
Epigenetic Mechanisms as Emerging Therapeutic Targets and Microfluidic Chips Application in Pulmonary Arterial Hypertension



Abstract
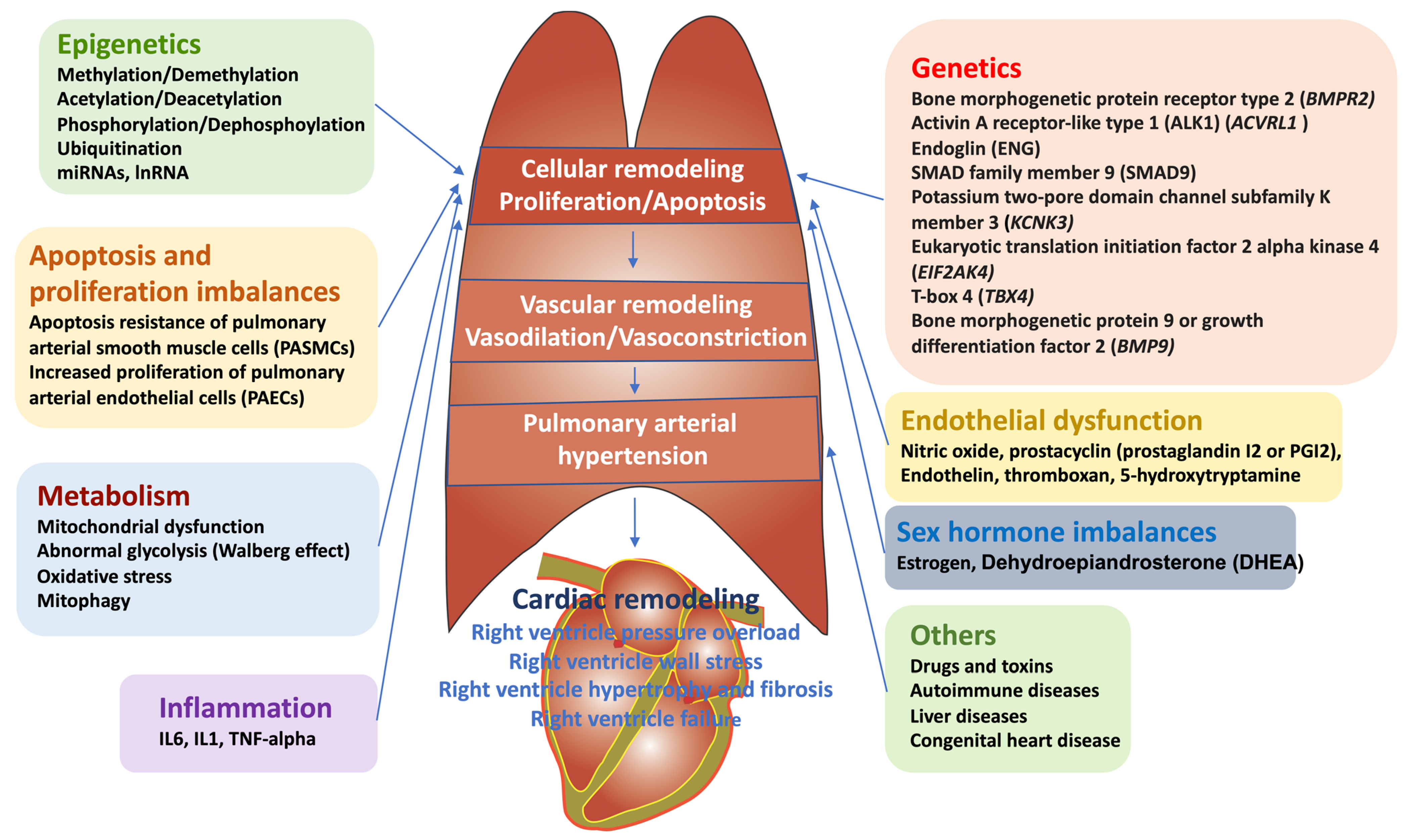
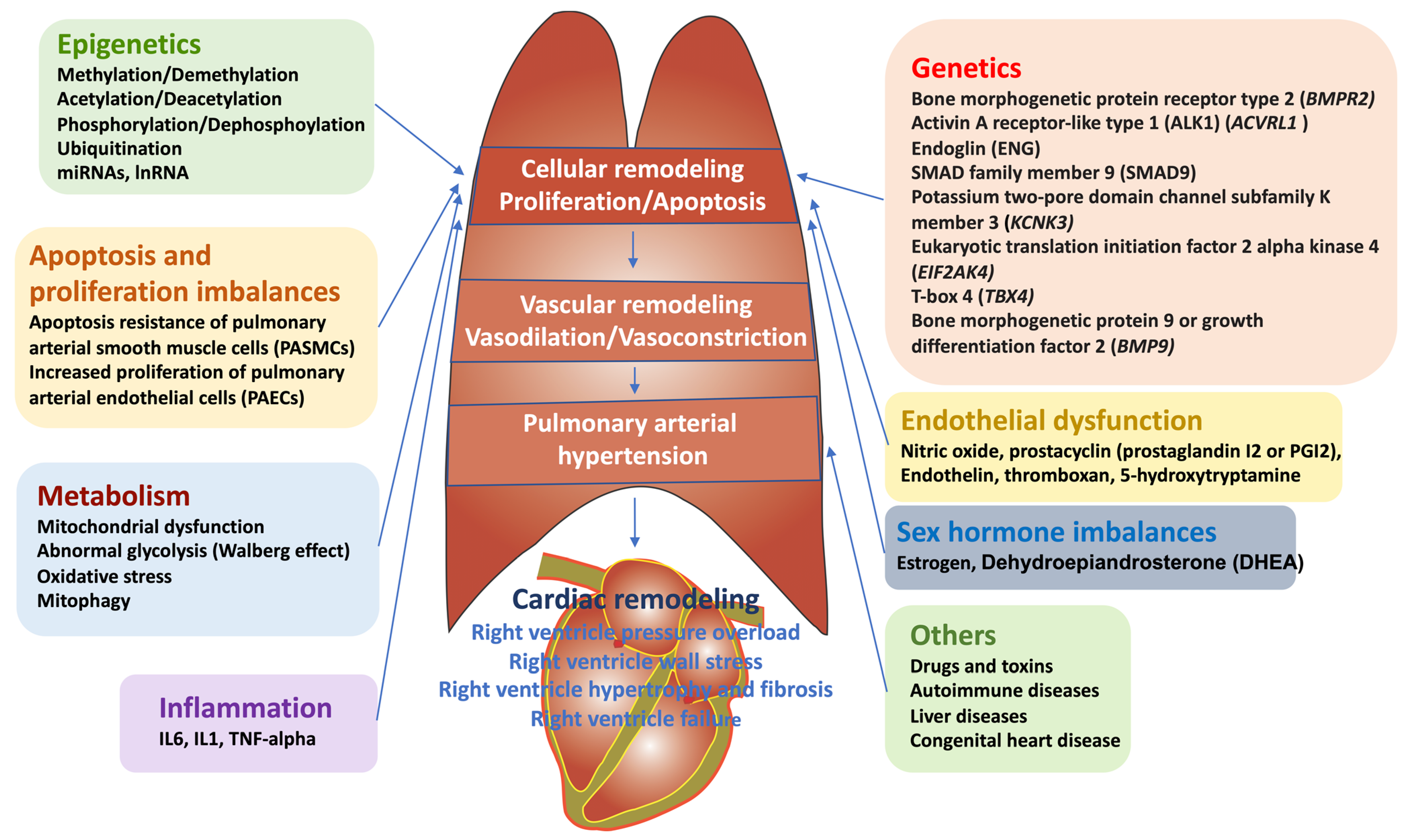
:1. Introduction: Definition, Epidemiology, and Clinical Symptoms of PAH
- (1)
- Pulmonary arterial hypertension (PAH) or Group I
- i.
- Idiopathic PAH (IPAH: primary pulmonary hypertension) or inherited PAH
- ii.
- PAH induced by drug or toxins
- iii.
- Associated PAH (APAH): PAH associated with connective tissue and heart disorders, infection of human immunodeficiency virus portal hypertension, congenital tissue and heart diseases, schistosomiasis, chronic hemolytic anemia, pulmonary capillary hemangiomatosis, pulmonary veno-occlusive disease, and newborn PH
- (2)
- PH associated with left-heart disease (venous PH) or group II (ex. mitral valve)
- (3)
- PH associated with lung disease (hypoxic PH) or group III (ex. COPD, interstitial lung disease, sleep apnea)
- (4)
- PH associated with thromboembolic diseases (thromboembolic PH) or group IV
- (5)
- PH with multifactorial mechanisms (miscellaneous PH) or group V (ex. increased red blood cell production, metabolic abnormalities)
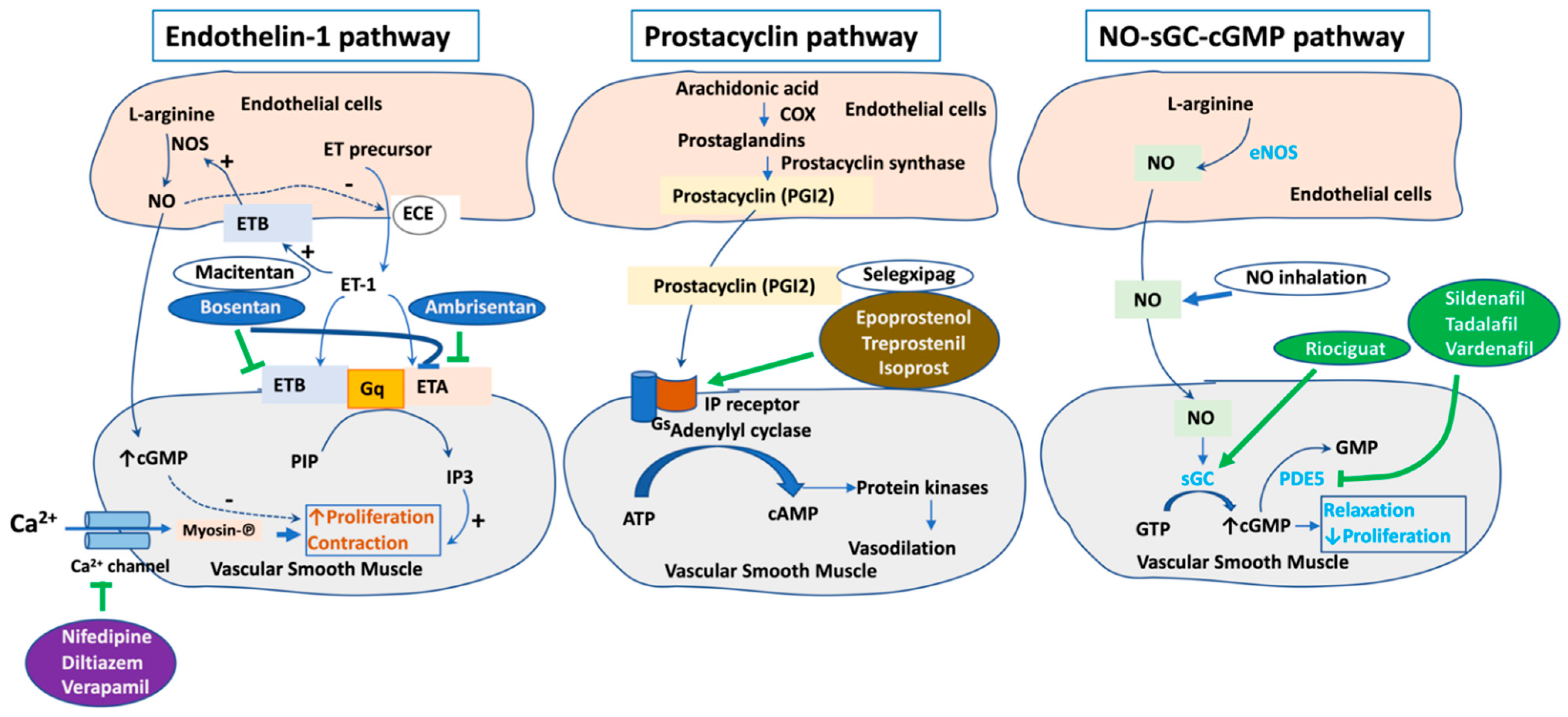
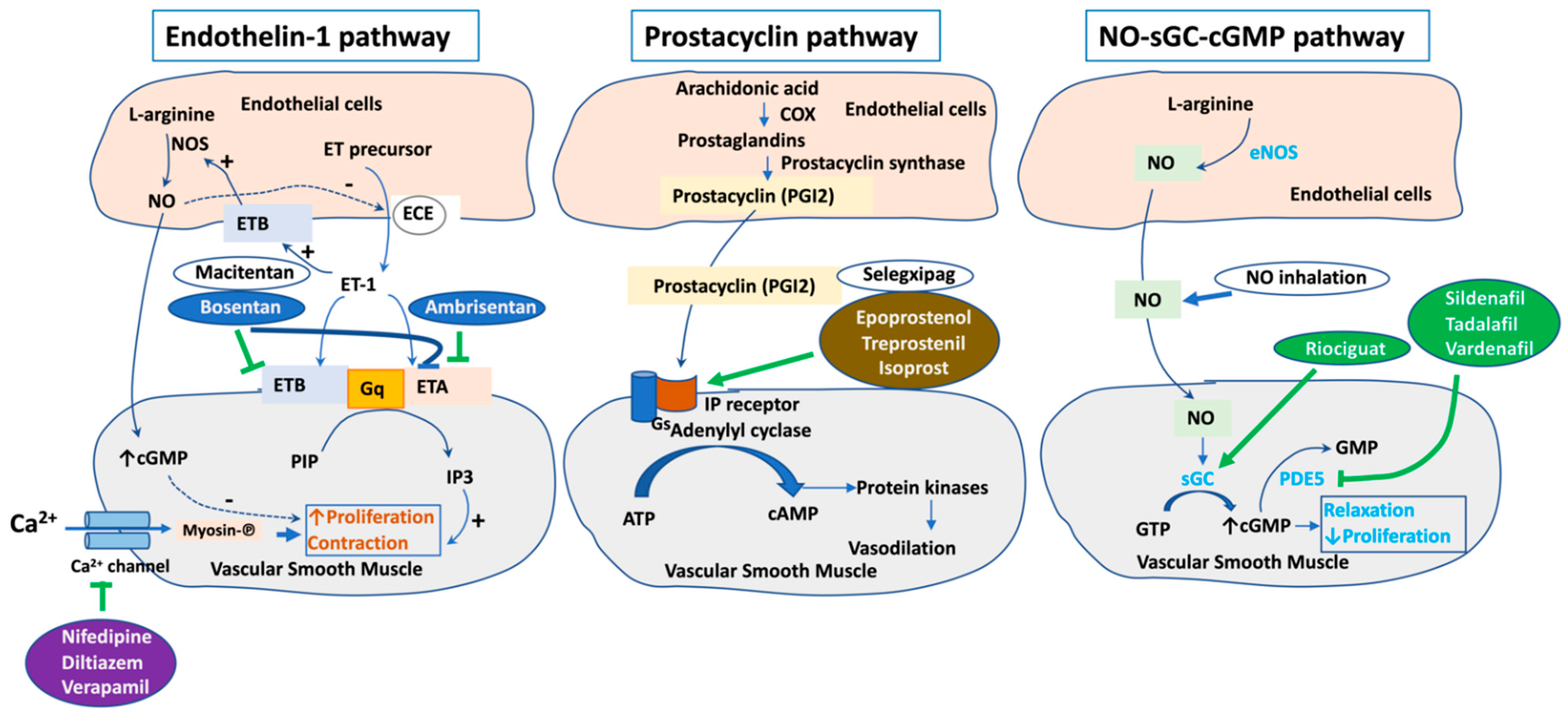
2. Current Therapeutic Targets for PAH
3. Sex Disparity in PAH and Microfluidic Chip Model Mimicking PAH-Insulted Artery to Study Sex Discrepancy in PAH Pathophysiology
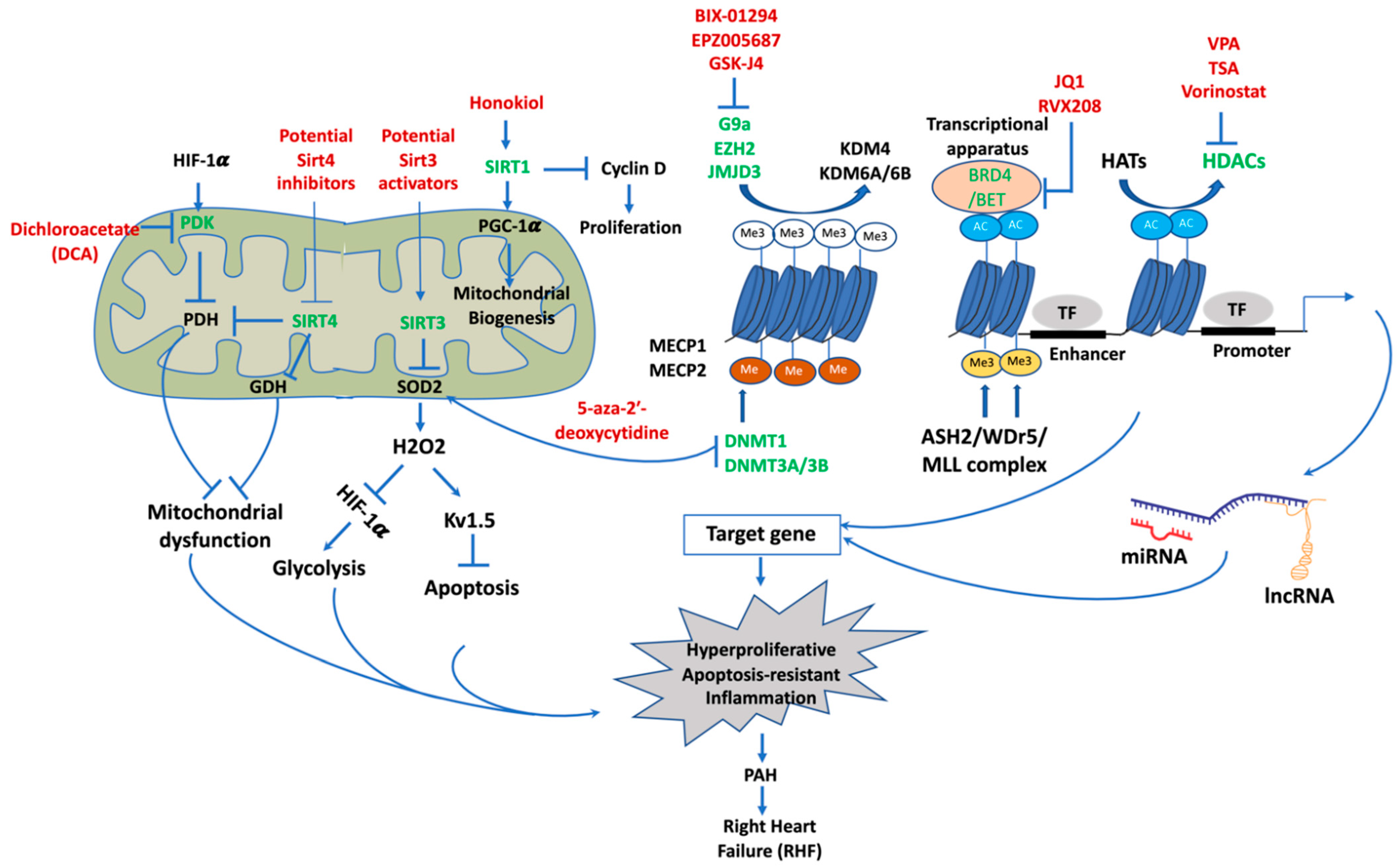
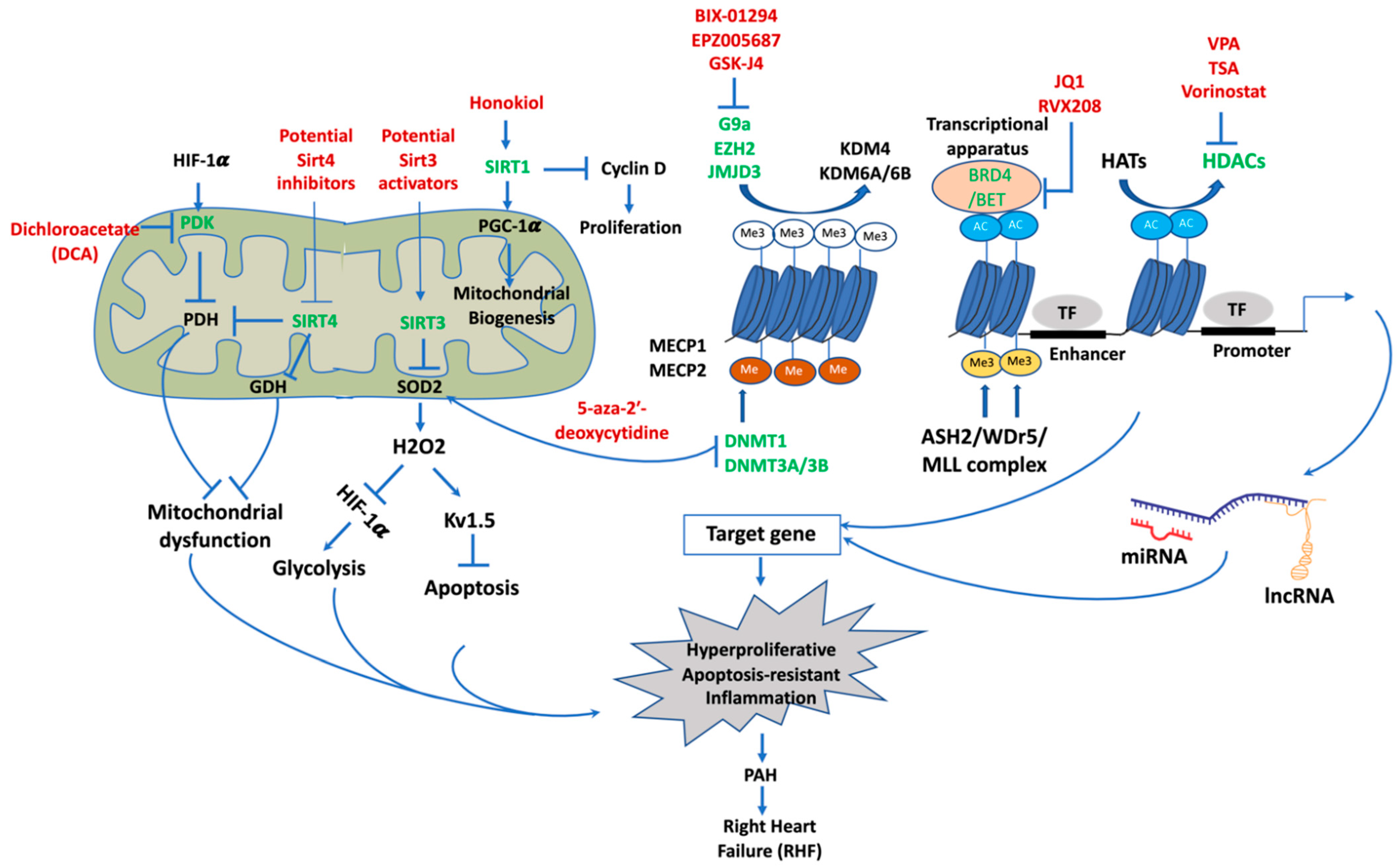
4. Epigenetic Mechanisms of Pathogenesis in PAH
4.1. DNA Methylation
4.2. Histone Post-Translational Modifications
4.2.1. Histone Methylation and Demethylation and Targeting Histone Demethylase for Treatment of PAH
4.2.2. Histone Acetylation and Deacetylation
Targeting Histone Deacetylase for PAH Therapy
Targeting Bromodomains as Histone Acetylation Readers for PAH Therapy
4.3. Sirtuins and Targeting Sirtuins for PAH Therapy
4.3.1. Noncoding Ribonucleic Acids (ncRNAs)
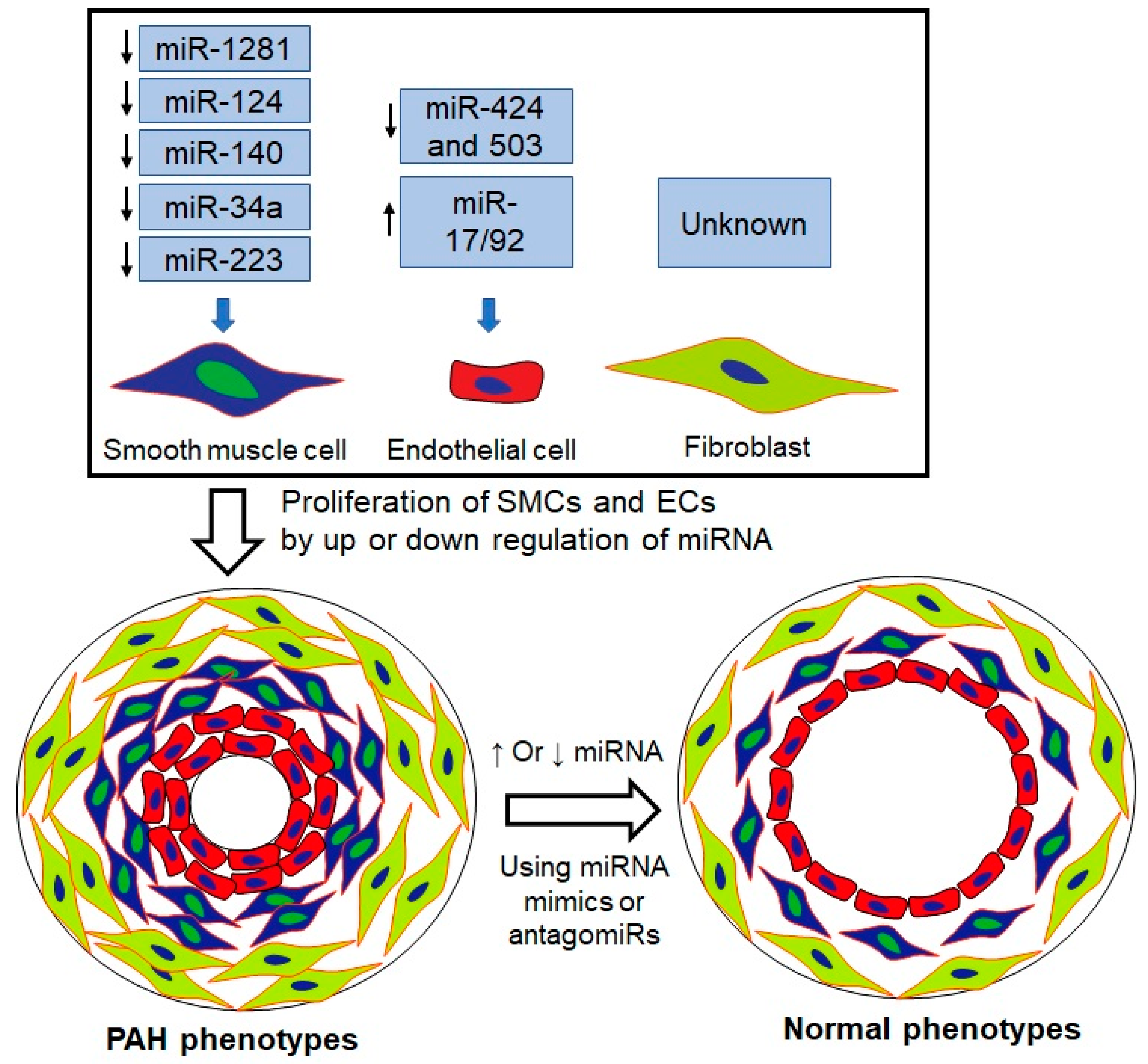
4.3.2. Role of miRNAs in PAH
4.3.3. Role of lncRNAs in PAH
4.3.4. Therapeutic Potential of miRNAs and lncRNAs
miRNAs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Types/Animal Models | miRNA | Target mRNA | Function of miRNA | Expression | References |
|---|---|---|---|---|---|
| Human and rat PASMCs | miR-1281 | Phosphatidylinositol 3-kinase–DNA methyltransferase 1–miR-1281–histone deacetylase 4 | Antiproliferation |  | Y Li et al., J Am Heart Assoc. 2018 [167] |
| HPAH and IPAH BOECs Rat SUGEN-hypoxia model of severe PAH | miR-124 | PTPB1 and PKM2 | Proliferation |  | P Caruso et al., Circulation 2017 [168] |
| Hypoxic human PASMCs | miR-140 | miR-140-5p–DNMT1–SOD2 | Proliferation |  | Y Zhang et al., Biochem Biophys Res Commun. 2016 [169] |
| Human PASMCs PAH patients and in preclinical models of PAH. | miR-34a | miR-34a-3p–MiD-DRP1 | Proliferation and anti-apoptosis. | 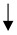 | KH Chen et al., Circulation 2018 [170] |
| Human PASMCs Sugen/hypoxia rat model | miR-204 | miR-204–BRD4 | Proliferation | 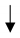 | M Meloche et al., Circ Res. 2015 [132] |
| Endothelial cells (PAECs) | miR-424 and 503 | FGF2 and FGFR1 | Proliferation | 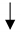 | J Kim et al., Nat Med. 2013 [171] |
| Human PAH lungs, distal PAs, and isolated PASMCs | miR-223 | PARP-1 | PASMC proliferation | 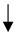 | J Meloche et al., Am J Physiol Cell Physiol 2015 [172] |
| Human PAH PAECs | miR-17/92 | BMPR2 | PAEC survival |  | M Brock et al., Circ Res. 2009 [173] |
lncRNAs
| lncRNAs | Cells/Animal Models | Targets | Functions | Expression | References |
|---|---|---|---|---|---|
| MEG3 | Human PASMCs | miR-21/PTEN; p53 pathway | Inhibits proliferation and migration | | Zhu B et al., Biochem. Biophys. Res. Commun. 2018 Sun Z et al., Cell Physiol. Biochem. 2017 [175,176] |
| MEG3 | Human PASMCs | miR-328- 3p/IGF1R | Proliferation of PASMC under hypoxia | | Xing Y et al., Mol. Ther. 2019 [177] |
| LNCRNA-ANG362 | Human PASMCs | NF-κB-miR-221 and miR-222 | Proliferation and migration of HPASMCs | | Wang H et al., SHOCK 2020 [178] |
| TYKRIL | Human PASMCs | p53/PDGFR axis | Proliferation and anti-apoptosis | | Zehendner CM et al., Am. J. Respir. Crit. Care Med. 2020 [179] |
| LnRPT | PDGF-BB-induced hyperproliferation of rat PASMCs | Notch signaling pathway | Proliferation | | Chen J et al., Am. J. Respir. Cell Mol. Biol. 2018 [180] |
| SMILR | Human PASMCs, MCT-induced PH in Rats | RhoA/ROCK/ miR-141signaling | Vascular remodeling and PAH | | Lei S et al., Am. J. Physiol. Heart Circ. Physiol. 2020 [181] |
| MANTIS | MCT-induced PH rat models | BRG1 | Angiogenesis and apoptosis | | Leisegang MS et al., Circulation 2017 [182] |
| CASC2 | PASMCs Hypoxic PAH in rats | α-SMA | Inhibits proliferation and migration | | Gong J et al., Respir. Res. 2019 [183] |
5. Microfluidics for Epigenetic Analysis
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PH | pulmonary hypertension |
| PA | pulmonary artery |
| PAH | pulmonary arterial hypertension |
| PVR | pulmonary vascular resistance |
| RV | right ventricular |
| RHF | right-heart failure |
| RVH | right-ventricular hypertrophy |
| mPAP | mean pulmonary artery pressure |
| LVEDP | left-ventricular end-diastolic pressure |
| WU | Wood units |
| WHO | World Health Organization |
| COPD | chronic obstructive pulmonary disease |
| IPAH | idiopathic PAH |
| HPAH | heritable PAH |
| APAH | associated PAH |
| PASMCs | pulmonary arterial smooth muscle cells |
| PAECs | pulmonary arterial endothelial cells |
| PAADCs | pulmonary arterial adventitial cells |
| HDMs | histone demethylases |
| HDACs | histone deacetylases |
| Sirt1 | Sirtuin-1 |
| Sirt3 | Sirtuin-3 |
| Sirt4 | Sirtuin-4 |
| BRD4 | bromodomain-containing protein 4 |
| 5-HT | 5-hydroxytryptamine |
| ERAs | endothelin receptor antagonists |
| PCAs | prostacyclin analogues |
| PDE-Is | phosphodiesterase 5 inhibitors |
| sGCs | soluble guanylate cyclase stimulators |
| FDA | the US Food and Drug Administration |
| CCBs | calcium channel blockers |
| NO | nitric oxide |
| cGMP | cyclic guanosine monophosphate |
| sGC | soluble guanylyl cyclase |
| PDE5 | phosphodiesterase-5 |
| cGMP | cyclic guanosine monophosphate |
| GMP | guanosine monophosphate |
| PGI2 | prostacyclin |
| cAMP | cyclic adenosine monophosphate |
| PKA | protein kinase A, |
| SC | subcutaneous |
| IV | infusion |
| ECE | endothelin-converting enzyme |
| IP3 | inositol triphosphate |
| E2 | estradiol |
| DHEA-S | dehydroepiandrosterone sulfate |
| Treg | regulatory T cells |
| Erα | estrogen receptor alpha |
| Erβ | estrogen receptor beta |
| TFAM | mitochondrial transcription factors |
| NFAT | nuclear factor of activated T-cells |
| NLPR3FSP-1 | NLR (NOD like receptor) family pyrin domain-containing 3 fibroblast-specific protein 1 |
| CD31 | platelet endothelial cell adhesion molecule (PECAM-1) also known as cluster of differentiation 31 |
| CYP1B1 | cytochrome P450 family 1 subfamily B member 1 |
| ncRNAs | noncoding RNAs |
| lncRNAs | long noncoding RNAs |
| circRNAs | circular RNAs |
| CpG | cytosine-phosphate-guanine |
| DNMTs | DNA methyltransferases |
| MTase | methyltransferase |
| H3K3me3 | histone 3 lysine 4 trimethylation |
| H3K27me3 | histone 3 lysine 27 trimethylation |
| K4me3 | lysine 4 trimethylation |
| MeCP1 | methylated CpG-binding protein 1 |
| MeCP2 | Methylated CpG-binding protein 2 |
| MBDs | methyl-CpG-binding domain proteins |
| SOD2 | superoxide dismutase 2 |
| FHR | Fawn-hooded rats |
| HIF-1α | hypoxia-inducible factor |
| Kv1.5 | voltage-gated K+ channels |
| siRNA | small interfering RNA |
| BMPR2 | bone morphogenetic protein receptor 2 |
| TBX4 | T-box 4 |
| ACVRL1 | activin A receptor-like type 1 |
| ENG | endoglin |
| SMAD9 | small mothers against decapentaplegic homolog 9 |
| CAV1 | caveolin-1 |
| KCNK3 | potassium channel subfamily K member 3 |
| ATP13A3 | ATPase 13A3 |
| SOX17 | SRY-box 17 |
| AQP1 | aquaporin 1 |
| BMP9 (GDF2) | growth differentiation factor 2 |
| EIF2AK4 | eukaryotic translation initiation factor 2 alpha kinase 4 |
| TGF-β | transforming growth factor beta |
| BMP/TGF | bone morphogenic protein/transforming growth factor |
| WES | whole genome sequencing |
| SPS | small-patella syndrome |
| HMTs | histone methyltransferases |
| JmjC | Jumonji C family |
| LSD | lysine-specific demethylases |
| FAD | flavin adenine dinucleotide |
| α-KG | α-ketoglutarate |
| CAM | cell adhesion molecules |
| ICAMs | intercellular adhesion molecules |
| VCAMs | vascular cell adhesion molecules |
| MRTF-A | myocardin-related transcription factor A |
| NF-κB | nuclear factor kappa B |
| ASH2 | small homeotic 2 |
| WDR5 | WD repeat domain 5 |
| EZH2 | enhancer of zeste homolog 2 |
| TAC | transverse aortic constriction |
| ROS | reactive oxygen species |
| SOD1 | superoxide dismutase type 1 |
| GSK-J4 | a KDM6B inhibitor |
| LSD1 | lysine-specific histone demethylase 1A |
| HATs | histone acetyltransferases |
| HDACs | histone deacetylases |
| BRDPs | bromodomain-containing proteins |
| eNOS | endothelial nitric oxide synthase |
| PPHN | persistent PH of the newborn |
| PAB | pulmonary artery banding |
| MCT | monocrotaline |
| TSA | trichostatin A |
| VPA | valproic acid |
| MEF2 | myocyte enhancer factor 2 |
| IGF-1/pAKT | insulin-like growth factor-1/phosphorylation of protein kinase B |
| RVSP | RV systolic pressure |
| CO | cardiac output |
| TPR | pulmonary vascular resistance |
| BRD | bromodomain |
| BET | bromodomain and extra-terminal domain |
| BCL2 | B-cell lymphoma 2 |
| FOXM1 | Forkhead Box M1 |
| NAD+ | nicotinamide adenine dinucleotide |
| FOXO1 | Forkhead box protein O1 |
| CR | calorie restriction |
| MnSOD | manganese superoxide dismutase |
| SOD2 | superoxide dismutase 2 |
| Cat | catalase |
| Sirt3KO | Sirtuin-3 knockout |
| PDK | pyruvate dehydrogenase kinase |
| PDH | pyruvate dehydrogenase complex |
| GDH | glutamate dehydrogenase |
| DCA | dichloroacetate |
| UCP2 | uncoupling protein 2 |
| PI3K/AKT | phosphoinositide 3-kinase/protein kinase B |
| Sphk1 | sphingosine kinase 1 |
| SP1 | specificity protein 1 |
| Pri-miRNAs | primary miRNAs |
| RISC | RNA-induced silencing complex |
References
- Montani, D.; Gunther, S.; Dorfmuller, P.; Perros, F.; Girerd, B.; Garcia, G.; Jais, X.; Savale, L.; Artaud-Macari, E.; Price, L.C.; et al. Pulmonary arterial hypertension. Orphanet. J. Rare Dis. 2013, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Condon, D.F.; Nickel, N.P.; Anderson, R.; Mirza, S.; de Jesus Perez, V.A. The 6th World Symposium on Pulmonary Hypertension: What’s old is new. F1000Research 2019, 8, 888. [Google Scholar] [CrossRef]
- Kirson, N.Y.; Birnbaum, H.G.; Ivanova, J.I.; Waldman, T.; Joish, V.; Williamson, T. Prevalence of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension in the United States. Curr. Med. Res. Opin. 2011, 27, 1763–1768. [Google Scholar] [CrossRef]
- Peacock, A.J.; Murphy, N.F.; McMurray, J.J.; Caballero, L.; Stewart, S. An epidemiological study of pulmonary arterial hypertension. Eur. Respir. J. 2007, 30, 104–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef]
- Swinnen, K.; Quarck, R.; Godinas, L.; Belge, C.; Delcroix, M. Learning from registries in pulmonary arterial hypertension: Pitfalls and recommendations. Eur. Respir. Rev. 2019, 28, 190050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeper, M.M.; Kramer, T.; Pan, Z.; Eichstaedt, C.A.; Spiesshoefer, J.; Benjamin, N.; Olsson, K.M.; Meyer, K.; Vizza, C.D.; Vonk-Noordegraaf, A.; et al. Mortality in pulmonary arterial hypertension: Prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur. Respir. J. 2017, 50, 1700740. [Google Scholar] [CrossRef] [Green Version]
- D’Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A.; Maron, B.A. Molecular Mechanisms of Pulmonary Vascular Remodeling in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2016, 17, 761. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, K.R.; Frid, M.G.; Graham, B.B.; Tuder, R.M. Dynamic and diverse changes in the functional properties of vascular smooth muscle cells in pulmonary hypertension. Cardiovasc. Res. 2018, 114, 551–564. [Google Scholar] [CrossRef]
- Schermuly, R.T.; Ghofrani, H.A.; Wilkins, M.R.; Grimminger, F. Mechanisms of disease: Pulmonary arterial hypertension. Nat. Rev. Cardiol. 2011, 8, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Nozik-Grayck, E.; Gerasimovskaya, E.; Anwar, A.; Li, M.; Riddle, S.; Frid, M. The adventitia: Essential role in pulmonary vascular remodeling. Compr. Physiol. 2011, 1, 141–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eddahibi, S.; Guignabert, C.; Barlier-Mur, A.M.; Dewachter, L.; Fadel, E.; Dartevelle, P.; Humbert, M.; Simonneau, G.; Hanoun, N.; Saurini, F.; et al. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: Critical role for serotonin-induced smooth muscle hyperplasia. Circulation 2006, 113, 1857–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Launay, J.M.; Herve, P.; Peoc’h, K.; Tournois, C.; Callebert, J.; Nebigil, C.G.; Etienne, N.; Drouet, L.; Humbert, M.; Simonneau, G.; et al. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat. Med. 2002, 8, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Ghofrani, H.A. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax 2016, 71, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Camerini, F.; Alberti, E.; Klugmann, S.; Salvi, A. Primary pulmonary hypertension: Effects of nifedipine. Br. Heart J. 1980, 44, 352–356. [Google Scholar] [CrossRef]
- Douglas, J.S., Jr. Hemodynamic effects of nifedipine in primary pulmonary hypertension. J. Am. Coll. Cardiol. 1983, 2, 174–179. [Google Scholar] [CrossRef]
- Packer, M. Therapeutic application of calcium-channel antagonists for pulmonary hypertension. Am. J. Cardiol. 1985, 55, 196B–201B. [Google Scholar] [CrossRef]
- Rich, S.; Kaufmann, E.; Levy, P.S. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N. Engl. J. Med. 1992, 327, 76–81. [Google Scholar] [CrossRef]
- Joseph, T.; DiPiro, R.L.T.; Gary, C.; Yee, G.R.M.; Barbara, G.; Wells, L. Michael Posey. Pharmacotherapy: A Pathophysiologic Approach, 11th ed.; McGraw-Hill Education: New York, NY, USA, 2020. [Google Scholar]
- Rubin, L.J.; Galie, N.; Grimminger, F.; Grunig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.; Langleben, D.; Fritsch, A.; Menezes, F.; et al. Riociguat for the treatment of pulmonary arterial hypertension: A long-term extension study (PATENT-2). Eur. Respir. J. 2015, 45, 1303–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghofrani, H.A.; Galie, N.; Grimminger, F.; Grunig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef]
- Christman, B.W.; McPherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N. Engl. J. Med. 1992, 327, 70–75. [Google Scholar] [CrossRef]
- Montani, D.; Chaumais, M.C.; Guignabert, C.; Gunther, S.; Girerd, B.; Jais, X.; Algalarrondo, V.; Price, L.C.; Savale, L.; Sitbon, O.; et al. Targeted therapies in pulmonary arterial hypertension. Pharmacol. Ther. 2014, 141, 172–191. [Google Scholar] [CrossRef]
- Parikh, V.; Bhardwaj, A.; Nair, A. Pharmacotherapy for pulmonary arterial hypertension. J. Thorac. Dis. 2019, 11, S1767–S1781. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Simonneau, G. Treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1425–1436. [Google Scholar] [CrossRef]
- Davie, N.; Haleen, S.J.; Upton, P.D.; Polak, J.M.; Yacoub, M.H.; Morrell, N.W.; Wharton, J. ET(A) and ET(B) receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am. J. Respir. Crit. Care Med. 2002, 165, 398–405. [Google Scholar] [CrossRef]
- Rivera-Lebron, B.N.; Risbano, M.G. Ambrisentan: A review of its use in pulmonary arterial hypertension. Ther. Adv. Respir. Dis. 2017, 11, 233–244. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galie, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.C.; Le Brun, F.O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galie, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Badesch, D.B.; Raskob, G.E.; Elliott, C.G.; Krichman, A.M.; Farber, H.W.; Frost, A.E.; Barst, R.J.; Benza, R.L.; Liou, T.G.; Turner, M.; et al. Pulmonary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest 2010, 137, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Kawut, S.M.; Al-Naamani, N.; Agerstrand, C.; Berman Rosenzweig, E.; Rowan, C.; Barst, R.J.; Bergmann, S.; Horn, E.M. Determinants of right ventricular ejection fraction in pulmonary arterial hypertension. Chest 2009, 135, 752–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010, 122, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, S.; Traiger, G.L.; Turner, M.; McGoon, M.D.; Wason, P.; Barst, R.J. Sex differences in the diagnosis, treatment, and outcome of patients with pulmonary arterial hypertension enrolled in the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Chest 2012, 141, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Domsic, R.T.; Lingala, B.; Alkassab, F.; Bolster, M.; Csuka, M.E.; Derk, C.; Fischer, A.; Frech, T.; Furst, D.E.; et al. Survival and predictors of mortality in systemic sclerosis-associated pulmonary arterial hypertension: Outcomes from the pulmonary hypertension assessment and recognition of outcomes in scleroderma registry. Arthritis Care Res. (Hoboken) 2014, 66, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, W.; van de Veerdonk, M.C.; Trip, P.; de Man, F.; Heymans, M.W.; Marcus, J.T.; Kawut, S.M.; Bogaard, H.J.; Boonstra, A.; Vonk Noordegraaf, A. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014, 145, 1230–1236. [Google Scholar] [CrossRef] [Green Version]
- Ventetuolo, C.E.; Hess, E.; Austin, E.D.; Baron, A.E.; Klinger, J.R.; Lahm, T.; Maddox, T.M.; Plomondon, M.E.; Thompson, L.; Zamanian, R.T.; et al. Sex-based differences in veterans with pulmonary hypertension: Results from the veterans affairs-clinical assessment reporting and tracking database. PLoS ONE 2017, 12, e0187734. [Google Scholar] [CrossRef]
- Gabler, N.B.; French, B.; Strom, B.L.; Liu, Z.; Palevsky, H.I.; Taichman, D.B.; Kawut, S.M.; Halpern, S.D. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest 2012, 141, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Mair, K.M.; Wright, A.F.; Duggan, N.; Rowlands, D.J.; Hussey, M.J.; Roberts, S.; Fullerton, J.; Nilsen, M.; Loughlin, L.; Thomas, M.; et al. Sex-dependent influence of endogenous estrogen in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 456–467. [Google Scholar] [CrossRef] [Green Version]
- Lahm, T.; Albrecht, M.; Fisher, A.J.; Selej, M.; Patel, N.G.; Brown, J.A.; Justice, M.J.; Brown, M.B.; Van Demark, M.; Trulock, K.M.; et al. 17beta-Estradiol attenuates hypoxic pulmonary hypertension via estrogen receptor-mediated effects. Am. J. Respir. Crit. Care Med. 2012, 185, 965–980. [Google Scholar] [CrossRef]
- Tofovic, S.P. Estrogens and development of pulmonary hypertension: Interaction of estradiol metabolism and pulmonary vascular disease. J. Cardiovasc. Pharmacol. 2010, 56, 696–708. [Google Scholar] [CrossRef] [Green Version]
- Badlam, J.B.; Austin, E.D. Beyond oestrogens: Towards a broader evaluation of the hormone profile in pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1801058. [Google Scholar] [CrossRef]
- Ventetuolo, C.E.; Baird, G.L.; Barr, R.G.; Bluemke, D.A.; Fritz, J.S.; Hill, N.S.; Klinger, J.R.; Lima, J.A.; Ouyang, P.; Palevsky, H.I.; et al. Higher Estradiol and Lower Dehydroepiandrosterone-Sulfate Levels Are Associated with Pulmonary Arterial Hypertension in Men. Am. J. Respir. Crit. Care Med. 2016, 193, 1168–1175. [Google Scholar] [CrossRef]
- Osman, M.S.; Michelakis, E.D. Immunity Comes to Play in the “Sex Paradox” of Pulmonary Arterial Hypertension. Circ. Res. 2018, 122, 1635–1637. [Google Scholar] [CrossRef]
- Tamosiuniene, R.; Tian, W.; Dhillon, G.; Wang, L.; Sung, Y.K.; Gera, L.; Patterson, A.J.; Agrawal, R.; Rabinovitch, M.; Ambler, K.; et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ. Res. 2011, 109, 867–879. [Google Scholar] [CrossRef]
- Huertas, A.; Tu, L.; Gambaryan, N.; Girerd, B.; Perros, F.; Montani, D.; Fabre, D.; Fadel, E.; Eddahibi, S.; Cohen-Kaminsky, S.; et al. Leptin and regulatory T-lymphocytes in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2012, 40, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Huertas, A.; Phan, C.; Bordenave, J.; Tu, L.; Thuillet, R.; Le Hiress, M.; Avouac, J.; Tamura, Y.; Allanore, Y.; Jovan, R.; et al. Regulatory T Cell Dysfunction in Idiopathic, Heritable and Connective Tissue-Associated Pulmonary Arterial Hypertension. Chest 2016, 149, 1482–1493. [Google Scholar] [CrossRef]
- Tamosiuniene, R.; Manouvakhova, O.; Mesange, P.; Saito, T.; Qian, J.; Sanyal, M.; Lin, Y.C.; Nguyen, L.P.; Luria, A.; Tu, A.B.; et al. Dominant Role for Regulatory T Cells in Protecting Females Against Pulmonary Hypertension. Circ. Res. 2018, 122, 1689–1702. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. Estrogenic control of mitochondrial function. Redox. Biol. 2020, 31, 101435. [Google Scholar] [CrossRef]
- Farha, S.; Hu, B.; Comhair, S.; Zein, J.; Dweik, R.; Erzurum, S.C.; Aldred, M.A. Mitochondrial Haplogroups and Risk of Pulmonary Arterial Hypertension. PLoS ONE 2016, 11, e0156042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef] [Green Version]
- Kepp, O.; Galluzzi, L.; Kroemer, G. Mitochondrial control of the NLRP3 inflammasome. Nat. Immunol. 2011, 12, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.; Lochner, M.; Berod, L.; Sparwasser, T. Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol. 2016, 28, 514–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Rocha, S.T.; Gendrel, A.V. The influence of DNA methylation on monoallelic expression. Essays Biochem. 2019, 63, 663–676. [Google Scholar] [CrossRef]
- Aldred, M.A.; Comhair, S.A.; Varella-Garcia, M.; Asosingh, K.; Xu, W.; Noon, G.P.; Thistlethwaite, P.A.; Tuder, R.M.; Erzurum, S.C.; Geraci, M.W.; et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1153–1160. [Google Scholar] [CrossRef]
- Low, L.A.; Tagle, D.A. Tissue chips-innovative tools for drug development and disease modeling. Lab Chip 2017, 17, 3026–3036. [Google Scholar] [CrossRef]
- Al-Hilal, T.A.; Keshavarz, A.; Kadry, H.; Lahooti, B.; Al-Obaida, A.; Ding, Z.; Li, W.; Kamm, R.; McMurtry, I.F.; Lahm, T.; et al. Pulmonary-arterial-hypertension (PAH)-on-a-chip: Fabrication, validation and application. Lab Chip 2020, 20, 3334–3345. [Google Scholar] [CrossRef]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Gillette, T.G.; Hill, J.A. Readers, writers, and erasers: Chromatin as the whiteboard of heart disease. Circ. Res. 2015, 116, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, M.U.; Lam, M.T.; Glass, C.K. Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, C.; Ronn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Song, M.; Qu, J.; Liu, G.H. Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 2018, 123, 773–786. [Google Scholar] [CrossRef]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, H.; Wakabayashi, M.; Hattori, N.; Yamashita, S.; Ushijima, T. Identification of coexistence of DNA methylation and H3K27me3 specifically in cancer cells as a promising target for epigenetic therapy. Carcinogenesis 2015, 36, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.; Umit Kaniskan, H.; Jin, J.; Gozani, O. Epigenetics and beyond: Targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discov. 2021, 20, 265–286. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.Y.; Huang, H.W.; Shu, C.W.; Hou, M.F.; Yuan, S.S.; Wang, H.R.; Chang, Y.T.; Farooqi, A.A.; Tang, J.Y.; Chang, H.W. DNA methylation, histone acetylation and methylation of epigenetic modifications as a therapeutic approach for cancers. Cancer Lett. 2016, 373, 185–192. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermann, A.; Schmitt, S.; Jeltsch, A. The human Dnmt2 has residual DNA-(cytosine-C5) methyltransferase activity. J. Biol. Chem. 2003, 278, 31717–31721. [Google Scholar] [CrossRef] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Huppke, P.; Laccone, F.; Kramer, N.; Engel, W.; Hanefeld, F. Rett syndrome: Analysis of MECP2 and clinical characterization of 31 patients. Hum. Mol. Genet. 2000, 9, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Medvedeva, Y.A.; Khamis, A.M.; Kulakovskiy, I.V.; Ba-Alawi, W.; Bhuyan, M.S.; Kawaji, H.; Lassmann, T.; Harbers, M.; Forrest, A.R.; Bajic, V.B.; et al. Effects of cytosine methylation on transcription factor binding sites. BMC Genom. 2014, 15, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Lee, T.H. Effects of DNA methylation on the structure of nucleosomes. J. Am. Chem. Soc. 2012, 134, 173–175. [Google Scholar] [CrossRef] [Green Version]
- Ballestar, E.; Paz, M.F.; Valle, L.; Wei, S.; Fraga, M.F.; Espada, J.; Cigudosa, J.C.; Huang, T.H.; Esteller, M. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J. 2003, 22, 6335–6345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thebaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [Green Version]
- Austin, E.D.; Loyd, J.E. The genetics of pulmonary arterial hypertension. Circ Res 2014, 115, 189–202. [Google Scholar] [CrossRef] [Green Version]
- Quarck, R.; Perros, F. Rescuing BMPR2-driven endothelial dysfunction in PAH: A novel treatment strategy for the future? Stem. Cell Investig. 2017, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Yan, Y.; Chen, J.W.; Yuan, P.; Wang, X.J.; Jiang, R.; Wang, L.; Zhao, Q.H.; Wu, W.H.; Simonneau, G.; et al. Hypermethylation of BMPR2 Promoter Occurs in Patients with Heritable Pulmonary Arterial Hypertension and Inhibits BMPR2 Expression. Am. J. Respir. Crit. Care Med. 2017, 196, 925–928. [Google Scholar] [CrossRef]
- Pousada, G.; Baloira, A.; Valverde, D. Methylation Analysis of the BMPR2 Gene Promoter Region in Patients With Pulmonary Arterial Hypertension. Arch. Bronconeumol. 2016, 52, 293–298. [Google Scholar] [CrossRef]
- Cutter, A.R.; Hayes, J.J. A brief review of nucleosome structure. FEBS Lett. 2015, 589, 2914–2922. [Google Scholar] [CrossRef] [Green Version]
- Marino-Ramirez, L.; Kann, M.G.; Shoemaker, B.A.; Landsman, D. Histone structure and nucleosome stability. Expert Rev. Proteom. 2005, 2, 719–729. [Google Scholar] [CrossRef]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [Green Version]
- Stillman, B. Histone Modifications: Insights into Their Influence on Gene Expression. Cell 2018, 175, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Barnes, C.E.; English, D.M.; Cowley, S.M. Acetylation & Co: An expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 2019, 63, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Andreoli, F.; Del Rio, A. Physicochemical modifications of histones and their impact on epigenomics. Drug Discov. Today 2014, 19, 1372–1379. [Google Scholar] [CrossRef]
- Meng, Y.; Li, H.; Liu, C.; Zheng, L.; Shen, B. Jumonji domain-containing protein family: The functions beyond lysine demethylation. J. Mol. Cell Biol. 2018, 10, 371–373. [Google Scholar] [CrossRef]
- Gamen, E.; Seeger, W.; Pullamsetti, S.S. The emerging role of epigenetics in pulmonary hypertension. Eur. Respir. J. 2016, 48, 903–917. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Yang, Y.; Cheng, X.; Fang, F.; Xu, G.; Yuan, Z.; Xia, J.; Kong, H.; Xie, W.; Wang, H.; et al. Megakaryocytic leukemia 1 directs a histone H3 lysine 4 methyltransferase complex to regulate hypoxic pulmonary hypertension. Hypertension 2015, 65, 821–833. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Lu, Z.; Singh, D.; Raj, J.U. BIX-01294 treatment blocks cell proliferation, migration and contractility in ovine foetal pulmonary arterial smooth muscle cells. Cell Prolif 2012, 45, 335–344. [Google Scholar] [CrossRef]
- Aljubran, S.A.; Cox, R., Jr.; Tamarapu Parthasarathy, P.; Kollongod Ramanathan, G.; Rajanbabu, V.; Bao, H.; Mohapatra, S.S.; Lockey, R.; Kolliputi, N. Enhancer of zeste homolog 2 induces pulmonary artery smooth muscle cell proliferation. PLoS ONE 2012, 7, e37712. [Google Scholar] [CrossRef]
- Shi, Z.L.; Fang, K.; Li, Z.H.; Ren, D.H.; Zhang, J.Y.; Sun, J. EZH2 Inhibition Ameliorates Transverse Aortic Constriction-Induced Pulmonary Arterial Hypertension in Mice. Can. Respir. J. 2018, 2018, 9174926. [Google Scholar] [CrossRef] [Green Version]
- Gambaryan, N.; Meng, C.; Humbert, M.; Adcock, I.; Wort, S. H3K27 histone lysine methylation as potential therapeutic target in pulmonary arterial hypertension. Eur. Respir. J. 2013, 42, 5157. [Google Scholar]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 2019, 12, 129. [Google Scholar] [CrossRef] [Green Version]
- Pojoga, L.H.; Williams, J.S.; Yao, T.M.; Kumar, A.; Raffetto, J.D.; do Nascimento, G.R.; Reslan, O.M.; Adler, G.K.; Williams, G.H.; Shi, Y.; et al. Histone demethylase LSD1 deficiency during high-salt diet is associated with enhanced vascular contraction, altered NO-cGMP relaxation pathway, and hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1862–H1871. [Google Scholar] [CrossRef] [Green Version]
- Klinger, J.R. The nitric oxide/cGMP signaling pathway in pulmonary hypertension. Clin. Chest. Med. 2007, 28, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, I.; Poreba, E.; Kamieniarz, K.; Schneider, R. Histone modifiers in cancer: Friends or foes? Genes Cancer 2011, 2, 631–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, A.; Lambert, C.; Habbout, K.; Ranchoux, B.; Paquet-Marceau, S.; Trinh, I.; Breuils-Bonnet, S.; Paradis, R.; Nadeau, V.; Paulin, R.; et al. FOXM1 promotes pulmonary artery smooth muscle cell expansion in pulmonary arterial hypertension. J. Mol Med. (Berl.) 2018, 96, 223–235. [Google Scholar] [CrossRef]
- Wang, S.; Pike, A.M.; Lee, S.S.; Strong, M.A.; Connelly, C.J.; Greider, C.W. BRD4 inhibitors block telomere elongation. Nucleic Acids Res. 2017, 45, 8403–8410. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Dulak, A.M.; Hattersley, M.M.; Willis, B.S.; Nikkilä, J.; Wang, A.; Lau, A.; Reimer, C.; Zinda, M.; Fawell, S.E.; et al. BRD4 facilitates replication stress-induced DNA damage response. Oncogene 2018, 37, 3763–3777. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, C.; Hajji, N.; Oliver, E.; Cotroneo, E.; Wharton, J.; Wang, D.; Li, M.; McKinsey, T.A.; Stenmark, K.R.; et al. Histone deacetylation inhibition in pulmonary hypertension: Therapeutictherapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012, 126, 455–467. [Google Scholar] [CrossRef]
- Cho, Y.K.; Eom, G.H.; Kee, H.J.; Kim, H.S.; Choi, W.Y.; Nam, K.I.; Ma, J.S.; Kook, H. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ. J. 2010, 74, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Lan, B.; Hayama, E.; Kawaguchi, N.; Furutani, Y.; Nakanishi, T. Therapeutic efficacy of valproic acid in a combined monocrotaline and chronic hypoxia rat model of severe pulmonary hypertension. PLoS ONE 2015, 10, e0117211. [Google Scholar] [CrossRef] [Green Version]
- Bogaard, H.J.; Mizuno, S.; Hussaini, A.A.; Toldo, S.; Abbate, A.; Kraskauskas, D.; Kasper, M.; Natarajan, R.; Voelkel, N.F. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am. J. Respir. Crit. Care Med. 2011, 183, 1402–1410. [Google Scholar] [CrossRef]
- Chen, F.; Li, X.; Aquadro, E.; Haigh, S.; Zhou, J.; Stepp, D.W.; Weintraub, N.L.; Barman, S.A.; Fulton, D.J.R. Inhibition of histone deacetylase reduces transcription of NADPH oxidases and ROS production and ameliorates pulmonary arterial hypertension. Free Radic. Biol. Med. 2016, 99, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hwangbo, C.; Hu, X.; Kang, Y.; Papangeli, I.; Mehrotra, D.; Park, H.; Ju, H.; McLean, D.L.; Comhair, S.A.; et al. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation 2015, 131, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavasin, M.A.; Demos-Davies, K.; Horn, T.R.; Walker, L.A.; Lemon, D.D.; Birdsey, N.; Weiser-Evans, M.C.; Harral, J.; Irwin, D.C.; Anwar, A.; et al. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ. Res. 2012, 110, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Sun, M.; Ramchandran, R.; Raj, J.U. IGF-1 signaling in neonatal hypoxia-induced pulmonary hypertension: Role of epigenetic regulation. Vasc. Pharmacol. 2015, 73, 20–31. [Google Scholar] [CrossRef] [Green Version]
- Boucherat, O.; Chabot, S.; Paulin, R.; Trinh, I.; Bourgeois, A.; Potus, F.; Lampron, M.C.; Lambert, C.; Breuils-Bonnet, S.; Nadeau, V.; et al. HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7, 4546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef] [Green Version]
- Meloche, J.; Potus, F.; Vaillancourt, M.; Bourgeois, A.; Johnson, I.; Deschamps, L.; Chabot, S.; Ruffenach, G.; Henry, S.; Breuils-Bonnet, S.; et al. Bromodomain-Containing Protein 4: The Epigenetic Origin of Pulmonary Arterial Hypertension. Circ. Res. 2015, 117, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Van der Feen, D.E.; Kurakula, K.; Tremblay, E.; Boucherat, O.; Bossers, G.P.L.; Szulcek, R.; Bourgeois, A.; Lampron, M.C.; Habbout, K.; Martineau, S.; et al. Multicenter Preclinical Validation of BET Inhibition for the Treatment of Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 200, 910–920. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD(+) in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, C.; Satterstrom, F.K.; Haigis, M.C.; Mostoslavsky, R. From sirtuin biology to human diseases: An update. J. Biol. Chem. 2012, 287, 42444–42452. [Google Scholar] [CrossRef] [Green Version]
- Poulose, N.; Raju, R. Sirtuin regulation in aging and injury. Biochim. Biophys. Acta 2015, 1852, 2442–2455. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Auwerx, J. Protein deacetylation by SIRT1: An emerging key post-translational modification in metabolic regulation. Pharmacol. Res. 2010, 62, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Y.; Hirschey, M.D.; Shimazu, T.; Ho, L.; Verdin, E. Mitochondrial sirtuins. Biochim. Biophys. Acta 2010, 1804, 1645–1651. [Google Scholar] [CrossRef]
- Zurlo, G.; Piquereau, J.; Moulin, M.; Pires Da Silva, J.; Gressette, M.; Ranchoux, B.; Garnier, A.; Ventura-Clapier, R.; Fadel, E.; Humbert, M.; et al. Sirtuin 1 regulates pulmonary artery smooth muscle cell proliferation: Role in pulmonary arterial hypertension. J. Hypertens. 2018, 36, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Lei, J.; Qu, Y.; Zhang, H.; Xin, W.; Ma, F.; Liu, S.; Li, Z.; Jin, F.; Fu, E. Calorie Restriction Attenuates Monocrotaline-induced Pulmonary Arterial Hypertension in Rats. J. Cardiovasc. Pharmacol. 2015, 65, 562–570. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.D.; Bazan, I.; Zhang, Y.; Fares, W.H.; Lee, P.J. Mitochondrial dysfunction and pulmonary hypertension: Cause, effect, or both. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L782–L796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulin, R.; Dromparis, P.; Sutendra, G.; Gurtu, V.; Zervopoulos, S.; Bowers, L.; Haromy, A.; Webster, L.; Provencher, S.; Bonnet, S.; et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014, 20, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waypa, G.B.; Osborne, S.W.; Marks, J.D.; Berkelhamer, S.K.; Kondapalli, J.; Schumacker, P.T. Sirtuin 3 deficiency does not augment hypoxia-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2013, 49, 885–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583. [Google Scholar] [CrossRef] [Green Version]
- Hogan, S.E.; Rodriguez Salazar, M.P.; Cheadle, J.; Glenn, R.; Medrano, C.; Petersen, T.H.; Ilagan, R.M. Mesenchymal stromal cell-derived exosomes improve mitochondrial health in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L723–L737. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathias, R.A.; Greco, T.M.; Oberstein, A.; Budayeva, H.G.; Chakrabarti, R.; Rowland, E.A.; Kang, Y.; Shenk, T.; Cristea, I.M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Xue, J.; Meng, X.; Slutzky, J.L.; Calvert, A.E.; Chicoine, L.G. Resveratrol prevents hypoxia-induced arginase II expression and proliferation of human pulmonary artery smooth muscle cells via Akt-dependent signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L317–L325. [Google Scholar] [CrossRef] [Green Version]
- Vazquez-Garza, E.; Bernal-Ramirez, J.; Jerjes-Sanchez, C.; Lozano, O.; Acuna-Morin, E.; Vanoye-Tamez, M.; Ramos-Gonzalez, M.R.; Chapoy-Villanueva, H.; Perez-Plata, L.; Sanchez-Trujillo, L.; et al. Resveratrol Prevents Right Ventricle Remodeling and Dysfunction in Monocrotaline-Induced Pulmonary Arterial Hypertension with a Limited Improvement in the Lung Vasculature. Oxid. Med. Cell Longev. 2020, 2020, 1841527. [Google Scholar] [CrossRef]
- Csiszar, A.; Labinskyy, N.; Olson, S.; Pinto, J.T.; Gupte, S.; Wu, J.M.; Hu, F.; Ballabh, P.; Podlutsky, A.; Losonczy, G.; et al. Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats. Hypertension 2009, 54, 668–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paffett, M.L.; Lucas, S.N.; Campen, M.J. Resveratrol reverses monocrotaline-induced pulmonary vascular and cardiac dysfunction: A potential role for atrogin-1 in smooth muscle. Vasc. Pharmacol. 2012, 56, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Li, M.T.; Jia, Y.Y.; Liu, J.J.; Wang, Q.; Tian, Z.; Liu, Y.T.; Chen, H.Z.; Liu, D.P.; Zeng, X.F. Regulation of Cell Cycle Regulators by SIRT1 Contributes to Resveratrol-Mediated Prevention of Pulmonary Arterial Hypertension. Biomed. Res. Int. 2015, 2015, 762349. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Tu, Y.; Jia, X.; Fang, K.; Liu, L.; Wan, L.; Xiang, C.; Wang, Y.; Sun, X.; Liu, T.; et al. Resveratrol Protects Against Pulmonary Arterial Hypertension in Rats via Activation of Silent Information Regulator 1. Cell Physiol. Biochem. 2017, 42, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Zhai, C.; Feng, W.; Wang, J.; Zhu, Y.; Li, S.; Wang, Q.; Zhang, Q.; Yan, X.; Chai, L.; et al. Resveratrol inhibits monocrotaline-induced pulmonary arterial remodeling by suppression of SphK1-mediated NF-kappaB activation. Life Sci. 2018, 210, 140–149. [Google Scholar] [CrossRef]
- Wilson, D.N.; Schacht, S.E.; Al-Nakkash, L.; Babu, J.R.; Broderick, T.L. Resveratrol prevents pulmonary trunk remodeling but not right ventricular hypertrophy in monocrotaline-induced pulmonary hypertension. Pathophysiology 2016, 23, 243–250. [Google Scholar] [CrossRef]
- Yang, D.L.; Zhang, H.G.; Xu, Y.L.; Gao, Y.H.; Yang, X.J.; Hao, X.Q.; Li, X.H. Resveratrol inhibits right ventricular hypertrophy induced by monocrotaline in rats. Clin. Exp. Pharmacol. Physiol. 2010, 37, 150–155. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Itani, H.A.; Nazarewicz, R.R.; McMaster, W.G.; Flynn, C.R.; Uzhachenko, R.; Fessel, J.P.; Gamboa, J.L.; Harrison, D.G.; Dikalov, S.I. Sirt3 Impairment and SOD2 Hyperacetylation in Vascular Oxidative Stress and Hypertension. Circ. Res. 2017, 121, 564–574. [Google Scholar] [CrossRef]
- Lander, E.S. Initial impact of the sequencing of the human genome. Nature 2011, 470, 187–197. [Google Scholar] [CrossRef]
- Jin, Q.; Zhao, Z.; Zhao, Q.; Yu, X.; Yan, L.; Zhang, Y.; Luo, Q.; Liu, Z. Long noncoding RNAs: Emerging roles in pulmonary hypertension. Heart Fail. Rev. 2020, 25, 795–815. [Google Scholar] [CrossRef]
- Santos-Ferreira, C.A.; Abreu, M.T.; Marques, C.I.; Goncalves, L.M.; Baptista, R.; Girao, H.M. Micro-RNA Analysis in Pulmonary Arterial Hypertension: Current Knowledge and Challenges. JACC Basic Transl. Sci. 2020, 5, 1149–1162. [Google Scholar] [CrossRef]
- Grant, J.S.; White, K.; MacLean, M.R.; Baker, A.H. MicroRNAs in pulmonary arterial remodeling. Cell Mol. Life Sci. 2013, 70, 4479–4494. [Google Scholar] [CrossRef] [Green Version]
- Kocken, J.M.M.; da Costa Martins, P.A. Epigenetic Regulation of Pulmonary Arterial Hypertension-Induced Vascular and Right Ventricular Remodeling: New Opportunities? Int. J. Mol. Sci. 2020, 21, 8901. [Google Scholar] [CrossRef]
- Han, Y.; Ali, M.K.; Dua, K.; Spiekerkoetter, E.; Mao, Y. Role of Long Non-Coding RNAs in Pulmonary Arterial Hypertension. Cells 2021, 10, 1892. [Google Scholar] [CrossRef]
- Gupta, S.; Li, L. Modulation of miRNAs in Pulmonary Hypertension. Int. J. Hypertens. 2015, 2015, 169069. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Qian, Z.; Lin, B.; Chen, J.; Luo, Y.; Qu, J.; Raj, J.U.; Gou, D. Phosphatidylinositol 3-Kinase-DNA Methyltransferase 1-miR-1281-Histone Deacetylase 4 Regulatory Axis Mediates Platelet-Derived Growth Factor-Induced Proliferation and Migration of Pulmonary Artery Smooth Muscle Cells. J. Am. Heart Assoc. 2018, 7, e007572. [Google Scholar] [CrossRef]
- Caruso, P.; Dunmore, B.J.; Schlosser, K.; Schoors, S.; Dos Santos, C.; Perez-Iratxeta, C.; Lavoie, J.R.; Zhang, H.; Long, L.; Flockton, A.R.; et al. Identification of MicroRNA-124 as a Major Regulator of Enhanced Endothelial Cell Glycolysis in Pulmonary Arterial Hypertension via PTBP1 (Polypyrimidine Tract Binding Protein) and Pyruvate Kinase M2. Circulation 2017, 136, 2451–2467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J. MiR-140-5p regulates hypoxia-mediated human pulmonary artery smooth muscle cell proliferation, apoptosis and differentiation by targeting Dnmt1 and promoting SOD2 expression. Biochem. Biophys. Res. Commun. 2016, 473, 342–348. [Google Scholar] [CrossRef]
- Chen, K.H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications. Circulation 2018, 138, 287–304. [Google Scholar] [CrossRef]
- Kim, J.; Kang, Y.; Kojima, Y.; Lighthouse, J.K.; Hu, X.; Aldred, M.A.; McLean, D.L.; Park, H.; Comhair, S.A.; Greif, D.M.; et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 2013, 19, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Meloche, J.; Le Guen, M.; Potus, F.; Vinck, J.; Ranchoux, B.; Johnson, I.; Antigny, F.; Tremblay, E.; Breuils-Bonnet, S.; Perros, F.; et al. miR-223 reverses experimental pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2015, 309, C363–C372. [Google Scholar] [CrossRef] [Green Version]
- Brock, M.; Trenkmann, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Fischler, M.; Ulrich, S.; Speich, R.; Huber, L.C. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ. Res. 2009, 104, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Smaldone, M.C.; Davies, B.J. BC-819, a plasmid comprising the H19 gene regulatory sequences and diphtheria toxin A, for the potential targeted therapy of cancers. Curr. Opin. Mol. Ther. 2010, 12, 607–616. [Google Scholar]
- Zhu, B.; Gong, Y.; Yan, G.; Wang, D.; Qiao, Y.; Wang, Q.; Liu, B.; Hou, J.; Li, R.; Tang, C. Down-regulation of lncRNA MEG3 promotes hypoxia-induced human pulmonary artery smooth muscle cell proliferation and migration via repressing PTEN by sponging miR-21. Biochem. Biophys. Res. Commun. 2018, 495, 2125–2132. [Google Scholar] [CrossRef]
- Sun, Z.; Nie, X.; Sun, S.; Dong, S.; Yuan, C.; Li, Y.; Xiao, B.; Jie, D.; Liu, Y. Long Non-Coding RNA MEG3 Downregulation Triggers Human Pulmonary Artery Smooth Muscle Cell Proliferation and Migration via the p53 Signaling Pathway. Cell Physiol. Biochem. 2017, 42, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Zheng, X.; Fu, Y.; Qi, J.; Li, M.; Ma, M.; Wang, S.; Li, S.; Zhu, D. Long Noncoding RNA-Maternally Expressed Gene 3 Contributes to Hypoxic Pulmonary Hypertension. Mol. Ther. 2019, 27, 2166–2181. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qin, R.; Cheng, Y. LncRNA-Ang362 Promotes Pulmonary Arterial Hypertension by Regulating miR-221 and miR-222. Shock 2020, 53, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Zehendner, C.M.; Valasarajan, C.; Werner, A.; Boeckel, J.N.; Bischoff, F.C.; John, D.; Weirick, T.; Glaser, S.F.; Rossbach, O.; Jae, N.; et al. Long Noncoding RNA TYKRIL Plays a Role in Pulmonary Hypertension via the p53-mediated Regulation of PDGFRbeta. Am. J. Respir. Crit. Care Med. 2020, 202, 1445–1457. [Google Scholar] [CrossRef]
- Chen, J.; Guo, J.; Cui, X.; Dai, Y.; Tang, Z.; Qu, J.; Raj, J.U.; Hu, Q.; Gou, D. The Long Noncoding RNA LnRPT Is Regulated by PDGF-BB and Modulates the Proliferation of Pulmonary Artery Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2018, 58, 181–193. [Google Scholar] [CrossRef]
- Lei, S.; Peng, F.; Li, M.L.; Duan, W.B.; Peng, C.Q.; Wu, S.J. LncRNA-SMILR modulates RhoA/ROCK signaling by targeting miR-141 to regulate vascular remodeling in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H377–H391. [Google Scholar] [CrossRef]
- Leisegang, M.S.; Fork, C.; Josipovic, I.; Richter, F.M.; Preussner, J.; Hu, J.; Miller, M.J.; Epah, J.; Hofmann, P.; Gunther, S.; et al. Long Noncoding RNA MANTIS Facilitates Endothelial Angiogenic Function. Circulation 2017, 136, 65–79. [Google Scholar] [CrossRef]
- Gong, J.; Chen, Z.; Chen, Y.; Lv, H.; Lu, H.; Yan, F.; Li, L.; Zhang, W.; Shi, J. Long non-coding RNA CASC2 suppresses pulmonary artery smooth muscle cell proliferation and phenotypic switch in hypoxia-induced pulmonary hypertension. Respir. Res. 2019, 20, 53. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Chidambara, V.A.; Andreasen, S.Z.; Golabi, M.; Huynh, V.N.; Linh, Q.T.; Bang, D.D.; Wolff, A. Point-of-care devices for pathogen detections: The three most important factors to realise towards commercialization. TrAC Trends Anal. Chem. 2020, 131, 116004. [Google Scholar] [CrossRef]
- Nguyen, T.; Zoëga Andreasen, S.; Wolff, A.; Duong Bang, D. From Lab on a Chip to Point of Care Devices: The Role of Open Source Microcontrollers. Micromachines 2018, 9, 403. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Lu, C. Microfluidic Chromatin Immunoprecipitation for Analysis of Epigenomic Regulations. In Microfluidic Methods for Molecular Biology; Lu, C., Verbridge, S.S., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 349–363. [Google Scholar] [CrossRef]
- Wu, A.R.; Hiatt, J.B.; Lu, R.; Attema, J.L.; Lobo, N.A.; Weissman, I.L.; Clarke, M.F.; Quake, S.R. Automated microfluidic chromatin immunoprecipitation from 2000 cells. Lab Chip 2009, 9, 1365–1370. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Lu, C. A Microfluidic Device with Integrated Sonication and Immunoprecipitation for Sensitive Epigenetic Assays. Anal. Chem. 2016, 88, 1965–1972. [Google Scholar] [CrossRef] [Green Version]
- De, A.; Sparreboom, W.; van den Berg, A.; Carlen, E.T. Rapid microfluidic solid-phase extraction system for hyper-methylated DNA enrichment and epigenetic analysis. Biomicrofluidics 2014, 8, 054119. [Google Scholar] [CrossRef] [Green Version]
- Sina, A.A.; Carrascosa, L.G.; Trau, M. DNA Methylation-Based Point-of-Care Cancer Detection: Challenges and Possibilities. Trends Mol. Med. 2019, 25, 955–966. [Google Scholar] [CrossRef]
- Beltran-Garcia, J.; Osca-Verdegal, R.; Mena-Molla, S.; Garcia-Gimenez, J.L. Epigenetic IVD Tests for Personalized Precision Medicine in Cancer. Front. Genet. 2019, 10, 621. [Google Scholar] [CrossRef]

| Sirtuin Members/Molecular Pathway | Experimental Animal Model | Modulators | Summary | References and Year |
|---|---|---|---|---|
| Sirt1 (Sirt1 activator) | MCT, rat | Resveratrol | Resveratrol inhibits RV remodeling and dysregulation in MCT-induced PAH. | Vazquez-Garza E et al., Oxid Med Cell Longev. 2020 [151] |
| SphK1/S1P signaling | MCT, rat | Resveratrol | Resveratrol improves pulmonary vascular remodeling and attenuates the PAH development by inhibiting SphK1/S1P-mediated NF-kappaB activation and subsequent cyclin D1 expression. | Shi W et al., Life Sci. 2018 [156] |
| Sirt1 | Human, rat | Resveratrol, SRT1720 | Resveratrol and SRT1720 alleviate RVSP and RVH, significantly reduce PASMC proliferation, and promote PASMC apoptosis via mediating Sirt1. | Yu L et al., Cell Physiol Biochem. 2017 [155] |
| Rat | Resveratrol | Resveratrol is not responsible for attenuation of RV remodeling caused by MCT; however, it inhibits PASMC hypertrophy in the pulmonary vessels. | Wilson DN et al., Pathophysiology. 2016 [157] | |
| Sirt1, eNOS | MCT, rat, hypoxia, CR | SIRT1 induction inhibits induced PAH in experimental animal models by targeting eNOS pathway. | Ding M et al., J Cardiovasc Pharmacol. 2015 [141] | |
| Arginase II, PI3K/Akt signaling pathway | Neonatal rat model of chronic hypoxia-induced pulmonary hypertension | Resveratrol | Resveratrol normalizes RV hypertrophy and pulmonary artery remodeling by inhibiting hypoxia-induced arginase II expression mediated via the PI3K/Akt signaling pathway. | Chen B et al., Am J Physiol Lung Cell Mol Physiol. 2014 [150] |
| Sirt1, atrophic ubiquitin ligases atrogin-1 | MCT, rat | Resveratrol | Resveratrol ameliorates medial thickening of intrapulmonary arteries and phenotypes of pulmonary hypertension induced by MCT, such as RVSP and RVH. | Paffett ML et al., Vascul Pharmacol. 2012 [153] |
| Sirt1 | MCT, rat | Resveratrol | Resveratrol attenuates hypertrophy of right-ventricle heart. | Yang DL et al., Clin Exp Pharmacol Physiol 2010 [158] |
| Sirtuins | Human, bovine, MCT-rat | Resveratrol | Resveratrol treatment ameliorates RV systolic pressure and pulmonary arterial remodeling by exerting anti-inflammation, antioxidant, and antiproliferation effects in the pulmonary arteries. | Csiszar A et al., Hypertension 2009 [152] |
| Sirt1, PGC-1alpha, and its downstream effectors | Human, rat, chronic hypoxia | Stac-3 | Sirt1 inhibition exacerbates remodeling of pulmonary vessels. SIRT1 upregulation inhibits PASMC proliferation. | Zurlo G et al., J Hypertension 2008 [140] |
| Sirt3 | Human, mice | Angiotensin II | Downregulation of Sirt3 inactivates SOD2 causing PH. Hypertension is significantly increased in Sirt3KO mice responding to angiotensin II. | Dikalova AE et al., Circulation research 2017 [159] |
| Sirt3 | Human, rat | Sirt3 is downregulated in PAH, and its induction reverses PAH phenotype. Sirtuin-3 loss-of-function SNP rs11246020 is correlated with clinical IPAH. | Paulin R et al., Cell Metab. 2014 [144] | |
| Sirt4/PDH/GDH | Human, mouse, rat | Bone marrow-derived exosomes | SIRT4 expression is increased in prolonged hypoxia. A decrease in Sirt4 expression is correlated with enhanced expression of PDH and GDH, and mitochondrial dysfunction of PAH was reversed by bone marrow-derived exosomes. | Hogan S E et al., J Physiol Lung Cell Mol Physiol. 2019 [147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, L.; Hossen, N.; Nguyen, T.; Vo, A.; Ahsan, F. Epigenetic Mechanisms as Emerging Therapeutic Targets and Microfluidic Chips Application in Pulmonary Arterial Hypertension. Biomedicines 2022, 10, 170. https://doi.org/10.3390/biomedicines10010170
Ho L, Hossen N, Nguyen T, Vo A, Ahsan F. Epigenetic Mechanisms as Emerging Therapeutic Targets and Microfluidic Chips Application in Pulmonary Arterial Hypertension. Biomedicines. 2022; 10(1):170. https://doi.org/10.3390/biomedicines10010170
Chicago/Turabian StyleHo, Linh, Nazir Hossen, Trieu Nguyen, Au Vo, and Fakhrul Ahsan. 2022. "Epigenetic Mechanisms as Emerging Therapeutic Targets and Microfluidic Chips Application in Pulmonary Arterial Hypertension" Biomedicines 10, no. 1: 170. https://doi.org/10.3390/biomedicines10010170
APA StyleHo, L., Hossen, N., Nguyen, T., Vo, A., & Ahsan, F. (2022). Epigenetic Mechanisms as Emerging Therapeutic Targets and Microfluidic Chips Application in Pulmonary Arterial Hypertension. Biomedicines, 10(1), 170. https://doi.org/10.3390/biomedicines10010170