
Current Status and Future Directions of Immunotherapies in Soft Tissue Sarcomas


 and
and
Abstract
:1. Introduction
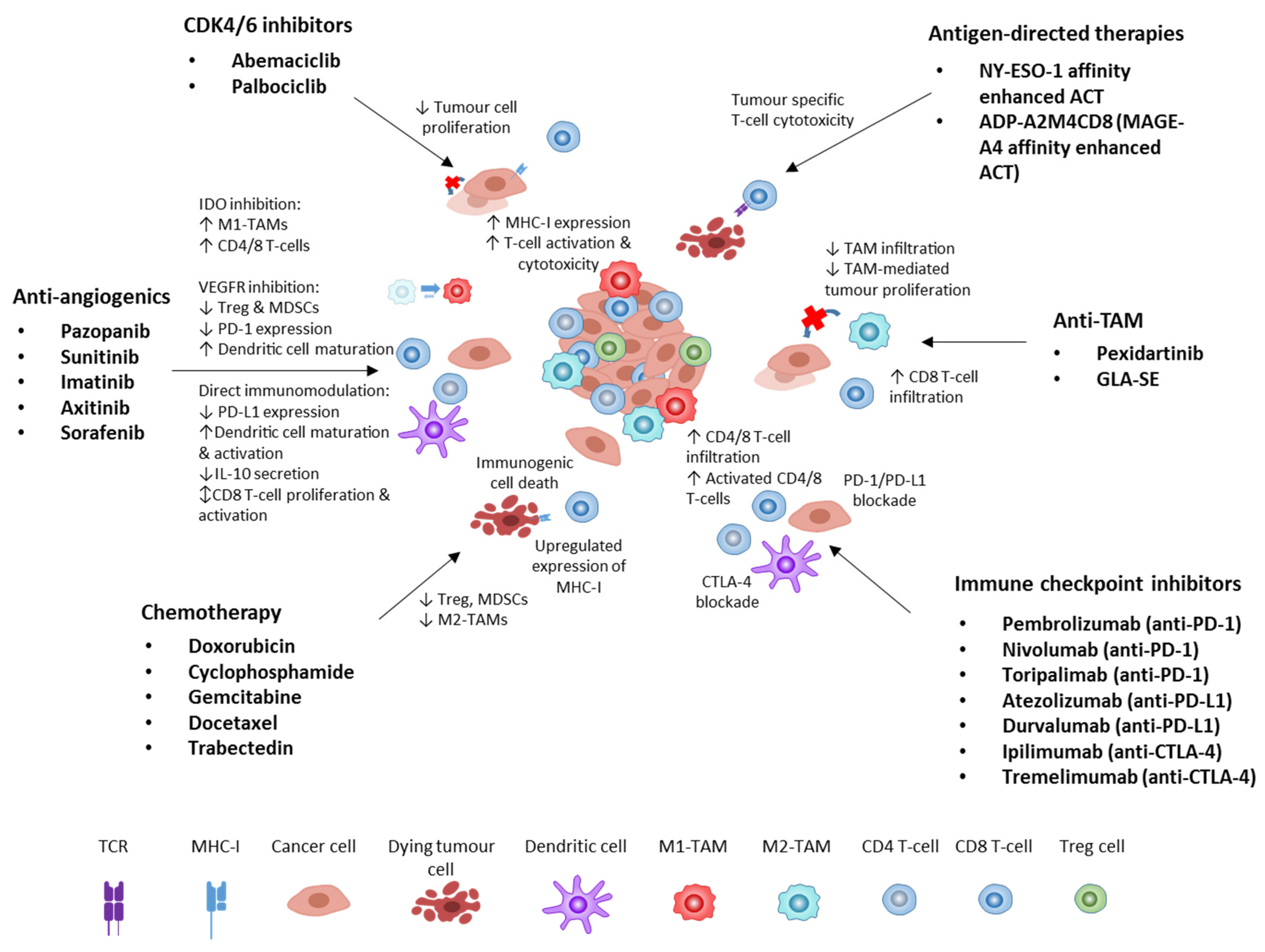
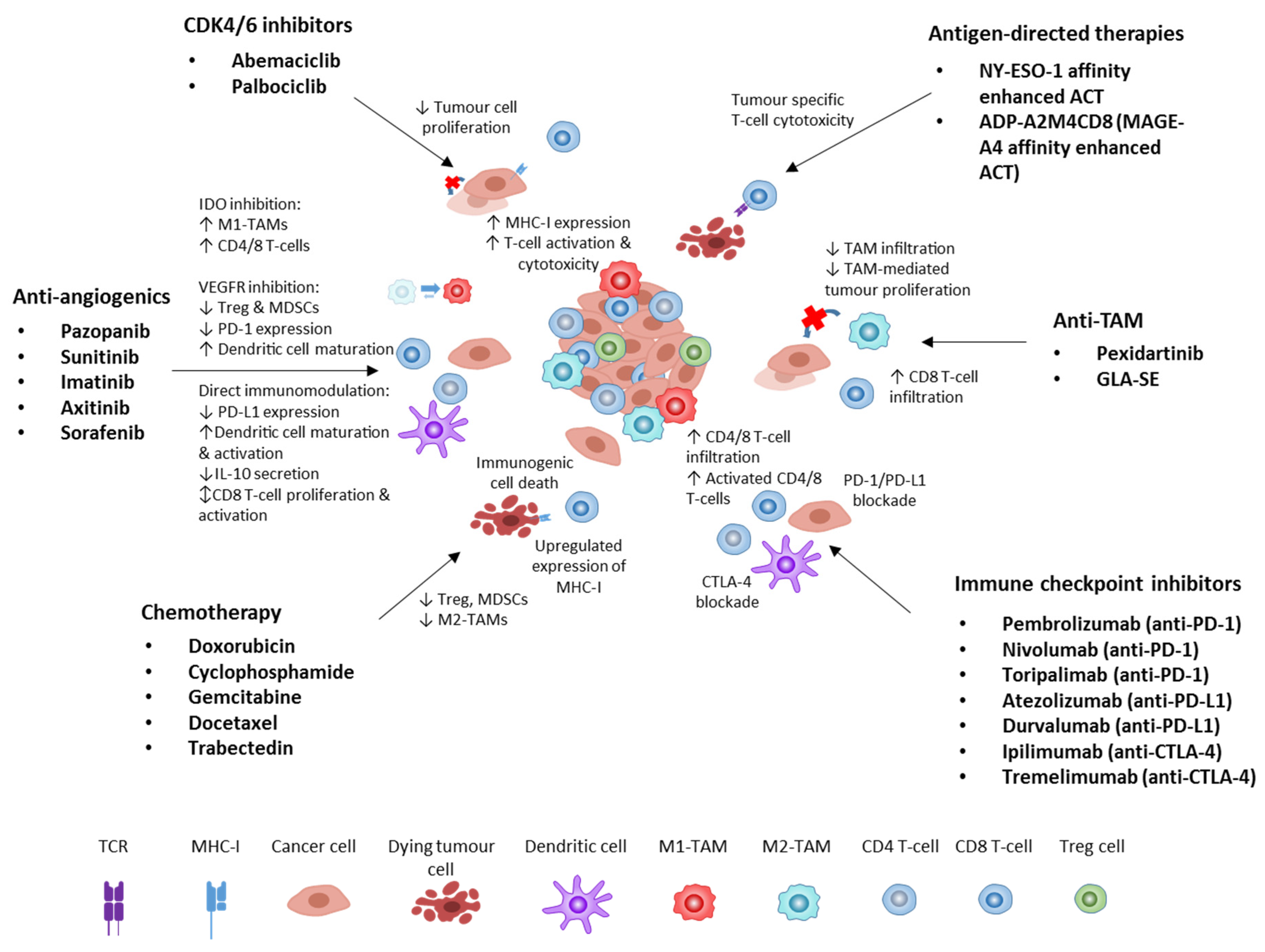
2. Immune Checkpoint Inhibitors in STS
2.1. Clinical Trials of CPIs in Mixed STS Cohorts

{kind=link}
| Year Reported | Study | Intervention | Evaluable STS Patients (n) | ORR | Subtype Specific Objective Responses | Other Responses | ||
|---|---|---|---|---|---|---|---|---|
| UPS (n) | LMS (n) | DDLPS (n) | ||||||
| 2013 Maki [13] | MSKCC Synovial Sarcoma pilot | Ipilimumab | 6 | 0% | NA | NA | NA | 0/6 SS |
| 2017 Ben Ami [14] | Dana Farber uterine LMS phase II | Nivolumab | 12 | 0% | NA | 0/12 | NA | |
| 2015 Patnaik [15] | KEYNOTE-001 | Pembrolizumab | 1 | 0% | NA | 0/1 | NA | |
| 2017 Groisberg [26] | MD Anderson series | CPIs in various combos | 40 | 5% | 0/1 | 0/12 | 0/6 | |
| 2017 Tawbi [16] | SARC028 | Pembrolizumab | 40 | 18% | 4/10 | 0/10 | 2/10 | 1/10 SS |
| 2019 Burgess [17] | SARC028 expansion cohort | Pembrolizumab | 79 | 16% | 9/40 | NA | 4/39 | |
| 2018 D’Angelo [18] | ALLIANCE A091401 | Nivolumab | 37 | 5% | 0/5 | 1/15 | 0/3 | 1/1 ASPS |
| 2018 D’Angelo [18] | ALLIANCE A091401 | Nivolumab and ipilimumab | 37 | 16% | 2/6 1/6 unconfirmed | 2/14 | 0/2 | 1/1 MFS 1/3 AS |
| 2020 Chen [19] | GIST/DDLPS/UPS expansions of ALLIANCE | Nivolumab | 36 | 6% | 1/12 | NA | 1/12 | 0/9 GIST |
| 2020 Chen [19] | GIST/DDLPS/UPS expansions of ALLIANCE | Nivolumab and ipilimumab | 36 | 11% | 2/12 | NA | 2/12 | 0/9 GIST |
| 2020 Quiroga [20] | Ohio State series | Nivolumab or pembrolizumab | 25 | 12% | 0/3 | 0/5 | 1/6 | 1 inflammatory myofibroblastic sarcoma |
| 2020 Somaiah [21] | MD Anderson phase II | Durvalumab and tremelimumab | 57 | 14% | 1/5 | 0/5 | 0/6 | 5/10 ASPS 1/5 Chordoma 1/5 AS |
| 2020 Monga [22] | US multi-institute | PD1 +/− CTLA4 or other CPIs (inc. anti-CSF1R) | 88 | 24% | 8/25 | 9/20 | NR | 1 fibroblastic sarcoma 1 SEF 1 MFS |
| 2020 Zhou [23] | Stanford series | Nivolumab and ipilimumab | 38 | 15% | 1/8 (classed as sarcoma NOS) | 0/9 | 1/6 | 2/5 MFS 1/2 SFT 1/3 MPNST |
| 2021 Chen [24] | Chinese multi-institute | Nivolumab | 76 | 7% | NR | NR | NR | |
| 2021 Chen [24] | Chinese multi-institute | Nivolumab and ipilimumab | 74 | 13% | NR | NR | NR | |
| 2020 Italiano [8] | Pooled analysis of phase II data | PD1/PDL1 +/− other CPIs | 384 | 15% | 16/103 | 5/82 | 4/61 | |
2.2. Immune-Based Biomarkers in Mixed STS Cohorts
3. CPI Effect in Rarer STS Subtypes
3.1. Alveolar Soft Part Sarcoma
3.2. SWI/SNF-Deficient Sarcomas
3.3. Clear Cell Sarcoma of Soft Tissue
| Year/Author | Trial | Agents | Rare Subtypes Evaluable (n) | ORR of Rare Subtype | Other Outcomes |
|---|---|---|---|---|---|
| 2020 Geoerger [63] | KEYNOTE-051 phase I-II | Pembrolizumab | MRT (2) EPS (1) | 50% MRT 100% EPS | |
| 2021 Blay [52] | AcSé Pembrolizumab phase II | Pembrolizumab | MRT (11) | 27% | 18.2% 12-month PFS rate (MRT) |
| 2019 Jones [69] | MD Anderson case series | CPI | CCS (11) | NR | 1/11 Durable response (41.8 months) 10/11 <6 months response duration |
| 2020 Painter [70] | Angiosarcoma project series | CPI | AS (6) | 33% | 2 CR (2/3 HNFS AG) |
| 2020 Somaiah [21] | MD Anderson phase II | Durvalumab and tremelimumab | AS (5) | 20% | 1 PR (1/1 cutaneous AS) |
| 2019 Florou [71] | Miami Miller case series | Pembrolizumab, AGEN1884, Pembrolizumab and axitinib | AS (7) | NR | 71% PR rate at 12 weeks 1/2 CR to AGEN1884 |
| 2021 Wagner [72] | DART phase II | Ipilimumab and nivolumab | AS (16) | 25% | 60% HNFS AS ORR 2 SD 38% 6-month PFS rate |
3.4. Angiosarcoma
4. CPI Combinations
4.1. Anti-Angiogenics
4.2. Chemotherapy and Radiotherapy
| Year/Author | Trial | Agents | Evaluable Patients (n) | ORR | Other Outcomes |
|---|---|---|---|---|---|
| 2019 Wilky [85] | Miami single-centre phase II | Pembrolizumab and axitinib | 30 | 27% (55% ASPS ORR) | 8 PR (6 ASPS, 1 ES, 1 LMS) 9 SD 3 minor responses (1 LMS, 1 SS, 1 UPS) 4.7-month mPFS |
| 2020 Martin-Broto [86] | IMMUNOSARC multicentre phase Ib/II | Nivolumab and sunitinib | 58 (12 phase Ib, 46 phase II) | 21% (57% ASPS ORR) | Phase II: 1 CR 5 PR 33 SD 7 PD |
| 2017 Toulmonde [93] | PEMBROSARC phase II | Pembrolizumab + metronomic cyclophosphamide | 50 | 2% | 1 PR 16 SD 31 PD 2 minor responses |
| 2021 Italiano [47] | Tertiary lymphoid structure selected PEMBROSARC phase II | Pembrolizumab + metronomic cyclophosphamide | 30 | 27% | 5 SD 4.1-month mPFS |
| 2017 Weiss [95] | PembroPlus phase 1b | Pembrolizumab and doxorubicin/gemcitabine/docetaxel | 6 | 0% | 1 SD 5 PD |
| 2020 Pollack [96] | Phase I/II | Pembrolizumab and doxorubicin | 37 | 19% | 7 PR 2 unconfirmed PR 11 SD 8.1-month mPFS |
4.3. CDK4/6 Inhibitors
5. Anti-Tumour-Associated Macrophage Approaches
6. Antigen-Directed Therapies
7. Future Directions
7.1. Immunomodulation Approaches
7.1.1. Immunogenic Cell Death
7.1.2. LXR Agonists
7.1.3. EZH2 Inhibitors
7.1.4. BO-112
7.1.5. Oncolytic Virus
7.2. Targeting Alternative Immune Checkpoints
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fletcher, C.D.M. The evolving classification of soft tissue tumours—An update based on the new 2013 WHO classification. Histopathology 2014, 64, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Linch, M.; Miah, A.B.; Thway, K.; Judson, I.R.; Benson, C. Systemic treatment of soft-tissue sarcoma—Gold standard and novel therapies. Nat. Rev. Clin. Oncol. 2014, 11, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Frezza, A.M.; Stacchiotti, S.; Gronchi, A. Systemic treatment in advanced soft tissue sarcoma: What is standard, what is new. BMC Med. 2017, 15, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dancsok, A.R.; Setsu, N.; Gao, D.; Blay, J.Y.; Thomas, D.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Expression of lymphocyte immunoregulatory biomarkers in bone and soft-tissue sarcomas. Mod. Pathol. 2019, 32, 1772–1785. [Google Scholar] [CrossRef] [PubMed]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolle, M.A.; Herbsthofer, L.; Granegger, B.; Goda, M.; Brcic, I.; Bergovec, M.; Scheipl, S.; Prietl, B.; Pichler, M.; Gerger, A.; et al. T-regulatory cells predict clinical outcome in soft tissue sarcoma patients: A clinico-pathological study. Br. J. Cancer 2021, 125, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Espinosa, I.; Vrijaldenhoven, S.; Subramanian, S.; Montgomery, K.D.; Zhu, S.; Marinelli, R.J.; Peterse, J.L.; Poulin, N.; Nielsen, T.O.; et al. Prognostic significance of macrophage infiltration in leiomyosarcomas. Clin. Cancer Res. 2008, 14, 1423–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A.; Bellera, C.; D’Angelo, S. PD1/PD-L1 targeting in advanced soft-tissue sarcomas: A pooled analysis of phase II trials. J. Hematol. Oncol. 2020, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, R.G.; Jungbluth, A.A.; Gnjatic, S.; Schwartz, G.K.; D’Adamo, D.R.; Keohan, M.L.; Wagner, M.J.; Scheu, K.; Chiu, R.; Ritter, E.; et al. A pilot study of anti-CTLA4 antibody ipilimumab in patients with synovial sarcoma. Sarcoma 2013, 2013, e168145. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Barysauskas, C.M.; Solomon, S.; Tahlil, K.; Malley, R.; Hohos, M.; Polson, K.; Loucks, M.; Severgnini, M.; Patel, T.; et al. Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study. Cancer 2017, 123, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patnaik, A.; Kang, S.P.; Rasco, D.; Papadopoulos, K.P.; Elassaiss-Schaap, J.; Beeram, M.; Drengler, R.; Chen, C.; Smith, L.; Espino, G.; et al. Phase I Study of Pembrolizumab (MK-3475, Anti–PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 4286–4293. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Burgess, M.A.; Bolejack, V.; Schuetze, S.; Van Tine, B.A.; Attia, S.; Riedel, R.F.; Hu, J.S.; Davis, L.E.; Okuno, S.H.; Priebat, D.A.; et al. Clinical activity of pembrolizumab (P) in undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated/pleomorphic liposarcoma (LPS): Final results of SARC028 expansion cohorts. J. Clin. Oncol. 2019, 37, 11015. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Chen, J.L.; Mahoney, M.R.; George, S.; Antonescu, C.R.; Liebner, D.A.; Van Tine, B.A.; Milhem, M.M.; Tap, W.D.; Streicher, H.; Schwartz, G.K.; et al. A multicenter phase II study of nivolumab +/− ipilimumab for patients with metastatic sarcoma (Alliance A091401): Results of expansion cohorts. J. Clin. Oncol. 2020, 38, 11511. [Google Scholar] [CrossRef]
- Quiroga, D.; Liebner, D.A.; Philippon, J.S.; Hoffman, S.; Tan, Y.; Chen, J.L.; Lenobel, S.; Wakely, P.E.; Pollock, R.; Tinoco, G. Activity of PD1 inhibitor therapy in advanced sarcoma: A single-center retrospective analysis. BMC Cancer 2020, 20, 527. [Google Scholar] [CrossRef] [PubMed]
- Somaiah, N.; Conley, A.P.; Lin, H.Y.; Amini, B.; Sabir, S.H.; Araujo, D.M.; Benjamin, R.S.; Livingston, J.A.; Patel, S.; Ratan, R.; et al. A phase II multi-arm study of durvalumab and tremelimumab for advanced or metastatic sarcomas. J. Clin. Oncol. 2020, 38, 11509. [Google Scholar] [CrossRef]
- Monga, V.; Skubitz, K.M.; Maliske, S.; Mott, S.L.; Dietz, H.; Hirbe, A.C.; Van Tine, B.A.; Oppelt, P.; Okuno, S.; Robinson, S.; et al. A Retrospective Analysis of the Efficacy of Immunotherapy in Metastatic Soft-Tissue Sarcomas. Cancers 2020, 12, 1873. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Bui, N.; Bolleddu, S.; Lohman, M.; Becker, H.C.; Ganjoo, K. Nivolumab plus ipilimumab for soft tissue sarcoma: A single institution retrospective review. Immunotherapy 2020, 12, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, X.; Liu, J.; Liang, D.; Zhao, M.; Yu, W.; Chen, P. Nivolumab plus ipilimumab versus nivolumab in individuals with treatment-naive programmed death-ligand 1 positive metastatic soft tissue sarcomas: A multicentre retrospective study. BMC Cancer 2021, 21, 108. [Google Scholar] [CrossRef] [PubMed]
- Roland, C.L.; Keung, E.Z.Y.; Lazar, A.J.; Torres, K.E.; Wang, W.L.; Guadagnolo, A.; Bishop, A.J.; Lin, H.Y.; Hunt, K.; Feig, B.W.; et al. Preliminary results of a phase II study of neoadjuvant checkpoint blockade for surgically resectable undifferentiated pleomorphic sarcoma (UPS) and dedifferentiated liposarcoma (DDLPS). J. Clin. Oncol. 2020, 38, 11505. [Google Scholar] [CrossRef]
- Groisberg, R.; Hong, D.S.; Behrang, A.; Hess, K.; Janku, F.; Piha-Paul, S.; Naing, A.; Fu, S.; Benjamin, R.; Patel, S.; et al. Characteristics and outcomes of patients with advanced sarcoma enrolled in early phase immunotherapy trials. J. Immunother. Cancer 2017, 5, 100. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Gooden, M.J.M.; De Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark. Res. 2020, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, S.P.; Shoushtari, A.N.; Agaram, N.P.; Kuk, D.; Qin, L.X.; Carvajal, R.D.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Schwartz, G.K.; et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum. Pathol. 2015, 46, 357–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Kim, E.K.; Jung, H.; Chon, H.J.; Han, J.W.; Shin, K.H.; Hu, H.; Kim, K.S.; Choi, Y.D.; Kim, S.; et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer 2016, 16, 434. [Google Scholar] [CrossRef] [Green Version]
- Movva, S.; Wen, W.; Chen, W.; Millis, S.Z.; Gatalica, Z.; Reddy, S.; von Mehren, M.; Tine, B.A. Multi-platform profiling of over 2000 sarcomas: Identification of biomarkers and novel therapeutic targets. Oncotarget 2015, 6, 12234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boxberg, M.; Steiger, K.; Lenze, U.; Rechl, H.; von Eisenhart-Rothe, R.; Wörtler, K.; Weichert, W.; Langer, R.; Specht, K. PD-L1 and PD-1 and characterization of tumor-infiltrating lymphocytes in high grade sarcomas of soft tissue-prognostic implications and rationale for immunotherapy. Oncoimmunology 2017, 7, e1389366. [Google Scholar] [CrossRef] [Green Version]
- Tazzari, M.; Bergamaschi, L.; De Vita, A.; Collini, P.; Barisella, M.; Bertolotti, A.; Ibrahim, T.; Pasquali, S.; Castelli, C.; Vallacchi, V. Molecular Determinants of Soft Tissue Sarcoma Immunity: Targets for Immune Intervention. Int. J. Mol. Sci 2021, 22, 7518. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.Z.; Burgess, M.; Salazar, R.; Parra, E.R.; Rodrigues-Canales, J.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Attia, S.; Riedel, R.F.; et al. Correlative analyses of the SARC028 trial reveal an association between sarcoma-associated immune infiltrate and response to pembrolizumab. Clin. Cancer Res. 2020, 26, 1258–1266. [Google Scholar] [CrossRef]
- Italiano, A.; Bessede, A.; Bompas, E.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Bertucci, F.; Toulmonde, M.; Bellera, C.A.; Guegan, J.P.; et al. PD1 inhibition in soft-tissue sarcomas with tertiary lymphoid structures: A multicenter phase II trial. J. Clin. Oncol. 2021, 39, 11507. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Maki, R.G. Diagnosis, Prognosis, and Treatment of Alveolar Soft-Part Sarcoma: A Review. JAMA Oncol. 2019, 5, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, M.; Lui, M.Y.; Antonescu, C.R.; Krause-Boehm, A.; Meindl, A.; Argani, P.; Healey, J.H.; Ueda, T.; Yoshikawa, H.; Meloni-Ehrig, A.; et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 2001, 20, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naqash, A.R.; Coyne, G.H.O.; Moore, N.; Sharon, E.; Takebe, N.; Fino, K.K.; Ferry-Galow, K.V.; Hu, J.S.; Van Tine, B.A.; Burgess, M.A.; et al. Phase II study of atezolizumab in advanced alveolar soft part sarcoma (ASPS). J. Clin. Oncol. 2021, 39, 11519. [Google Scholar] [CrossRef]
- Blay, J.Y.; Penel, N.; Ray-Coquard, I.L.; Schott, R.; Saada-Bouzid, E.; Bertucci, F.; Chevreau, C.M.; Bompas, E.; Coquan, E.; Cousin, S.; et al. High clinical benefit rates of pembrolizumab in very rare sarcoma histotypes: First results of the AcSé pembrolizumab study. Ann. Oncol. 2019, 30, v517. [Google Scholar] [CrossRef]
- Blay, J.Y.; Penel, N.; Ray-Coquard, I.L.; Cousin, S.; Bertucci, F.; Bompas, E.; Eymard, J.C.; Saada-Bouzid, E.; Soulie, P. High clinical activity of pembrolizumab in chordoma, alveolar soft part sarcoma (ASPS) and other rare sarcoma histotypes: The French AcSé pembrolizumab study from Unicancer. J. Clin. Oncol. 2021, 39, 11520. [Google Scholar] [CrossRef]
- Yang, J.; Dong, L.; Yang, S.; Han, X.; Han, Y.; Jiang, S.; Yao, J.; Zhang, Z.; Zhang, S.; Liu, P.; et al. Safety and clinical efficacy of toripalimab, a PD-1 mAb, in patients with advanced or recurrent malignancies in a phase I study. Eur. J. Cancer 2020, 130, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Cai, Q.; Jiang, Y.; Huang, G.; Bi, M.; Wang, B.; Zhou, Y.; Wang, G.; Ying, H.; Tao, Z.; et al. Activity and Safety of Geptanolimab (GB226) for Patients with Unresectable, Recurrent, or Metastatic Alveolar Soft Part Sarcoma: A Phase II, Single-arm Study. Clin. Cancer Res. 2020, 26, 6445–6452. [Google Scholar] [CrossRef]
- Huan, C.; Kelly, M.L.; Steele, R.; Shapira, I.; Gottesman, S.R.S.; Roman, C.A.J. Transcription factors TFE3 and TFEB are critical for CD40 ligand expression and thymus-dependent humoral immunity. Nat. Immunol. 2006, 7, 1082–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, X.; Miller, Z.A.; Benchabane, H.; Wrana, J.L.; Lodish, H.F. Synergism between Transcription Factors TFE3 and Smad3 in Transforming Growth Factor-β-induced Transcription of theSmad7 Gene *. J. Biol. Chem. 2000, 275, 33205–33208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan Coyne, G.; Naqash, A.R.; Sankaran, H.; Chen, A.P. Advances in the management of alveolar soft part sarcoma. Curr. Probl. Cancer 2021, 45, 100775. [Google Scholar] [CrossRef] [PubMed]
- Lewin, J.; Davidson, S.; Anderson, N.D.; Lau, B.Y.; Kelly, J.; Tabori, U.; Salah, S.; Butler, M.O.; Aung, K.L.; Shlien, A.; et al. Response to immune checkpoint inhibition in two patients with alveolar soft-part sarcoma. Cancer Immunol. Res. 2018, 6, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Salah, S.; Lewin, J.H.; Davidson, S.; Anderson, N.; Periera, S.L.; Lau, B.; Shlien, A.; Dickson, B.; Ferguson, P.; Wunder, J.; et al. Immunoprofiling in alveolar soft part sarcoma. J. Clin. Oncol. 2017, 35, 11059. [Google Scholar] [CrossRef]
- Conley, A.P.; Trinh, V.A.; Zobniw, C.M.; Posey, K.; Martinez, J.D.; Arrieta, O.G.; Wang, W.L.; Lazar, A.J.; Somaiah, N.; Roszik, J.; et al. Positive Tumor Response to Combined Checkpoint Inhibitors in a Patient With Refractory Alveolar Soft Part Sarcoma: A Case Report. J. Glob. Oncol. 2018, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Leruste, A.; Tosello, J.; Ramos, R.N.; Tauziède-Espariat, A.; Brohard, S.; Han, Z.Y.; Beccaria, K.; Andrianteranagna, M.; Caudana, P.; Nikolic, J.; et al. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 2019, 36, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Kang, H.J.; Yalon-Oren, M.; Marshall, L.V.; Vezina, C.; Pappo, A.; Laetsch, T.W.; Petrilli, A.S.; Ebinger, M.; Toporski, J.; et al. Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): Interim analysis of an open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 121–133. [Google Scholar] [CrossRef]
- Iijima, Y.; Sakakibara, R.; Ishizuka, M.; Honda, T.; Shirai, T.; Okamoto, T.; Tateishi, T.; Sakashita, H.; Tamaoka, M.; Takemoto, A.; et al. Notable response to nivolumab during the treatment of SMARCA4-deficient thoracic sarcoma: A case report. Immunotherapy 2020, 12, 563–569. [Google Scholar] [CrossRef]
- Takada, K.; Sugita, S.; Murase, K.; Kikuchi, T.; Oomori, G.; Ito, R.; Hayasaka, N.; Miyanishi, K.; Iyama, S.; Ikeda, H.; et al. Exceptionally rapid response to pembrolizumab in a SMARCA4-deficient thoracic sarcoma overexpressing PD-L1: A case report. Thorac. Cancer 2019, 10, 2312–2315. [Google Scholar] [CrossRef]
- Henon, C.; Blay, J.Y.; Massard, C.; Mir, O.; Bahleda, R.; Dumont, S.; Postel-Vinay, S.; Adam, J.; Soria, J.C.; Le Cesne, A. Long lasting major response to pembrolizumab in a thoracic malignant rhabdoid-like SMARCA4-deficient tumor. Ann. Oncol. 2019, 30, 1401–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zucman, J.; Delattre, O.; Desmaze, C.; Epstein, A.L.; Stenman, G.; Speleman, F.; Fletchers, C.D.M.; Aurias, A.; Thomas, G. EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat. Genet. 1993, 4, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.J.; Kim, J.J.; Ozsolak, F.; Widlund, H.R.; Rozenblatt-Rosen, O.; Granter, S.R.; Du, J.; Fletcher, J.A.; Denny, C.T.; Lessnick, S.L.; et al. Oncogenic MITF dysregulation in clear cell sarcoma: Defining the MiT family of human cancers. Cancer Cell 2006, 9, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, A.L.; Joon, A.; Haydu, L.E.; Lazar, A.J.; Tetzlaff, M.T.; Ravi, V.; Davies, M.A. Outcomes of melanoma soft parts/clear cell sarcoma (MSP/CCS) patients (pts) with immune and targeted therapies. J. Clin. Oncol. 2019, 37, e21046. [Google Scholar] [CrossRef]
- Painter, C.A.; Jain, E.; Tomson, B.N.; Dunphy, M.; Stoddard, R.E.; Thomas, B.S.; Damon, A.L.; Shah, S.; Kim, D.; Gómez Tejeda Zañudo, J.; et al. The Angiosarcoma Project: Enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat. Med. 2020, 26, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Florou, V.; Rosenberg, A.E.; Wieder, E.; Komanduri, K.V.; Kolonias, D.; Uduman, M.; Castle, J.C.; Buell, J.S.; Trent, J.C.; Wilky, B.A. Angiosarcoma patients treated with immune checkpoint inhibitors: A case series of seven patients from a single institution. J. Immunother. Cancer 2019, 7, 213. [Google Scholar]
- Wagner, M.J.; Othus, M.; Patel, S.P.; Ryan, C.; Sangal, A.; Powers, B.; Budd, G.T.; Victor, A.I.; Hsueh, C.T.; Chugh, R.; et al. Multicenter phase II trial (SWOG S1609, cohort 51) of ipilimumab and nivolumab in metastatic or unresectable angiosarcoma: A substudy of dual anti-CTLA-4 and anti-PD-1 blockade in rare tumors (DART). J. Immunother. Cancer 2021, 9, e002990. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Al-Saleem, T.; Brooks, J.J.; Rogatko, A.; Kraybill, W.G.; Eisenberg, B. Vascular Endothelial Growth Factor and Soft Tissue Sarcomas: Tumor Expression Correlates With Grade. Ann. Surg. Oncol. 2001, 8, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, X.; Dikov, M.M.; Novitskiy, S.V.; Mosse, C.A.; Yang, L.; Carbone, D.P. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood 2007, 110, 624–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8++ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN promotes resistance to T cell–mediated immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [Green Version]
- Tazzari, M.; Negri, T.; Rini, F.; Vergani, B.; Huber, V.; Villa, A.; Dagrada, P.; Colombo, C.; Fiore, M.; Gronchi, A.; et al. Adaptive immune contexture at the tumour site and downmodulation of circulating myeloid-derived suppressor cells in the response of solitary fibrous tumour patients to anti-angiogenic therapy. Br. J. Cancer 2014, 111, 1350–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazzari, M.; Indio, V.; Vergani, B.; De Cecco, L.; Rini, F.; Negri, T.; Camisaschi, C.; Fiore, M.; Stacchiotti, S.; Dagrada, G.P.; et al. Adaptive Immunity in Fibrosarcomatous Dermatofibrosarcoma Protuberans and Response to Imatinib Treatment. J. Investig. Dermatol. 2017, 137, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Stehle, F.; Schulz, K.; Fahldieck, C.; Kalich, J.; Lichtenfels, R.; Riemann, D.; Seliger, B. Reduced immunosuppressive properties of axitinib in comparison with other tyrosine kinase inhibitors. J. Biol. Chem. 2013, 288, 16334–16347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zizzari, I.G.; Napoletano, C.; Botticelli, A.; Caponnetto, S.; Calabrò, F.; Gelibter, A.; Rughetti, A.; Ruscito, I.; Rahimi, H.; Rossi, E.; et al. TK inhibitor pazopanib primes DCs by downregulation of the β-catenin pathway. Cancer Immunol. Res. 2018, 6, 711–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judson, I.; Morden, J.P.; Kilburn, L.; Leahy, M.; Benson, C.; Bhadri, V.; Campbell-Hewson, Q.; Cubedo, R.; Dangoor, A.; Fox, L.; et al. Cediranib in patients with alveolar soft-part sarcoma (CASPS): A double-blind, placebo-controlled, randomised, phase 2 trial. Lancet Oncol. 2019, 20, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Wilky, B.A.; Trucco, M.M.; Subhawong, T.K.; Florou, V.; Park, W.; Kwon, D.; Wieder, E.D.; Kolonias, D.; Rosenberg, A.E.; Kerr, D.A.; et al. Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: A single-centre, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 837–848. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Hindi, N.; Grignani, G.; Martinez-Trufero, J.; Redondo, A.; Valverde, C.; Stacchiotti, S.; Lopez-Pousa, A.; D’Ambrosio, L.; Gutierrez, A.; et al. Nivolumab and sunitinib combination in advanced soft tissue sarcomas: A multicenter, single-arm, phase Ib/II trial. J. Immunother. Cancer 2020, 8, e001561. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Zitvogel, L.; Kroemer, G. The secret ally: Immunostimulation by anticancer drugs. Nat. Rev. Drug Discov. 2012, 11, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.M.; Fowler, D.W.; Smith, P.; Dalgleish, A.G. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br. J. Cancer 2010, 102, 115–123. [Google Scholar] [CrossRef]
- Kodumudi, K.N.; Woan, K.; Gilvary, D.L.; Sahakian, E.; Wei, S.; Djeu, J.Y. A novel chemoimmunomodulating property of docetaxel: Suppression of myeloid-derived suppressor cells in tumor bearers. Clin. Cancer Res. 2010, 16, 4583–4594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, A.K.; Lake, R.A.; Marzo, A.L.; Scott, B.; Heath, W.R.; Collins, E.J.; Frelinger, J.A.; Robinson, B.W.S. Induction of Tumor Cell Apoptosis In Vivo Increases Tumor Antigen Cross-Presentation, Cross-Priming Rather than Cross-Tolerizing Host Tumor-Specific CD8 T Cells. J. Immunol. 2003, 170, 4905–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgiovine, C.; Frapolli, R.; Liguori, M.; Digifico, E.; Colombo, F.S.; Meroni, M.; Allavena, P.; D’Incalci, M. Inhibition of tumor-associated macrophages by trabectedin improves the antitumor adaptive immunity in response to anti-PD-1 therapy. Eur. J. Immunol. 2021, 51, 2677–2686. [Google Scholar] [CrossRef] [PubMed]
- De Vita, A.; Recine, F.; Miserocchi, G.; Pieri, F.; Spadazzi, C.; Cocchi, C.; Vanni, S.; Liverani, C.; Farnedi, A.; Fabbri, F.; et al. The potential role of the extracellular matrix in the activity of trabectedin in UPS and L-sarcoma: Evidences from a patie—Derived primary culture case series in tridimensional and zebrafish models. J. Exp. Clin. Cancer Res. 2021, 40, 165. [Google Scholar] [CrossRef] [PubMed]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.Y.; Le Cesne, A.; Bompas, E.; Piperno-Neumann, S.; Cousin, S.; Grellety, T.; et al. Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas a phase 2 clinical trial. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Toulmonde, M.; Grellety, T.; Blay, J.Y.; Le Cesne, A.; Penel, N.; Piperno-Neumann, S.; Bertucci, F.; Chevreau, C.; Bompas, E.; Cousin, S.; et al. PEMBROSARC combination of MK3475 and metronomic cyclophosphamide (mCP) in patients (pts) with advanced sarcomas a multicentre phase II trial with 3 new combination strategies. J. Clin. Immunol. 2018, 36, TPS11587. [Google Scholar] [CrossRef]
- Weiss, G.J.; Waypa, J.; Blaydorn, L.; Coats, J.; McGahey, K.; Sangal, A.; Niu, J.; Lynch, C.A.; Farley, J.H.; Khemka, V. A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PembroPlus). Br. J. Cancer 2017, 117, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, S.M.; Redman, M.W.; Baker, K.K.; Wagner, M.J.; Schroeder, B.A.; Loggers, E.T.; Trieselmann, K.; Copeland, V.C.; Zhang, S.; Black, G.; et al. Assessment of Doxorubicin and Pembrolizumab in Patients With Advanced Anthracycline-Naive Sarcoma: A Phase 1/2 Nonrandomized Clinical Trial. JAMA Oncol. 2020, 6, 1778–1782. [Google Scholar] [CrossRef]
- Marcrom, S.; De Los Santos, J.F.; Conry, R.M. Complete response of mediastinal clear cell sarcoma to pembrolizumab with radiotherapy. Clin. Sarcoma Res. 2017, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Seebacher, N.A.; Garbutt, C.; Ma, H.; Gao, P.; Xiao, T.; Hornicek, F.J.; Duan, Z. Inhibition of cyclin-dependent kinase 4 as a potential therapeutic strategy for treatment of synovial sarcoma. Cell Death Dis. 2018, 9, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameratunga, M.; Kipps, E.; Okines, A.F.C.; Lopez, J.S. To cycle or fight—CDK4/6 inhibitors at the crossroads of anticancer immunity. Clin. Cancer Res. 2019, 25, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; Decristo, M.J.; Watt, A.C.; Brinjones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Sorrentino, J.A.; Dragnev, K.H.; Weiss, J.M.; Owonikoko, T.K.; Rytlewski, J.A.; Hood, J.; Yang, Z.; Malik, R.K.; Strum, J.C.; et al. CDK4/6 inhibition enhances antitumor efficacy of chemotherapy and immune checkpoint inhibitor combinations in preclinical models and enhances T-cell activation in patients with SCLC receiving chemotherapy. J. Immunother. Cancer 2020, 8, 847. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [Green Version]
- Jerby-Arnon, L.; Neftel, C.; Shore, M.E.; Weisman, H.R.; Mathewson, N.D.; McBride, M.J.; Haas, B.; Izar, B.; Volorio, A.; Boulay, G.; et al. Opposing immune and genetic mechanisms shape oncogenic programs in synovial sarcoma. Nat. Med. 2021, 27, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Schwartz, G.K.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Chi, P.; Antonescu, C.R.; Landa, J.; Qin, L.X.; Crago, A.M.; et al. Progression-Free Survival Among Patients With Well-Differentiated or Dedifferentiated Liposarcoma Treated With CDK4 Inhibitor Palbociclib: A Phase 2 Clinical Trial. JAMA Oncol. 2016, 2, 937–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Poh, A.R.; Ernst, M. Targeting macrophages in cancer: From bench to bedside. Front. Oncol. 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, D.; Fujiwara, Y.; Horlad, H.; Saito, Y.; Iriki, T.; Tsuboki, J.; Cheng, P.; Nakagata, N.; Mizuta, H.; Bekki, H.; et al. CD163 is required for protumoral activation of macrophages in human and murine sarcoma. Cancer Res. 2018, 78, 3255–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Sun, C.; Li, C.; Jiao, X.; Griffin, B.B.; Dongol, S.; Wu, H.; Zhang, C.; Cao, W.; Dong, R.; et al. Upregulated MELK Leads to Doxorubicin Chemoresistance and M2 Macrophage Polarization via the miR-34a/JAK2/STAT3 Pathway in Uterine Leiomyosarcoma. Front. Oncol. 2020, 10, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patwardhan, P.P.; Surriga, O.; Beckman, M.J.; De Stanchina, E.; Dematteo, R.P.; Tap, W.D.; Schwartz, G.K. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clin. Cancer Res. 2014, 20, 3146–3158. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- West, R.B.; Rubin, B.P.; Miller, M.A.; Subramanian, S.; Kaygusuz, G.; Montgomery, K.; Zhu, S.; Marinelli, R.J.; De Luca, A.; Downs-Kelly, E.; et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc. Natl. Acad. Sci. USA 2006, 103, 690–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.Y.; Alcindor, T.; Ganjoo, K.; Martín-Broto, J.; Ryan, C.W.; et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Jungbluth, A.A.; Antonescu, C.R.; Busam, K.J.; Iversen, K.; Kolb, D.; Coplan, K.; Chen, Y.T.; Stockert, E.; Ladanyi, M.; Old, L.J. Monophasic and biphasic synovial sarcomas abundantly express cancer/testis antigen NY-ESO-1 but not MAGE-A1 or CT7. Int. J. Cancer 2001, 94, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, S.M.; Jungbluth, A.A.; Hoch, B.L.; Farrar, E.A.; Bleakley, M.; Schneider, D.J.; Loggers, E.T.; Rodler, E.; Eary, J.F.; Conrad, E.U.; et al. NY-ESO-1 is a ubiquitous immunotherapeutic target antigen for patients with myxoid/round cell liposarcoma. Cancer 2012, 118, 4564–4570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, S.M.; Jones, R.L.; Farrar, E.A.; Lai, I.P.; Lee, S.M.; Cao, J.; Pillarisetty, V.G.; Hoch, B.L.; Gullett, A.; Bleakley, M.; et al. Tetramer guided, cell sorter assisted production of clinical grade autologous NY-ESO-1 specific CD8+ T cells. J. Immunother. Cancer 2014, 2, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, S.P.; Druta, M.; Liebner, D.A.; Schuetze, S.; Somaiah, N.; Van Tine, B.A.; Tap, W.D.; Pulham, T.; Chagin, K.; Norry, E.; et al. Pilot study of NY-ESO-1 c259 T cells in advanced myxoid/round cell liposarcoma. J. Clin. Oncol. 2018, 36, 3005. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1c259T cells in synovial sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, I.; Lowther, D.E.; Dryer-Minnerly, R.; Wang, R.; Fayngerts, S.; Nunez, D.; Betts, G.; Bath, N.; Tipping, A.J.; Melchiori, L.; et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J. Immunother. Cancer 2019, 7, 276. [Google Scholar] [CrossRef]
- Iura, K.; Kohashi, K.; Ishii, T.; Maekawa, A.; Bekki, H.; Otsuka, H.; Yamada, Y.; Yamamoto, H.; Matsumoto, Y.; Iwamoto, Y.; et al. MAGEA4 expression in bone and soft tissue tumors: Its utility as a target for immunotherapy and diagnostic marker combined with NY-ESO-1. Virchows Arch. 2017, 471, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef] [PubMed]
- Vredevoogd, D.W.; Kuilman, T.; Ligtenberg, M.A.; Boshuizen, J.; Stecker, K.E.; de Bruijn, B.; Krijgsman, O.; Huang, X.; Kenski, J.C.N.; Lacroix, R.; et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 2019, 178, 585–599.e15. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.G.; Jamal, K.; Dayal, J.H.; Tenev, T.; Kyula-Currie, J.; Guppy, N.; Gazinska, P.; Roulstone, V.; Liccardi, G.; Davies, E.; et al. RIPK1-mediated immunogenic cell death promotes anti-tumour immunity against soft-tissue sarcoma. EMBO Mol. Med. 2020, 12, e10979. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-Talk between Myeloid-Derived Suppressor Cells and Macrophages Subverts Tumor Immunity toward a Type 2 Response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ribas, A.; Mucida, D.; Correspondence, S.F.T.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840.e18. [Google Scholar] [CrossRef] [Green Version]
- Pech, M.F.; Fong, L.E.; Villalta, J.E.; Chan, L.J.G.; Kharbanda, S.; O’brien, J.J.; McAllister, F.E.; Firestone, A.J.; Jan, C.H. Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance. eLife 2019, 8, e47362. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Sharma, S.; Rollins, R.A.; Paul, T.A. The complex role of EZH2 in the tumor microenvironment: Opportunities and challenges for immunotherapy combinations. Future Med. Chem. 2020, 12, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korfhage, J.; Lombard, D.B. Malignant Peripheral Nerve Sheath Tumors: From Epigenome to Bedside. Mol. Cancer Res. 2019, 17, 1417–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aznar, M.A.; Planelles, L.; Perez-Olivares, M.; Molina, C.; Garasa, S.; Etxeberría, I.; Perez, G.; Rodriguez, I.; Bolaños, E.; Lopez-Casas, P.; et al. Immunotherapeutic effects of intratumoral nanoplexed poly I:C. J. Immunother. Cancer 2019, 7, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Márquez-Rodas, I.; Longo, F.; Rodriguez-Ruiz, M.E.; Calles, A.; Ponce, S.; Jove, M.; Rubio-Viqueira, B.; Perez-Gracia, J.L.; Gómez-Rueda, A.; López-Tarruella, S.; et al. Intratumoral nanoplexed poly I:C BO-112 in combination with systemic anti-PD-1 for patients with anti-PD-1-refractory tumors. Sci. Transl. Med. 2020, 12, eabb0391. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Kelly, C.M.; Antonescu, C.R.; Bowler, T.; Munhoz, R.; Chi, P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Movva, S.; Dholakia, R.; et al. Objective Response Rate among Patients with Locally Advanced or Metastatic Sarcoma Treated with Talimogene Laherparepvec in Combination with Pembrolizumab: A Phase 2 Clinical Trial. JAMA Oncol. 2020, 6, 402–408. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Moon, A.; Burke, J.; Hwang, T.H.; Kirn, D.H. A phase 2, open-label, randomized study of Pexa-Vec (JX-594) administered by intratumoral injection in patients with unresectable primary hepatocellular carcinoma. Methods Mol. Biol. 2015, 1317, 343–357. [Google Scholar] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT co-inhibitory receptors with specialized functions in immune regulation. Immunity 2016, 44, 989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. Tim-3 as a target for cancer immunotherapy and mechanisms of action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, H.L.; Ohashi, P.S. Molecular pathways: Evaluating the potential for B7-H4 as an immunoregulatory target. Clin. Cancer Res. 2017, 23, 2934–2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cai, P.; Liang, T.; Wang, L.; Hu, L. TIM-3 is a potential prognostic marker for patients with solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 31705–31713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podojil, J.R.; Miller, S.D. Potential targeting of B7-H4 for the treatment of cancer. Immunol. Rev. 2017, 276, 40–51. [Google Scholar] [CrossRef] [PubMed]
| Year/Author | Trial | Agents | Evaluable ASPS Patients (n) | ORR | Other Outcomes |
|---|---|---|---|---|---|
| 2017 Groisberg [26] | MD Anderson series | Anti-PD1 | 4 | 50% | 2 PR 2 SD |
| 2018 D’Angelo [18] | ALLIANCE A091401 | Nivolumab | 1 | 100% (PR) | |
| 2018 D’Angelo [18] | ALLIANCE A091401 | Nivolumab and ipilimumab | 1 | 0% | |
| 2021 Naqash [50] | NCI single arm phase II | Atezolizumab | 43 | 37% | 1 CR 14 PR 1 unconfirmed PR 25 SD 16.5-month mDOR |
| 2021 Blay [52] | AcSe single arm phase II French multicentre | Pembrolizumab | 14 | 50% | 7.5-month mPFS 36% 12-month PFS rate |
| 2020 Yang [53] | Beijing phase I | Toripalimab | 12 | 25% | 1 CR 4 PR 11.1-month mPFS |
| 2020 Shi [54] | Gxplore-005 Multicentre Chinese single arm phase II | Geptanolimab | 37 | 38% | 14 PR 6.9-month mPFS 3-month PFS rate 70% 6-month PFS rate 56% |
| 2020 Somaiah [21] | MD Anderson phase II | Durvalumab and tremelimumab | 10 | 50% (PR) | 90% 3-month PFS rate |
| 2020 Italiano [8] | Pooled analysis of phase II data | PD1/PDL1 ± other CPIs | 41 | 49% |
| Year/Author | Trial | Agents | Evaluable Patients (n) | ORR | Other Outcomes |
|---|---|---|---|---|---|
| 2018 D’Angelo [118] | Phase I/II | NY-ESO-1c259 T cells | MRCLS (2) | 100% unconfirmed PR | |
| 2018 D’Angelo [119] | Phase I/II | NY-ESO-1c259 T cells | SS (12) | 50% | 1 CR 5 PR |
| 2015 Robbins [120] | Phase I/II | NY-ESO-1 affinity-enhanced T cells Adjuvant IL-2 | SS (18) | 61% | 2 CR |
| 2019 Ramachandran [121] | Phase I/II cohort expansion | NY-ESO-1c259 T cells | SS (42) | Cohort 1: 50% Cohort 2: 40% Cohort 3: 20% Cohort 4: 27% | 1 CR 14 PR 24 SD 3 PD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerrison, W.G.J.; Lee, A.T.J.; Thway, K.; Jones, R.L.; Huang, P.H. Current Status and Future Directions of Immunotherapies in Soft Tissue Sarcomas. Biomedicines 2022, 10, 573. https://doi.org/10.3390/biomedicines10030573
Kerrison WGJ, Lee ATJ, Thway K, Jones RL, Huang PH. Current Status and Future Directions of Immunotherapies in Soft Tissue Sarcomas. Biomedicines. 2022; 10(3):573. https://doi.org/10.3390/biomedicines10030573
Chicago/Turabian StyleKerrison, William G. J., Alexander T. J. Lee, Khin Thway, Robin L. Jones, and Paul H. Huang. 2022. "Current Status and Future Directions of Immunotherapies in Soft Tissue Sarcomas" Biomedicines 10, no. 3: 573. https://doi.org/10.3390/biomedicines10030573
APA StyleKerrison, W. G. J., Lee, A. T. J., Thway, K., Jones, R. L., & Huang, P. H. (2022). Current Status and Future Directions of Immunotherapies in Soft Tissue Sarcomas. Biomedicines, 10(3), 573. https://doi.org/10.3390/biomedicines10030573