
HMTM-Mediated Enhancement of Brain Bioenergetics in a Mouse Tauopathy Model Is Blocked by Chronic Administration of Rivastigmine

 ,
, 


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Transgenic Mice
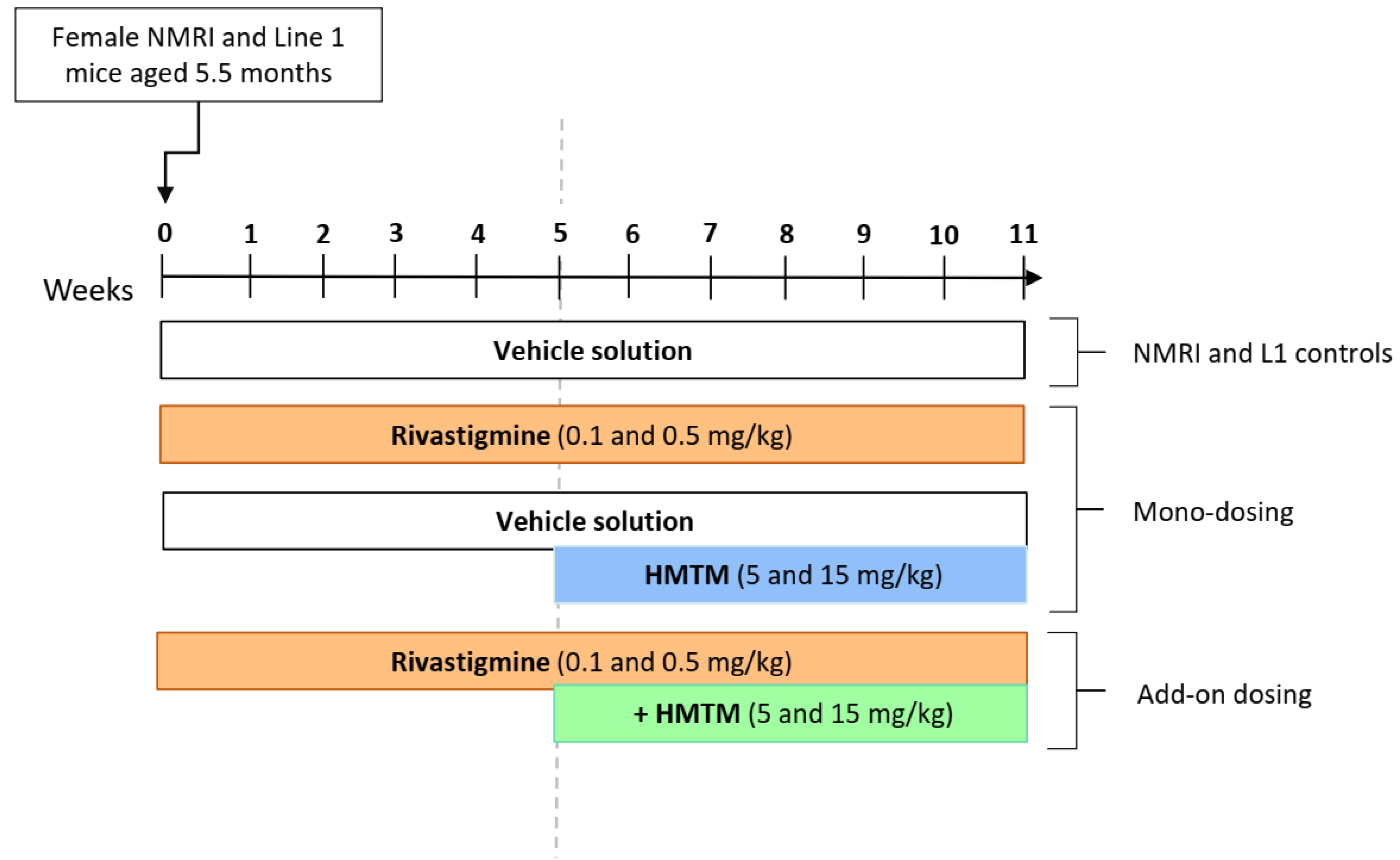
2.3. Experimental Design
2.4. Extraction and Quantification of Methylthionine (MT) in the Plasma and Brains of Treated Mice
2.5. Tissue Processing for Cell Biology and Metabolite Analysis
2.6. Total Protein Extraction for Immunoblotting
2.7. Protein Extraction for Determination of Enzyme Activities (Citrate Synthase; NADH Dehydrogenase; Cytochrome c Oxidase)
2.8. Tissue Processing for l-Lactate Quantification
2.9. Immunoblot Analysis
2.10. Citrate Synthase Activity
2.11. NADH Dehydrogenase Activity
2.12. Cytochrome c Oxidase Activity
2.13. l-Lactate Quantification
2.14. Statistical Analysis
3. Results
3.1. L1 Tau-Transgenic Mice Show Accumulation of l-Lactate in the Brain
3.2. Activity of ETC Complex I and Complex IV Is Facilitated by HMTM
3.3. Single and Combined Dosing of Rivastigmine and HMTM Do Not Change the Levels of Subunits of the Mitochondrial Electron Transport Chain
3.4. HMTM-Induced Reduction in the Levels of Brain l-Lactate Is Lowered Further by the Prior Chronic Administration of Rivastigmine
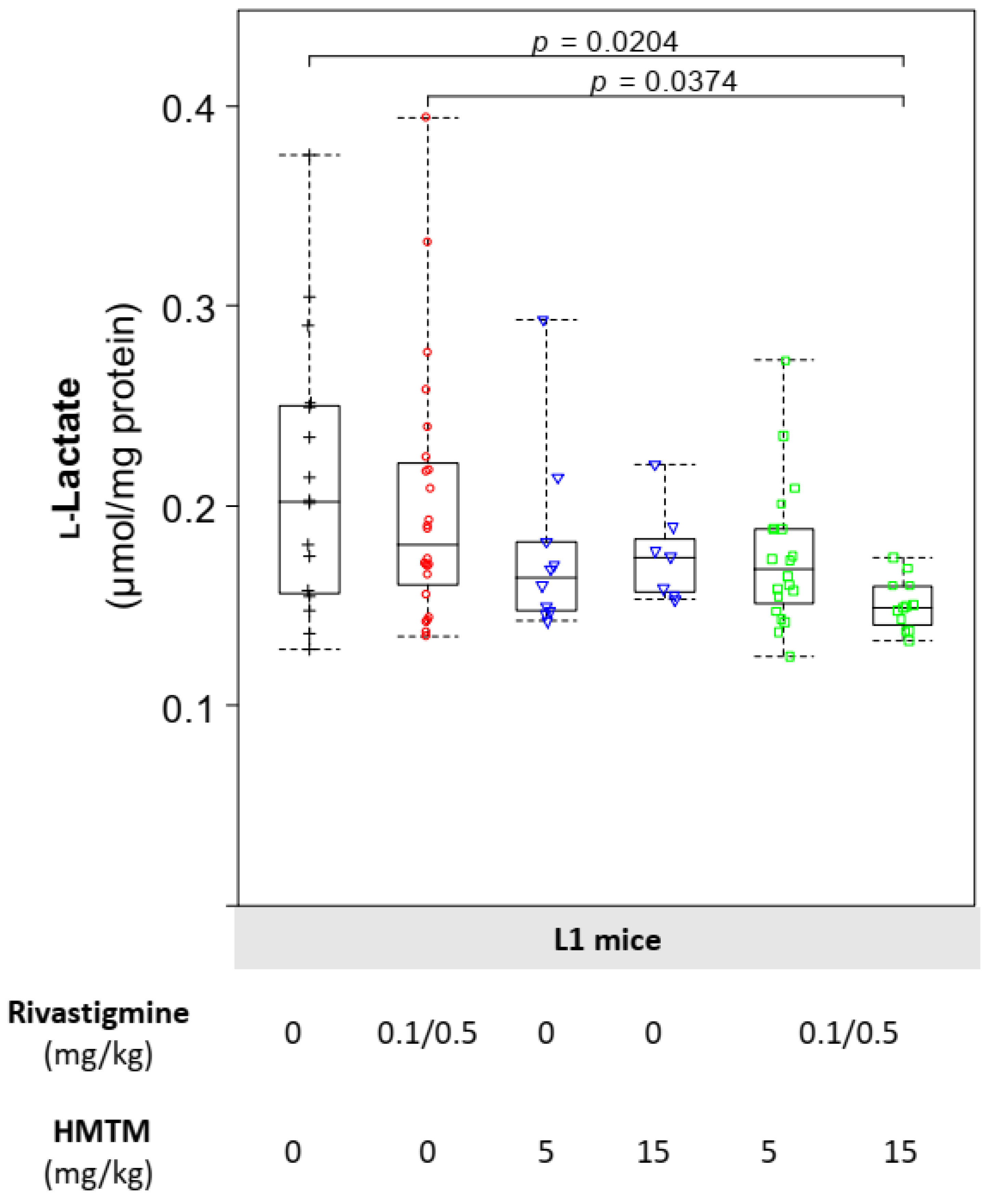
3.5. HMTM Monotherapy Increases the Levels of Astrocytic LDH-A Subunits

4. Discussion
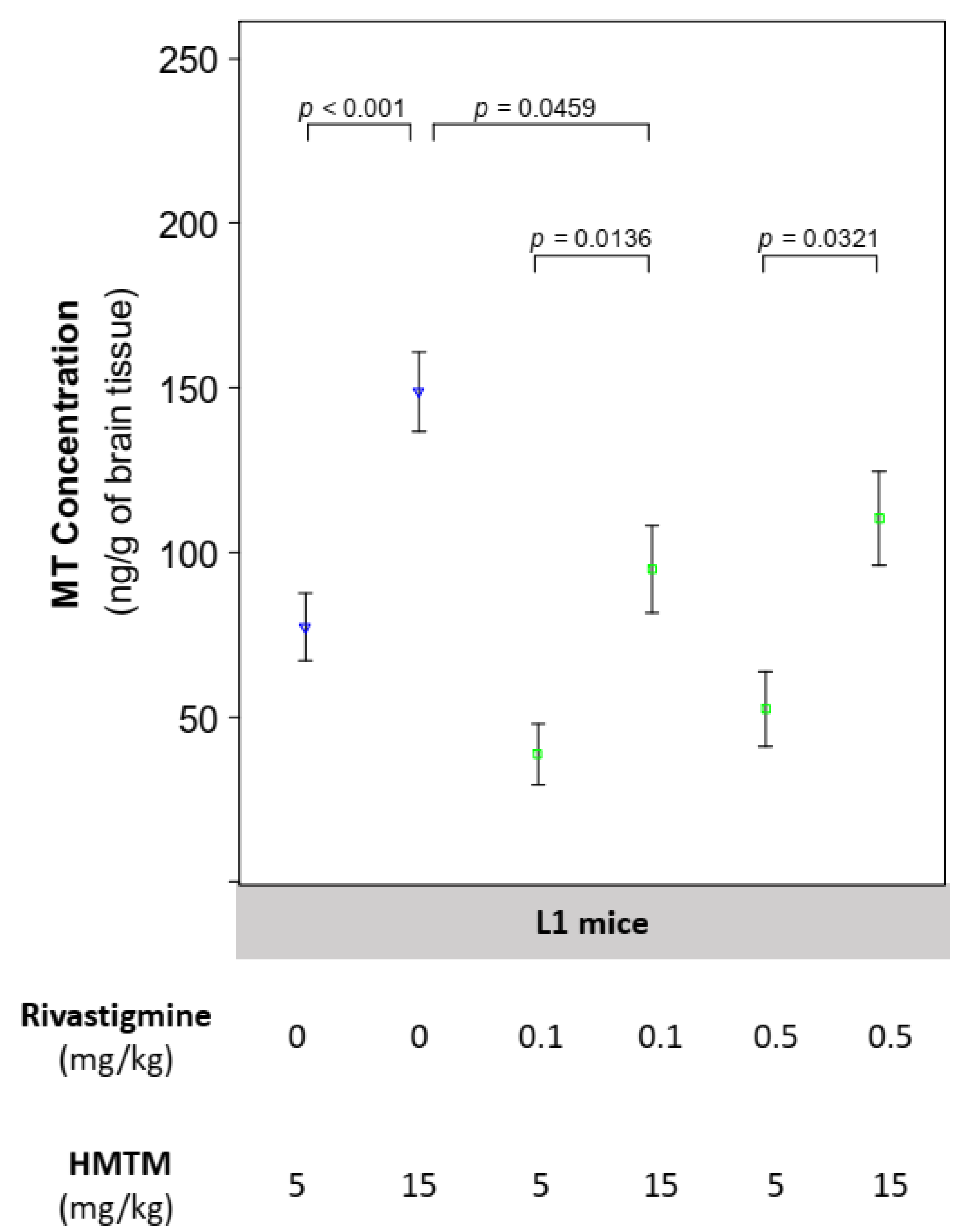
4.1. The Concentration of Brain MT Increases with Dose of HMTM and Is Reduced by Its Concomitant Administration with Rivastigmine
4.2. L1 Mice Are Relevant Models of Tauopathy for the Study of l-Lactate Metabolic Alterations despite Unchanged Mitochondrial Function
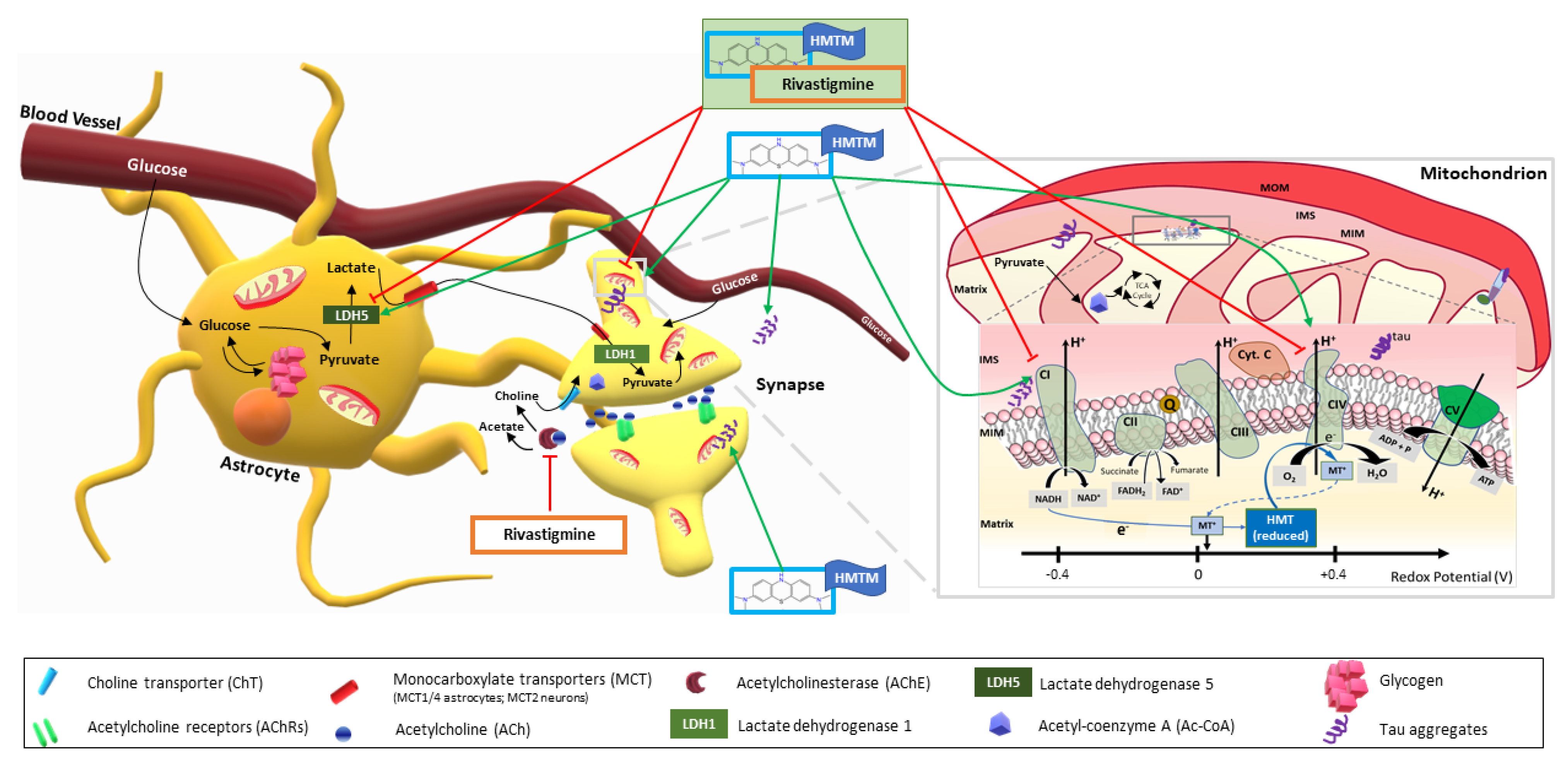
4.3. HMTM Monotherapy Enhances Energy Metabolism by Increasing Substrate Supply to More Active Mitochondria
4.4. HMTM Administered as an Add-On Drug to Mice Chronically Dosed with Rivastigmine Fails to Restore the Brain Energetic Profile in the L1 Transgenics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carvalho, K.M.; Winter, E.; de Souza Antunes, A.M. Analysis of technological developments in the treatment of Alzheimer’s disease through patent documents. Intell. Inf. Manag. 2015, 7, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Ezzat, S.M.; Salem, M.A.; El Mahdy, N.M.; Ragab, M.F. Rivastigmine. In Naturally Occurring Chemicals against Alzheimer’s Disease; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 93–108. [Google Scholar]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sánchez-López, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesús, G.; Beas-Zarate, C.; et al. Memantine for the treatment of dementia: A review on its current and future applications. J. Alzheimers Dis. 2018, 62, 1223–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer’s Association. 2020 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.C.; Knopman, D.S.; Emerson, S.S.; Ovbiagele, B.; Kryscio, R.J.; Perlmutter, J.S.; Kesselheim, A.S. Revisiting FDA approval of aducanumab. N. Engl. J. Med. 2021, 385, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D. Controversy and progress in Alzheimer’s disease—FDA approval of aducanumab. N. Engl. J. Med. 2021, 385, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Carman, M.D.; Fernandez-Madrid, I.J.; Power, M.D.; Lieberburg, I.; van Duinen, S.G.; Bots, G.T.A.M.; Luyendijk, W.; Frangione, B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wijsman, E.M.; Nemens, E.; Anderson, L.; Goddard, K.A.B.; Weber, J.L.; Bird, T.D.; Schellenberg, G.D. A familial Alzheimer’s disease locus on chromosome 1. Science 1995, 269, 970–973. [Google Scholar] [CrossRef]
- Schellenberg, G.D.; Bird, T.D.; Wijsman, E.M.; Orr, H.T.; Anderson, L.; Nemens, E.; White, J.A.; Bonnycastle, L.; Weber, J.L.; Alonso, M.E.; et al. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science 1992, 258, 668–671. [Google Scholar] [CrossRef]
- George-Hyslop, P.S.; Haines, J.; Rogaev, E.; Mortilla, M.; Vaula, G.; Pericak-Vance, M.; Foncin, J.-F.; Montesi, M.; Bruni, A.; Sorbi, S.; et al. Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat. Genet. 1992, 2, 330–334. [Google Scholar] [CrossRef]
- Cummings, J.L.; Goldman, D.P.; Simmons-Stern, N.R.; Ponton, E. The costs of developing treatments for Alzheimer’s disease: A retrospective exploration. Alzheimer’s Dement. 2021, 18, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, C.R. The molecular pathology of Alzheimer’s disease. Neuroimaging Clin. 2012, 22, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Schelter, B.O.; Wischik, D.J.; Storey, J.M.D.; Harrington, C.R. Modeling prion-like processing of tau protein in Alzheimer’s disease for pharmaceutical development. J. Alzheimers Dis. 2018, 62, 1287–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Melis, V.; Zabke, C.; Stamer, K.; Magbagbeolu, M.; Schwab, K.; Marschall, P.; Veh, R.W.; Bachmann, S.; Deiana, S.; Moreau, P.-H.; et al. Different pathways of molecular pathophysiology underlie cognitive and motor tauopathy phenotypes in transgenic models for Alzheimer’s disease and frontotemporal lobar degeneration. Cell Mol. Life Sci. 2015, 72, 2199–2222. [Google Scholar] [CrossRef] [Green Version]
- Grober, E.; Dickson, D.W.; Sliwinski, M.; Buschke, H.; Katz, M.; Crystal, H.; Lipton, R. Memory and mental status correlates of modified Braak staging. Neurobiol. Aging 1999, 20, 573–579. [Google Scholar] [CrossRef]
- Jellinger, K.; Braak, H.; Braak, E.; Fischer, P. Alzheimer lesions in the entorhinal region and isocortex in Parkinson’s and Alzheimer’s diseases. Ann. N. Y. Acad. Sci. 1991, 640, 203–209. [Google Scholar] [CrossRef]
- Mukaetova-Ladinska, E.B.; Garcia-Siera, F.; Hurt, J.; Gertz, H.J.; Xuereb, J.H.; Hills, R.; Brayne, C.; Huppert, F.A.; Paykel, E.S.; McGee, M.; et al. Staging of cytoskeletal and β-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am. J. Pathol. 2000, 157, 623–636. [Google Scholar] [CrossRef]
- Rauch, J.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Nathaniel, D.L.; Raghavan, P.; Nelson, M.; Tian, R.; Tse, E.; Hong, J.Y.; See, S.K.; Mok, S.-A.; Hein, M.Y.; et al. Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J. Biol. Chem. 2019, 294, 18952–18966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; Devos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Harrington, C.R.; Storey, J.M.D. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Congdon, E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Harrington, C.R.; Storey, J.M.D.; Clunas, S.; Harrington, K.A.; Horsley, D.; Ishaq, A.; Kemp, S.J.; Larch, C.P.; Marshall, C.; Nicoll, S.L.; et al. Cellular models of aggregation-dependent template-directed proteolysis to characterize tau aggregation inhibitors for treatment of Alzheimer disease. J. Biol. Chem. 2015, 290, 10862–10875. [Google Scholar] [CrossRef] [Green Version]
- Baddeley, T.C.; McCaffrey, J.; Storey, J.; Cheung, J.K.S.; Melis, V.; Horsley, D.; Harrington, C.; Wischik, C.M. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2014, 352, 110–118. [Google Scholar] [CrossRef]
- Atamna, H.; Mackey, J.; Dhahbi, J.M. Mitochondrial pharmacology: Electron transport chain bypass as strategies to treat mitochondrial dysfunction. BioFactors 2012, 38, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Atamna, H.; Nguyen, A.; Schultz, C.; Boyle, K.; Newberry, J.; Kato, H.; Ames, B.N. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2007, 22, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N.; Starkov, A.A. Methylene blue does not bypass Complex III antimycin block in mouse brain mitochondria. FEBS Lett. 2019, 593, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, D.X.; Caccamo, A.; Oddo, S. Methylene blue reduces Aβ levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011, 21, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.A.; Jové, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum. Mol. Genet. 2014, 23, 3716–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of low dose leuco-methylthioninium bis(hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: Cohort analysis as modified primary outcome in a phase III clinical trial. J. Alzheimers Dis. 2017, 61, 435–457. [Google Scholar] [CrossRef] [Green Version]
- Schelter, B.O.; Shiells, H.; Baddeley, T.C.; Rubino, C.M.; Ganesan, H.; Hammel, J.; Vuksanovic, V.; Staff, R.T.; Murray, A.D.; Bracoud, L.; et al. Concentration-dependent activity of hydromethylthionine on cognitive decline and brain atrophy in mild to moderate Alzheimer’s disease. J. Alzheimers Dis. 2019, 72, 931–946. [Google Scholar] [CrossRef] [Green Version]
- Riedel, G.; Klein, J.; Niewiadomska, G.; Kondak, C.; Schwab, K.; Lauer, D.; Magbagbeolu, M.; Steczkowska, M.; Zadrozny, M.; Wydrych, M.; et al. Mechanisms of anticholinesterase interference with tau aggregation inhibitor activity in a tau-transgenic mouse model. Curr. Alzheimer Res. 2020, 17, 285–296. [Google Scholar] [CrossRef]
- Bordone, M.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; De Freitas, A.E.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Chételat, G.; Arbizu, J.; Barthel, H.; Garibotto, V.; Law, I.; Morbelli, S.; van de Giessen, E.; Agosta, F.; Barkhof, F.; Brooks, D.J.; et al. Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of Alzheimer’s disease and other dementias. Lancet Neurol. 2020, 19, 951–962. [Google Scholar] [CrossRef]
- De Leon, M.J.; Convit, A.; Wolf, O.T.; Tarshish, C.Y.; DeSanti, S.; Rusinek, H.; Tsui, W.; Kandil, E.; Scherer, A.J.; Roche, A.; et al. Prediction of cognitive decline in normal elderly subjects with 2-[18F]fluoro-2-deoxy-d-glucose/positron-emission tomography (FDG/PET). Proc. Natl. Acad. Sci. USA 2001, 98, 10966–10971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groot, C.; Risacher, S.L.; Chen, J.A.; Dicks, E.; Saykin, A.J.; Mac Donald, C.L.; Mez, J.; Trittschuh, E.H.; Mukherjee, S.; Barkhof, F.; et al. Differential trajectories of hypometabolism across cognitively-defined Alzheimer’s disease subgroups. NeuroImage Clin. 2021, 31, 102725. [Google Scholar] [CrossRef] [PubMed]
- Nestor, P.; Fryer, T.D.; Smielewski, P.; Hodges, J.R. Limbic hypometabolism in Alzheimer’s disease and mild cognitive impairment. Ann. Neurol. 2003, 54, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Silverman, D.H.S.; Small, G.W.; Chang, C.Y.; Lu, C.S.; De Aburto, M.A.K.; Chen, W.; Czernin, J.; Rapoport, S.I.; Pietrini, P.; Alexander, G.E.; et al. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA J. Am. Med Assoc. 2001, 286, 2120–2127. [Google Scholar] [CrossRef] [Green Version]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical diagnosis of Alzheimer’s disease: Recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef]
- Correia, S.C.; Santos, R.X.; Perry, G.; Zhu, X.; Moreira, P.I.; Smith, M.A. Mitochondrial importance in Alzheimer’s, Huntington’s and Parkinson’s Diseases. Adv. Exp. Med. Biol. 2012, 724, 205–221. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective mitochondrial pyruvate flux affects cell bioenergetics in Alzheimer’s disease-related models. Cell Rep. 2020, 30, 2332–2348. [Google Scholar] [CrossRef] [Green Version]
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 2020, 94, e1592–e1604. [Google Scholar] [CrossRef]
- Peter, C.; Hongwan, D.; Küpfer, A.; Lauterburg, B.H. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur. J. Clin. Pharmacol. 2000, 56, 247–250. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous inference in general parametric models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogle, D.H.; Wheeler, P.; Dinno, A. FSA: Fisheries Stock Analysis; R Package Version 0.8.30; 2020. Available online: https://github.com/fishR-Core-Team/FSA (accessed on 9 March 2020).
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessières, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef] [PubMed]
- Calì, C.; Tauffenberger, A.; Magistretti, P. The strategic location of glycogen and lactate: From body energy reserve to brain plasticity. Front. Cell. Neurosci. 2019, 13, 82. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.M.; Öberg, J.; Brené, S.; Coppotelli, G.; Terzioglu, M.; Pernold, K.; Goiny, M.; Sitnikov, R.; Kehr, J.; Trifunovic, A.; et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc. Natl. Acad. Sci. USA 2010, 107, 20087–20092. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, K.-C.; Ryu, W.-I.; Amirault, K.M.; Healy, R.A.; Siegel, A.J.; McPhie, D.L.; Forester, B.; Cohen, B.M. Late-onset Alzheimer’s disease is associated with inherent changes in bioenergetics profiles. Sci. Rep. 2017, 7, 14038. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Oakley, S.S.; Maina, M.B.; Marshall, K.E.; Al-Hilaly, Y.K.; Harrington, C.R.; Wischik, C.M.; Serpell, L.C. Tau filament self-assembly and structure: Tau as a therapeutic target. Front. Neurol. 2020, 11, 590754. [Google Scholar] [CrossRef]
- Wischik, C.M.; Novak, M.; Edwards, P.C.; Klug, A.; Tichelaar, W.; Crowther, R.A. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.; Davidson, A.; Brown, A. Universal glia to neurone lactate transfer in the nervous system: Physiological functions and pathological consequences. Biosensors 2020, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Vezzoli, E.; Calì, C.; De Roo, M.; Ponzoni, L.; Sogne, E.; Gagnon, N.; Francolini, M.; Braida, D.; Sala, M.; Muller, D.; et al. Ultrastructural evidence for a role of astrocytes and glycogen-derived lactate in learning-dependent synaptic stabilization. Cereb. Cortex 2020, 30, 2114–2127. [Google Scholar] [CrossRef] [PubMed]
- Melis, V.; Magbagbeolu, M.; Rickard, J.E.; Horsley, D.; Davidson, K.; Harrington, K.A.; Goatman, K.; Goatman, E.A.; Deiana, S.; Close, S.P.; et al. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav. Pharmacol. 2015, 26, 353–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cranston, A.L.; Wysocka-Marijnissen, A.; Steczkowska, M.; Zadrożny, M.; Palasz, E.; Harrington, C.R.; Theuring, F.; Wischik, C.M.; Riedel, G.; Niewiadomska, G. Cholinergic and inflammatory phenotypes in transgenic tau mouse models of Alzheimer’s disease and frontotemporal lobar degeneration. Brain Commun. 2020, 2, fcaa033. [Google Scholar] [CrossRef]
- Bittar, P.G.; Charnay, Y.; Pellerin, L.; Bouras, C.; Magistretti, P.J. Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. J. Cereb. Blood Flow Metab. 1996, 16, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lin, X.; Wang, D.; Zhang, Z.; Guo, Y.; Ren, X.; Xu, B.; Yuan, J.; Liu, J.; Spencer, P.S.; et al. Mitochondrial molecular abnormalities revealed by proteomic analysis of hippocampal organelles of mice triple transgenic for Alzheimer disease. Front. Mol. Neurosci. 2018, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Kandimalla, R.; Manczak, M.; Fry, D.; Suneetha, Y.; Sesaki, H.; Reddy, P.H. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 4881–4897. [Google Scholar] [CrossRef] [Green Version]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 27, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mena, R.; Edwards, P.C.; Harrington, C.R.; Mukaetova-Ladinska, E.B.; Wischik, C.M. Staging the pathological assembly of truncated tau protein into paired helical filaments in Alzheimer’s disease. Acta Neuropathol. 1996, 91, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen-Wartiovaara, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2017, 19, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial respiratory chain complexes. In Membrane Protein Complexes: Structure and Function; Harris, J.R., Boekema, E.J., Eds.; Springer: Singapore, 2018; pp. 167–227. [Google Scholar]
- Schwab, K.; Melis, V.; Harrington, C.R.; Wischik, C.M.; Magbagbeolu, M.; Theuring, F.; Riedel, G. Proteomic analysis of hydromethylthionine in the line 66 model of frontotemporal dementia demonstrates actions on tau-dependent and tau-independent networks. Cells 2021, 10, 2162. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Groups | |||
|---|---|---|---|
| Genotype | Treatment (mg/kg) | N | |
| Weeks 1–5 | Weeks 6–11 | ||
| NMRI | Vehicle | 14 | |
| NMRI | Riva 0.5 | +Vehicle | 14 |
| NMRI | Vehicle | +HMTM 15 | 14 |
| NMRI | Riva 0.5 | +HMTM 15 | 12 |
| L1 | Vehicle | 16 | |
| L1 | Riva 0.1 | +Vehicle | 12 |
| L1 | Riva 0.5 | +Vehicle | 12 |
| L1 | Vehicle | +HMTM 5 | 10 |
| L1 | Vehicle | +HMTM 15 | 7 |
| L1 | Riva 0.1 | +HMTM 5 | 12 |
| L1 | Riva 0.5 | +HMTM 5 | 8 |
| L1 | Riva 0.1 | +HMTM 15 | 6 |
| L1 | Riva 0.5 | +HMTM 15 | 6 |
| NMRI (Median, IQR, n = 14) | L1 (Median, IQR, n = 16) | p | |
|---|---|---|---|
| l-Lactate (µmol/mg protein) | 0.144 (0.108–0.160) | 0.202 (0.157–0.250) | 0.00149 |
| LDH-A (10−2; a.u.) | 0.343 (0.230–0.518) | 0.559 (0.449–0.660) | 0.0275 |
| LDH-B (10−2; a.u.) | 0.165 (0.126–0.246) | 0.252 (0.214–0.274) | 0.0925 |
| NDUFB8-CI (10−5; a.u.) | 3.01 (2.41–4.06) | 3.33 (2.54–3.93) | 0.919 |
| SDHB-CII (10−5; a.u.) | 2.26 (1.51–3.31) | 2.64 (1.74–3.75) | 0.473 |
| UQCRC2C-CIII (10−5; a.u.) | 4.62 (4.35–5.85) | 4.82 (4.29–5.88) | 0.790 |
| MTCOI-CIV (10−5; a.u.) | 3.26 (2.40–4.05) | 3.28 (3.08–4.58) | 0.448 |
| ATP5A-CV (10−5; a.u.) | 3.01 (2.38–3.28) | 3.77 (2.96–4.36) | 0.142 |
| Complex I activity (normalised to citrate synthase) | 0.457 (0.303–0.664) | 0.408 (0.226–0.563) | 0.401 |
| Complex IV activity (normalised to citrate synthase) | 1.41 (1.05–2.23) | 1.62 (1.13–2.09) | 0.886 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, R.X.; Melis, V.; Goatman, E.A.; Leith, M.; Baddeley, T.C.; Storey, J.M.D.; Riedel, G.; Wischik, C.M.; Harrington, C.R. HMTM-Mediated Enhancement of Brain Bioenergetics in a Mouse Tauopathy Model Is Blocked by Chronic Administration of Rivastigmine. Biomedicines 2022, 10, 867. https://doi.org/10.3390/biomedicines10040867
Santos RX, Melis V, Goatman EA, Leith M, Baddeley TC, Storey JMD, Riedel G, Wischik CM, Harrington CR. HMTM-Mediated Enhancement of Brain Bioenergetics in a Mouse Tauopathy Model Is Blocked by Chronic Administration of Rivastigmine. Biomedicines. 2022; 10(4):867. https://doi.org/10.3390/biomedicines10040867
Chicago/Turabian StyleSantos, Renato X., Valeria Melis, Elizabeth A. Goatman, Michael Leith, Thomas C. Baddeley, John M. D. Storey, Gernot Riedel, Claude M. Wischik, and Charles R. Harrington. 2022. "HMTM-Mediated Enhancement of Brain Bioenergetics in a Mouse Tauopathy Model Is Blocked by Chronic Administration of Rivastigmine" Biomedicines 10, no. 4: 867. https://doi.org/10.3390/biomedicines10040867
APA StyleSantos, R. X., Melis, V., Goatman, E. A., Leith, M., Baddeley, T. C., Storey, J. M. D., Riedel, G., Wischik, C. M., & Harrington, C. R. (2022). HMTM-Mediated Enhancement of Brain Bioenergetics in a Mouse Tauopathy Model Is Blocked by Chronic Administration of Rivastigmine. Biomedicines, 10(4), 867. https://doi.org/10.3390/biomedicines10040867