
Macrophages Modulate Hepatic Injury Involving NLRP3 Inflammasome: The Example of Efavirenz


 ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Drugs
2.2. Cell Culture and Treatments
2.3. In Vivo Procedures
2.4. Protein Extraction and Western Blot (WB) Analysis
2.5. Electrophoretic Mobility Shift Assay (EMSA)
2.6. RNA Isolation and Real Time RT-PCR
2.7. Real Time PCR Assays
2.8. Fluorescence Microscopy and Static Fluorescence
2.9. Confocal Microscopy
2.10. ELISA Assay
2.11. Caspase-1 Activity Assay
2.12. Immunohistochemistry
2.13. MPO Activity
2.14. Data and Statistical Analyses
3. Results
3.1. Effects of EFV on Inflammatory Pathways in Hep3B Cells
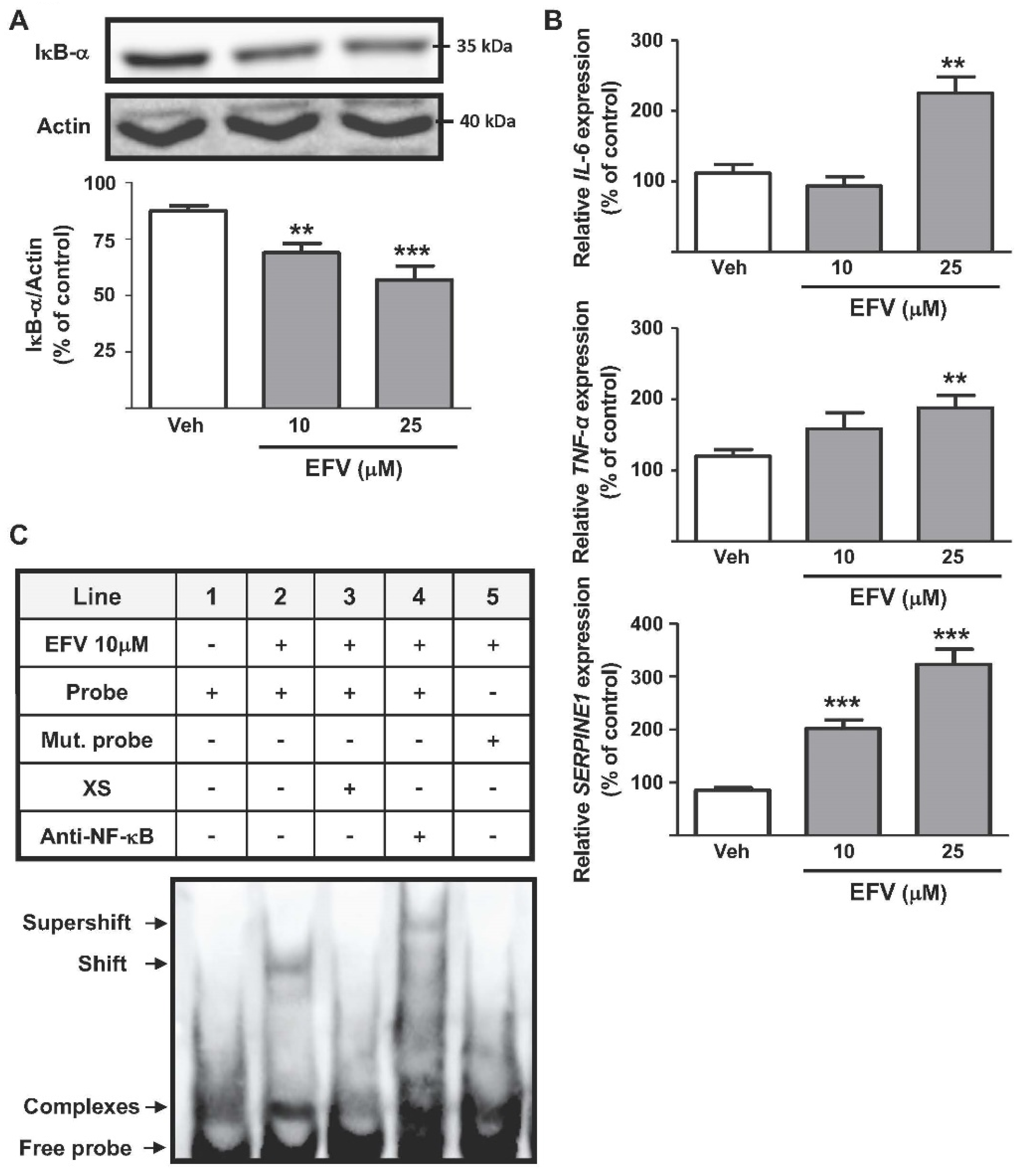
3.1.1. Activation of the NF-κB Signal Pathway
3.1.2. Activation of the NLRP3 Inflammasome
3.2. Effects of EFV on Activation and Inflammatory Pathways in LX2 Cells
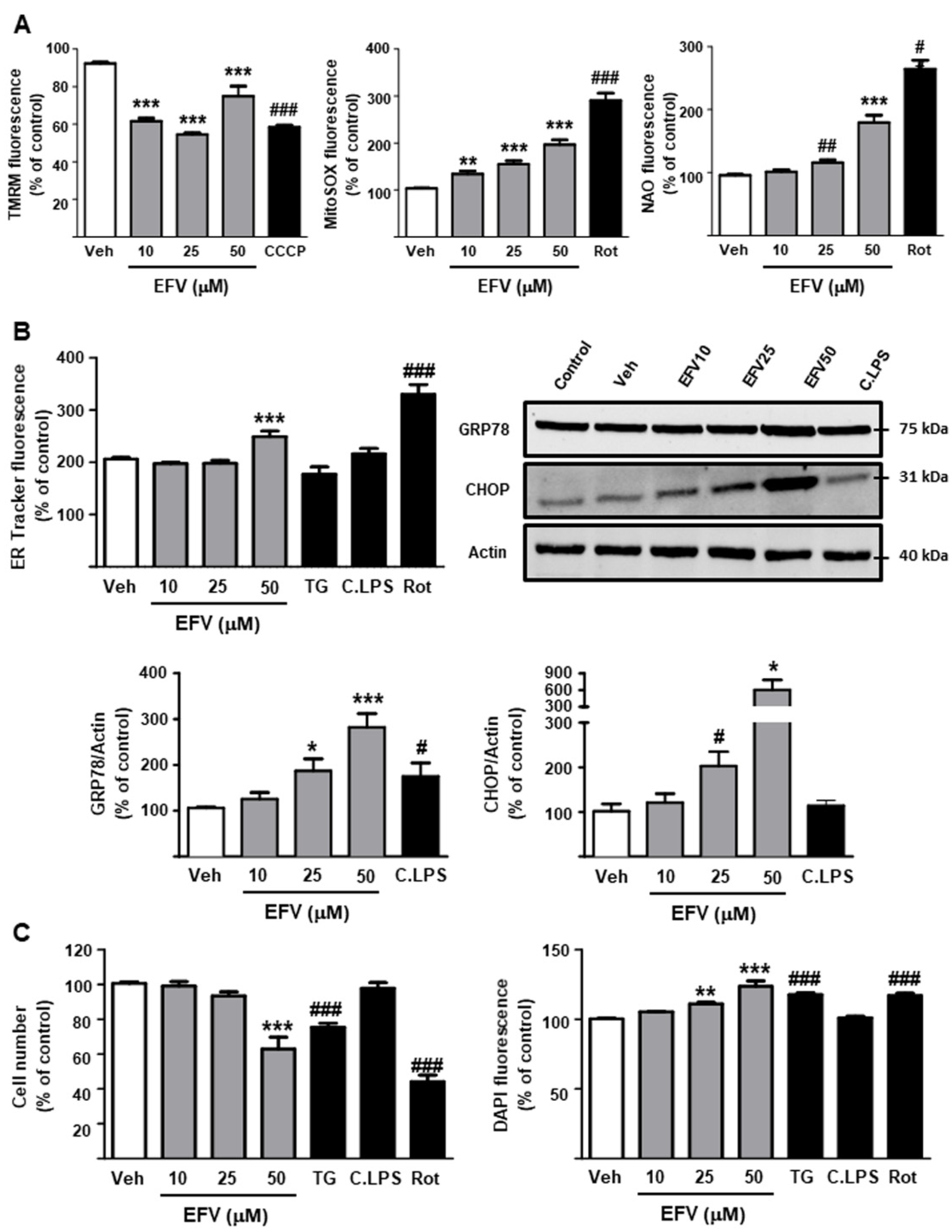
3.2.1. Undermining of Cellular Function: Mitochondria and ER
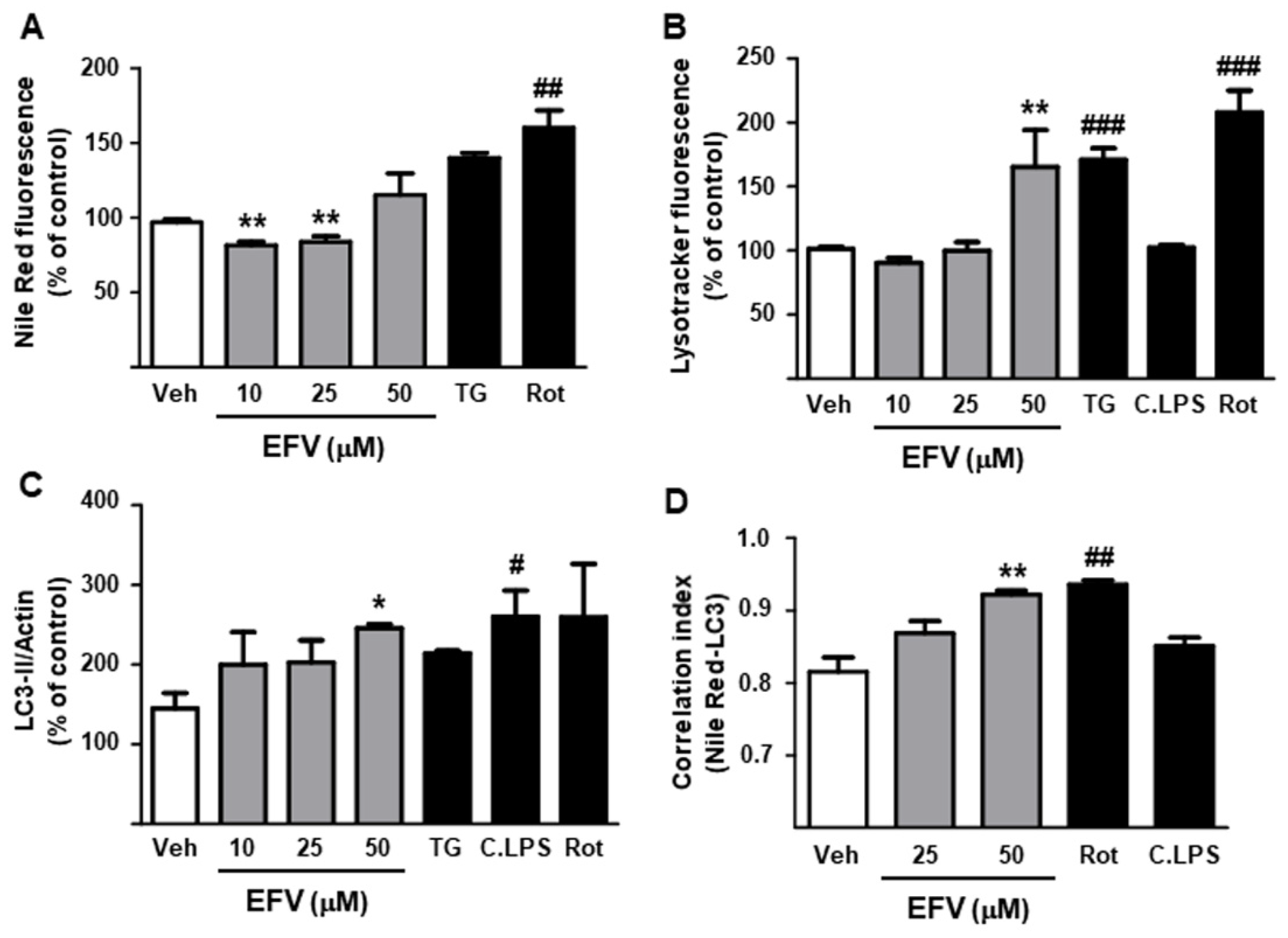
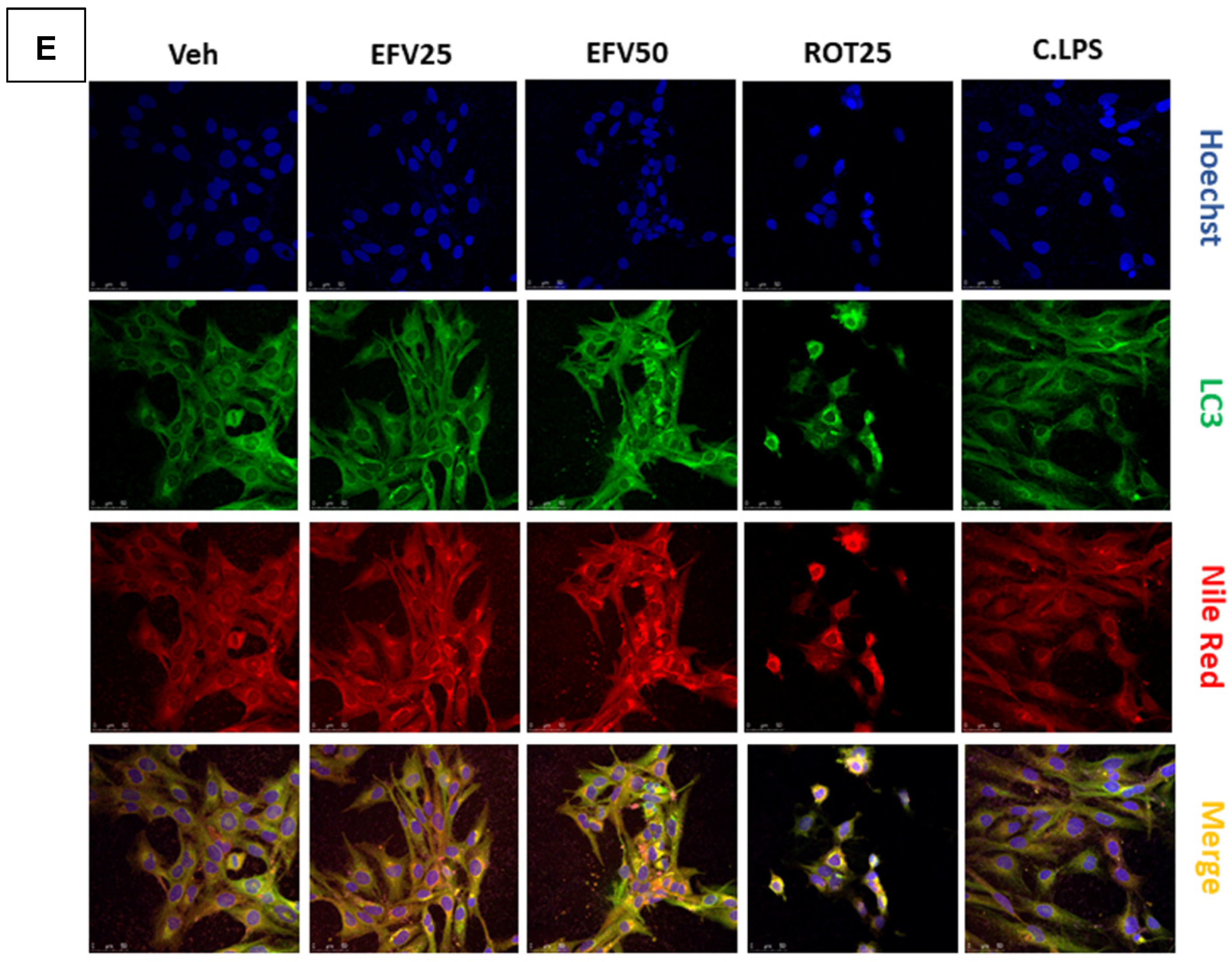
3.2.2. Degradation of Intracellular Lipid Droplets by Autophagy
3.2.3. Modulation of the Inflammatory Response in LX2 Cells
3.2.4. Activation of Fibrogenic Pathways
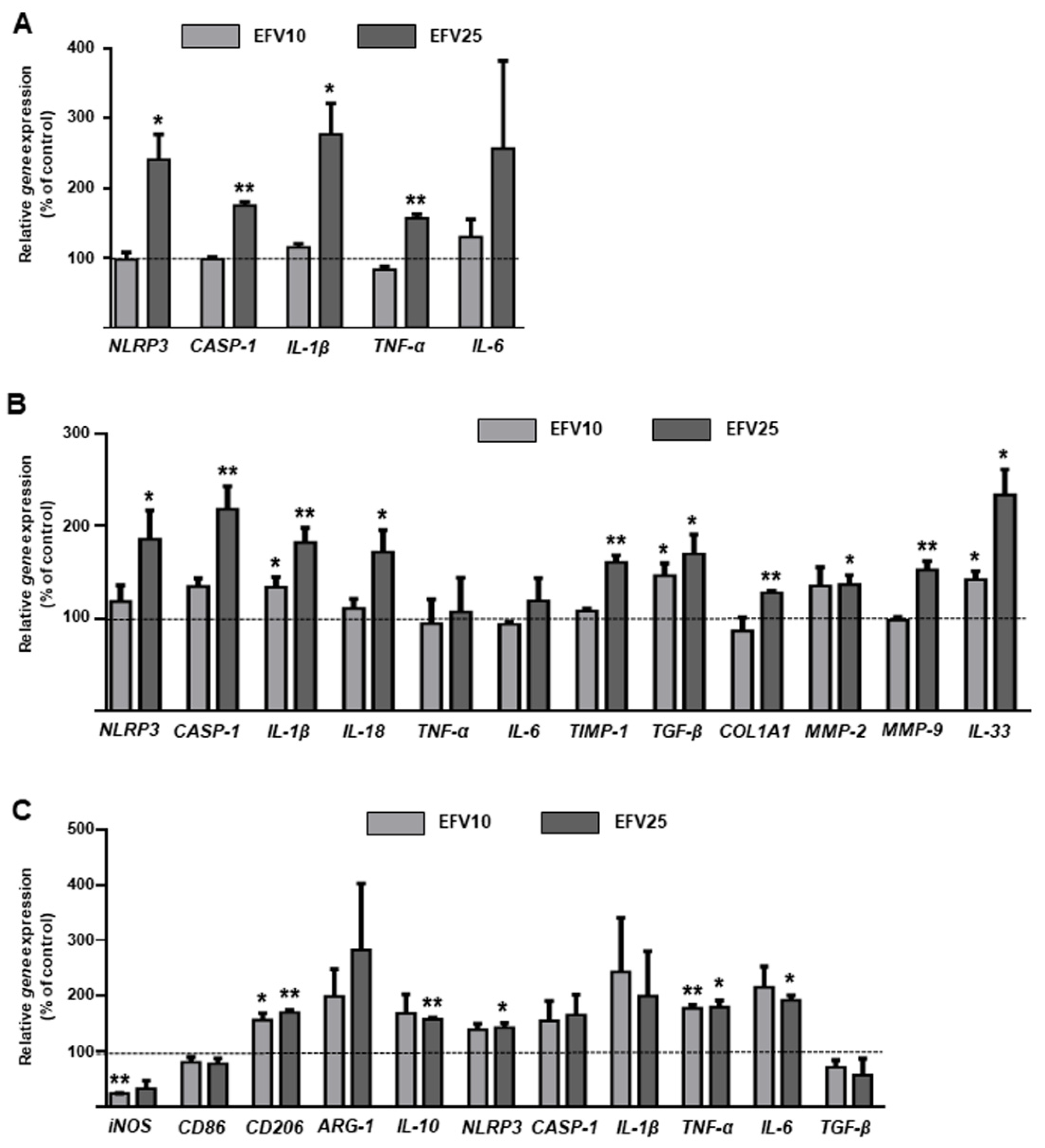
3.3. Effects of EFV on Macrophage Function and Phenotype
3.4. EFV Induces Similar Results in Primary Hepatic Cells
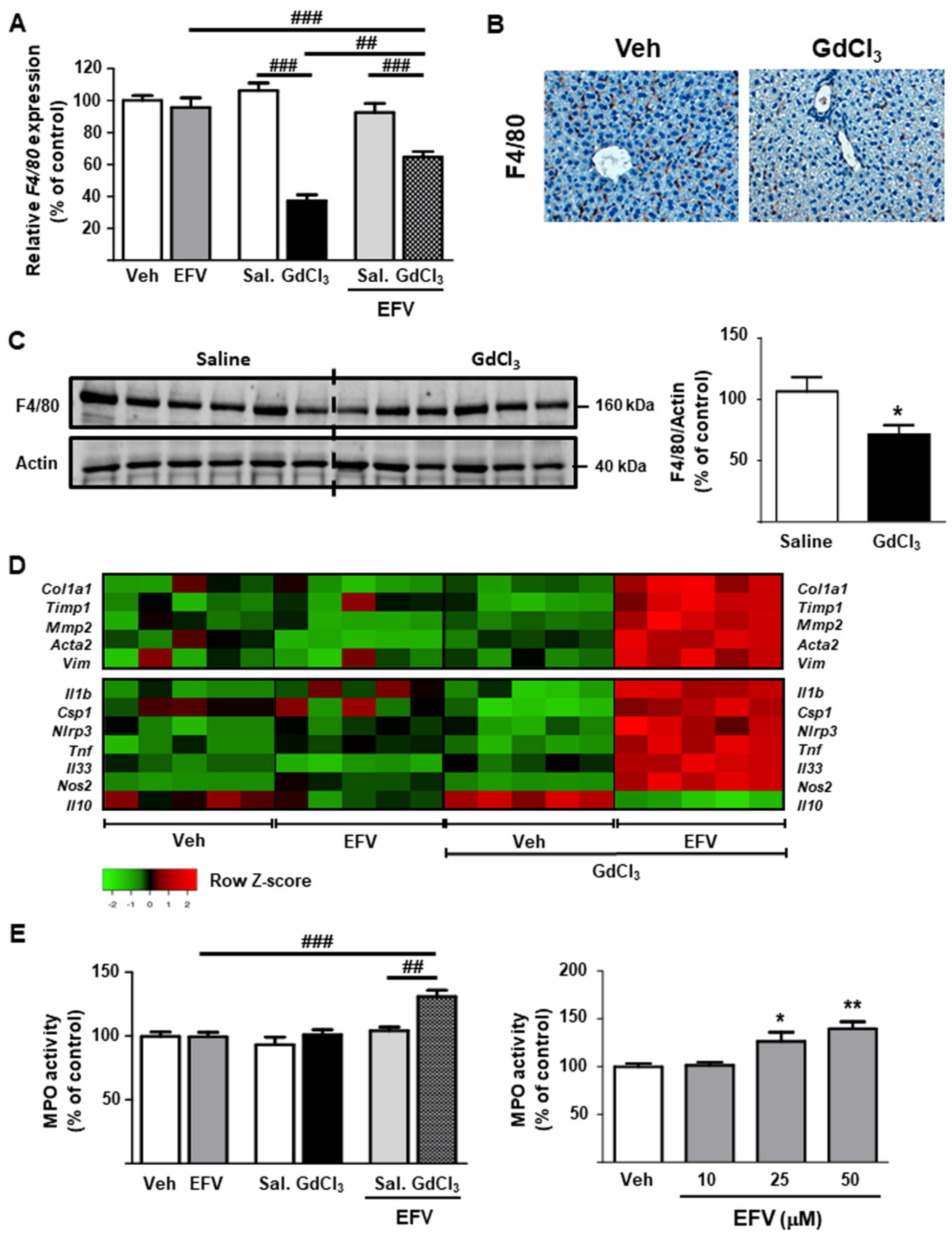
3.5. Depletion of Kupffer Cells Modulates the Hepatic Effects of EFV In Vivo
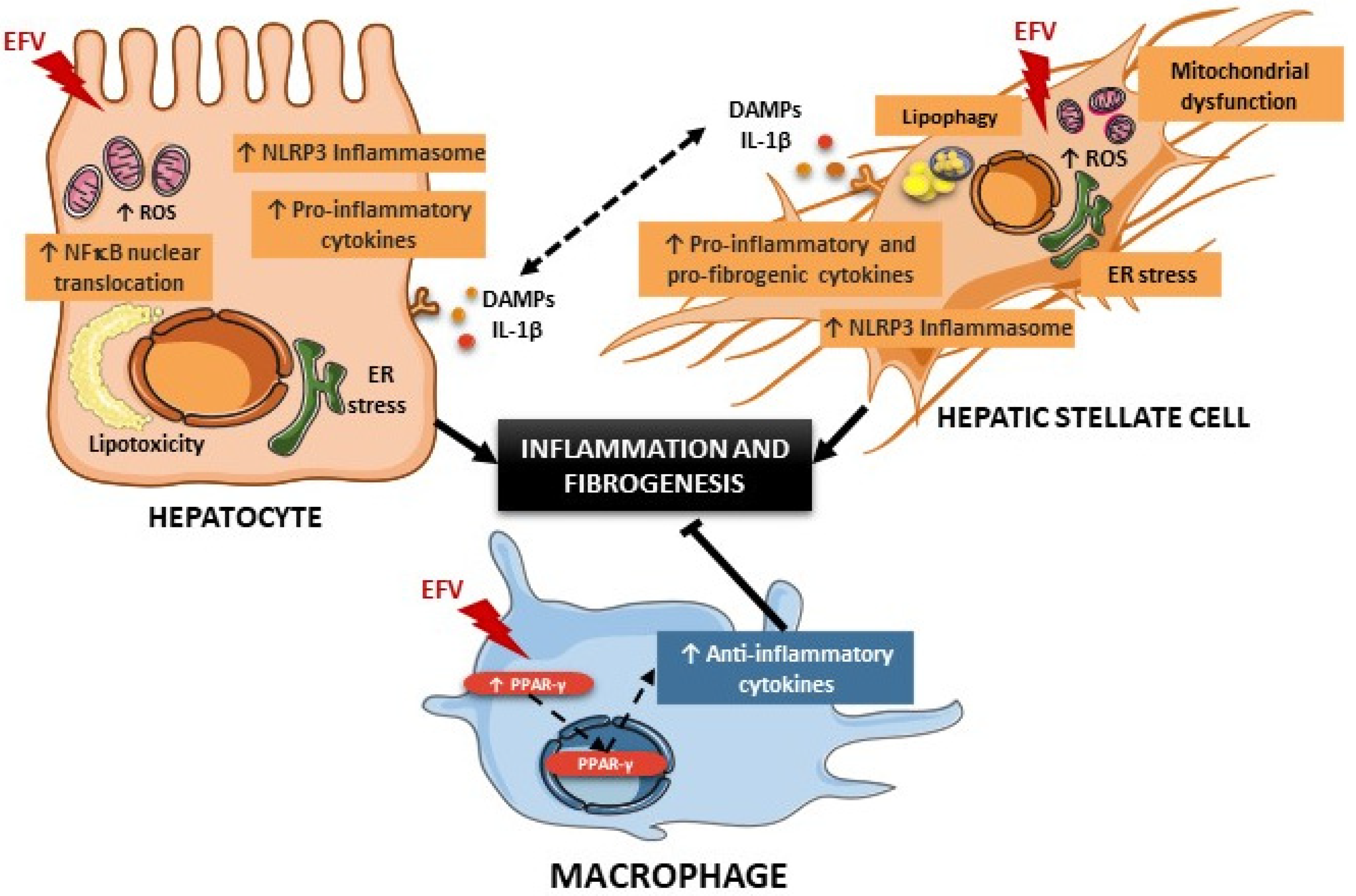
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.-D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2014, 21, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 6, 407–420. [Google Scholar] [CrossRef]
- Netea, M.G.; van der Meer, J.W. Immunodeficiency and genetic defects of pattern-recognition receptors. N. Engl. J. Med. 2011, 364, 60–70. [Google Scholar] [CrossRef]
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 inflammasome in alcoholic and non-alcoholic steatohepatitis. Semin. Liver Dis. 2020, 40, 298–306. [Google Scholar] [CrossRef]
- Gómez-Sucerquia, L.J.; Blas-García, A.; Martí-Cabrera, M.; Esplugues, J.V.; Apostolova, N. Profile of stress and toxicity gene expression in human hepatic cells treated with efavirenz. Antivir. Res. 2012, 94, 232–241. [Google Scholar] [CrossRef]
- Blas-García, A.; Apostolova, N.; Ballesteros, D.; Monleon, D.; Morales, J.M.; Rocha, M.; Victor, V.M.; Esplugues, J.V. Inhibition of mitochondrial function by efavirenz increases lipid content in hepatic cells. Hepatology 2010, 52, 115–125. [Google Scholar] [CrossRef]
- Apostolova, N.; Gómez-Sucerquia, L.J.; Morán, A.; Álvarez, A.; Blas-García, A.; Esplugues, J.V. Enhanced oxidative stress and increased mitochondrial mass during efavirenz-induced apoptosis in human hepatic cells. Br. J. Pharmacol. 2010, 160, 2069–2084. [Google Scholar] [CrossRef] [Green Version]
- Apostolova, N.; Gómez-Sucerquia, L.J.; Gortat, A.; Blas-García, A.; Esplugues, J.V. Compromising mitochondrial function with the antiretroviral efavirenz induces cell survival-promoting autophagy. Hepatology 2011, 54, 1009–1019. [Google Scholar] [CrossRef]
- Apostolova, N.; Gómez-Sucerquia, L.J.; Alegre, F.; Funes, H.A.; Victor, V.M.; Barrachina, M.D.; Blas-Garcia, A.; Esplugues, J.V. ER stress in human hepatic cells treated with efavirenz: Mitochondria again. J. Hepatol. 2013, 59, 780–789. [Google Scholar] [CrossRef]
- Seki, E.; Schwabe, R. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Gea, V.; Hilscher, M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Tuyama, A.C.; Hong, F.; Salman, Y.; Wang, C.; Ozkok, D.; Mosoian, A.; Chen, P.; Chen, B.K.; Klotman, M.E.; Bansal, M.B. Human immunodeficiency virus (HIV-1) infects human hepatic stellate cells and promotes collagen I and monocyte chemoattractant protein-1 expression: Implications for the pathogenesis if HIV/hepatitis C virus-induced liver fibrosis. Hepatology 2010, 52, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Gwag, T.; Meng, Z.; Sui, Y.; Helsley, R.N.; Park, S.H.; Wang, S.; Greenberg, R.N.; Zhou, C. Non-nucleoside reverse transcriptase inhibitor efavirenz activates PXR to induce hypercholesterolemia and hepatic steatosis. J. Hepatol. 2019, 70, 930–940. [Google Scholar] [CrossRef]
- Curran, A.; Rull, A.; Navarro, J.; Vidal-González, J.; Martin-Castillo, M.; Burgos, J.; Falcó, V.; Ribera, E.; Torrella, A.; Planas, B.; et al. Lipidomics reveals reduced inflammatory lipid species and storage lipids after switching from EFV/FTC/TDF to RPV/FTC/TDF: A randomized open-label trial. J. Clin. Med. 2020, 9, 1246. [Google Scholar] [CrossRef]
- Van Lunzen, J.; Antinori, A.; Cohen, C.J.; Arribas, J.R.; Wohl, D.A.; Rieger, A.; Rachlis, A.; Bloch, M.; Segal-Maurer, S.; Garner, W.; et al. Rilpivirine vs.efavirenz-based single tablet regimens in treatment-naive adults: Week 96 efficacy and safety from a randomized phase 3b study. AIDS 2016, 30, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Taramasso, L.; Tatarelli, P.; Ricci, E.; Madeddu, G.; Menzaghi, B.; Squillace, N.; De Socio, G.V.; Martinelli, C.; Gulminetti, R.; Maggi, P.; et al. Improvement of lipid profile after switching from efavirenz or ritonavir-boosted protease inhibitors to rilpivirine or once-daily integrase inhibitors: Results from a large observational cohort study (SCOLTA). BMC Infect. Dis. 2018, 18, 357. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Kim, S.J.; Lee, S.M. Activation of NLRP3 and AIM2 inflammasomes in Kupffer cells in hepatic ischemia/reperfusion. FEBS J. 2015, 282, 259–270. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Blas-García, A.; Apostolova, N.; Valls-Bellés, V.; Esplugues, J.V. Endoplasmic reticulum and mitochondria: Independent roles and crosstalk in fatty liver diseases and hepatic inflammation. Curr. Pharm. Des. 2016, 22, 2607–2618. [Google Scholar] [CrossRef]
- Sookoian, S.; Castaño, G.O.; Burgueño, A.L.; Rosselli, M.S.; Gianotti, T.F.; Mallardi, P.; San Martino, J.; Pirola, C.J. Circulating levels and hepatic expression of molecular mediators of atherosclerosis in nonalcoholic fatty liver disease. Atherosclerosis 2010, 209, 585–591. [Google Scholar] [CrossRef]
- Inzaugarat, M.E.; Johnson, C.D.; Holtmann, T.M.; McGeough, M.D.; Trautwein, C.; Papouchado, B.G.; Schwabe, R.; Hoffman, H.M.; Wree, A.; Feldstein, A.E. NLR family pyrin domain-containing 3 inflammasome activation in hepatic stellate cells induces liver fibrosis in mice. Hepatology 2019, 69, 845–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, G.; Csak, T. Inflammasomes in liver diseases. J. Hepatol. 2012, 57, 642–654. [Google Scholar] [CrossRef] [Green Version]
- Wree, A.; Eguchi, A.; McGeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation and fibrosis. Hepatology 2014, 59, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Liu, Q.; Jiang, R.; Lv, L.; Shoto, S.S.; Maillet, I.; Quesniaux, V.; Tang, J.; Zhang, W.; Sun, B.; et al. Interleukin-33 drives hepatic fibrosis through activation of hepatic stellate cells. Cell. Mol. Immunol. 2018, 15, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Iracheta-Vellve, A.; Petrasek, J.; Satishchandran, A.; Gyongyosi, B.; Saha, B.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J. Hepatol. 2015, 63, 1147–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaki, M.H.; Vogel, P.; Malireddi, R.K.; Body-Malapel, M.; Anand, P.K.; Bertin, J.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 2011, 20, 649–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukens, J.R.; Gurung, P.; Shaw, P.J.; Barr, M.J.; Zaki, M.H.; Brown, S.A.; Vogel, P.; Chi, H.; Kanneganti, T.D. The NLRP12 sensor negatively regulates autoinflammatory disease by modulating interleukin-4 production in T cells. Immunity 2015, 42, 654–664. [Google Scholar] [CrossRef] [Green Version]
- Morán-Salvador, E.; Titos, E.; Rius, B.; González-Périz, A.; García-Alonso, V.; López-Vicario, C.; Miquel, R.; Barak, Y.; Arroyo, V.; Clària, J. Cell-specific PPARγ deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J. Hepatol. 2013, 59, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Cohen, P.L. The PPAR-γ antagonist GW9662 elicits differentiation of M2c-like cells and upregulation of the MerTK/Gas 6 axis: A key role for PPAR-γ in human macrophage polarization. J. Inflamm. 2015, 12, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bumpus, N.N. Efavirenz and 8-hydroxyefavirenz induce cell death via a JNK- and BimEL-dependent mechanism in primary human hepatocytes. Toxicol. Appl. Pharmacol. 2011, 257, 227–234. [Google Scholar] [CrossRef]
- Imaizumi, N.; Kwang Lee, K.; Zhang, C.; Boelsterli, U.A. Mechanisms of cell death pathway activation following drug-induced inhibition of mitochondrial complex I. Redox Biol. 2015, 4, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echenique, I.A.; Rich, J.D. EFV/FTC/TDF-associated hepatotoxicity: A case report and review. AIDS Patient Care STDS 2013, 27, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.; Ona, M.A.; Papafragkakis, H.; Carey, J.; Moshenyat, Y.; Alhaddad, A.; Anand, S. Acute liver toxicity due to efavirenz/emtricitabine/tenofovir. Case Reports Hepatol. 2015, 2015, 280353. [Google Scholar] [CrossRef]
- Elsharkawy, A.M.; Schwab, U.; McCarron, B.; Burt, A.D.; Daly, A.K.; Hudson, M.; Masson, S. Efavirenz induced acute liver failure requiring liver transplantation in a slow drug metaboliser. J. Clin. Virol. 2013, 58, 331–333. [Google Scholar] [CrossRef]
- Sonderup, M.W.; Wainwright, H.; Hall, P.; Hairwadzi, H.; Spearman, C.W. A clinicopathological cohort study of liver pathology in 301 patients with human immunodeficiency virus/acquire immune deficiency syndrome. Hepatology 2015, 61, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Dragovic, G.; Nikolic, K.; Dimitrijevic, B.; Jevtovic, D.; Salemovic, D.; Tomanovic, N.; Boricic, I. Severe hepatotoxicity induced by efavirenz in a treatment naïve, low body mass index HIV-infected, female patient with no hepatitis and other virus co-infections. Ultrastruct. Pathol. 2019, 43, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Macías, J.; Berenguer, J.; Japón, M.A.; Girón-González, J.A.; Rivero, A.; López-Cortés, L.F.; Moreno, A.; Márquez, M.; Iribarren, J.A.; Ortega, E.; et al. Hepatic steatosis and steatohepatitis in human immunodeficiency virus/hepatitis C virus-coinfected patients. Hepatology 2012, 56, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 2007, 13, 851–856. [Google Scholar] [CrossRef]
- Imaeda, A.B.; Watanabe, A.; Sohail, M.A.; Mahmood, S.; Mohamadnejad, M.; Sutterwala, F.S.; Flavell, R.A.; Mehal, W.Z. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J. Clin. Investig. 2009, 119, 305–314. [Google Scholar] [CrossRef]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Wu, G.; Li, S.; Weinberg, E.M.; Kumthip, K.; Peng, L.F.; Méndez-Navarro, J.; Chen, W.C.; Jilg, N.; Zhao, H.; et al. HIV and HCV cooperatively promote hepatic fibrogenesis via induction of reactive oxygen species and NFκB. J. Biol. Chem. 2011, 286, 2665–2674. [Google Scholar] [CrossRef] [Green Version]
- Nasi, M.; Pinti, M.; Mussini, C.; Cossarizza, A. Persistent inflammation in HIV infection: Established concepts, new perspectives. Immunol. Lett. 2014, 161, 184–188. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alegre, F.; Martí-Rodrigo, A.; Polo, M.; Ortiz-Masiá, D.; Bañuls, C.; Pinti, M.; Álvarez, Á.; Apostolova, N.; Esplugues, J.V.; Blas-García, A. Macrophages Modulate Hepatic Injury Involving NLRP3 Inflammasome: The Example of Efavirenz. Biomedicines 2022, 10, 109. https://doi.org/10.3390/biomedicines10010109
Alegre F, Martí-Rodrigo A, Polo M, Ortiz-Masiá D, Bañuls C, Pinti M, Álvarez Á, Apostolova N, Esplugues JV, Blas-García A. Macrophages Modulate Hepatic Injury Involving NLRP3 Inflammasome: The Example of Efavirenz. Biomedicines. 2022; 10(1):109. https://doi.org/10.3390/biomedicines10010109
Chicago/Turabian StyleAlegre, Fernando, Alberto Martí-Rodrigo, Miriam Polo, Dolores Ortiz-Masiá, Celia Bañuls, Marcello Pinti, Ángeles Álvarez, Nadezda Apostolova, Juan V. Esplugues, and Ana Blas-García. 2022. "Macrophages Modulate Hepatic Injury Involving NLRP3 Inflammasome: The Example of Efavirenz" Biomedicines 10, no. 1: 109. https://doi.org/10.3390/biomedicines10010109
APA StyleAlegre, F., Martí-Rodrigo, A., Polo, M., Ortiz-Masiá, D., Bañuls, C., Pinti, M., Álvarez, Á., Apostolova, N., Esplugues, J. V., & Blas-García, A. (2022). Macrophages Modulate Hepatic Injury Involving NLRP3 Inflammasome: The Example of Efavirenz. Biomedicines, 10(1), 109. https://doi.org/10.3390/biomedicines10010109