] [CrossRef]
- Dávalos, A.; Blanco, M.; Pedraza, S.; Leira, R.; Castellanos, M.; Pumar, J.M.; Silva, Y.; Serena, J.; Castillo, J. The clinical-DWI mismatch: A new diagnostic approach to the brain tissue at risk of infarction. Neurology 2004, 62, 2187–2192. [Google Scholar] [CrossRef]
- Wang, R.Y.; Wang, P.S.; Yang, Y.R. Effect of age in rats following middle cerebral artery occlusion. Gerontology 2003, 49, 27–32. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart disease and stroke statistics—2018 update: A report from the American Heart Association. Circulation 2018, 137, e69–e492. [Google Scholar] [CrossRef]
- Fischer, U.; Arnold, M.; Nedeltchev, K.; Schoenenberger, N.A.; Kapeller, L.; Höllinger, P.; Schroth, G.; Ballinari, P.; Mattle, H.P. Impact of comorbidity on ischemic stroke outcome. Acta Neurol. Scand. 2006, 113, 108–113. [Google Scholar] [CrossRef]
- Schwartz, R.H.; Bayley, M.; Lanctôt, K.L.; Murray, B.J.; Cayley, M.L.; Lien, K.; Sicard, M.N.; Thorpe, K.E.; Dowlatshahi, D.; Mandzia, J.L.; et al. Post-stroke depression, obstructive sleep apnea, and cognitive impairment: Rationale for, and barriers to, routine screening. Int. J. Stroke 2016, 11, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Myint, P.K.; Luben, R.N.; Welch, A.A.; Bingham, S.A.; Wareham, N.J.; Khaw, K.T. Plasma vitamin C concentrations predict risk of incident stroke over 10 y in 20,649 participants of the European Prospective Investigation into Cancer-Norfolk prospective population study. Am. J. Clin. Nutr. 2008, 87, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Lei, H.; Liu, A.J.; Zou, Y.X.; Shen, F.M.; Su, D.F. Increased oxidative stress is responsible for severer cerebral infarction in stroke-prone spontaneously hypertensive rats. CNS Neurosci. Ther. 2011, 17, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Ducruet, A.F.; Mack, W.J.; Mocco, J.; Hoh, D.J.; Coon, A.L.; D’Ambrosio, A.L.; Winfree, C.J.; Pinsky, D.J.; Connolly, E.S., Jr. Preclinical evaluation of postischemic dehydroascorbic acid administration in a large-animal stroke model. Transl. Stroke Res. 2011, 2, 399–403. [Google Scholar] [CrossRef]
- Rabadi, M.H.; Kristal, B.S. Effect of vitamin C supplementation on stroke recovery: A case-control study. Clin. Interv. Aging 2007, 2, 147–151. [Google Scholar] [CrossRef]
- Schürks, M.; Glynn, R.J.; Rist, P.M.; Tzourio, C.; Kurth, T. Effects of vitamin E on stroke subtypes: Meta-analysis of randomised controlled trials. Br. J. Med. 2010, 341, c5702. [Google Scholar] [CrossRef]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forro, L.; Schlegel, W.; Krause, K.H. Nox4 activity is determined by MRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef]
- Shirley, R.; Ord, E.N.J.; Work, L.M. Oxidative stress and the use of antioxidants in stroke. Antioxidants 2014, 3, 472–501. [Google Scholar] [CrossRef]
- Chen, H.; Song, Y.S.; Chan, P.H. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J. Cerebr. Blood Flow Metab. 2009, 29, 1262–1272. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwartz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef]
- Sun, Q.-A.; Hess, D.T.; Wang, B.; Miyagi, M.; Stamler, J.S. Off-target thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl) thio-1,2,3-triazolo[4,5-d] pyrimidine (VAS2870). Free Radic. Biol. Med. 2012, 52, 1897–1902. [Google Scholar] [CrossRef]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ. Res. 2001, 89, 408–414. [Google Scholar] [CrossRef]
- Laleu, B.; Gaggini, F.; Orchard, M.; Fioraso-Cartier, L.; Cagnon, L.; Houngninou-Molango, S.; Gradia, A.; Duboux, G.; Merlot, C.; Heitz, F.; et al. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox 4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2010, 53, 7715–7730. [Google Scholar] [CrossRef]
- Itoh, T.; Kawakami, M.; Yamauchi, Y.; Shimizu, S.; Nakamura, M. Effect of allopurinol on ischemia and reperfusion-induced cerebral injury in spontaneously hypertensive rats. Stroke 1986, 17, 1284–1287. [Google Scholar] [CrossRef]
- Dawson, J.; Quinn, T.J.; Harrow, C.; Lees, K.R.; Walters, M.R. The effect of allopurinol on the cerebral vasculature of patients with subcortical stroke; a randomized trial. Br. J. Clin. Pharmacol. 2009, 68, 662–668. [Google Scholar] [CrossRef]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Kondo, S.; Pai, E.F.; Nishino, T. An extremely potent inhibitor of xanthine oxidase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J. Biol. Chem. 2003, 278, 1848–1855. [Google Scholar] [CrossRef]
- Naito, S.; Nishimura, M.; Tamao, Y. Evaluation of the pharmacological actions and pharmacokinetics of BOF-4272, a xanthine oxidase inhibitor, in mouse liver. J. Pharm. Pharmacol. 2000, 52, 173–179. [Google Scholar] [CrossRef]
- Becker, M.A.; Kisicki, J.; Khosravan, R.; Wu, J.; Mulford, D.; Hunt, B.; MacDonald, P.; Joseph-Ridge, N. Febuxostat (TMX-67), a novel, non-purine, selective inhibitor of xanthine oxidase, is safe and decreases serum urate in healthy volunteers. Nucleosides Nucleotides Nucleic Acids 2004, 23, 1111–1116. [Google Scholar] [CrossRef]
- Iadecola, C.; Niwa, K.; Nogawa, S.; Zhao, X.; Nagayama, M.; Araki, E.; Morham, S.; Ross, E. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 1294–1299. [Google Scholar] [CrossRef]
- Nagayama, M.; Niwa, K.; Nagayama, T.; Ross, M.E.; Iadecola, C. The cyclooxygenase-2 inhibitor NS-398 ameliorates ischemic brain injury in wild-type mice but not in mice with deletion of the inducible nitric oxide synthase gene. J. Cereb. Blood Flow Metab. 1999, 19, 1213–1219. [Google Scholar] [CrossRef]
- Yigitkanli, K.; Pekcek, A.; Karatas, H.; Pallast, S.; Mandeville, E.; Joshi, N.; Smirnova, N.; Gazaryan, I.; Ratan, R.R.; Witztum, J.L.; et al. Inhibition of 12/15-lipoxygenase as a therapeutic strategy to treat stroke. Ann. Neurol. 2013, 73, 129–135. [Google Scholar] [CrossRef]
- Lapchak, P.A.; Maher, P.; Schubert, D.; Zivin, J.A. Baicalein, an antioxidant 12/15-lipoxygenase inhibitor improves clinical rating scores following multiple infarct embolic strokes. Neuroscience 2007, 150, 585–591. [Google Scholar] [CrossRef]
- Packer, L.; Witt, E.H.; Tritschler, H.J. Alpha-lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Zhang, J.-F.; Zhang, Y.-L.; Wu, Y.-C. The role of SIRT1 in ischemic stroke: Pathogenesis and therapeutic strategies. Front. Neurosci. 2018, 12, 833. [Google Scholar] [CrossRef]
- Panigrahi, M.; Sadguna, Y.; Shivakumar, B.R.; Kolluri, S.V.; Roy, S.; Packer, L.; Ravindranath, V. Alpha-lipoic acid protects against reperfusion injury following cerebral ischemia in rats. Brain Res. 1996, 717, 184–188. [Google Scholar] [CrossRef]
- Choi, K.-H.; Park, M.-S.; Kim, H.-S.; Kim, K.-T.; Kim, H.-S.; Kim, J.-T.; Kim, B.-C.; Kim, M.-K.; Park, J.-T.; Cho, K.-H. Alpha-lipoic acid treatment is neurorestorative and promotes functional recovery after stroke in rats. Mol. Brain 2015, 8, 9. [Google Scholar] [CrossRef]
- Choi, K.-H.; Park, M.-S.; Kim, J.-T.; Kim, H.-Y.; Kim, J.-H.; Nam, T.-S.; Choi, S.-M.; Lee, S.-H.; Kim, B.-C.; Kim, M.-K.; et al. Lipoic acid use and functional outcomes after thrombolysis in patients with acute ischemic stroke and diabetes. PLoS ONE 2016, 11, e0163484. [Google Scholar] [CrossRef]
- Homepage on the Internet. Available online: www.clinicaltrials.gov (accessed on 29 June 2021).
- Zhao, Z.; Cheng, M.; Maples, K.R.; Ma, J.Y.; Buchan, A.M. NXY-059, a novel free radical trapping compound, reduces cortical infarction after permanent focal cerebral ischemia in the rat. Brain Res. 2001, 909, 46–50. [Google Scholar] [CrossRef]
- Marshall, J.W.; Duffin, K.J.; Green, A.R.; Ridley, R.M. NXY-059, a free radical-trapping agent, substantially lessens the functional disability resulting from cerebral ischemia in a primate species. Stroke 2001, 32, 190–198. [Google Scholar] [CrossRef]
- Lees, K.R.; Zivin, J.A.; Ashwood, T.; Davalos, A.; Davis, S.M.; Diener, H.C.; Grotta, J.; Lyden, P.; Shuaib, A.; Hårdemark, H.-G.; et al. NXY-059 for acute ischemic stroke. N. Engl. J. Med. 2006, 354, 588–600. [Google Scholar] [CrossRef]
- Shuaib, A.; Lees, K.R.; Lyden, P.; Grotta, J.; Davalos, A.; Davis, S.M.; Diener, H.-C.; Ashwood, T.; Wasiewski, W.W.; Emeribe, U. NXY-059 for the treatment of acute ischemic stroke. N. Engl. J. Med. 2007, 357, 562–571. [Google Scholar] [CrossRef]
- Fisher, M.; Lees, K.; Papadakis, M.; Buchan, A.M. NXY-059: Brain or vessel protection. Stroke 2006, 37, 2189–2190. [Google Scholar] [CrossRef]
- Koziol, J.A.; Feng, A.C. On the analysis and interpretation of outcome measures in stroke clinical trials: Lessons from the SAINT I study of NXY-059 for acute ischemic stroke. Stroke 2006, 37, 2644–2647. [Google Scholar] [CrossRef]
- Fleishaker, J.C.; Peters, G.R.; Cathcart, K.S.; Steenwyk, R.C. Evaluation of the pharmacokinetics and tolerability of tirilazad mesylate, a 21-aminosteroid free radical scavenger: II. Multiple-dose administration. J. Clin. Pharmacol. 1993, 33, 182–190. [Google Scholar] [CrossRef]
- Sena, E.; Wheble, P.; Sandercock, P.; Macleod, M. Systematic review and meta-analysis of the efficacy of tirilazad in experimental stroke. Stroke 2007, 38, 388–394. [Google Scholar] [CrossRef]
- The RANTTAS Investigators. A randomized trial of tirilazad mesylate in patients with acute stroke (RANTTAS). Stroke 1996, 27, 1453–1458. [Google Scholar] [CrossRef]
- Haley, E.C.; on behalf of the RANTTAS II Investigators. High-dose tirilazad for acute stroke (RANTTAS II). Stroke 1998, 29, 1256–1257. [Google Scholar] [CrossRef][Green Version]
- Tirilazad International Steering Committee. Tirilazad mesylate in acute ischemic stroke: A systematic review. Stroke 2000, 31, 2257–2262. [Google Scholar] [CrossRef]
- Aruoma, O.J.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Khan, M.; Sekhon, B.; Jatana, M.; Giri, S.; Gilg, A.G.; Sekhon, C.; Singh, I.; Singh, A.K. Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J. Neurosci. Res. 2004, 76, 519–527. [Google Scholar] [CrossRef]
- Sabetghadam, M.; Mazdeh, M.; Abolfathi, P.; Mohammadi, Y.; Mehrpooya, M. Evidence for a beneficial effect of oral N-acetylcysteine on functional outcomes and inflammatory biomarkers in patients with acute ischemic stroke. Neuropsychiatr. Dis. Treat. 2020, 16, 1265–1278. [Google Scholar] [CrossRef]
- Trovarelli, G.; de Medio, G.E.; Dorman, R.V.; Piccini, G.L.; Horrocks, L.A.; Porcellati, G. Effect of cytidine diphosphate choline (CDP-choline) on ischemia-induced alterations of brain lipid in the gerbil. Neurochem. Res. 1981, 6, 821–833. [Google Scholar] [CrossRef]
- Bustamante, A.; Giralt, D.; Garcia-Bonilla, L.; Campos, M.; Rosell, A.; Montaner, J. Citicoline in pre-clinical animal models of stroke: A meat-analysis shows the optimal neuroprotective profile and the missing steps for jumping into a stroke clinical trial. J. Neurochem. 2012, 123, 217–225. [Google Scholar] [CrossRef]
- Dávalos, A.; Alvarez-Sabín, J.; Castillo, J.; Díez-Tejedor, E.; Ferro, J.; Martínez-Vila, E.; Serena, J.; Segura, T.; Cruz, V.T.; Masjuan, J.; et al. International Citicoline Trial on acUte Stroke (ICTUS) trial investigators. Citicoline in the treatment of acute ischaemic stroke: An international, randomised, multicentre, placebo-controlled study (ICTUS trial). Lancet 2012, 380, 349–357. [Google Scholar] [CrossRef]
- Higashi, Y. Edaravone for the treatment of acute cerebral infarction: Role of endothelium-derived nitric oxide and oxidative stress. Exp. Opin. Pharmacother. 2009, 10, 323–331. [Google Scholar] [CrossRef]
- Lukic-Panin, V.; Deguchi, K.; Yamashita, T.; Shang, J.; Zhang, X.; Tian, F.; Liu, N.; Kawai, H.; Matsuura, T.; Abe, K. Free radical scavenger edaravone administration protects against tissue plasminogen activator induced oxidative stress and blood brain barrier damage. Curr. Neurovasc. Res. 2010, 7, 319–329. [Google Scholar] [CrossRef]
- Kimura, K.; Aoki, J.; Sakamoto, Y.; Kobayashi, K.; Sakai, K.; Inoue, T.; Iguchi, Y.; Shibazaki, K. Administration of edaravone, a free radical scavenger, during t-PA infusion can enhance early recanalization in acute stroke patients—A preliminary study. J. Neurol. Sci. 2012, 313, 132–136. [Google Scholar] [CrossRef]
- Enomoto, M.; Yatsushige, H.; Fushimi, K.; Otomo, Y. Clinical effects of early edaravone use in acute ischemic stroke patients treated by endovascular reperfusion therapy. Stroke 2019, 50, 652–658. [Google Scholar] [CrossRef]
- Reiter, R.J.; Mayo, J.C.; Tan, D.-X.; Sainz, R.M.; Alatorre-Jimenez, M.; Quin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–268. [Google Scholar] [CrossRef]
- Fischer, T.W.; Kleszczynski, K.; Hardkop, L.H.; Kruse, N.; Zillikens, D. Melatonin enhances antioxidative enzyme gene expression (CAT, GPx, SOD), prevents their UVB-induced depletion, and protects against the formation of DNA damage (8-hydroxy-2′-deoxyguanosine) in ex vivo human skin. J Pineal Res. 2013, 54, 303–312. [Google Scholar] [CrossRef]
- Namura, S.; Nagata, I.; Takami, S.; Masayasu, H.; Kikuchi, H. Ebselen reduces cytochrome c release from mitochondria and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke 2001, 32, 1906–1911. [Google Scholar] [CrossRef]
- Takasago, T.; Peters, E.E.; Graham, D.I.; Masayasu, H.; Macrae, I.M. Neuroprotective efficacy of ebselen, an anti-oxidant with anti-inflammatory actions, in a rodent model of permanent middle cerebral artery occlusion. Br. J. Pharmacol. 1997, 122, 1251–1256. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in acute ischemic stroke: A placebo-controlled, double-blind clinical trial. Ebselen Study Group. Stroke 1998, 29, 12–17. [Google Scholar] [CrossRef]
- Lesage, A.S.; Peeters, L.; Leysen, J.E. Lubeluzole, a novel long-term neuroprotectant, inhibits the glutamate-activated nitric oxide synthase pathway. J. Pharmacol. Exp. Ther. 1996, 279, 759–766. [Google Scholar]
- Aronowski, J.; Strong, R.; Grotta, J.C. Treatment of experimental focal ischemia in rats with lubeluzole. Neuropharmacology 1996, 35, 689–693. [Google Scholar] [CrossRef]
- Diener, H.C.; Cortens, M.; Ford, G.; Grotta, J.; Hacke, W.; Kaste, M.; Koudstaal, P.J.; Wessel, T. Lubeluzole in acute ischemic stroke treatment: A double-blind study with an 8-h inclusion window comparing a 10-mg daily dose of lubeluzole with placebo. Stroke 2000, 31, 2543–2551. [Google Scholar] [CrossRef]
- Gandolfo, C.; Sandercock, P.; Conti, M. Lubeluzole for acute ischaemic stroke. Cochrane Database Syst. Rev. 2002, CD001924. [Google Scholar] [CrossRef]
- Murphy, M.P. Antioxidants as therapies: Can we improve on nature? Free Radic. Biol. Med. 2014, 66, 20–23. [Google Scholar] [CrossRef]
- Parkinson Study Group QE Investigators; Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Jurcau, A. Insights into the Pathogenesis of Neurodegenerative Diseases: Focus on Mitochondrial Dysfunction and Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 11847. [Google Scholar] [CrossRef]
- Plotnikov, E.Y.; Silachev, D.N.; Jankauskas, S.S.; Rokitskaya, T.I.; Chupyrkina, A.A.; Pevzner, I.B.; Zorova, L.D.; Isaev, N.K.; Antonenko, Y.N.; Skulatchev, V.P.; et al. Mild uncoupling of respiration and phosphorylation as a mechanism providing nephron- and neuroprotective effects of penetrating cations of the SkQ family. Biochemistry 2012, 77, 1029–1037. [Google Scholar]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Argaud, L.; Gateauroesch, O.; Muntean, D.; Chalabreysse, L.; Loufouat, J.; Robert, D.; Ovize, M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J. Mol. Cell. Cardiol. 2005, 38, 367–374. [Google Scholar] [CrossRef]
- Ohsawa, I.; Ishikawa, M.; Takahashi, K.; Watanabe, M.; Nishimaki, K.; Yamagata, K.; Katsura, K.-L.; Katayama, Y.; Asoh, S.; Ohta, S. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat. Med. 2007, 13, 688–694. [Google Scholar] [CrossRef]
- Esposito, E.; Mandeville, E.T.; Hayakawa, K.; Singhal, A.B.; Lo, E.H. Effects of normobaric oxygen on the progression of focal cerebral ischemia in rats. Exp. Neurol. 2013, 249, 33–38. [Google Scholar] [CrossRef]
- Terpolilli, N.A.; Kim, S.-W.; Thal, S.C.; Kataoka, H.; Zeisig, V.; Nitzsche, B.; Klaesner, B.; Zhu, C.; Schwarzmaier, S.; Meissner, L.; et al. Inhalation of nitric oxide prevents ischemic brain damage in experimental stroke by selective dilatation of collateral arterioles. Circ. Res. 2012, 110, 727–738. [Google Scholar] [CrossRef]
- Warner, D.S.; Sheng, H.; Batinic-Haberle, I. Oxidants, antioxidants and the ischemic brain. J. Exp. Biol. 2004, 207, 3221–3231. [Google Scholar] [CrossRef]
- Fujimura, M.; Morita-Fujimura, Y.; Noshita, N.; Sugawara, T.; Kawase, M.; Chan, P.H. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J. Neurosci. 2000, 20, 2817–2824. [Google Scholar] [CrossRef]
- Davis, A.S.; Zhao, H.; Sun, G.H.; Sapolsky, R.M.; Steinberg, G.K. Gene therapy using SOD1 protects striatal neurons from experimental stroke. Neurosci. Lett. 2007, 411, 32–36. [Google Scholar] [CrossRef]
- Leonardo, C.C.; Doré, S. Dietary flavonoids are neuroprotective through Nrf2-coordinated induction of endogenous cytoprotective proteins. Nutr. Neurosci. 2011, 14, 226–236. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26. [Google Scholar] [CrossRef]
- Fuse, Y.; Endo, Y.; Araoi, S.; Daitoku, H.; Suzuki, H.; Kato, M.; Kobayashi, M. The possible repositioning of an oral anti-arthritic drug, auranofin, for Nfr2-activating therapy: The demonstration of Nrf2-dependent anti-oxidative action using a zebrafish model. Free Radic. Biol. Med. 2011, 14, 405–411. [Google Scholar]
- Jurcau, A. The Role of Natural Antioxidants in the Prevention of Dementia—Where Do We Stand and Future Perspectives. Nutrients 2021, 13, 282. [Google Scholar] [CrossRef]
- Tanaka, N.; Ikeda, Y.; Ohta, Y.; Deguchi, K.; Tian, F.; Shang, J.; Matsuura, T.; Abe, K. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011, 1370, 246–253. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. SIRT1 in neurodevelopment and brain senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef]
- Shin, J.A.; Lee, K.E.; Kim, H.S.; Park, E.M. Acute resveratrol treatment modulates multiple signaling pathways in the ischemic brain. Neurochem. Res. 2012, 37, 2686–2696. [Google Scholar] [CrossRef]
- Koronowski, K.B.; Dave, K.R.; Saul, I.; Camarena, V.; Thompson, J.W.; Neumann, J.T.; Young, J.I.; Perez-Pinzon, M.A. Resveratrol preconditioning induces a novel extended window of ischemic tolerance in the mouse brain. Stroke 2015, 46, 2293–2298. [Google Scholar] [CrossRef]
- Ray, A.; Cleary, M.P. The potential role of leptin in tumor invasion and metastasis. Cytokine Growth Factor Rev. 2017, 38, 80–97. [Google Scholar] [CrossRef]
- Nuñez-Figueredo, Y.; Pardo-Andreu, G.L.; Ramírez-Sánchez, J.; Delgado-Hernández, R.; Ochoa-Rodríguez, E.; Verdecia-Reyes, Y.; Naal, Z.; Muller, A.P.; Portela, L.V.; Souza, D.O. Antioxidant effects of JM-20 on rat brain mitochondria and synaptosomes: Mitoprotection against Ca2+-induced mitochondrial impairment. Brain Res. Bull. 2014, 109, 68–76. [Google Scholar] [CrossRef]
- Ye, R.; Yang, Q.; Kong, X.; Li, N.; Zhang, Y.; Han, J.; Xiong, L.; Liu, X.; Zhao, G. Sevoflurane preconditioning improves mitochondrial function and long-term neurologic sequelae after transient cerebral ischemia: Role of mitochondrial permeability transition. Crit. Care Med. 2012, 40, 2685–2693. [Google Scholar] [CrossRef]
- Agrawal, S.; Gollapudi, S.; Su, H.; Gupta, S. Leptin activates human B cells to secrete TNF-α, IL-6, and IL-10 via JAK2/STAT3 and p38MAPK/ERK1/2 signaling pathway. J. Clin. Immunol. 2011, 31, 472–478. [Google Scholar] [CrossRef]
- Nowzari, Z.; Masoumi, M.; Nazari-Robati, M.; Akbari, H.; Shahrokhi, N.; Asadikaram, G. Association of polymorphisms of leptin, leptin receptor and apelin receptor genes with susceptibility to coronary artery disease and hypertension. Life Sci. 2018, 207, 166–171. [Google Scholar] [CrossRef]
- Matsuyama, H.; Suzuki, H.I. Systems and synthetic microRNA biology: From biogenesis to disease pathogenesis. Int. J. Mol. Sci. 2019, 21, 132. [Google Scholar] [CrossRef]
- Yu, S.; Zhai, J.; Yu, J.; Yang, Q.; Yang, J. MiR-98-5p protects against cerebral ischemia/reperfusion injury through anti-apoptosis and anti-oxidative stress in mice. J. Biochem. 2021, 169, 195–206. [Google Scholar] [CrossRef]
- Xin, H.; Buller, B.; Katakowski, M.; Zhang, Y.; Wang, X.; Shang, X.; Zhang, Z.G.; Chopp, M. Exosome-mediated transfer of miR-133bfrom multipotent mesenchymal stromal cells to neural cells contributes to neurite outgrowth. Stem Cells 2012, 30, 1556–1564. [Google Scholar] [CrossRef]
- Huang, S.; Ge, X.; Yu, J.; Han, Z.; Yin, Z.; Li, Y.; Chen, F.; Wang, H.; Zhang, J.; Lei, P. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J. 2018, 32, 512–528. [Google Scholar] [CrossRef]
- Dabrowska, S.; Andrzejewska, A.; Lukomska, B.; Janowski, M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflamm. 2019, 16, 178. [Google Scholar] [CrossRef]
- Jurcau, A.; Simion, A. Neuroinflammation in Cerebral Ischemia and Ischemia/Reperfusion Injuries: From Pathophysiology to Therapeutic Strategies. Int. J. Mol. Sci. 2022, 23, 14. [Google Scholar] [CrossRef]
- Dulamea, A.O. The potential use of mesenchymal stem cells in stroke therapy—From bench to bedside. J. Neurol. Sci. 2015, 352, 1–11. [Google Scholar] [CrossRef]
- Hess, D.C.; Wechsler, L.R.; Clark, W.M.; Savitz, S.I.; Ford, G.A.; Chiu, D.; Yavagal, D.R.; Uchino, K.; Liebeskind, D.S.; Auchus, A.P.; et al. Safety and efficacy of multipotent adult progenitor cells in acute ischaemic stroke (MASTERS): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2017, 16, 360–368. [Google Scholar] [CrossRef]
- Amani, H.; Habibey, R.; Hajmiresmail, S.J.; Latifi, S.; Pazoki-Toroudi, H.; Akhavan, O. Antioxidant nanomaterials in advanced diagnoses and treatments of ischemia reperfusion injuries. J. Mater. Chem. B 2017, 5, 9452–9476. [Google Scholar] [CrossRef]
- Sandhir, R.; Yadav, A.; Sunkaria, A.; Singhal, N. Nano-antioxidants: An emerging strategy for intervention against neurodegenerative conditions. Neurochem. Int. 2015, 98, 209–226. [Google Scholar] [CrossRef]
- Alkaff, S.A.; Radhakrishnan, K.; Nedumaran, A.M.; Liao, P.; Czarny, B. Nanocarriers for stroke therapy: Advances and obstacles in translating animal studies. Int. J. Nanomed. 2020, 15, 445–464. [Google Scholar] [CrossRef]
- Song, G.; Zhao, M.; Chen, H.; Lenahan, C.; Zhou, X.; Ou, Y.; He, Y. The role of nanomaterials in stroke treatment: Targeting oxidative stress. Oxid. Med. Cell. Longev. 2021, 2021, 8857486. [Google Scholar] [CrossRef]
- Li, Q.; Tang, G.; Xue, S.; He, X.; Miao, P.; Li, Y.; Wang, J.; Xiong, L.; Wang, Y.; Zhang, C.; et al. Silica-coated superparamagnetic iron oxide nanoparticles targeting of EPCs in ischemic brain injury. Biomaterials 2013, 34, 4982–4992. [Google Scholar] [CrossRef]
- Murta, V.; Schilrreff, P.; Rosciszewski, G.; Morilla, M.J.; Ramos, A.J. G5G2.5 core-shell tecto-dendrimer specifically targets reactive glia in brain ischemia. J. Neurochem. 2018, 144, 748–760. [Google Scholar] [CrossRef]
- Dong, X.; Gao, J.; Su, Y.; Wang, Z. Nanomedicine for ischemic stroke. Int. J. Mol. Sci. 2020, 21, 7600. [Google Scholar] [CrossRef]
- Takamiya, M.; Miyamoto, Y.; Yamashita, T.; Deguchi, K.; Ohta, Y.; Abe, K. Strong neuroprotection with a novel platinum nanoparticle against ischemic stroke- and tissue plasminogen activator-related brain damages in mice. Neuroscience 2012, 221, 47–51. [Google Scholar] [CrossRef]
- Clark, A.; Zhu, A.; Sun, K.; Petty, H.R. Cerium oxide and platinum nanoparticles protect cells from oxidant-mediated apoptosis. J. Nanopart. Res. 2011, 13, 5547–5555. [Google Scholar] [CrossRef]
- Kim, C.K.; Kim, T.; Choi, I.Y.; Soh, M.; Kim, D.; Kim, Y.J.; Jang, H.; Yang, H.S.; Kim, J.Y.; Park, H.K.; et al. Ceria nanoparticles that can protect against ischemic stroke. Angew. Chem. Int. Ed. Engl. 2012, 51, 11039–11043. [Google Scholar] [CrossRef]
- Bao, Q.; Hu, P.; Xu, Y.; Cheng, T.; Wei, C.; Pan, L.; Shi, J. Simultaneous Blood-Brain Barrier Crossing and Protection for Stroke Treatment Based on Edaravone-Loaded Ceria Nanoparticles. ACS Nano 2018, 12, 6794–6805. [Google Scholar] [CrossRef]
- Al-Jamal, K.T.; Gherardini, L.; bardi, G.; Nunes, A.; Guo, C.; Bussy, C.; Herrero, M.A.; Bianco, A.; Prato, M.; Kostarelos, K.; et al. Functional motor recovery from brain ischemic insult by carbonnanotube-mediated siRNA silencing. Proc. Natl. Acad. Sci. USA 2011, 108, 10953–10957. [Google Scholar] [CrossRef]
- Tiebosch, I.A.; Crielaard, B.J.; Bouts, M.J.; Zwartbol, R.; Salas-Perdomo, A.; Lammers, T.; Planas, A.M.; Storm, G.; Dijkhuizen, R.M. Combined treatment with recombinant tissue plasminogen activator and dexamethasone phosphate-containing liposomes improves neurological outcome and restricts lesion progression after embolic stroke in rats. J. Neurochem. 2012, 123, 65–74. [Google Scholar] [CrossRef]
- Zhao, Y.; Xin, Z.; Li, N.; Chang, S.; Chen, Y.; Geng, L.; Chang, H.; Shi, H.; Chang, Y.Z. Nano-liposomes of lycopene reduces ischemic brain damage in rodents by regulating iron metabolism. Free Radic. Biol. Med. 2018, 124, 1–11. [Google Scholar] [CrossRef]
- Sinha, J.; Das, N.; Basu, M.K. Liposomal antioxidants in combating ischemia-reperfusion injury in rat brain. Biomed. Pharmacother. 2001, 55, 264–271. [Google Scholar] [CrossRef]
- Jin, Q.; Cai, Y.; Li, S.; Liu, H.; Zhou, X.; Lu, C.; Gao, X.; Qian, J.; Zhang, J.; Ju, S.; et al. Edaravone-Encapsulated Agonistic Micelles Rescue Ischemic Brain Tissue by Tuning Blood-Brain Barrier Permeability. Theranostics 2017, 7, 884–898. [Google Scholar] [CrossRef]
- Reddy, M.K.; Labhasetwar, V. Nanoparticle-mediated delivery of superoxide dismutase to the brain: An effective strategy to reduce ischemia-reperfusion injury. FASEB J. 2009, 23, 1384–1395. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sarkar, S.; Jana, S.; Swarnakar, S.; Das, N. neuroprotective role of nanoencapsulated curcumin against cerebral ischemia-reperfusion induced oxidative injury. Brain Res. 2019, 1704, 164–173. [Google Scholar] [CrossRef]
- Lu, X.; Dong, J.; Zheng, D.; Li, X.; Ding, D.; Xu, H. Reperfusion combined with intraarterial administration of resveratrol-loaded nanoparticles improved cerebral ischemia-reperfusion injury in rats. Nanomed. Nanotechnol. Biol. Med. 2020, 28, 102208. [Google Scholar] [CrossRef]
- Karimi Zarchi, A.A.; Abbasi, S.; Faramarzi, M.A.; Gilani, K.; Ghazi-Khansari, M.; Amani, A. Development and optimization of N-Acetylcysteine-loaded poly (lactic-co-glycolic acid) nanoparticles by electrospray. Int. J. Biol. Macromol. 2015, 72, 764–770. [Google Scholar] [CrossRef]
- Hosoo, H.; Marushima, A.; Nagasaki, Y.; Hirayama, A.; Ito, H.; Puentes, S.; Mujagic, A.; Tsurushima, H.; Tsuruta, W.; Suzuki, K.; et al. Neurovascular unit protection from cerebral ischemia-reperfusion injury by radical-containing nanoparticles in mice. Stroke 2017, 48, 2238–2247. [Google Scholar] [CrossRef]
- Ghosh, A.; Sarkar, S.; Mandal, A.K.; Das, N. Neuroprotective role of nanoencapsulated quercetin in combating ischemia-reperfusion induced neuronal damage in young and aged rats. PLoS ONE 2013, 8, e57735. [Google Scholar] [CrossRef]
- Oh, J.; Lee, M.S.; Jeong, J.H.; Lee, M. Deoxycholic Acid-Conjugated Polyethylenimine for Delivery of Heme Oxygenase-1 Gene in Rat Ischemic Stroke Model. J. Pharm. Sci. 2017, 106, 3524–3532. [Google Scholar] [CrossRef]
- Lee, J.; Hyun, H.; Kim, J.; Ryu, J.H.; Kim, H.A.; Park, J.H.; Lee, M. Dexamethasone-loaded peptide micelles for delivery of the hemeoxigenase-1 gene to ischemic brain. J. Control. Release 2012, 158, 131–138. [Google Scholar] [CrossRef]
- Rajkovic, O.; Gourmel, C.; d’Arcy, R.; Wong, R.; Rajkovic, I.; Tirelli, N.; Pinteaux, E. Reactive oxygen species-responsive nanoparticles for the treatment of ischemic stroke. Adv. Ther. 2019, 2, 1900038. [Google Scholar] [CrossRef]
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Howells, D.W. Evaluation of Combination Therapy in Animal Models of Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2012, 32, 585–597. [Google Scholar] [CrossRef]
- Jelinek, M.; Jurajda, M.; Duris, K. Oxidative Stress in the Brain: Basic Concepts and Treatment Strategies in Stroke. Antioxidants 2021, 10, 1886. [Google Scholar] [CrossRef]
- He, J.; Liu, J.; Huang, Y.; Tang, X.; Xiao, H.; Hu, Z. Oxidative Stress, Inflammation, and Autophagy: Potential Targets of Mesenchymal Stem Cells-Based Therapies in Ischemic Stroke. Front. Neurosci. 2021, 15, 641157. [Google Scholar] [CrossRef]
- Boshuizen, M.C.S.; Steinberg, G.K. Stem Cell–Based Immunomodulation After Stroke: Effects on Brain Repair Processes. Stroke 2018, 49, 1563–1570. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Pathways of Cerebral Tissue Protection Following Ischemia | Sirtuins Involved |
|---|---|
| Reduces oxidative stress | SIRT 1, 3, 6 |
| Promotes glutamate uptake | SIRT 4 |
| Increased calcium buffering | SIRT 3 |
| Preserves mitochondrial respiration | SIRT 1, 3, 5 |
| Maintains ATP levels | SIRT 3, 4 |
| Promotes neural stem cell proliferation | SIRT 1, 2 |
| Promotes neural stem cell differentiation | SIRT 1, 2, 6 |
| Preserves blood flow | SIRT 1 |
| Maintains blood brain barrier integrity | SIRT 3 |
| DNA repair | SIRT 1 |
| Reduces inflammation | SIRT 1 |
| Mediates neuroprotective effects of NAD+ | SIRT 1, 5 |
| Pathways of Cerebral Injury Following Ischemia | Sirtuins Involved |
| Increases oxidative stress | SIRT 2, 3, 6 |
| Damages mitochondrial respiration | SIRT 3 |
| Increases autophagy | SIRT 6 |
| Induces axonal injury | SIRT 2 |
| Nanoparticles | Drugs | Therapeutic Effect | References |
|---|---|---|---|
| PtNPs | - | Reduces superoxide generation and MMP9 activation, diminishes infarct volume | [220] |
| Nanoceria | - or Edaravone | Diminishes oxidative stress, scavenges ROS, reduces neuronal apoptosis, downregulates iNOS | [216,221,222,223] |
| Carbon nanotubes | Caspase-3 siRNA | Gene silencing of caspase-3, improved neuronal survival | [224] |
| Liposomes | Dexamethasone + tPA | Reduces infarct volume | [225] |
| lycopene | Inhibits NOX2, reduces the levels of NO | [226] | |
| Vitamin C, E | Reduces oxidative stress | [227] | |
| Polymer nanoparticles | edaravone | Free radical scavenging | [228] |
| SOD | Reduces oxidative stress, diminishes infarct size | [229] | |
| curcumin | Reduces oxidative injury | [230] | |
| resveratrol | Diminishes lipid peroxidation | [231] | |
| N-acetylcysteine | Increases antioxidant activity | [232] | |
| Amino-TEMPO | Reduces oxidative injury and BBB disruption | [233] | |
| Quercetin | Reduces mitochondrial damage | [234] | |
| Heme oxygenase- 1 +/− dexamethasone | Reduces oxidative stress, diminishes ischemic brain damage | [235,236] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurcau, A.; Ardelean, A.I. Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke. Biomedicines 2022, 10, 574. https://doi.org/10.3390/biomedicines10030574
Jurcau A, Ardelean AI. Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke. Biomedicines. 2022; 10(3):574. https://doi.org/10.3390/biomedicines10030574
Chicago/Turabian StyleJurcau, Anamaria, and Adriana Ioana Ardelean. 2022. "Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke" Biomedicines 10, no. 3: 574. https://doi.org/10.3390/biomedicines10030574
APA StyleJurcau, A., & Ardelean, A. I. (2022). Oxidative Stress in Ischemia/Reperfusion Injuries following Acute Ischemic Stroke. Biomedicines, 10(3), 574. https://doi.org/10.3390/biomedicines10030574