
Alternative Pharmacological Strategies for the Treatment of Alzheimer’s Disease: Focus on Neuromodulator Function

Abstract
1. Introduction
2. Neuromodulator Function in the Central Nervous System and Dysregulation in AD
2.1. Neuropeptides
2.2. Neuropeptides; Dysfunction in AD
2.3. Hormones
2.4. Hormones; Dysfunction in AD
2.5. Neurotrophins
2.6. Neurotrophins; Dysfunction in AD
2.7. ATP
2.8. ATP; Dysfunction in AD
2.9. Metal Ions
2.10. Metal Ions; Dysfunction in AD
3. Targeting Neuromodulator Function for AD Treatment
3.1. Neuropeptide Targeting
3.2. Hormone Targeting
3.3. Neurotrophin Targeting
3.4. ATP Targeting
3.5. Metal Ion Targeting
4. Boosting Efficacy and Reducing Limitations of AD Therapeutics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grossberg, G.T.; Tong, G.; Burke, A.D.; Tariot, P.N. Present Algorithms and Future Treatments for Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 67, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the Global Burden of Alzheimer’s Disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.J.; Wimo, A.; Guerchet, M.M.; Ali, G.C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015—The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International (ADI): London, UK, 2015. [Google Scholar]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. Caveolin: A New Link Between Diabetes and AD. Cell. Mol. Neurobiol. 2020, 40, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J.F. Selective loss of central cholinergic neurons in alzheimer’s disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Perry, E.K.; Gibson, P.H.; Blessed, G.; Perry, R.H.; Tomlinson, B.E. Neurotransmitter Enzyme Abnormalities in Senile Dementia. Choline Acetyltransferase and Glutamic Acid Decarboxylase Activities in Necropsy Brain Tissue. J. Neurol. Sci. 1977, 34, 247–265. [Google Scholar] [CrossRef]
- Perry, E.K.; Tomlinson, B.E.; Blessed, G.; Bergmann, K.; Gibson, P.H.; Perry, R.H. Correlation of Cholinergic Abnormalities with Senile Plaques and Mental Test Scores in Senile Dementia. BMJ 1978, 2, 1457–1459. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S.; Cholinergic System Working Group, and for the (APMI). Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimer’s Dis. 2019, 6, 2–15. [Google Scholar] [CrossRef]
- Perry, E.K. The cholinergic hypothesis—Ten years on. Br. Med. Bull. 1986, 42, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The Cholinergic Hypothesis of Alzheimer’s Disease: A Review of Progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Olajide, O.J.; Gbadamosi, I.T.; Yawson, E.O.; Arogundade, T.; Lewu, F.S.; Ogunrinola, K.Y.; Adigun, O.O.; Bamisi, O.; Lambe, E.; Arietarhire, L.O.; et al. Hippocampal Degeneration and Behavioral Impairment During Alzheimer-Like Pathogenesis Involves Glutamate Excitotoxicity. J. Mol. Neurosci. 2021, 71, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.A.; Antimisiaris, D.; O’Brien, J. Drugs for Alzheimer’s Disease: Are They Effective? Pharm. Ther. 2010, 35, 208–211. [Google Scholar]
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic Effects of Tacrine Administration in Patients With Alzheimer’s Disease. JAMA 1994, 271, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities At Synapses of Alzheimer’s Disease Neurons. J. Alzheimer’s Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid Beta, Mitochondrial Dysfunction and Synaptic Damage: Implications for Cognitive Decline in Aging and Alzheimer’s Disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Alzheimer’s Disease Pathologic Cascades: Who Comes First, What Drives What. Neurotox. Res. 2012, 22, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Chong, F.P.; Ng, K.Y.; Koh, R.Y.; Chye, S.M. Tau Proteins and Tauopathies in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2018, 38, 965–980. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between Amyloid-β and Tau in Alzheimer’s Disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Bestard-Lorigados, I.; Song, W. The Synapse as a Treatment Avenue for Alzheimer’s Disease. Mol. Psychiatry 2022, 27, 2940–2949. [Google Scholar] [CrossRef]
- Asher, S.; Priefer, R. Alzheimer’s Disease Failed Clinical Trials. Life Sci. 2022, 306, 120861. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Liu, K.Y.; Howard, R. Can We Learn Lessons from the FDA’s Approval of Aducanumab? Nat. Rev. Neurol. 2021, 17, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Aducanumab: European Agency Rejects Alzheimer’s Drug over Efficacy and Safety Concerns. BMJ 2021, 375, n3127. [Google Scholar] [CrossRef] [PubMed]
- Tonda-Turo, C.; Origlia, N.; Mattu, C.; Accorroni, A.; Chiono, V. Current Limitations in the Treatment of Parkinson’s and Alzheimer’s Diseases: State-of-the-Art and Future Perspective of Polymeric Carriers. Curr. Med. Chem. 2018, 25, 5755–5771. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Mazurek, M.F.; Chattha, G.K.; Svendsen, C.N.; Bird, E.D.; Martin, J.B. Neuropeptide Y Immunoreactivity Is Reduced in Cerebral Cortex in Alzheimer’s Disease. Ann. Neurol. 1986, 20, 282–288. [Google Scholar] [CrossRef]
- Koide, S.; Onishi, H.; Hashimoto, H.; Kai, T.; Yamagami, S. Plasma Neuropeptide Y Is Reduced in Patients with Alzheimer’s Disease. Neurosci. Lett. 1995, 198, 149–151. [Google Scholar] [CrossRef]
- Li, C.; Wu, X.; Liu, S.; Zhao, Y.; Zhu, J.; Liu, K. Roles of Neuropeptide Y in Neurodegenerative and Neuroimmune Diseases. Front. Neurosci. 2019, 13, 869. [Google Scholar] [CrossRef]
- Mazurek, M.F.; Beal, M.F.; Bird, E.D.; Martin, J.B. Oxytocin in Alzheimer’s Disease: Postmortem Brain Levels. Neurology 1987, 37, 1001–1003. [Google Scholar] [CrossRef]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in Insulin and Insulin Signaling in Alzheimer’s Disease: Cause or Consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-N.; Hofman, M.A.; Swaab, D.F. VIP Neurons in the Human SCN in Relation to Sex, Age, and Alzheimer’s Disease. Neurobiol. Aging 1995, 16, 571–576. [Google Scholar] [CrossRef]
- Han, P.; Liang, W.; Baxter, L.C.; Yin, J.; Tang, Z.; Beach, T.G.; Caselli, R.J.; Reiman, E.M.; Shi, J. Pituitary Adenylate Cyclase–Activating Polypeptide Is Reduced in Alzheimer Disease. Neurology 2014, 82, 1724–1728. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Almqvist, E.G.; Wallin, A.; Johansson, J.-O.; Andreasson, U.; Blennow, K.; Zetterberg, H.; Svensson, J. Cerebrospinal Fluid Substance P Concentrations Are Elevated in Patients with Alzheimer’s Disease. Neurosci. Lett. 2015, 609, 58–62. [Google Scholar] [CrossRef] [PubMed]
- De Souza, E.B.; Battaglia, G. Increased Corticotropin-Releasing Factor Receptors in Rat Cerebral Cortex Following Chronic Atropine Treatment. Brain Res. 1986, 397, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Vale, W.W.; Zweig, R.M.; Singer, H.S.; Mayeux, R.; Kuhar, M.J.; Price, D.L.; Souza, E.B.D. Reductions in Corticotropin Releasing Factor-like Immunoreactivity in Cerebral Cortex in Alzheimer’s Disease, Parkinson’s Disease, and Progressive Supranuclear Palsy. Neurology 1987, 37, 905. [Google Scholar] [CrossRef] [PubMed]
- Muhlbauer, M.; Metcalf, J.C., Jr.; Robertson, J.T.; Fridland, G.; Desiderio, D.M. Opioid Peptides in the Cerebrospinal Fluid of Alzheimer Patients. Biomed. Chromatogr. 1986, 1, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Poljak, A.; McLean, C.A.; Sachdev, P.; Brodaty, H.; Smythe, G.A. Quantification of Hemorphins in Alzheimer’s Disease Brains. J. Neurosci. Res. 2004, 75, 704–714. [Google Scholar] [CrossRef]
- Yakovleva, T.; Marinova, Z.; Kuzmin, A.; Seidah, N.G.; Haroutunian, V.; Terenius, L.; Bakalkin, G. Dysregulation of Dynorphins in Alzheimer Disease. Neurobiol. Aging 2007, 28, 1700–1708. [Google Scholar] [CrossRef]
- Jiang, H.K.; Owyang, V.V.; Hong, J.S.; Gallagher, M. Elevated Dynorphin in the Hippocampal Formation of Aged Rats: Relation to Cognitive Impairment on a Spatial Learning Task. Proc. Natl. Acad. Sci. USA 1989, 86, 2948–2951. [Google Scholar] [CrossRef]
- Meilandt, W.J.; Yu, G.-Q.; Chin, J.; Roberson, E.D.; Palop, J.J.; Wu, T.; Scearce-Levie, K.; Mucke, L. Enkephalin Elevations Contribute to Neuronal and Behavioral Impairments in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2008, 28, 5007–5017. [Google Scholar] [CrossRef]
- Dauvilliers, Y.A.; Lehmann, S.; Jaussent, I.; Gabelle, A. Hypocretin and Brain β-Amyloid Peptide Interactions in Cognitive Disorders and Narcolepsy. Front. Aging Neurosci. 2014, 6, 119. [Google Scholar] [CrossRef]
- Deuschle, M.; Schilling, C.; Leweke, F.M.; Enning, F.; Pollmächer, T.; Esselmann, H.; Wiltfang, J.; Frölich, L.; Heuser, I. Hypocretin in Cerebrospinal Fluid Is Positively Correlated with Tau and PTau. Neurosci. Lett. 2014, 561, 41–45. [Google Scholar] [CrossRef]
- Liguori, C.; Romigi, A.; Nuccetelli, M.; Zannino, S.; Sancesario, G.; Martorana, A.; Albanese, M.; Mercuri, N.B.; Izzi, F.; Bernardini, S.; et al. Orexinergic System Dysregulation, Sleep Impairment, and Cognitive Decline in Alzheimer Disease. JAMA Neurol. 2014, 71, 1498–1505. [Google Scholar] [CrossRef]
- Liguori, C. Orexin and Alzheimer’s Disease. In Behavioral Neuroscience of Orexin/Hypocretin; Lawrence, A.J., de Lecea, L., Eds.; Current Topics in Behavioral Neurosciences; Springer International Publishing: Cham, Switzerland, 2017; pp. 305–322. ISBN 978-3-319-57535-3. [Google Scholar]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, Iron and Zinc in Alzheimer’s Disease Senile Plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Faller, P. Copper and Zinc Binding to Amyloid-β: Coordination, Dynamics, Aggregation, Reactivity and Metal-Ion Transfer. ChemBioChem 2009, 10, 2837–2845. [Google Scholar] [CrossRef]
- Karr, J.W.; Szalai, V.A. Cu(II) Binding to Monomeric, Oligomeric, and Fibrillar Forms of the Alzheimer’s Disease Amyloid-β Peptide. Biochemistry 2008, 47, 5006–5016. [Google Scholar] [CrossRef]
- Lyubartseva, G.; Smith, J.L.; Markesbery, W.R.; Lovell, M.A. Alterations of Zinc Transporter Proteins ZnT-1, ZnT-4 and ZnT-6 in Preclinical Alzheimer’s Disease Brain. Brain Pathol. 2010, 20, 343–350. [Google Scholar] [CrossRef]
- Rottkamp, C.A.; Raina, A.K.; Zhu, X.; Gaier, E.; Bush, A.I.; Atwood, C.S.; Chevion, M.; Perry, G.; Smith, M.A. Redox-Active Iron Mediates Amyloid-β Toxicity. Free Radic. Biol. Med. 2001, 30, 447–450. [Google Scholar] [CrossRef]
- Mayes, J.; Tinker-Mill, C.; Kolosov, O.; Zhang, H.; Tabner, B.J.; Allsop, D. β-Amyloid Fibrils in Alzheimer Disease Are Not Inert When Bound to Copper Ions but Can Degrade Hydrogen Peroxide and Generate Reactive Oxygen Species. J. Biol. Chem. 2014, 289, 12052–12062. [Google Scholar] [CrossRef]
- McCord, M.C.; Aizenman, E. The Role of Intracellular Zinc Release in Aging, Oxidative Stress, and Alzheimer’s Disease. Front. Aging Neurosci. 2014, 6, 77. [Google Scholar] [CrossRef]
- Smith, M.A.; Harris, P.L.R.; Sayre, L.M.; Perry, G. Iron Accumulation in Alzheimer Disease Is a Source of Redox-Generated Free Radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef]
- Traxler, L.; Herdy, J.R.; Stefanoni, D.; Eichhorner, S.; Pelucchi, S.; Szücs, A.; Santagostino, A.; Kim, Y.; Agarwal, R.K.; Schlachetzki, J.C.M.; et al. Warburg-like Metabolic Transformation Underlies Neuronal Degeneration in Sporadic Alzheimer’s Disease. Cell Metab. 2022, 34, 1248–1263.e6. [Google Scholar] [CrossRef]
- Pan, R.-Y.; He, L.; Zhang, J.; Liu, X.; Liao, Y.; Gao, J.; Liao, Y.; Yan, Y.; Li, Q.; Zhou, X.; et al. Positive Feedback Regulation of Microglial Glucose Metabolism by Histone H4 Lysine 12 Lactylation in Alzheimer’s Disease. Cell Metab. 2022, 34, 634–648.e6. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Orellana, F.; Godoy, P.A.; Bastidas, C.Y.; Silva-Grecchi, T.; Guzmán, L.; Aguayo, L.G.; Fuentealba, J. ATP Leakage Induces P2XR Activation and Contributes to Acute Synaptic Excitotoxicity Induced by Soluble Oligomers of β-Amyloid Peptide in Hippocampal Neurons. Neuropharmacology 2016, 100, 116–123. [Google Scholar] [CrossRef]
- Hattori, N.; Kitagawa, K.; Higashida, T.; Yagyu, K.; Shimohama, S.; Wataya, T.; Perry, G.; Smith, M.A.; Inagaki, C. Cl−-ATPase and Na+/K+-ATPase Activities in Alzheimer’s Disease Brains. Neurosci. Lett. 1998, 254, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Orellana, F.; Fuentes-Fuentes, M.C.; Godoy, P.A.; Silva-Grecchi, T.; Panes, J.D.; Guzmán, L.; Yévenes, G.E.; Gavilán, J.; Egan, T.M.; Aguayo, L.G.; et al. P2X Receptor Overexpression Induced by Soluble Oligomers of Amyloid Beta Peptide Potentiates Synaptic Failure and Neuronal Dyshomeostasis in Cellular Models of Alzheimer’s Disease. Neuropharmacology 2018, 128, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.A.; Ramírez-Molina, O.; Fuentealba, J. Exploring the Role of P2X Receptors in Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 1330. [Google Scholar] [CrossRef]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF MRNA Is Decreased in the Hippocampus of Individuals with Alzheimer’s Disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef]
- Laske, C.; Stransky, E.; Leyhe, T.; Eschweiler, G.W.; Wittorf, A.; Richartz, E.; Bartels, M.; Buchkremer, G.; Schott, K. Stage-Dependent BDNF Serum Concentrations in Alzheimer’s Disease. J. Neural Transm. 2006, 113, 1217–1224. [Google Scholar] [CrossRef]
- Jiao, S.-S.; Shen, L.-L.; Zhu, C.; Bu, X.-L.; Liu, Y.-H.; Liu, C.-H.; Yao, X.-Q.; Zhang, L.-L.; Zhou, H.-D.; Walker, D.G.; et al. Brain-Derived Neurotrophic Factor Protects against Tau-Related Neurodegeneration of Alzheimer’s Disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef]
- Kaminari, A.; Giannakas, N.; Tzinia, A.; Tsilibary, E.C. Overexpression of Matrix Metalloproteinase-9 (MMP-9) Rescues Insulin-Mediated Impairment in the 5XFAD Model of Alzheimer’s Disease. Sci. Rep. 2017, 7, 683. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Wakabayashi, K.; Tsuji, S.; Takahashi, H.; Nawa, H. Regional Specificity of Alterations in NGF, BDNF and NT-3 Levels in Alzheimer’s Disease. Neuroreport 1996, 7, 2925–2928. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, R.; Gericke, C.A.; Jendroska, K.; Hartung, H.; Cervós-Navarro, J. NGF Content in the Cerebral Cortex of Non-Dementedpatients with Amyloid-Plaques and in SymptomaticALZHEIMERfn2fn2s Disease. Int. J. Dev. Neurosci. 1998, 16, 787–794. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Müller-Spahn, F.; Hulette, C.; Rosenberg, C.; Otten, U. Decreased TrkA Neurotrophin Receptor Expression in the Parietal Cortex of Patients with Alzheimer’s Disease. Neurosci. Lett. 1998, 241, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Bennett, D.A.; Cuello, A.C. The Human Brain NGF Metabolic Pathway Is Impaired in the Pre-Clinical and Clinical Continuum of Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 6023–6037. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-Specific Neurotrophin Imbalances in Alzheimer Disease: Decreased Levels of Brain-Derived Neurotrophic Factor and Increased Levels of Nerve Growth Factor in Hippocampus and Cortical Areas. Arch. Neurol. 2000, 57, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Cao, J.; Yang, S.-S.; Fu, Z.-Q.; Zeng, P.; Chu, J.; Ning, L.-N.; Zhang, T.; Shi, Y.; Tian, Q.; et al. Endoplasmic Reticulum Stress Induces Spatial Memory Deficits by Activating GSK-3. J. Cell. Mol. Med. 2018, 22, 3489–3502. [Google Scholar] [CrossRef]
- Tan, X.; Liang, Z.; Li, Y.; Zhi, Y.; Yi, L.; Bai, S.; Forest, K.H.; Nichols, R.A.; Dong, Y.; Li, Q.X. Isoorientin, a GSK-3β Inhibitor, Rescues Synaptic Dysfunction, Spatial Memory Deficits and Attenuates Pathological Progression in APP/PS1 Model Mice. Behav. Brain Res. 2021, 398, 112968. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen Synthase Kinase-3 Signaling in Alzheimer’s Disease. Biochim. Biophys. Acta BBA Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef]
- Patnaik, A.; Zagrebelsky, M.; Korte, M.; Holz, A. Signaling via the P75 Neurotrophin Receptor Facilitates Amyloid-β-Induced Dendritic Spine Pathology. Sci. Rep. 2020, 10, 13322. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.-F.; Ho, Y.-S.; Hung, C.H.-L.; Wuwongse, S.; Poon, C.-H.; Chiu, K.; Yang, X.; Chu, L.-W.; Chang, R.C.-C. Protective Effects of Testosterone on Presynaptic Terminals against Oligomeric β-Amyloid Peptide in Primary Culture of Hippocampal Neurons. BioMed Res. Int. 2014, 2014, 103906. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Kang, L.; Geng, D.; Han, S.; Li, S.; Du, J.; Wang, C.; Cui, H. Testosterone Modulates Structural Synaptic Plasticity of Primary Cultured Hippocampal Neurons through ERK-CREB Signalling Pathways. Mol. Cell. Endocrinol. 2020, 503, 110671. [Google Scholar] [CrossRef] [PubMed]
- Beauchet, O. Testosterone and Cognitive Function: Current Clinical Evidence of a Relationship. Eur. J. Endocrinol. 2006, 155, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Cui, C.; Yan, X.; Zhang, B.; Song, W.; Huo, D.; Wang, H.; Yang, Z. Effects of Testosterone on Synaptic Plasticity Mediated by Androgen Receptors in Male SAMP8 Mice. J. Toxicol. Environ. Health A 2016, 79, 849–855. [Google Scholar] [CrossRef]
- Bianchi, V.E. Impact of Testosterone on Alzheimer’s Disease. World J. Mens Health 2022, 40, 243–256. [Google Scholar] [CrossRef]
- Green, K.N.; Billings, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids Increase Amyloid-β and Tau Pathology in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2006, 26, 9047–9056. [Google Scholar] [CrossRef]
- Canet, G.; Chevallier, N.; Zussy, C.; Desrumaux, C.; Givalois, L. Central Role of Glucocorticoid Receptors in Alzheimer’s Disease and Depression. Front. Neurosci. 2018, 12, 739. [Google Scholar] [CrossRef]
- Sotiropoulos, I.; Catania, C.; Riedemann, T.; Fry, J.P.; Breen, K.C.; Michaelidis, T.M.; Almeida, O.F.X. Glucocorticoids Trigger Alzheimer Disease-like Pathobiochemistry in Rat Neuronal Cells Expressing Human Tau. J. Neurochem. 2008, 107, 385–397. [Google Scholar] [CrossRef]
- Pedrazzoli, M.; Losurdo, M.; Paolone, G.; Medelin, M.; Jaupaj, L.; Cisterna, B.; Slanzi, A.; Malatesta, M.; Coco, S.; Buffelli, M. Glucocorticoid Receptors Modulate Dendritic Spine Plasticity and Microglia Activity in an Animal Model of Alzheimer’s Disease. Neurobiol. Dis. 2019, 132, 104568. [Google Scholar] [CrossRef]
- Short, R.A.; O’Brien, P.C.; Graff-Radford, N.R.; Bowen, R.L. Elevated Gonadotropin Levels in Patients With Alzheimer Disease. Mayo Clin. Proc. 2001, 76, 906–909. [Google Scholar] [CrossRef]
- Casadesus, G.; Milliken, E.L.; Webber, K.M.; Bowen, R.L.; Lei, Z.; Rao, C.V.; Perry, G.; Keri, R.A.; Smith, M.A. Increases in Luteinizing Hormone Are Associated with Declines in Cognitive Performance. Mol. Cell. Endocrinol. 2007, 269, 107–111. [Google Scholar] [CrossRef]
- Berry, A.; Tomidokoro, Y.; Ghiso, J.; Thornton, J. Human Chorionic Gonadotropin (a Luteinizing Hormone Homologue) Decreases Spatial Memory and Increases Brain Amyloid-β Levels in Female Rats. Horm. Behav. 2008, 54, 143–152. [Google Scholar] [CrossRef]
- Xiong, J.; Kang, S.S.; Wang, Z.; Liu, X.; Kuo, T.-C.; Korkmaz, F.; Padilla, A.; Miyashita, S.; Chan, P.; Zhang, Z.; et al. FSH Blockade Improves Cognition in Mice with Alzheimer’s Disease. Nature 2022, 603, 470–476. [Google Scholar] [CrossRef]
- Rao, C.V. Involvement of Luteinizing Hormone in Alzheimer Disease Development in Elderly Women. Reprod. Sci. 2017, 24, 355–368. [Google Scholar] [CrossRef]
- Russo, A.F. Overview of Neuropeptides: Awakening the Senses? Headache 2017, 57, 37–46. [Google Scholar] [CrossRef]
- Yeo, X.Y.; Cunliffe, G.; Ho, R.C.; Lee, S.S.; Jung, S. Potentials of Neuropeptides as Therapeutic Agents for Neurological Diseases. Biomedicines 2022, 10, 343. [Google Scholar] [CrossRef]
- Abounoori, M.; Maddah, M.M.; Ardeshiri, M.R. Orexin Neuropeptides Modulate the Hippocampal-Dependent Memory through Basolateral Amygdala Interconnections. Cereb. Circ. Cogn. Behav. 2022, 3, 100035. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.H.; Viola, H. ERK1/2: A Key Cellular Component for the Formation, Retrieval, Reconsolidation and Persistence of Memory. Front. Mol. Neurosci. 2018, 11, 361. [Google Scholar] [CrossRef]
- Zhao, F.; Siu, J.J.; Huang, W.; Askwith, C.; Cao, L. Insulin Modulates Excitatory Synaptic Transmission and Synaptic Plasticity in the Mouse Hippocampus. Neuroscience 2019, 411, 237–254. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, B.; Cao, H.; Liu, W.; Guo, F.; Shen, F.; Ye, B.; Liu, H.; Li, Y.; Liu, Z. Oxytocin Exerts Antidepressant-like Effect by Potentiating Dopaminergic Synaptic Transmission in the MPFC. Neuropharmacology 2020, 162, 107836. [Google Scholar] [CrossRef]
- Dou, D.; Liang, J.; Zhai, X.; Li, G.; Wang, H.; Han, L.; Lin, L.; Ren, Y.; Liu, S.; Liu, C.; et al. Oxytocin Signalling in Dendritic Cells Regulates Immune Tolerance in the Intestine and Alleviates DSS-Induced Colitis. Clin. Sci. 2021, 135, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Ciranna, L.; Costa, L. Pituitary Adenylate Cyclase-Activating Polypeptide Modulates Hippocampal Synaptic Transmission and Plasticity: New Therapeutic Suggestions for Fragile X Syndrome. Front. Cell. Neurosci. 2019, 13, 524. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Reis, D.; Caulino-Rocha, A. VIP Modulation of Hippocampal Synaptic Plasticity: A Role for VIP Receptors as Therapeutic Targets in Cognitive Decline and Mesial Temporal Lobe Epilepsy. Front. Cell. Neurosci. 2020, 14, 153. [Google Scholar] [CrossRef] [PubMed]
- Abel, T.; Nguyen, P.V. Regulation of Hippocampus-Dependent Memory by Cyclic AMP-Dependent Protein Kinase. Prog. Brain Res. 2008, 169, 97–115. [Google Scholar] [CrossRef]
- Whittaker, E.; Vereker, E.; Lynch, M.A. Neuropeptide Y Inhibits Glutamate Release and Long-Term Potentiation in Rat Dentate Gyrus. Brain Res. 1999, 827, 229–233. [Google Scholar] [CrossRef]
- Smith, N.K.; Kondev, V.; Hunt, T.R.; Grueter, B.A. Neuropeptide Y Modulates Excitatory Synaptic Transmission and Promotes Social Behavior in the Mouse Nucleus Accumbens. Neuropharmacology 2022, 217, 109201. [Google Scholar] [CrossRef]
- Kash, T.L.; Winder, D.G. Neuropeptide Y and Corticotropin-Releasing Factor Bi-Directionally Modulate Inhibitory Synaptic Transmission in the Bed Nucleus of the Stria Terminalis. Neuropharmacology 2006, 51, 1013–1022. [Google Scholar] [CrossRef]
- Wang, X.F.; Ge, T.T.; Fan, J.; Yang, W.; Cui, R.J. The Role of Substance P in Epilepsy and Seizure Disorders. Oncotarget 2017, 8, 78225–78233. [Google Scholar] [CrossRef] [PubMed]
- Śmiałowska, M.; Domin, H.; Zięba, B.; Koźniewska, E.; Michalik, R.; Piotrowski, P.; Kajta, M. Neuroprotective Effects of Neuropeptide Y-Y2 and Y5 Receptor Agonists in Vitro and in Vivo. Neuropeptides 2009, 43, 235–249. [Google Scholar] [CrossRef]
- Saklani, P.; Khan, H.; Gupta, S.; Kaur, A.; Singh, T.G. Neuropeptides: Potential Neuroprotective Agents in Ischemic Injury. Life Sci. 2022, 288, 120186. [Google Scholar] [CrossRef] [PubMed]
- Peineau, S.; Rabiant, K.; Pierrefiche, O.; Potier, B. Synaptic Plasticity Modulation by Circulating Peptides and Metaplasticity: Involvement in Alzheimer’s Disease. Pharmacol. Res. 2018, 130, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Colmers, W.F.; Saggau, P. Inhibition of Synaptic Transmission by Neuropeptide Y in Rat Hippocampal Area CA1: Modulation of Presynaptic Ca2+ Entry. J. Neurosci. 1997, 17, 8169–8177. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dong, C.; Li, W.; Bu, W.; Wu, J.; Zhao, W. Neuropeptide Y Protects Cerebral Cortical Neurons by Regulating Microglial Immune Function. Neural Regen. Res. 2014, 9, 959–967. [Google Scholar] [CrossRef]
- Howell, O.W.; Doyle, K.; Goodman, J.H.; Scharfman, H.E.; Herzog, H.; Pringle, A.; Beck-Sickinger, A.G.; Gray, W.P. Neuropeptide Y Stimulates Neuronal Precursor Proliferation in the Post-Natal and Adult Dentate Gyrus. J. Neurochem. 2005, 93, 560–570. [Google Scholar] [CrossRef]
- Mahar, I.; Albuquerque, M.S.; Mondragon-Rodriguez, S.; Cavanagh, C.; Davoli, M.A.; Chabot, J.-G.; Williams, S.; Mechawar, N.; Quirion, R.; Krantic, S. Phenotypic Alterations in Hippocampal NPY- and PV-Expressing Interneurons in a Presymptomatic Transgenic Mouse Model of Alzheimer’s Disease. Front. Aging Neurosci. 2017, 8, 327. [Google Scholar] [CrossRef]
- Cai, Z.; Ratka, A. Opioid System and Alzheimer’s Disease. NeuroMolecular Med. 2012, 14, 91–111. [Google Scholar] [CrossRef]
- Ménard, C.; Herzog, H.; Schwarzer, C.; Quirion, R. Possible Role of Dynorphins in Alzheimer’s Disease and Age-Related Cognitive Deficits. Neurodegener. Dis. 2014, 13, 82–85. [Google Scholar] [CrossRef]
- Xu, Y.; Zhi, F.; Balboni, G.; Yang, Y.; Xia, Y. Opposite Roles of δ- and μ-Opioid Receptors in BACE1 Regulation and Alzheimer’s Injury. Front. Cell. Neurosci. 2020, 14, 88. [Google Scholar] [CrossRef]
- Manuel, I.; Lombardero, L.; Llorente-Ovejero, A.; Rodríguez-Puertas, R. Neuropeptides and Neurolipids: What They Are and How They Relate to Alzheimer’s Disease. In Genetics, Neurology, Behavior, and Diet in Dementia; Martin, C.R., Preedy, V.R., Eds.; Academic Press: Cambridge, MA, USA, 2020; Chapter 27; pp. 423–439. ISBN 978-0-12-815868-5. [Google Scholar]
- Ishii, M.; Hiller, A.J.; Pham, L.; McGuire, M.J.; Iadecola, C.; Wang, G. Amyloid-Beta Modulates Low-Threshold Activated Voltage-Gated L-Type Calcium Channels of Arcuate Neuropeptide Y Neurons Leading to Calcium Dysregulation and Hypothalamic Dysfunction. J. Neurosci. 2019, 39, 8816–8825. [Google Scholar] [CrossRef]
- Kyrou, I.; Tsigos, C. Stress Hormones: Physiological Stress and Regulation of Metabolism. Curr. Opin. Pharmacol. 2009, 9, 787–793. [Google Scholar] [CrossRef]
- Owen, J.A., Jr. Physiology of the Menstrual Cycle. Am. J. Clin. Nutr. 1975, 28, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Barry, J.A.; Owens, R. From Fetuses to Boys to Men: The Impact of Testosterone on Male Lifespan Development. In The Palgrave Handbook of Male Psychology and Mental Health; Barry, J.A., Kingerlee, R., Seager, M., Sullivan, L., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 3–24. ISBN 978-3-030-04384-1. [Google Scholar]
- Ali, S.A.; Begum, T.; Reza, F. Hormonal Influences on Cognitive Function. Malays. J. Med. Sci. MJMS 2018, 25, 31–41. [Google Scholar] [CrossRef]
- Lupien, S.J.; Maheu, F.; Tu, M.; Fiocco, A.; Schramek, T.E. The Effects of Stress and Stress Hormones on Human Cognition: Implications for the Field of Brain and Cognition. Brain Cogn. 2007, 65, 209–237. [Google Scholar] [CrossRef]
- De Quervain, D.; Wolf, O.T.; Roozendaal, B. Glucocorticoid-Induced Enhancement of Extinction—From Animal Models to Clinical Trials. Psychopharmacology 2019, 236, 183–199. [Google Scholar] [CrossRef]
- Taxier, L.R.; Gross, K.S.; Frick, K.M. Oestradiol as a Neuromodulator of Learning and Memory. Nat. Rev. Neurosci. 2020, 21, 535–550. [Google Scholar] [CrossRef]
- Barth, C.; Villringer, A.; Sacher, J. Sex Hormones Affect Neurotransmitters and Shape the Adult Female Brain during Hormonal Transition Periods. Front. Neurosci. 2015, 9, 37. [Google Scholar] [CrossRef]
- Nathan, L.; Shi, W.; Dinh, H.; Mukherjee, T.K.; Wang, X.; Lusis, A.J.; Chaudhuri, G. Testosterone Inhibits Early Atherogenesis by Conversion to Estradiol: Critical Role of Aromatase. Proc. Natl. Acad. Sci. USA 2001, 98, 3589–3593. [Google Scholar] [CrossRef]
- Höfer, P.; Lanzenberger, R.; Kasper, S. Testosterone in the Brain: Neuroimaging Findings and the Potential Role for Neuropsychopharmacology. Eur. Neuropsychopharmacol. 2013, 23, 79–88. [Google Scholar] [CrossRef]
- Hawley, W.R.; Grissom, E.M.; Martin, R.C.; Halmos, M.B.; Bart, C.L.S.; Dohanich, G.P. Testosterone Modulates Spatial Recognition Memory in Male Rats. Horm. Behav. 2013, 63, 559–565. [Google Scholar] [CrossRef]
- Muthu, S.J.; Lakshmanan, G.; Shimray, K.W.; Kaliyappan, K.; Sathyanathan, S.B.; Seppan, P. Testosterone Influence on Microtubule-Associated Proteins and Spine Density in Hippocampus: Implications on Learning and Memory. Dev. Neurosci. 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Gao, G.; Huang, W. A Study on Co-Localization of FSH and Its Receptor in Rat Hippocampus. J. Mol. Histol. 2008, 39, 49–55. [Google Scholar] [CrossRef]
- Burnham, V.; Sundby, C.; Laman-Maharg, A.; Thornton, J. Luteinizing Hormone Acts at the Hippocampus to Dampen Spatial Memory. Horm. Behav. 2017, 89, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bryan, K.J.; Mudd, J.C.; Richardson, S.L.; Chang, J.; Lee, H.; Zhu, X.; Smith, M.A.; Casadesus, G. Down-Regulation of Serum Gonadotropins Is as Effective as Estrogen Replacement at Improving Menopause-Associated Cognitive Deficits. J. Neurochem. 2010, 112, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.A.; Bhatta, S.; McGee, H.; Casadesus, G. Luteinizing Hormone: Evidence for Direct Action in the CNS. Horm. Behav. 2015, 76, 57–62. [Google Scholar] [CrossRef]
- Rodrigues, M.A.; Verdile, G.; Foster, J.K.; Hogervorst, E.; Joesbury, K.; Dhaliwal, S.; Corder, E.H.; Laws, S.M.; Hone, E.; Prince, R.; et al. Gonadotropins and Cognition in Older Women. J. Alzheimer’s Dis. 2008, 13, 267–274. [Google Scholar] [CrossRef]
- Woolley, C.S.; McEwen, B.S. Roles of Estradiol and Progesterone in Regulation of Hippocampal Dendritic Spine Density during the Estrous Cycle in the Rat. J. Comp. Neurol. 1993, 336, 293–306. [Google Scholar] [CrossRef]
- Joshi, S.; Sun, H.; Rajasekaran, K.; Williamson, J.; Perez-Reyes, E.; Kapur, J. A Novel Therapeutic Approach for Treatment of Catamenial Epilepsy. Neurobiol. Dis. 2018, 111, 127–137. [Google Scholar] [CrossRef]
- Kapur, J.; Joshi, S. Progesterone Modulates Neuronal Excitability Bidirectionally. Neurosci. Lett. 2021, 744, 135619. [Google Scholar] [CrossRef]
- Rasmuson, S.; Andrew, R.; Näsman, B.; Seckl, J.R.; Walker, B.R.; Olsson, T. Increased Glucocorticoid Production and Altered Cortisol Metabolism in Women with Mild to Moderate Alzheimer’s Disease. Biol. Psychiatry 2001, 49, 547–552. [Google Scholar] [CrossRef]
- Giubilei, F.; Patacchioli, F.R.; Antonini, G.; Monti, M.S.; Tisei, P.; Bastianello, S.; Monnazzi, P.; Angelucci, L. Altered Circadian Cortisol Secretion in Alzheimer’s Disease: Clinical and Neuroradiological Aspects. J. Neurosci. Res. 2001, 66, 262–265. [Google Scholar] [CrossRef]
- Elgh, E.; Lindqvist Åstot, A.; Fagerlund, M.; Eriksson, S.; Olsson, T.; Näsman, B. Cognitive Dysfunction, Hippocampal Atrophy and Glucocorticoid Feedback in Alzheimer’s Disease. Biol. Psychiatry 2006, 59, 155–161. [Google Scholar] [CrossRef]
- Sambamurti, K.; Kinsey, R.; Maloney, B.; Ge, Y.-W.; Lahiri, D.K. Gene Structure and Organization of the Human β-Secretase (BACE) Promoter. FASEB J. 2004, 18, 1034–1036. [Google Scholar] [CrossRef]
- Lesuis, S.L.; Lucassen, P.J.; Krugers, H.J. Early Life Stress Impairs Fear Memory and Synaptic Plasticity; a Potential Role for GluN2B. Neuropharmacology 2019, 149, 195–203. [Google Scholar] [CrossRef]
- Klyubin, I.; Ondrejcak, T.; Hu, N.-W.; Rowan, M.J. Glucocorticoids, Synaptic Plasticity and Alzheimer’s Disease. Curr. Opin. Endocr. Metab. Res. 2022, 25, 100365. [Google Scholar] [CrossRef]
- Lesuis, S.L.; Weggen, S.; Baches, S.; Lucassen, P.J.; Krugers, H.J. Targeting Glucocorticoid Receptors Prevents the Effects of Early Life Stress on Amyloid Pathology and Cognitive Performance in APP/PS1 Mice. Transl. Psychiatry 2018, 8, 53. [Google Scholar] [CrossRef]
- Dhikav, V.; Anand, K.S. Glucocorticoids May Initiate Alzheimer’s Disease: A Potential Therapeutic Role for Mifepristone (RU-486). Med. Hypotheses 2007, 68, 1088–1092. [Google Scholar] [CrossRef]
- Arsenault-Lapierre, G.; Chertkow, H.; Lupien, S. Seasonal Effects on Cortisol Secretion in Normal Aging, Mild Cognitive Impairment and Alzheimer’s Disease. Neurobiol. Aging 2010, 31, 1051–1054. [Google Scholar] [CrossRef]
- Sex and Gender Differences in Alzheimer’s Disease—1st Edition. Available online: https://www.elsevier.com/books/sex-and-gender-differences-in-alzheimers-disease/teresa-ferretti/978-0-12-819344-0 (accessed on 13 July 2022).
- Ferretti, M.T.; Santuccione Chadha, A. The Missing X Factor in Alzheimer Disease. Nat. Rev. Neurol. 2021, 17, 727–728. [Google Scholar] [CrossRef]
- Ratnakumar, A.; Zimmerman, S.E.; Jordan, B.A.; Mar, J.C. Estrogen Activates Alzheimer’s Disease Genes. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 906–917. [Google Scholar] [CrossRef]
- Yue, X.; Lu, M.; Lancaster, T.; Cao, P.; Honda, S.-I.; Staufenbiel, M.; Harada, N.; Zhong, Z.; Shen, Y.; Li, R. Brain Estrogen Deficiency Accelerates Aβ Plaque Formation in an Alzheimer’s Disease Animal Model. Proc. Natl. Acad. Sci. USA 2005, 102, 19198–19203. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Du, N.; Liu, Y.; Fan, X.; Wang, Y.; Jia, X.; Hou, X.; Wang, B. Low Testosterone Level and Risk of Alzheimer’s Disease in the Elderly Men: A Systematic Review and Meta-Analysis. Mol. Neurobiol. 2016, 53, 2679–2684. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, S.; Engert, S.; Behl, C. Testosterone Stimulates Rapid Secretory Amyloid Precursor Protein Release from Rat Hypothalamic Cells via the Activation of the Mitogen-Activated Protein Kinase Pathway. Neurosci. Lett. 2000, 296, 49–52. [Google Scholar] [CrossRef] [PubMed]
- McAllister, C.; Long, J.; Bowers, A.; Walker, A.; Cao, P.; Honda, S.-I.; Harada, N.; Staufenbiel, M.; Shen, Y.; Li, R. Genetic Targeting Aromatase in Male Amyloid Precursor Protein Transgenic Mice Down-Regulates β-Secretase (BACE1) and Prevents Alzheimer-Like Pathology and Cognitive Impairment. J. Neurosci. 2010, 30, 7326–7334. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Rosario, E.R.; Soper, J.C.; Pike, C.J. Androgens Regulate Tau Phosphorylation Through Phosphatidylinositol 3-Kinase–Protein Kinase B–Glycogen Synthase Kinase 3β Signaling. Neuroscience 2022, in press. [Google Scholar] [CrossRef]
- Muthu, S.J.; Seppan, P. Apoptosis in Hippocampal Tissue Induced by Oxidative Stress in Testosterone Deprived Male Rats. Aging Male 2020, 23, 1598–1610. [Google Scholar] [CrossRef]
- Yan, X.-S.; Yang, Z.-J.; Jia, J.-X.; Song, W.; Fang, X.; Cai, Z.-P.; Huo, D.-S.; Wang, H. Protective Mechanism of Testosterone on Cognitive Impairment in a Rat Model of Alzheimer’s Disease. Neural Regen. Res. 2019, 14, 649–657. [Google Scholar] [CrossRef]
- Wang, L.; Pei, J.-H.; Jia, J.-X.; Wang, J.; Song, W.; Fang, X.; Cai, Z.-P.; Huo, D.-S.; Wang, H.; Yang, Z.-J. Inhibition of Oxidative Stress by Testosterone Improves Synaptic Plasticity in Senescence Accelerated Mice. J. Toxicol. Environ. Health A 2019, 82, 1061–1068. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in Neuronal Development and Function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef]
- Miller, F.D.; Kaplan, D.R. Neurotrophin Signalling Pathways Regulating Neuronal Apoptosis. Cell. Mol. Life Sci. CMLS 2001, 58, 1045–1053. [Google Scholar] [CrossRef]
- Rösch, H.; Schweigreiter, R.; Bonhoeffer, T.; Barde, Y.-A.; Korte, M. The Neurotrophin Receptor P75NTR Modulates Long-Term Depression and Regulates the Expression of AMPA Receptor Subunits in the Hippocampus. Proc. Natl. Acad. Sci. USA 2005, 102, 7362–7367. [Google Scholar] [CrossRef]
- Minichiello, L.; Calella, A.M.; Medina, D.L.; Bonhoeffer, T.; Klein, R.; Korte, M. Mechanism of TrkB-Mediated Hippocampal Long-Term Potentiation. Neuron 2002, 36, 121–137. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and Their Receptors: A Convergence Point for Many Signalling Pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Balkowiec, A.; Katz, D.M. Cellular Mechanisms Regulating Activity-Dependent Release of Native Brain-Derived Neurotrophic Factor from Hippocampal Neurons. J. Neurosci. 2002, 22, 10399–10407. [Google Scholar] [CrossRef]
- Minichiello, L. TrkB Signalling Pathways in LTP and Learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef]
- Ortega, F.; Pérez-Sen, R.; Morente, V.; Delicado, E.G.; Miras-Portugal, M.T. P2X7, NMDA and BDNF Receptors Converge on GSK3 Phosphorylation and Cooperate to Promote Survival in Cerebellar Granule Neurons. Cell. Mol. Life Sci. 2010, 67, 1723–1733. [Google Scholar] [CrossRef]
- Caffino, L.; Mottarlini, F.; Fumagalli, F. Born to Protect: Leveraging BDNF Against Cognitive Deficit in Alzheimer’s Disease. CNS Drugs 2020, 34, 281–297. [Google Scholar] [CrossRef]
- Proenca, C.C.; Song, M.; Lee, F.S. Differential Effects of BDNF and Neurotrophin 4 (NT4) on Endocytic Sorting of TrkB Receptors. J. Neurochem. 2016, 138, 397–406. [Google Scholar] [CrossRef]
- Lessmann, V.; Gottmann, K.; Heumann, R. BDNF and NT-4/5 Enhance Glutamatergic Synaptic Transmission in Cultured Hippocampal Neurones. Neuroreport 1994, 6, 21–25. [Google Scholar] [CrossRef]
- Conner, J.M.; Franks, K.M.; Titterness, A.K.; Russell, K.; Merrill, D.A.; Christie, B.R.; Sejnowski, T.J.; Tuszynski, M.H. NGF Is Essential for Hippocampal Plasticity and Learning. J. Neurosci. 2009, 29, 10883–10889. [Google Scholar] [CrossRef]
- Lohof, A.M.; Ip, N.Y.; Poo, M. Potentiation of Developing Neuromuscular Synapses by the Neurotrophins NT-3 and BDNF. Nature 1993, 363, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Wang, T.; Olafsson, P.; Lu, B. Neurotrophin 3 Potentiates Neuronal Activity and Inhibits Gamma-Aminobutyratergic Synaptic Transmission in Cortical Neurons. Proc. Natl. Acad. Sci. USA 1994, 91, 12341–12345. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Schuman, E.M. Long-Lasting Neurotrophin-Induced Enhancement of Synaptic Transmission in the Adult Hippocampus. Science 1995, 267, 1658–1662. [Google Scholar] [CrossRef] [PubMed]
- Schuman, E.M. Neurotrophin Regulation of Synaptic Transmission. Curr. Opin. Neurobiol. 1999, 9, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, V.; Ducatenzeiler, A.; Dowd, E.; Jänne, J.; Grant, S.M.; Szyf, M.; Wandosell, F.; Avila, J.; Grimm, H.; Dunnett, S.B.; et al. Altered Mitogen-Activated Protein Kinase Signaling, Tau Hyperphosphorylation and Mild Spatial Learning Dysfunction in Transgenic Rats Expressing the β-Amyloid Peptide Intracellularly in Hippocampal and Cortical Neurons. Neuroscience 2004, 129, 583–592. [Google Scholar] [CrossRef]
- Devi, L.; Ohno, M. TrkB Reduction Exacerbates Alzheimer’s Disease-like Signaling Aberrations and Memory Deficits without Affecting β-Amyloidosis in 5XFAD Mice. Transl. Psychiatry 2015, 5, e562. [Google Scholar] [CrossRef]
- Subramaniam, S.; Unsicker, K. ERK and Cell Death: ERK1/2 in Neuronal Death. FEBS J. 2010, 277, 22–29. [Google Scholar] [CrossRef]
- Murase, K.; Igarashi, K.; Hayashi, K. Neurotrophin-3 (NT-3) Levels in the Developing Rat Nervous System and in Human Samples. Clin. Chim. Acta 1994, 227, 23–36. [Google Scholar] [CrossRef]
- Durany, N.; Michel, T.; Kurt, J.; Cruz-Sánchez, F.F.; Cervós-Navarro, J.; Riederer, P. Brain-Derived Neurotrophic Factor and Neurotrophin-3 Levels in Alzheimer’s Disease Brains. Int. J. Dev. Neurosci. 2000, 18, 807–813. [Google Scholar] [CrossRef]
- Skou, J. Chr. The Influence of Some Cations on an Adenosine Triphosphatase from Peripheral Nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Biology, Structure and Mechanism of P-Type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295. [Google Scholar] [CrossRef]
- Horisberger, J.; Lemas, V.; Kraehenbuhl, J.; Rossier, B.C. Structure-Function Relationship of Na, K-ATPase. Annu. Rev. Physiol. 1991, 53, 565–584. [Google Scholar] [CrossRef]
- Shiroya, T.; Fukunaga, R.; Akashi, K.; Shimada, N.; Takagi, Y.; Nishino, T.; Hara, M.; Inagaki, C. An ATP-Driven Cl− Pump in the Brain. J. Biol. Chem. 1989, 264, 17416–17421. [Google Scholar] [CrossRef]
- Webb, T.E.; Simon, J.; Krishek, B.J.; Bateson, A.N.; Smart, T.G.; King, B.F.; Burnstock, G.; Barnard, E.A. Cloning and Functional Expression of a Brain G-Protein-Coupled ATP Receptor. FEBS Lett. 1993, 324, 219–225. [Google Scholar] [CrossRef]
- Valera, S.; Hussy, N.; Evans, R.J.; Adami, N.; North, R.A.; Surprenant, A.; Buell, G. A New Class of Ligand-Gated Ion Channel Defined by P2X Receptor for Extracellular ATP. Nature 1994, 371, 516–519. [Google Scholar] [CrossRef]
- Hattori, M.; Gouaux, E. Molecular Mechanism of ATP Binding and Ion Channel Activation in P2X Receptors. Nature 2012, 485, 207–212. [Google Scholar] [CrossRef]
- Housley, G.D.; Raybould, N.P.; Thorne, P.R. Fluorescence Imaging of Na+ Influx via P2X Receptors in Cochlear Hair Cells. Hear. Res. 1998, 119, 1–13. [Google Scholar] [CrossRef]
- Egan, T.M.; Khakh, B.S. Contribution of Calcium Ions to P2X Channel Responses. J. Neurosci. 2004, 24, 3413–3420. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, H.-B. ATP Activates P2X Receptors to Mediate Gap Junctional Coupling in the Cochlea. Biochem. Biophys. Res. Commun. 2012, 426, 528–532. [Google Scholar] [CrossRef]
- Gordon, G.R.J.; Baimoukhametova, D.V.; Hewitt, S.A.; Rajapaksha, W.R.A.K.J.S.; Fisher, T.E.; Bains, J.S. Norepinephrine Triggers Release of Glial ATP to Increase Postsynaptic Efficacy. Nat. Neurosci. 2005, 8, 1078–1086. [Google Scholar] [CrossRef]
- Pougnet, J.-T.; Toulme, E.; Martinez, A.; Choquet, D.; Hosy, E.; Boué-Grabot, E. ATP P2X Receptors Downregulate AMPA Receptor Trafficking and Postsynaptic Efficacy in Hippocampal Neurons. Neuron 2014, 83, 417–430. [Google Scholar] [CrossRef]
- Boué-Grabot, E.; Pankratov, Y. Modulation of Central Synapses by Astrocyte-Released ATP and Postsynaptic P2X Receptors. Neural Plast. 2017, 2017, 9454275. [Google Scholar] [CrossRef]
- Lalo, U.; Palygin, O.; Verkhratsky, A.; Grant, S.G.N.; Pankratov, Y. ATP from Synaptic Terminals and Astrocytes Regulates NMDA Receptors and Synaptic Plasticity through PSD-95 Multi-Protein Complex. Sci. Rep. 2016, 6, 33609. [Google Scholar] [CrossRef]
- Guzman, S.J.; Gerevich, Z. P2Y Receptors in Synaptic Transmission and Plasticity: Therapeutic Potential in Cognitive Dysfunction. Neural Plast. 2016, 2016, e1207393. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef]
- Pickett, E.K.; Rose, J.; McCrory, C.; McKenzie, C.-A.; King, D.; Smith, C.; Gillingwater, T.H.; Henstridge, C.M.; Spires-Jones, T.L. Region-Specific Depletion of Synaptic Mitochondria in the Brains of Patients with Alzheimer’s Disease. Acta Neuropathol. 2018, 136, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The Role of Mitochondrial Dysfunction in Alzheimer’s Disease Pathogenesis. Alzheimer’s Dement. 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Joo, I.L.; Lam, W.W.; Oakden, W.; Hill, M.E.; Koletar, M.M.; Morrone, C.D.; Stanisz, G.J.; McLaurin, J.; Stefanovic, B. Early Alterations in Brain Glucose Metabolism and Vascular Function in a Transgenic Rat Model of Alzheimer’s Disease. Prog. Neurobiol. 2022, 217, 102327. [Google Scholar] [CrossRef] [PubMed]
- Bigl, M.; Brückner, M.K.; Arendt, T.; Bigl, V.; Eschrich, K. Activities of Key Glycolytic Enzymes in the Brains of Patients with Alzheimer’s Disease. J. Neural Transm. 1999, 106, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of Mitochondrial F1FO-ATP Synthase via OSCP in Alzheimer’s Disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c Oxidase and Mitochondrial F1F0-ATPase (ATP Synthase) Activities in Platelets and Brain from Patients with Alzheimer’s Disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Han, J.; Hyun, J.; Park, J.; Jung, S.; Oh, Y.; Kim, Y.; Ryu, S.-H.; Kim, S.-H.; Jeong, E.I.; Jo, D.-G.; et al. Aberrant Role of Pyruvate Kinase M2 in the Regulation of Gamma-Secretase and Memory Deficits in Alzheimer’s Disease. Cell Rep. 2021, 37, 110102. [Google Scholar] [CrossRef]
- DiChiara, T.; DiNunno, N.; Clark, J.; Bu, R.L.; Cline, E.N.; Rollins, M.G.; Gong, Y.; Brody, D.L.; Sligar, S.G.; Velasco, P.T.; et al. Alzheimer’s Toxic Amyloid Beta Oligomers: Unwelcome Visitors to the Na/K ATPase Alpha3 Docking Station. Yale J. Biol. Med. 2017, 90, 45–61. [Google Scholar]
- Verkhratsky, A.; Pankratov, Y.; Lalo, U.; Nedergaard, M. P2X Receptors in Neuroglia. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 151–161. [Google Scholar] [CrossRef]
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 Receptor Signalling Mediates Astrocytic Hyperactivity in Vivo in an Alzheimer’s Disease Mouse Model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Snow, W.M.; Stortz, G.; Perez, C.; Djordjevic, J.; Goertzen, A.L.; Ko, J.H.; Albensi, B.C. Regional Hypometabolism in the 3xTg Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2019, 127, 264–277. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial Bioenergetic Deficit Precedes Alzheimer’s Pathology in Female Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.-F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 Signalling Rescues Amyloid Pathology and Mitochondrial Dysfunction in Alzheimer’s Disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef]
- DeBenedictis, C.A.; Raab, A.; Ducie, E.; Howley, S.; Feldmann, J.; Grabrucker, A.M. Concentrations of Essential Trace Metals in the Brain of Animal Species—A Comparative Study. Brain Sci. 2020, 10, 460. [Google Scholar] [CrossRef]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper Signaling in the Mammalian Nervous System: Synaptic Effects. J. Neurosci. Res. 2013, 91, 2–19. [Google Scholar] [CrossRef]
- Peters, C.; Muñoz, B.; Sepúlveda, F.J.; Urrutia, J.; Quiroz, M.; Luza, S.; De Ferrari, G.V.; Aguayo, L.G.; Opazo, C. Biphasic Effects of Copper on Neurotransmission in Rat Hippocampal Neurons. J. Neurochem. 2011, 119, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Weiser, T.; Wienrich, M. The Effects of Copper Ions on Glutamate Receptors in Cultured Rat Cortical Neurons. Brain Res. 1996, 742, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Sharonova, I.N.; Vorobjev, V.S.; Haas, H.L. High-Affinity Copper Block of GABAA Receptor-Mediated Currents in Acutely Isolated Cerebellar Purkinje Cells of the Rat. Eur. J. Neurosci. 1998, 10, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Leiva, J.; Palestini, M.; Infante, C.; Goldschmidt, A.; Motles, E. Copper Suppresses Hippocampus LTP in the Rat, but Does Not Alter Learning or Memory in the Morris Water Maze. Brain Res. 2009, 1256, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Weber, N.L.; Smith, J.P. Copper Inhibits NMDA Receptor-Independent LTP and Modulates the Paired-Pulse Ratio after LTP in Mouse Hippocampal Slices. Int. J. Alzheimer’s Dis. 2011, 2011, e864753. [Google Scholar] [CrossRef]
- Colledge, M.; Snyder, E.M.; Crozier, R.A.; Soderling, J.A.; Jin, Y.; Langeberg, L.K.; Lu, H.; Bear, M.F.; Scott, J.D. Ubiquitination Regulates PSD-95 Degradation and AMPA Receptor Surface Expression. Neuron 2003, 40, 595–607. [Google Scholar] [CrossRef]
- Acevedo, K.; Masaldan, S.; Opazo, C.M.; Bush, A.I. Redox Active Metals in Neurodegenerative Diseases. J. Biol. Inorg. Chem. JBIC Publ. Soc. Biol. Inorg. Chem. 2019, 24, 1141–1157. [Google Scholar] [CrossRef]
- Marreiro, D.D.N.; Cruz, K.J.C.; Morais, J.B.S.; Beserra, J.B.; Severo, J.S.; de Oliveira, A.R.S. Zinc and Oxidative Stress: Current Mechanisms. Antioxidants 2017, 6, 24. [Google Scholar] [CrossRef]
- Brzóska, M.M.; Kozłowska, M.; Rogalska, J.; Gałażyn-Sidorczuk, M.; Roszczenko, A.; Smereczański, N.M. Enhanced Zinc Intake Protects against Oxidative Stress and Its Consequences in the Brain: A Study in an In Vivo Rat Model of Cadmium Exposure. Nutrients 2021, 13, 478. [Google Scholar] [CrossRef]
- Takeda, A. Movement of Zinc and Its Functional Significance in the Brain. Brain Res. Rev. 2000, 34, 137–148. [Google Scholar] [CrossRef]
- Kay, A.R.; Neyton, J.; Paoletti, P. A Startling Role for Synaptic Zinc. Neuron 2006, 52, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Koh, J.-Y.; Bush, A.I. The Neurobiology of Zinc in Health and Disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef]
- Palmiter, R.D.; Cole, T.B.; Quaife, C.J.; Findley, S.D. ZnT-3, a Putative Transporter of Zinc into Synaptic Vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 14934–14939. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Koh, J.-Y.; Aizenman, E.; Bush, A.I.; Hershfinkel, M. The Neurophysiology and Pathology of Brain Zinc. J. Neurosci. 2011, 31, 16076–16085. [Google Scholar] [CrossRef]
- Smart, T.G.; Hosie, A.M.; Miller, P.S. Zn2+ Ions: Modulators of Excitatory and Inhibitory Synaptic Activity. Neuroscientist 2004, 10, 432–442. [Google Scholar] [CrossRef]
- Qian, J.; Noebels, J.L. Exocytosis of Vesicular Zinc Reveals Persistent Depression of Neurotransmitter Release during Metabotropic Glutamate Receptor Long-Term Depression at the Hippocampal CA3–CA1 Synapse. J. Neurosci. 2006, 26, 6089–6095. [Google Scholar] [CrossRef]
- Kodirov, S.A.; Takizawa, S.; Joseph, J.; Kandel, E.R.; Shumyatsky, G.P.; Bolshakov, V.Y. Synaptically Released Zinc Gates Long-Term Potentiation in Fear Conditioning Pathways. Proc. Natl. Acad. Sci. USA 2006, 103, 15218–15223. [Google Scholar] [CrossRef] [PubMed]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the Physiology and Pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Spence, H.; McNeil, C.J.; Waiter, G.D. The Impact of Brain Iron Accumulation on Cognition: A Systematic Review. PLoS ONE 2020, 15, e0240697. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimer’s Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef]
- Muñoz, P.; Humeres, A.; Elgueta, C.; Kirkwood, A.; Hidalgo, C.; Núñez, M.T. Iron Mediates N-Methyl-d-Aspartate Receptor-Dependent Stimulation of Calcium-Induced Pathways and Hippocampal Synaptic Plasticity. J. Biol. Chem. 2011, 286, 13382–13392. [Google Scholar] [CrossRef]
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.; Saboury, A.A. Role of Copper in the Onset of Alzheimer’s Disease Compared to Other Metals. Front. Aging Neurosci. 2018, 9, 446. [Google Scholar] [CrossRef]
- Kaden, D.; Bush, A.I.; Danzeisen, R.; Bayer, T.A.; Multhaup, G. Disturbed Copper Bioavailability in Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2011, 2011, e345614. [Google Scholar] [CrossRef] [PubMed]
- Exley, C.; House, E.; Polwart, A.; Esiri, M.M. Brain Burdens of Aluminum, Iron, and Copper and Their Relationships with Amyloid-β Pathology in 60 Human Brains. J. Alzheimer’s Dis. 2012, 31, 725–730. [Google Scholar] [CrossRef]
- Gerber, H.; Wu, F.; Dimitrov, M.; Osuna, G.M.G.; Fraering, P.C. Zinc and Copper Differentially Modulate Amyloid Precursor Protein Processing by γ-Secretase and Amyloid-β Peptide Production. J. Biol. Chem. 2017, 292, 3751–3767. [Google Scholar] [CrossRef]
- Kong, G.K.-W.; Miles, L.A.; Crespi, G.A.N.; Morton, C.J.; Ng, H.L.; Barnham, K.J.; McKinstry, W.J.; Cappai, R.; Parker, M.W. Copper Binding to the Alzheimer’s Disease Amyloid Precursor Protein. Eur. Biophys. J. 2008, 37, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Luo, X.; Xu, H.; Ma, Q.; Yuan, J.; Li, X.; Chang, R.C.-C.; Qu, Z.; Huang, X.; Zhuang, Z.; et al. Identification of the Key Molecules Involved in Chronic Copper Exposure-Aggravated Memory Impairment in Transgenic Mice of Alzheimer’s Disease Using Proteomic Analysis. J. Alzheimer’s Dis. 2015, 44, 455–469. [Google Scholar] [CrossRef]
- Brewer, G.J. Alzheimer’s Disease Causation by Copper Toxicity and Treatment with Zinc. Front. Aging Neurosci. 2014, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Sasanian, N.; Bernson, D.; Horvath, I.; Wittung-Stafshede, P.; Esbjörner, E.K. Redox-Dependent Copper Ion Modulation of Amyloid-β (1-42) Aggregation In Vitro. Biomolecules 2020, 10, 924. [Google Scholar] [CrossRef]
- Hung, Y.H.; Bush, A.I.; Cherny, R.A. Copper in the Brain and Alzheimer’s Disease. JBIC J. Biol. Inorg. Chem. 2010, 15, 61–76. [Google Scholar] [CrossRef]
- Kalita, J.; Kumar, V.; Misra, U.K.; Bora, H.K. Memory and Learning Dysfunction Following Copper Toxicity: Biochemical and Immunohistochemical Basis. Mol. Neurobiol. 2018, 55, 3800–3811. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F.M. A possible role of zinc in the pathology of dementia. Lancet 1981, 317, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; Paradis, M.D.; Vonsattel, J.-P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid Induction of Alzheimer Aβ Amyloid Formation by Zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Tõugu, V.; Karafin, A.; Palumaa, P. Binding of Zinc(II) and Copper(II) to the Full-Length Alzheimer’s Amyloid-β Peptide. J. Neurochem. 2008, 104, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Jensen, K.B.; Jensen, M.S.; Silva, D.S.; Kesslak, P.J.; Danscher, G.; Frederickson, C.J. Histochemically-Reactive Zinc in Amyloid Plaques, Angiopathy, and Degenerating Neurons of Alzheimer’s Diseased Brains. Brain Res. 2000, 852, 274–278. [Google Scholar] [CrossRef]
- Björkdahl, C.; Sjögren, M.J.; Winblad, B.; Pei, J.-J. Zinc Induces Neurofilament Phosphorylation Independent of P70 S6 Kinase in N2a Cells. NeuroReport 2005, 16, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Park, E.J.; Seo, J.; Ko, S.J.; Lee, J.; Kim, C.H. Zinc Stimulates Tau S214 Phosphorylation by the Activation of Raf/Mitogen-Activated Protein Kinase-Kinase/Extracellular Signal-Regulated Kinase Pathway. Neuroreport 2011, 22, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive Loss in Zinc Transporter-3 Knock-Out Mice: A Phenocopy for the Synaptic and Memory Deficits of Alzheimer’s Disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef]
- Van Duijn, S.; Bulk, M.; van Duinen, S.G.; Nabuurs, R.J.A.; van Buchem, M.A.; van der Weerd, L.; Natté, R. Cortical Iron Reflects Severity Of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 60, 1533–1545. [Google Scholar] [CrossRef]
- Hwang, E.M.; Kim, S.-K.; Sohn, J.-H.; Lee, J.Y.; Kim, Y.; Kim, Y.S.; Mook-Jung, I. Furin Is an Endogenous Regulator of α-Secretase Associated APP Processing. Biochem. Biophys. Res. Commun. 2006, 349, 654–659. [Google Scholar] [CrossRef]
- Silvestri, L.; Camaschella, C. A Potential Pathogenetic Role of Iron in Alzheimer’s Disease. J. Cell. Mol. Med. 2008, 12, 1548–1550. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wang, P.; Zhong, M.-L.; Wang, T.; Huang, X.-S.; Li, J.-Y.; Wang, Z.-Y. Deferoxamine Inhibits Iron Induced Hippocampal Tau Phosphorylation in the Alzheimer Transgenic Mouse Brain. Neurochem. Int. 2013, 62, 165–172. [Google Scholar] [CrossRef]
- Muñoz, P.; Zavala, G.; Castillo, K.; Aguirre, P.; Hidalgo, C.; Núñez, M.T. Effect of Iron on the Activation of the MAPK/ERK Pathway in PC12 Neuroblastoma Cells. Biol. Res. 2006, 39, 189–190. [Google Scholar] [CrossRef][Green Version]
- Dos Santos, V.V.; Santos, D.B.; Lach, G.; Rodrigues, A.L.S.; Farina, M.; De Lima, T.C.M.; Prediger, R.D. Neuropeptide Y (NPY) Prevents Depressive-like Behavior, Spatial Memory Deficits and Oxidative Stress Following Amyloid-β (Aβ1–40) Administration in Mice. Behav. Brain Res. 2013, 244, 107–115. [Google Scholar] [CrossRef]
- Santos-Carvalho, A.; Elvas, F.; Álvaro, A.R.; Ambrósio, A.F.; Cavadas, C. Neuropeptide Y Receptors Activation Protects Rat Retinal Neural Cells against Necrotic and Apoptotic Cell Death Induced by Glutamate. Cell Death Dis. 2013, 4, e636. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Santos, T.; Viegas, M.; Cortes, L.; Bernardino, L.; Vieira, O.V.; Malva, J.O. Neuropeptide Y Inhibits Interleukin-1β-Induced Phagocytosis by Microglial Cells. J. Neuroinflamm. 2011, 8, 169. [Google Scholar] [CrossRef]
- Spencer, B.; Potkar, R.; Metcalf, J.; Thrin, I.; Adame, A.; Rockenstein, E.; Masliah, E. Systemic Central Nervous System (CNS)-Targeted Delivery of Neuropeptide Y (NPY) Reduces Neurodegeneration and Increases Neural Precursor Cell Proliferation in a Mouse Model of Alzheimer Disease. J. Biol. Chem. 2016, 291, 1905–1920. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, M.; Du, Y.; Zhang, W.; Bai, M.; Zhang, Z.; Li, Z.; Miao, J. Inhibition of C-Jun N-Terminal Kinase Activation Reverses Alzheimer Disease Phenotypes in APPswe/PS1dE9 Mice. Ann. Neurol. 2015, 77, 637–654. [Google Scholar] [CrossRef]
- Ramin, M.; Azizi, P.; Motamedi, F.; Haghparast, A.; Khodagholi, F. Inhibition of JNK Phosphorylation Reverses Memory Deficit Induced by β-Amyloid (1–42) Associated with Decrease of Apoptotic Factors. Behav. Brain Res. 2011, 217, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Kojro, E.; Postina, R.; Buro, C.; Meiringer, C.; Gehrig-Burger, K.; Fahrenholz, F. The Neuropeptide PACAP Promotes α-Secretase Pathway for Processing Alzheimer Amyloid Precursor Protein. FASEB J. 2006, 20, 512–514. [Google Scholar] [CrossRef]
- Rat, D.; Schmitt, U.; Tippmann, F.; Dewachter, I.; Theunis, C.; Wieczerzak, E.; Postina, R.; van Leuven, F.; Fahrenholz, F.; Kojro, E. Neuropeptide Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Slows down Alzheimer’s Disease-like Pathology in Amyloid Precursor Protein-Transgenic Mice. FASEB J. 2011, 25, 3208–3218. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, O.T.; Ay, H.; Aytan, N.; Carreras, I.; Kowall, N.W.; Dedeoglu, A.; Tuncel, N. Vasoactive Intestinal Peptide Decreases β-Amyloid Accumulation and Prevents Brain Atrophy in the 5xFAD Mouse Model of Alzheimer’s Disease. J. Mol. Neurosci. 2019, 68, 389–396. [Google Scholar] [CrossRef] [PubMed]
- El-Ganainy, S.O.; Soliman, O.A.; Ghazy, A.A.; Allam, M.; Elbahnasi, A.I.; Mansour, A.M.; Gowayed, M.A. Intranasal Oxytocin Attenuates Cognitive Impairment, β-Amyloid Burden and Tau Deposition in Female Rats with Alzheimer’s Disease: Interplay of ERK1/2/GSK3β/Caspase-3. Neurochem. Res. 2022, 47, 2345–2356. [Google Scholar] [CrossRef]
- Ye, C.; Cheng, M.; Ma, L.; Zhang, T.; Sun, Z.; Yu, C.; Wang, J.; Dou, Y. Oxytocin Nanogels Inhibit Innate Inflammatory Response for Early Intervention in Alzheimer’s Disease. ACS Appl. Mater. Interfaces 2022, 14, 21822–21835. [Google Scholar] [CrossRef] [PubMed]
- Jolivalt, C.G.; Lee, C.A.; Beiswenger, K.K.; Smith, J.L.; Orlov, M.; Torrance, M.A.; Masliah, E. Defective Insulin Signaling Pathway and Increased Glycogen Synthase Kinase-3 Activity in the Brain of Diabetic Mice: Parallels with Alzheimer’s Disease and Correction by Insulin. J. Neurosci. Res. 2008, 86, 3265–3274. [Google Scholar] [CrossRef]
- Vandal, M.; White, P.J.; Tremblay, C.; St-Amour, I.; Chevrier, G.; Emond, V.; Lefrançois, D.; Virgili, J.; Planel, E.; Giguere, Y.; et al. Insulin Reverses the High-Fat Diet-Induced Increase in Brain Aβ and Improves Memory in an Animal Model of Alzheimer Disease. Diabetes 2014, 63, 4291–4301. [Google Scholar] [CrossRef]
- Na, H.; Gan, Q.; Mcparland, L.; Yang, J.B.; Yao, H.; Tian, H.; Zhang, Z.; Qiu, W.Q. Characterization of the Effects of Calcitonin Gene-Related Peptide Receptor Antagonist for Alzheimer’s Disease. Neuropharmacology 2020, 168, 108017. [Google Scholar] [CrossRef]
- Bitencourt, R.M.; Guerra de Souza, A.C.; Bicca, M.A.; Pamplona, F.A.; de Mello, N.; Passos, G.F.; Medeiros, R.; Takahashi, R.N.; Calixto, J.B.; Prediger, R.D. Blockade of Hippocampal Bradykinin B1 Receptors Improves Spatial Learning and Memory Deficits in Middle-Aged Rats. Behav. Brain Res. 2017, 316, 74–81. [Google Scholar] [CrossRef]
- Duncan, M.J.; Farlow, H.; Tirumalaraju, C.; Yun, D.-H.; Wang, C.; Howard, J.A.; Sanden, M.N.; O’Hara, B.F.; McQuerry, K.J.; Bachstetter, A.D. Effects of the Dual Orexin Receptor Antagonist DORA-22 on Sleep in 5XFAD Mice. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 70–80. [Google Scholar] [CrossRef]
- Lanté, F.; Chafai, M.; Raymond, E.F.; Salgueiro Pereira, A.R.; Mouska, X.; Kootar, S.; Barik, J.; Bethus, I.; Marie, H. Subchronic Glucocorticoid Receptor Inhibition Rescues Early Episodic Memory and Synaptic Plasticity Deficits in a Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2015, 40, 1772–1781. [Google Scholar] [CrossRef]
- Pineau, F.; Canet, G.; Desrumaux, C.; Hunt, H.; Chevallier, N.; Ollivier, M.; Belanoff, J.K.; Givalois, L. New Selective Glucocorticoid Receptor Modulators Reverse Amyloid-β Peptide–Induced Hippocampus Toxicity. Neurobiol. Aging 2016, 45, 109–122. [Google Scholar] [CrossRef]
- Baglietto-Vargas, D.; Medeiros, R.; Martinez-Coria, H.; LaFerla, F.M.; Green, K.N. Mifepristone Alters Amyloid Precursor Protein Processing to Preclude Amyloid Beta and Also Reduces Tau Pathology. Biol. Psychiatry 2013, 74, 357–366. [Google Scholar] [CrossRef]
- Christensen, A.; Pike, C.J. Age-Dependent Regulation of Obesity and Alzheimer-Related Outcomes by Hormone Therapy in Female 3xTg-AD Mice. PLoS ONE 2017, 12, e0178490. [Google Scholar] [CrossRef]
- Shang, X.-L.; Zhao, J.-H.; Cao, Y.-P.; Xue, Y.-X. Effects of Synaptic Plasticity Regulated by 17β-Estradiol on Learning and Memory in Rats with Alzheimer’s Disease. Neurosci. Bull. 2010, 26, 133–139. [Google Scholar] [CrossRef][Green Version]
- Erickson, K.I.; Colcombe, S.J.; Raz, N.; Korol, D.L.; Scalf, P.; Webb, A.; Cohen, N.J.; McAuley, E.; Kramer, A.F. Selective Sparing of Brain Tissue in Postmenopausal Women Receiving Hormone Replacement Therapy. Neurobiol. Aging 2005, 26, 1205–1213. [Google Scholar] [CrossRef]
- Jovanovic, H.; Kocoska-Maras, L.; Rådestad, A.F.; Halldin, C.; Borg, J.; Hirschberg, A.L.; Nordström, A.-L. Effects of Estrogen and Testosterone Treatment on Serotonin Transporter Binding in the Brain of Surgically Postmenopausal Women—A PET Study. NeuroImage 2015, 106, 47–54. [Google Scholar] [CrossRef]
- Hunter, C.L.; Bimonte-Nelson, H.A.; Nelson, M.; Eckman, C.B.; Granholm, A.-C. Behavioral and Neurobiological Markers of Alzheimer’s Disease in Ts65Dn Mice: Effects of Estrogen. Neurobiol. Aging 2004, 25, 873–884. [Google Scholar] [CrossRef]
- Costa, A.J.; Oliveira, R.B.; Wachilewski, P.; Nishino, M.S.; Bassani, T.B.; Stilhano, R.S.; Cerutti, J.M.; Nozima, B.; Porto, C.S.; da Silva Pereira, G.J.; et al. Membrane Estrogen Receptor ERα Activation Improves Tau Clearance via Autophagy Induction in a Tauopathy Cell Model. Brain Res. 2022, 1795, 148079. [Google Scholar] [CrossRef]
- Gray, N.E.; Zweig, J.A.; Kawamoto, C.; Quinn, J.F.; Copenhaver, P.F. STX, a Novel Membrane Estrogen Receptor Ligand, Protects Against Amyloid-β Toxicity. J. Alzheimer’s Dis. 2016, 51, 391–403. [Google Scholar] [CrossRef]
- Quinn, J.F.; Kelly, M.J.; Harris, C.J.; Hack, W.; Gray, N.E.; Kulik, V.; Bostick, Z.; Brumbach, B.H.; Copenhaver, P.F. The Novel Estrogen Receptor Modulator STX Attenuates Amyloid-β Neurotoxicity in the 5XFAD Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2022, 174, 105888. [Google Scholar] [CrossRef]
- Palm, R.; Chang, J.; Blair, J.; Garcia-Mesa, Y.; Lee, H.; Castellani, R.J.; Smith, M.A.; Zhu, X.; Casadesus, G. Down-Regulation of Serum Gonadotropins but Not Estrogen Replacement Improves Cognition in Aged-Ovariectomized 3xTg AD Female Mice. J. Neurochem. 2014, 130, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.-L.; Zhuo, S.; Mei, H.; Chen, X.-F.; Li, N.; Zhu, T.-F.; Chen, S.-T.; Wang, J.-M.; Hou, R.-X.; Le, Y.-Y. Androgen Alleviates Neurotoxicity of β-Amyloid Peptide (Aβ) by Promoting Microglial Clearance of Aβ and Inhibiting Microglial Inflammatory Response to Aβ. CNS Neurosci. Ther. 2017, 23, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, F.; Gao, J.; Dong, Y.; Zhang, Y.; Yang, G.; Soriano, S.G.; Feng, H.-J.; Xie, Z. Testosterone Attenuates Sevoflurane-Induced Tau Phosphorylation and Cognitive Impairment in Neonatal Male Mice. Br. J. Anaesth. 2021, 127, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Carteri, R.B.; Kopczynski, A.; Rodolphi, M.S.; Strogulski, N.R.; Sartor, M.; Feldmann, M.; De Bastiani, M.A.; Duval Wannmacher, C.M.; de Franceschi, I.D.; Hansel, G.; et al. Testosterone Administration after Traumatic Brain Injury Reduces Mitochondrial Dysfunction and Neurodegeneration. J. Neurotrauma 2019, 36, 2246–2259. [Google Scholar] [CrossRef]
- Jia, J.; Kang, L.; Li, S.; Geng, D.; Fan, P.; Wang, L.; Cui, H. Amelioratory Effects of Testosterone Treatment on Cognitive Performance Deficits Induced by Soluble Aβ1–42 Oligomers Injected into the Hippocampus. Horm. Behav. 2013, 64, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF Reduce Aβ Production by Enhancing α-Secretase Processing of APP. J. Neurochem. 2017, 142, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Kazim, S.F.; Blanchard, J.; Dai, C.-L.; Tung, Y.-C.; LaFerla, F.M.; Iqbal, I.-G.; Iqbal, K. Disease Modifying Effect of Chronic Oral Treatment with a Neurotrophic Peptidergic Compound in a Triple Transgenic Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2014, 71, 110–130. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Massa, S.M.; Tran, K.C.; Simmons, D.A.; Rajadas, J.; Zeng, A.Y.; Jang, T.; Carsanaro, S.; Longo, F.M. A Small Molecule TrkB/TrkC Neurotrophin Receptor Co-Activator with Distinctive Effects on Neuronal Survival and Process Outgrowth. Neuropharmacology 2016, 110, 343–361. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.; McHugh, T.L.M.; Yang, T.; Syriani, W.; Massa, S.M.; Longo, F.M.; Simmons, D.A. Small Molecule Modulation of TrkB and TrkC Neurotrophin Receptors Prevents Cholinergic Neuron Atrophy in an Alzheimer’s Disease Mouse Model at an Advanced Pathological Stage. Neurobiol. Dis. 2022, 162, 105563. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Wang, L.; Liu, X.; Yan, S.; Yan, S.S.; Wang, Y.; Zhang, W. Multi-Faced Neuroprotective Effects of Geniposide Depending on the RAGE-Mediated Signaling in an Alzheimer Mouse Model. Neuropharmacology 2015, 89, 175–184. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, X.; Xu, P.; Ji, X.; Chi, T.; Liu, P.; Zou, L. Tolfenamic Acid Inhibits GSK-3β and PP2A Mediated Tau Hyperphosphorylation in Alzheimer’s Disease Models. J. Physiol. Sci. 2020, 70, 29. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Kügler, S.; Lastres-Becker, I. Pharmacological Targeting of GSK-3 and NRF2 Provides Neuroprotection in a Preclinical Model of Tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Sirevaag, A.; Wiegand, S.J.; Lindsay, R.M.; Björklund, A. Reversal of Spatial Memory Impairments in Aged Rats by Nerve Growth Factor and Neurotrophins 3 and 4/5 but Not by Brain-Derived Neurotrophic Factor. Proc. Natl. Acad. Sci. USA 1994, 91, 8607–8611. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Baumann, T.L.; Bakay, R.A.E.; Ostrove, J.M.; Siffert, J.; Fleisher, A.S.; Herzog, C.D.; Barba, D.; Pay, M.; Salmon, D.P.; et al. A Phase1 Study of Stereotactic Gene Delivery of AAV2-NGF for Alzheimer’s Disease. Alzheimer’s Dement. 2014, 10, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Wang, S.; Ackermann, M.; Neufurth, M.; Steffen, R.; Mecja, E.; Muñoz-Espí, R.; Feng, Q.; Schröder, H.C.; Wang, X. Rebalancing β-Amyloid-Induced Decrease of ATP Level by Amorphous Nano/Micro Polyphosphate: Suppression of the Neurotoxic Effect of Amyloid β-Protein Fragment 25-35. Int. J. Mol. Sci. 2017, 18, 2154. [Google Scholar] [CrossRef]
- Emmanuel, I.A.; Olotu, F.A.; Agoni, C.; Soliman, M.E.S. Deciphering the ‘Elixir of Life’: Dynamic Perspectives into the Allosteric Modulation of Mitochondrial ATP Synthase by J147, a Novel Drug in the Treatment of Alzheimer’s Disease. Chem. Biodivers. 2019, 16, e1900085. [Google Scholar] [CrossRef]
- Prior, M.; Dargusch, R.; Ehren, J.L.; Chiruta, C.; Schubert, D. The Neurotrophic Compound J147 Reverses Cognitive Impairment in Aged Alzheimer’s Disease Mice. Alzheimer’s Res. Ther. 2013, 5, 25. [Google Scholar] [CrossRef]
- He, Y.; Jia, K.; Li, L.; Wang, Q.; Zhang, S.; Du, J.; Du, H. Salvianolic Acid B Attenuates Mitochondrial Stress against Aβ Toxicity in Primary Cultured Mouse Neurons. Biochem. Biophys. Res. Commun. 2018, 498, 1066–1072. [Google Scholar] [CrossRef]
- Lee, Y.W.; Kim, D.H.; Jeon, S.J.; Park, S.J.; Kim, J.M.; Jung, J.M.; Lee, H.E.; Bae, S.G.; Oh, H.K.; Ho Son, K.H.; et al. Neuroprotective Effects of Salvianolic Acid B on an Aβ25–35 Peptide-Induced Mouse Model of Alzheimer’s Disease. Eur. J. Pharmacol. 2013, 704, 70–77. [Google Scholar] [CrossRef]
- Tang, Y.; Huang, D.; Zhang, M.-H.; Zhang, W.-S.; Tang, Y.-X.; Shi, Z.-X.; Deng, L.; Zhou, D.-H.; Lu, X.-Y. Salvianolic Acid B Inhibits Aβ Generation by Modulating BACE1 Activity in SH-SY5Y-APPsw Cells. Nutrients 2016, 8, 333. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; Guo, J.; Sun, J.; Sun, Q. Salvianolic Acid B Improves Cognitive Impairment by Inhibiting Neuroinflammation and Decreasing Aβ Level in Porphyromonas Gingivalis-Infected Mice. Aging 2020, 12, 10117–10128. [Google Scholar] [CrossRef]
- Chen, X.; Hu, J.; Jiang, L.; Xu, S.; Zheng, B.; Wang, C.; Zhang, J.; Wei, X.; Chang, L.; Wang, Q. Brilliant Blue G Improves Cognition in an Animal Model of Alzheimer’s Disease and Inhibits Amyloid-β-Induced Loss of Filopodia and Dendrite Spines in Hippocampal Neurons. Neuroscience 2014, 279, 94–101. [Google Scholar] [CrossRef]
- Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; León-Otegui, M.; Hontecillas-Prieto, L.; del Puerto, A.; Trejo, J.L.; Lucas, J.J.; Garrido, J.J.; Gualix, J.; Miras-Portugal, M.T.; et al. In Vivo P2X7 Inhibition Reduces Amyloid Plaques in Alzheimer’s Disease through GSK3β and Secretases. Neurobiol. Aging 2012, 33, 1816–1828. [Google Scholar] [CrossRef]
- Pradhan, K.; Das, G.; Kar, C.; Mukherjee, N.; Khan, J.; Mahata, T.; Barman, S.; Ghosh, S. Rhodamine-Based Metal Chelator: A Potent Inhibitor of Metal-Catalyzed Amyloid Toxicity. ACS Omega 2020, 5, 18958–18967. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.-X.; Tammer, A.; et al. Metal-Protein Attenuation With Iodochlorhydroxyquin (Clioquinol) Targeting Aβ Amyloid Deposition and Toxicity in Alzheimer Disease: A Pilot Phase 2 Clinical Trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a Copper-Zinc Chelator Markedly and Rapidly Inhibits Beta-Amyloid Accumulation in Alzheimer’s Disease Transgenic Mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Barnham, K.J.; Ciccotosto, G.D.; Tickler, A.K.; Ali, F.E.; Smith, D.G.; Williamson, N.A.; Lam, Y.-H.; Carrington, D.; Tew, D.; Kocak, G.; et al. Neurotoxic, Redox-Competent Alzheimer’s β-Amyloid Is Released from Lipid Membrane by Methionine Oxidation *. J. Biol. Chem. 2003, 278, 42959–42965. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Friedman, J.E.; Angel, I.; Kozak, A.; Koh, J.-Y. The Lipophilic Metal Chelator DP-109 Reduces Amyloid Pathology in Brains of Human β-Amyloid Precursor Protein Transgenic Mice. Neurobiol. Aging 2004, 25, 1315–1321. [Google Scholar] [CrossRef]
- Shachar, D.B.; Kahana, N.; Kampel, V.; Warshawsky, A.; Youdim, M.B.H. Neuroprotection by a Novel Brain Permeable Iron Chelator, VK-28, against 6-Hydroxydopamine Lession in Rats. Neuropharmacology 2004, 46, 254–263. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Du, Y.-F.; Chen, L. Neuropeptides Exert Neuroprotective Effects in Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 11, 493. [Google Scholar] [CrossRef]
- Duarte-Neves, J.; Pereira de Almeida, L.; Cavadas, C. Neuropeptide Y (NPY) as a Therapeutic Target for Neurodegenerative Diseases. Neurobiol. Dis. 2016, 95, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Pain, S.; Brot, S.; Gaillard, A. Neuroprotective Effects of Neuropeptide Y against Neurodegenerative Disease. Curr. Neuropharmacol. 2022, 20, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. C-Jun N-Terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.; Ueta, Y.; Yamada, D.; Sasaki-Hamada, S.; Iwai, T.; Akita, T.; Yamashita, C.; Saitoh, A.; Oka, J.-I. Intracerebroventricular Administration of Oxytocin and Intranasal Administration of the Oxytocin Derivative Improve β-Amyloid Peptide (25–35)-Induced Memory Impairment in Mice. Neuropsychopharmacol. Rep. 2022. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.K.; Lakshmi, K.K.; Krishna, K.V.; Agrawal, M.; Singhvi, G.; Saha, R.N.; Saraf, S.; Saraf, S.; Shukla, R.; Alexander, A. Insulin Mediated Novel Therapies for the Treatment of Alzheimer’s Disease. Life Sci. 2020, 249, 117540. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Gupta, G.; Shrivastava, B.; Dahiya, R.; Tiwari, J.; Ashwathanarayana, M.; Sharma, R.K.; Agrawal, M.; Mishra, A.; Dua, K. Calcitonin Gene-Related Peptide (CGRP): A Novel Target for Alzheimer’s Disease. CNS Neurosci. Ther. 2017, 23, 457–461. [Google Scholar] [CrossRef]
- John, H.; Maronde, E.; Forssmann, W.-G.; Meyer, M.; Adermann, K. N-Terminal Acetylation Protects Glucagon-like Peptide GLP-1-(7-34)-Amide from DPP-IV-Mediated Degradation Retaining CAMP- and Insulin-Releasing Capacity. Eur. J. Med. Res. 2008, 13, 73–78. [Google Scholar]
- Räder, A.F.B.; Reichart, F.; Weinmüller, M.; Kessler, H. Improving Oral Bioavailability of Cyclic Peptides by N-Methylation. Bioorg. Med. Chem. 2018, 26, 2766–2773. [Google Scholar] [CrossRef]
- Chen, X.; Mietlicki-Baase, E.G.; Barrett, T.M.; McGrath, L.E.; Koch-Laskowski, K.; Ferrie, J.J.; Hayes, M.R.; Petersson, E.J. Thioamide Substitution Selectively Modulates Proteolysis and Receptor Activity of Therapeutic Peptide Hormones. J. Am. Chem. Soc. 2017, 139, 16688–16695. [Google Scholar] [CrossRef]
- Pomara, N.; Hernando, R.T.; de la Pena, C.B.; Sidtis, J.J.; Cooper, T.B.; Ferris, S. The Effect of Mifepristone (RU 486) on Plasma Cortisol in Alzheimer’s Disease. Neurochem. Res. 2006, 31, 585–588. [Google Scholar] [CrossRef]
- Ibrahim, A.W.; Sodipo, O.A.; Mshelia, I.A.; Machina, B.K. Hormone Replacement Therapy and Alzheimer’s Disease in Older Women: A Systematic Review of Literature. J. Neurosci. Behav. Health 2018, 10, 1–8. [Google Scholar] [CrossRef]
- Shumaker, S.A.; Legault, C.; Rapp, S.R.; Thal, L.; Wallace, R.B.; Ockene, J.K.; Hendrix, S.L.; Jones III, B.N.; Assaf, A.R.; Jackson, R.D.; et al. Estrogen Plus Progestin and the Incidence of Dementia and Mild Cognitive Impairment in Postmenopausal WomenThe Women’s Health Initiative Memory Study: A Randomized Controlled Trial. JAMA 2003, 289, 2651–2662. [Google Scholar] [CrossRef]
- Anastasio, T. Exploring the Contribution of Estrogen to Amyloid-Beta Regulation: A Novel Multifactorial Computational Modeling Approach. Front. Pharmacol. 2013, 4, 16. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Su Kang, S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping Yu, S.; et al. Delta-Secretase Cleaves Amyloid Precursor Protein and Regulates the Pathogenesis in Alzheimer’s Disease. Nat. Commun. 2015, 6, 8762. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, M.; Liu, X.; Kang, S.S.; Kwon, I.-S.; Duong, D.M.; Seyfried, N.T.; Hu, W.T.; Liu, Z.; Wang, J.-Z.; et al. Cleavage of Tau by Asparagine Endopeptidase Mediates the Neurofibrillary Pathology in Alzheimer’s Disease. Nat. Med. 2014, 20, 1254–1262. [Google Scholar] [CrossRef]
- Lin, J.; Li, X.; Yuan, F.; Lin, L.; Cook, C.L.; Rao, C.V.; Lei, Z. Genetic Ablation of Luteinizing Hormone Receptor Improves the Amyloid Pathology in a Mouse Model of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2010, 69, 253–261. [Google Scholar] [CrossRef]
- Ma, K.-G.; Qian, Y.-H. Alpha 7 Nicotinic Acetylcholine Receptor and Its Effects on Alzheimer’s Disease. Neuropeptides 2019, 73, 96–106. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F.; Tschirner, S.K.; Steiner, H. Secretases in Alzheimer’s Disease: Novel Insights into Proteolysis of APP and TREM2. Curr. Opin. Neurobiol. 2022, 72, 101–110. [Google Scholar] [CrossRef]
- Kazim, S.F.; Iqbal, K. Neurotrophic Factor Small-Molecule Mimetics Mediated Neuroregeneration and Synaptic Repair: Emerging Therapeutic Modality for Alzheimer’s Disease. Mol. Neurodegener. 2016, 11, 50. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease and Its Pharmaceutical Potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Gratwicke, J.; Kahan, J.; Zrinzo, L.; Hariz, M.; Limousin, P.; Foltynie, T.; Jahanshahi, M. The Nucleus Basalis of Meynert: A New Target for Deep Brain Stimulation in Dementia? Neurosci. Biobehav. Rev. 2013, 37, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Ebanks, B.; Ingram, T.L.; Chakrabarti, L. ATP Synthase and Alzheimer’s Disease: Putting a Spin on the Mitochondrial Hypothesis. Aging 2020, 12, 16647–16662. [Google Scholar] [CrossRef] [PubMed]
- Mark, R.J.; Hensley, K.; Butterfield, D.A.; Mattson, M.P. Amyloid Beta-Peptide Impairs Ion-Motive ATPase Activities: Evidence for a Role in Loss of Neuronal Ca2+ Homeostasis and Cell Death. J. Neurosci. 1995, 15, 6239–6249. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Yanazawa, M.; Sasahara, T.; Kitamura, Y.; Hiroaki, H.; Fukazawa, Y.; Kii, I.; Nishiyama, T.; Kakita, A.; Takeda, H.; et al. Na, K-ATPase A3 Is a Death Target of Alzheimer Patient Amyloid-β Assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E4465–E4474. [Google Scholar] [CrossRef]
- Woods, L.T.; Ajit, D.; Camden, J.M.; Erb, L.; Weisman, G.A. Purinergic Receptors as Potential Therapeutic Targets in Alzheimer’s Disease. Neuropharmacology 2016, 104, 169–179. [Google Scholar] [CrossRef]
- Jiang, L.-H.; Mackenzie, A.B.; North, R.A.; Surprenant, A. Brilliant Blue G Selectively Blocks ATP-Gated Rat P2X7 Receptors. Mol. Pharmacol. 2000, 58, 82–88. [Google Scholar] [CrossRef]
- Peng, W.; Cotrina, M.L.; Han, X.; Yu, H.; Bekar, L.; Blum, L.; Takano, T.; Tian, G.-F.; Goldman, S.A.; Nedergaard, M. Systemic Administration of an Antagonist of the ATP-Sensitive Receptor P2X7 Improves Recovery after Spinal Cord Injury. Proc. Natl. Acad. Sci. USA 2009, 106, 12489–12493. [Google Scholar] [CrossRef]
- Hegde, M.L.; Bharathi, P.; Suram, A.; Venugopal, C.; Jagannathan, R.; Poddar, P.; Srinivas, P.; Sambamurti, K.; Rao, K.J.; Scancar, J.; et al. Challenges Associated with Metal Chelation Therapy in Alzheimer’s Disease. J. Alzheimer’s Dis. 2009, 17, 457–468. [Google Scholar] [CrossRef]
- McLachlan, D.R.C.; Kruck, T.P.A.; Kalow, W.; Andrews, D.F.; Dalton, A.J.; Bell, M.Y.; Smith, W.L. Intramuscular Desferrioxamine in Patients with Alzheimer’s Disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Bergeron, R.J.; Brittenham, G.M. The Development of Iron Chelators for Clinical Use; CRC Press: Boca Raton, FL, USA, 1993; ISBN 978-0-8493-8679-4. [Google Scholar]
- Mishra, P.; Kumar, A.; Panda, G. Anti-Cholinesterase Hybrids as Multi-Target-Directed Ligands against Alzheimer’s Disease (1998–2018). Bioorg. Med. Chem. 2019, 27, 895–930. [Google Scholar] [CrossRef]
- Yang, A.; Liu, C.; Wu, J.; Kou, X.; Shen, R. A Review on α-Mangostin as a Potential Multi-Target-Directed Ligand for Alzheimer’s Disease. Eur. J. Pharmacol. 2021, 897, 173950. [Google Scholar] [CrossRef]
- Chi, X.-Q.; Hou, B.; Yang, L.; Zi, C.-T.; Lv, Y.-F.; Li, J.-Y.; Ren, F.-C.; Yuan, M.-Y.; Hu, J.-M.; Zhou, J. Design, Synthesis and Cholinesterase Inhibitory Activity of α-Mangostin Derivatives. Nat. Prod. Res. 2020, 34, 1380–1388. [Google Scholar] [CrossRef]
- Hao, X.-M.; Li, L.-D.; Duan, C.-L.; Li, Y.-J. Neuroprotective Effect of α-Mangostin on Mitochondrial Dysfunction and α-Synuclein Aggregation in Rotenone-Induced Model of Parkinson’s Disease in Differentiated SH-SY5Y Cells. J. Asian Nat. Prod. Res. 2017, 19, 833–845. [Google Scholar] [CrossRef]
- Wang, S.; Li, Q.; Jing, M.; Alba, E.; Yang, X.; Sabaté, R.; Han, Y.; Pi, R.; Lan, W.; Yang, X.; et al. Natural Xanthones from Garcinia Mangostana with Multifunctional Activities for the Therapy of Alzheimer’s Disease. Neurochem. Res. 2016, 41, 1806–1817. [Google Scholar] [CrossRef]
- Zou, W.; Yin, P.; Shi, Y.; Jin, N.; Gao, Q.; Li, J.; Liu, F. A Novel Biological Role of α-Mangostin via TAK1–NF-ΚB Pathway against Inflammatory. Inflammation 2019, 42, 103–112. [Google Scholar] [CrossRef]
- Spiazzi, C.C.; Soares, M.B.; Izaguirry, A.P.; Vargas, L.M.; Zanchi, M.M.; Pavin, N.F.; Affeld, R.F.; Ludtke, D.S.; Prigol, M.; Santos, F.W. Selenofuranoside Ameliorates Memory Loss in Alzheimer-like Sporadic Dementia: AChE Activity, Oxidative Stress, and Inflammation Involvement. Oxid. Med. Cell. Longev. 2015, 2015, 976908. [Google Scholar] [CrossRef]
- Ramalho, J.B.; Izaguirry, A.P.; Soares, M.B.; Spiazzi, C.C.; Pavin, N.F.; Affeldt, R.F.; Lüdtke, D.S.; Pinton, S.; Santos, F.W.; Prigol, M. Selenofuranoside Improves Long-Term Memory Deficits in Rats after Exposure to Monosodium Glutamate: Involvement of Na+, K+-ATPase Activity. Physiol. Behav. 2018, 184, 27–33. [Google Scholar] [CrossRef]
- Santos, M.A.; Chand, K.; Chaves, S. Recent Progress in Multifunctional Metal Chelators as Potential Drugs for Alzheimer’s Disease. Coord. Chem. Rev. 2016, 327–328, 287–303. [Google Scholar] [CrossRef]
- Carvalho, R.R.V.; Peres, T.V.; Liria, C.W.; Machini, M.T.; Aschner, M.; Espósito, B.P. Conjugates of Desferrioxamine and Aromatic Amines Improve Markers of Iron-Dependent Neurotoxicity. BioMetals 2021, 34, 259–275. [Google Scholar] [CrossRef]
- Xuan, Z.; Gu, X.; Yan, S.; Xie, Y.; Zhou, Y.; Zhang, H.; Jin, H.; Hu, S.; Mak, M.S.H.; Zhou, D.; et al. Dimeric Tacrine(10)-Hupyridone as a Multitarget-Directed Ligand To Treat Alzheimer’s Disease. ACS Chem. Neurosci. 2021, 12, 2462–2477. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Nakanishi, A.; Ogura, Y.; Matsuda, S. Dietary Regulation of PI3K/AKT/GSK-3β Pathway in Alzheimer’s Disease. Alzheimer’s Res. Ther. 2014, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Alcántara-González, F.; Mendoza-Perez, C.R.; Zaragoza, N.; Juarez, I.; Arroyo-García, L.E.; Gamboa, C.; De La Cruz, F.; Zamudio, S.; Garcia-Dolores, F.; Flores, G. Combined Administration of Cerebrolysin and Donepezil Induces Plastic Changes in Prefrontal Cortex in Aged Mice. Synapse 2012, 66, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.B.; Mauk, R.; Nelson, D.; Johnson, D.A. Donepezil Treatment Restores the Ability of Estradiol to Enhance Cognitive Performance in Aged Rats: Evidence for the Cholinergic Basis of the Critical Period Hypothesis. Horm. Behav. 2009, 56, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, G.; Magrì, D.; Gioria, S.; Medaglini, D.; Calzolai, L. Characterization of Nanoparticles-Based Vaccines for COVID-19. Nat. Nanotechnol. 2022, 17, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Santos, B.; Gremião, M.P.D.; Chorilli, M. Nanotechnology-Based Drug Delivery Systems for the Treatment of Alzheimer’s Disease. Int. J. Nanomedicine 2015, 10, 4981–5003. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Kanekiyo, T.; Singh, J. Functionalized Nanoparticles for Brain Targeted BDNF Gene Therapy to Rescue Alzheimer’s Disease Pathology in Transgenic Mouse Model. Int. J. Biol. Macromol. 2022, 208, 901–911. [Google Scholar] [CrossRef]
- Patel, P.A.; Patil, S.C.; Kalaria, D.R.; Kalia, Y.N.; Patravale, V.B. Comparative in Vitro and in Vivo Evaluation of Lipid Based Nanocarriers of Huperzine A. Int. J. Pharm. 2013, 446, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.-Y.; Zhou, H.-H.; Li, X.; Liu, Z.-Q. Huperzine A Alleviates Oxidative Glutamate Toxicity in Hippocampal HT22 Cells via Activating BDNF/TrkB-Dependent PI3K/Akt/MTOR Signaling Pathway. Cell. Mol. Neurobiol. 2016, 36, 915–925. [Google Scholar] [CrossRef]
- Wang, B.; Wang, H.; Wei, Z.; Song, Y.; Zhang, L.; Chen, H. Efficacy and Safety of Natural Acetylcholinesterase Inhibitor Huperzine A in the Treatment of Alzheimer’s Disease: An Updated Meta-Analysis. J. Neural Transm. 2009, 116, 457–465. [Google Scholar] [CrossRef]
- Liu, G.; Men, P.; Harris, P.L.R.; Rolston, R.K.; Perry, G.; Smith, M.A. Nanoparticle Iron Chelators: A New Therapeutic Approach in Alzheimer Disease and Other Neurologic Disorders Associated with Trace Metal Imbalance. Neurosci. Lett. 2006, 406, 189–193. [Google Scholar] [CrossRef]
- Schroeder, U.; Sommerfeld, P.; Ulrich, S.; Sabel, B.A. Nanoparticle Technology for Delivery of Drugs Across the Blood–Brain Barrier. J. Pharm. Sci. 1998, 87, 1305–1307. [Google Scholar] [CrossRef] [PubMed]


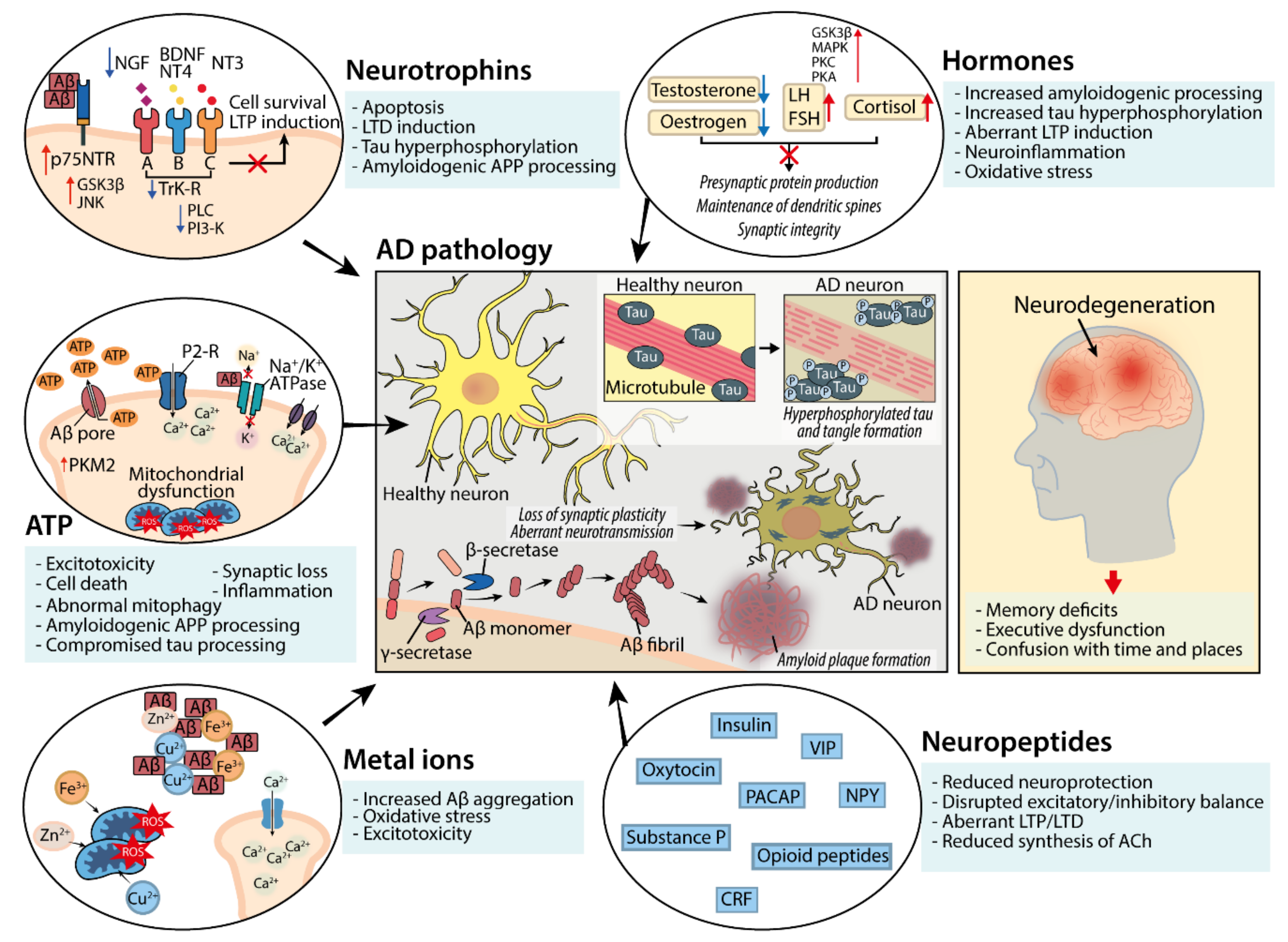{kind=link}
| Neuromodulator Group | Name of Treatment | Stage Tested | Impact on AD Pathology |
|---|---|---|---|
| Neuropeptide | Neuropeptide Y | Rodent models and cultured cells | Protects against Aβ-induced spatial memory deficits, oxidative stress [249], excitotoxicity [250], inflammation [251] and neurodegeneration [252]. |
| Nimodipine | Rodent models | Restores NPY-expressing neuronal function and associated neuroprotection via targeting of Aβ-mediated Ca2+ channel disruption [113]. | |
| SP600125 | Rodent models | Reduces Aβ plaque burden, tau hyperphosphorylation, synaptic loss [253] memory impairment and apoptosis [254] via inhibition of JNK phosphorylation. | |
| PACAP | Rodent models and cultured cells | Promotes α-secretase-dependent APP processing [255], improves cognition and reduced pathogenic Aβ burden [256]. | |
| VIP | Rodent models | Protects against Aβ accumulation, reduces brain atrophy [257]. | |
| Oxytocin | Rodent models | Improves memory retention, reduces Aβ and tau deposition, ERK1/2, GSK3β and AChE activity and neuronal death [258]. Reduces inflammatory cytokine release and hippocampal atrophy [259]. | |
| Insulin | Rodent models | Partially reduces levels of phosphorylated tau and improves learning ability [260]. Reduces pathological Aβ production and improves memory [261]. | |
| BIBN 4096 BS (BIBN) | Rodent models | CGRP antagonist, increases expression of PSD95, reduces neuroinflammation, Aβ and tau pathology [262]. | |
| des-Arg9-[Leu8]-bradykinin (DALBK) | Rodent models | Bradykinin 1 receptor antagonist, reverses spatial learning and memory deficits [263]. | |
| UFP-512 | Cultured cells | Specific agonist of δ-opioid receptors. Exerts neuroprotective actions by attenuating β-secretase expression [111]. | |
| Dual orexin receptor antagonist (DORA-22) | Rodent models | Improves sleep [264]. | |
| Hormone | Mifepristone | Rodent models | Glucocorticoid receptor antagonist, reduces β-secretase expression and Aβ production, improves learning and memory [140]. Reverses synaptic deficits [265,266], reduces tau pathology [267]. |
| CORT108297 and CORT113176 | Rodent models | Selective glucocorticoid receptor antagonists. Reverse Aβ production, neuroinflammation, hippocampal atrophy, synaptic deficits. Re-establish levels of synaptotagmin and PSD95 [266]. | |
| Oestrogen (hormone replacement therapy) | Rodent models and human AD patients | Commencement during early stages/before the menopause reduces Aβ plaque burden, improves behavioural performance [268], enhances LTP [269], reduces brain atrophy [270], improves neurotransmission [271,272], promotes tau clearance [273]. | |
| STX | Rodent models and cultured cells | Oestrogen receptor modulator, reduces Aβ levels, associated mitochondrial toxicity, and synaptic deficits and improves spatial memory [274,275]. | |
| FSH-Ab | Rodent models | Reduces FSH levels, blocks δ-secretase-mediated amyloidogenic APP processing and tau neurofibrillary tangle production. Increases dendritic spine and synapse number. Improves learning and memory [86]. | |
| Leuprolide acetate | Rodent models | Reduces LH levels, inhibits GSK3β signalling, increases BDNF transcription, rescues spatial memory [276]. | |
| Testosterone | Rodent models and cultured cells | Increases dendritic spine number, PSD95 [152], presynaptic protein levels [74], Aβ clearance [277]. Reduces tau hyperphosphorylation via GSK3β inhibition [278], preserves mitochondrial function [279]. Improves memory retention [280]. | |
| Neurotrophin | BDNF | Rodent models and cultured cells | Reduces neuron loss and synaptic degeneration, rescues working memory deficits [62]. Stimulates non-amyloidogenic APP processing [281]. |
| Peptide 021 | Rodent models | Increases BDNF signalling, inhibits GSK3β-mediated tau hyperphosphorylation, reduces synaptic deficits and associated cognitive decline [282]. | |
| LM22B-10 and PTX-BD10-2 | Rodent models and cultured cells | Activate TrKB and TrKC receptors. Promote survival and outgrowth of hippocampal neurons, reduce tau pathology and cholinergic neuron degeneration [283,284]. | |
| Geniposide | Rodent models | Suppresses ERK signalling, reduces inflammatory cytokine release and augments synaptic plasticity [285]. | |
| Tolfenamic acid | Rodent models | Inhibits GSK3β. Attenuates memory deficits, decreases tau hyperphosphorylation [286]. | |
| Isoorientin | Rodent models | Selective GSK3β inhibitor. Attenuates tau hyperphosphorylation, Aβ deposition and neuroinflammation. Improves LTP and spatial memory [70]. | |
| Dimethyl fumarate | Rodent models | Modulates GSK3β activity. Decreases tau phosphorylation and neuroinflammation, increases BDNF expression [287]. | |
| NGF | Rodent models and human AD patients | Reverses spatial memory deficits, reduces cholinergic atrophy [288]. Halts accelerated decline of neuropsychological test performance [289]. | |
| NT-3 | Rodent models | Reverses spatial memory deficits, reduces cholinergic atrophy [288]. | |
| NT-4 | Rodent models | Reverses spatial memory deficits [288]. | |
| ATP | Inorganic polyphosphates | Rodent models and cultured cells | Protect against Aβ-mediated neurotoxic effects by enhancing intracellular ATP levels [290]. |
| J147 | Rodent and human protein models | Modulates activity of ATP synthase [291], increases BDNF and NGF expression and learning and memory ability [292]. | |
| Salvianolic acid B | Rodent models and cultured cells | Rescues Aβ-mediated dysfunction of ATP synthase and mitochondrial stress [293], decreases Aβ accumulation and neuroinflammation and improves cognitive function [294,295,296]. | |
| PTEN-induced putative kinase 1 (PINK-1) | Rodent models | Mitophagy-associated protein, restores mitochondrial function and attenuates Aβ accumulation. Improves learning and memory [203]. | |
| Shikonin and compound 3K | Rodent models | PKM2 inhibitors. Reduce microglial glycolysis and pro-inflammatory activity and Aβ plaque content [54]. | |
| Brilliant Blue G (BBG) | Rodent models and cultured cells | P2X7 receptor antagonist. Rescues dendritic spine loss and memory impairments [297]. Attenuates Aβ plaque production via inhibition of GSK3β and activation of α-secretase [298]. | |
| Metal ion | Rhodamine-B-based compound (Rh-BT) | Rodent models and cultured cells | Copper chelator. Prevents formation of toxic amyloid aggregates and inhibits metal-induced ROS production [299]. |
| Iodochlorhydroxyquin (clioquinol) | Rodent models, cultured cells and human AD patients | Copper/zinc chelator. Slows cognitive decline and plasma Aβ accumulation in AD patients [300]. Reduces deposition of Aβ [301,302]. | |
| DP-109 | Rodent models | Zinc/copper chelator. Reduces amyloid beta plaque burden in the brain [303]. | |
| VK-28 | Rodent models | Iron chelator. Exerts neuroprotective effects to reduce neurodegeneration [304]. | |
| Deferoxamine (DFO) | Rodent models and cultured cells | Iron chelator. Decreases Aβ-associated neurotoxicity [49] and suppresses tau hyperphosphorylation by reducing activation of GSK-3β [247]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunliffe, G.; Lim, Y.T.; Chae, W.; Jung, S. Alternative Pharmacological Strategies for the Treatment of Alzheimer’s Disease: Focus on Neuromodulator Function. Biomedicines 2022, 10, 3064. https://doi.org/10.3390/biomedicines10123064
Cunliffe G, Lim YT, Chae W, Jung S. Alternative Pharmacological Strategies for the Treatment of Alzheimer’s Disease: Focus on Neuromodulator Function. Biomedicines. 2022; 10(12):3064. https://doi.org/10.3390/biomedicines10123064
Chicago/Turabian StyleCunliffe, Grace, Yi Tang Lim, Woori Chae, and Sangyong Jung. 2022. "Alternative Pharmacological Strategies for the Treatment of Alzheimer’s Disease: Focus on Neuromodulator Function" Biomedicines 10, no. 12: 3064. https://doi.org/10.3390/biomedicines10123064
APA StyleCunliffe, G., Lim, Y. T., Chae, W., & Jung, S. (2022). Alternative Pharmacological Strategies for the Treatment of Alzheimer’s Disease: Focus on Neuromodulator Function. Biomedicines, 10(12), 3064. https://doi.org/10.3390/biomedicines10123064