
Atomistic Descriptions of Gas-Surface Interactions on Tin Dioxide


Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
*
Author to whom correspondence should be addressed.
Chemosensors 2021, 9(9), 270; https://doi.org/10.3390/chemosensors9090270
Submission received: 27 July 2021
/
Revised: 30 August 2021
/
Accepted: 2 September 2021
/
Published: 18 September 2021
(This article belongs to the Special Issue Gas Sensors: Simulation, Modeling, and Characterization)
Abstract
:Historically, in gas sensing literature, the focus on “mechanisms” has been on oxygen species chemisorbed (ionosorbed) from the ambient atmosphere, but what these species actually represent and the location of the adsorption site on the surface of the solid are typically not well described. Recent advances in computational modelling and experimental surface science provide insights on the likely mechanism by which oxygen and other species interact with the surface of SnO2, providing insight into future directions for materials design and optimisation. This article reviews the proposed models of adsorption and reaction of oxygen on SnO2, including a summary of conventional evidence for oxygen ionosorption and recent operando spectroscopy studies of the atomistic interactions on the surface. The analysis is extended to include common target and interfering reducing gases, such as CO and H2, cross-interactions with H2O vapour, and NO2 as an example of an oxidising gas. We emphasise the importance of the surface oxygen vacancies as both the preferred adsorption site of many gases and in the self-doping mechanism of SnO2.
1. Introduction
Conductometric gas sensors (CGS) are small, easily fabricated devices that require relatively low power consumption and contain no moving parts. In their simplest form, they consist only of two electrodes interconnected by a layer of gas-sensitive material and can be printed as films directly onto a substrate [1,2]. These characteristics make CGS perfect for mass-scale production and portable device applications. Large scale deployment of such devices could, for example, screen for diabetes or detect imminent electric transformer failure by detecting gases characteristic to the processes [3,4]. However, despite their many advantages, CGS often suffer from cross-sensitivity to interfering gases and performance deterioration over time [5,6], and the main driving force in CGS research targets improvements in sensors’ sensitivity, selectivity and response time. Insufficient understanding of the mechanisms of surface reactions makes improvements possible only through empirical exploration.
Spectroscopic investigation of sensors under working conditions is often unattainable due to the UHV requirement of many analytical techniques, for example, X-ray photoelectron spectroscopy (XPS), ion scattering spectroscopy (ISS) or low-energy electron diffraction (LEED). In the catalytic community, this incompatibility is known as the “pressure gap”; this is why early sensor operation models were developed by applying prevailing chemistry principles to explain phenomenological observations. The most prevalent model is the oxygen ionosorption mechanism, which explains the sensor’s increased resistance on exposure to oxygen through dissociative adsorption of ambient dioxygen to form ions electrostatically stabilised by the surface [7,8,9,10,11,12,13].
It was conjectured that because of its electron affinity, oxygen would withdraw electrons from the conduction band of an n-type semiconductor, and “consequently, there will be no chemisorbed oxygen atoms, but oxygen ions, in the surface” [14]. In this description, the localisation of negative charge at the surface of SnO2 grains results in a charge depletion zone below the surface, which constitutes a potential barrier for electrons trying to cross the grain boundary, effectively increasing the sensor’s resistance. Usually, the process is only broadly described using the following reaction equation [7,8], which sometimes is extended to include intermediate O2 physisorbed and O2– chemisorbed species [10,15].
½ O2(gas) + e− → O−
O2(gas) → O2(phys) + e− → O2− + e– → 2 O−
The lack of reference to adsorption sites is not the only problem with ionosorption models, however, which also do not consider whether the oxygen dissociation is symmetric, and if asymmetric, the fate of the other oxygen atom. Moreover, the models often assume that there is no mass transport between the bulk of the sensor and the gas phase [7,10,12], which is not a valid assumption to make, as explained further below. Finally, there is also the question of the existence of O– ions adsorbed at the surface, for which there is no conclusive evidence after decades of research [16].
In the absence of adequate analytical techniques, early atomistic insight into the surface processes of gas sensing was provided by computational studies. The chemical state of the surface can be analysed either kinetically, modelling the temporal evolution of a system defined by a set of chemical reactions by optimising an array of parameters, or thermodynamically, estimating the most stable adsorption configurations from first-principles calculations on a model surface, such as a cluster or a slab. The former approach may be used to, for example, estimate the equilibrium density of oxygen adsorbates on O2 or predict sensor behaviour in varying oxygen pressure [7,15]. The latter is essential in discovering possible adsorbate species and investigating their effects on the surface [17,18]. However, analysis of sensors working under conditions close to ambient has become increasingly more available with the advent of operando spectroscopy. It offers microscopic insight into processes formerly studied only post-mortem in UHV after exposure to a target gas in a preparation chamber, thus presenting new possibilities. For example, changes in the electronic structure of a working sensor may be studied under dynamic pressure and temperature conditions through Near Ambient Pressure (NAP) XPS with simultaneous resistance measurements [19].
In this work, the authors attempt to summarise the proposed mechanisms for the interactions of SnO2 with oxygen and reducing and oxidising gases as understood from phenomenological and computational studies and highlight those supported by spectroscopic evidence. Since providing an atomistic description of such interactions is the aim of this review, a sensible place to start is by considering the atoms of the surface itself, which is the subject of the next section.
2. The Dynamic Surfaces of Tin Dioxide
Tin dioxide, SnO2, is among the most common sensitive materials used in CGS. It is abundant, inexpensive, non-toxic and shows excellent stability to reducing conditions. Since Taguchi introduced the first commercial SnO2-based gas sensors in 1972, and this oxide has been the most widely studied gas-sensitive material, with an initial focus on detecting reducing gases such as CO and volatile organic compounds (VOC). However, recently the focus is shifting towards low-temperature NO2 detection.
The crystalline form of SnO2, known as cassiterite, is a rutile-type tetragonal structure with lattice parameters a = b = 4.737 Å and c = 3.186 Å [20]. It is an intrinsic n-type semiconductor with a wide, direct bandgap of 3.6 eV [21], which also makes it transparent and of high interest in touchscreen electronics. Although the exact origin of the intrinsic conductivity is still debated, oxygen vacancies (VO) seem to play an important role [22,23,24,25]; therefore, a change in the density of VO will affect the sensor’s resistance.
The surface is the interface of oxygen exchange between the bulk and the ambient and a terminal for interaction with other gases and thus requires due consideration. Large SnO2 single crystals, which are notoriously challenging to grow, preferentially expose the (110), (101) and (100) surfaces [26]. Of these three, the (110) was shown to have the lowest energy and is the subject of most computational adsorption studies [17,18,27,28,29,30,31], though some results are also available for the (101) surface [32,33]. Both surfaces feature bridging oxygen sites (Obr), which protrude outwards from the surface determined by the plane of Sn atoms. The (110) surface has another type of exposed oxygen atom, the in-plane oxygen (Opl), which does not have a counterpart on the (101) surface, though the second layer (101) oxygen atoms (first subsurface oxygen) are sometimes referred to in that way.
Obr atoms can be removed to form vacancies in the process of surface reduction, facilitated by the dual valency of Sn2+/4+. The removal results in either a partially or fully reduced surface, the latter indicating one with all Obr removed, as presented in Figure 1. The surface vacancies can migrate via in-plane sites into the bulk in a thermally activated process, which becomes noticeable at temperatures above 250 °C [24]. Moreover, the formation energies of these vacancies mutually depend on the defects’ densities, as indicated by computational studies [34]. It means that while the initial reduction of the stoichiometric surface will proceed by mainly removing Obr, at a relative Vbr density of 0.5, the formation energy difference between bridging and in-plane defects is much smaller, so both should coexist at the surface. Therefore, the equilibrium density of surface vacancies should depend not only on the chemical potential of gaseous oxygen but also on the density of subsurface VO in bulk SnO2, assuming the temperature is high enough for the atoms to reach their equilibrium position. The role of vacancies in sensor operation mechanisms is two-fold; as a self-doping mechanism, which provides intrinsic conductivity in SnO2, and as the preferred adsorption site for oxygen, which does not adsorb onto stoichiometric surfaces of SnO2, as indicated by many computational studies [17,28,32] and explained in the following section.
3. Interactions with Oxygen
3.1. Insight from Computational Studies
The early computational work by Yamaguchi et al. on the surfaces of SnO2 explored the adsorption energies of oxygen adsorbates placed on a (110) surface bridging vacancy using a point charge model [36]. They found that adsorbate interaction with a vacancy yields negative adsorption energies for a range of both mono- and diatomic species. According to their results, a molecule of oxygen can adsorb both in side-on and end-on configurations and can be singly or doubly charged, i.e., O2− and O22−. The study also shows that an O− adsorbate is stabilised by the surface vacancy with an energy similar to that of typical bridging lattice oxygen, O2− (16.22 vs. 16.97 eV per 2O−). However, the authors acknowledge that the investigated configurations of oxygen adsorbates were guessed due to the lack of information on adsorption geometry.
In a follow-up letter, Yamaguchi considers the dissociation of oxygen on a fully reduced surface and concludes that the dissociation of O22− leads to the formation of O− monoatomic species, which are coupled with a Vbr and immobilised by a rather sizeable potential barrier, and that this adsorption site is more favourable than a neighbouring Sn5c site [27]. However, a more recent computational study suggests that the range of possible adsorbates is less diverse. Investigation of a similarly reduced surface led to the conclusion that the dissociation of O22− results in the healing of two vacancies, i.e., the formation of O2− ions indistinguishable from Obr [17]. The same study considers the adsorption of O2 onto an isolated Vbr, in which case they find that the dissociation of O2 leads to the healing of the vacancy by one of the atoms, while the other becomes an O− ion adsorbed onto a neighbouring Sn5c site, as there are no other nearby vacancies to interact within the model. Therefore, it seems that O− should only exist under particular circumstances as a remnant of the final steps of surface oxidation after nearly all vacancies have been healed. For both the reduced surface and an isolated vacancy, the dissociation has a relatively large activation energy (1.32 and 0.54 eV, respectively) [17], which is at variance with the results of Yamaguchi, who reports an activation barrier of only 0.09 eV [27]. Such a small barrier is unlikely as, experimentally, oxidation of the surface was determined to be a thermally activated process [37].
Prior to the dissociation, the molecular oxygen adsorbates may exist in a physisorbed state, where there is no charge transfer between the surface and the molecule, and in a chemisorbed state, where the molecule withdraws electron density from the surface. Currently, there is no agreement on whether the charge of the adsorbate is related to the adsorption geometry. While one DFT study finds that end-on and side-on configurations (Figure 2) correspond to superoxide and peroxide species, respectively [17], a different study only reports a side-on configuration for diatomic oxygen, which they assign to a superoxide [28]. In both studies, the adsorbate is bound to the Vbr on one side and Sn5c on the other, which seems to be the most favourable adsorption geometry; see O-lie in Figure 2.
The above descriptions were of the (110) surface, for which most computational studies are performed, and there are limited data on the adsorption of O2 onto other surfaces. An investigation of the (101) surface indicates that O2 interaction mechanisms are similar to those of the (110) surface [32]. Only weak interactions are found for a stoichiometric surface, and the vacancy is again an essential active site. Upon adsorption onto an isolated vacancy on a partially reduced surface, the molecule transforms into a peroxide-like species with an adsorption energy of 1.02 eV. Unlike for the (110) surface, this adsorption configuration should not lead to dissociation into O−, as the peroxide bond cannot break [32]. However, the adsorption of O2 onto two neighbouring vacancies leads to the adsorbate dissociation and healing of both defects, similar to the (110) surface.
Although the localisation of electrons in the antibonding π* orbitals of the peroxide adsorbates results in the formation of negatively charged species, this process should not lead to the accumulation of negative charge at the surface or a subsurface depletion layer. It was found that upon adsorption onto a vacancy, the oxidation state of a neighbouring Sn is changed from +2 to +4 [32]; therefore, the charge of the Vbr should be −2 rather than 0 for the surface to remain overall charge-neutral. Because of this, the peroxide species should not contribute to a dipole layer formation, rather than to the localisation of electrons on the vacancy site.
Recently, a different computational description of the adsorption of O2 onto SnO2 was proposed, where instead of creating a vacancy, the surface was enriched with extra electrons to enable adsorption. The study shows that an electron added to the system can stabilise it by lowering the energy of one antibonding π* orbital of O2− to just above the valence band maximum [38]. In such a configuration, the molecule is bound to two neighbouring Sn5c sites, and the negative charge of the adsorbate is not compensated by its location, as it lies outside of regular lattice anion sites. Therefore, this process should lead to the accumulation of charge at the surface. However, it is not entirely clear where the electrons come from in a system without oxygen vacancies or dopants, which could significantly alter the results by affecting the electronic structure of the surface [39]. The study also shows that introducing a second electron does not significantly affect the geometry of the adsorbate or the energy of the second π* orbital. Instead, the second and subsequent electrons occupy the conduction band; therefore, the authors concluded that O22– should not form on the (110), (100) and (101) surfaces of SnO2 [38]. This conclusion is at variance with the experimental evidence of peroxide O22– adsorbate [40], discussed in the following section.
The same study revisits the possibility of monoatomic oxygen adsorbates, pointed out by the authors as the species responsible for the observed resistance change [38]. Several adsorption configurations were found on the surface, which may be divided into two categories, “peroxide-like” and “lattice-like” species. The former is characterised by a peroxide bond to an Obr in addition to a bond to Sn5c; this adsorption configuration is equivalent to the adsorption of O22− onto a Vbr, and the authors conclude that it is charge neutral with respect to the lattice and should not affect the resistance of the sensor. The latter is bound to either one or two different Sn5c sites in a geometry not far removed from the local coordination of lattice oxygen atoms; in this case, stable adsorption configurations were found only on the (100) surface, which features a considerable distortion of the adsorption site. On the (110) and (101) surfaces, all monoatomic adsorbates were found unstable to transformation into O2 and O2−. Based on these results, the authors concluded that the resistance of SnO2 is defined by interactions of O2 with only the (100) surface. However, gas-sensitive phenomena were also observed on other surfaces, for example, an epitaxial (101) thin film [41], which shows that these results might be incomplete.
3.2. What Species Were Observed Spectroscopically?
Because of the aforementioned “pressure gap”, the choice of analytical techniques suitable for studying gas sensors is limited, so the evidence is scarce. The most widely accepted evidence of oxygen ionosorption on SnO2 comes from an ESR investigation published by Chang in 1980 [42]. The paper describes a reversible transformation of O2− into O− witnessed at temperatures between 100 and 150 °C, shown in Figure 3. However, it was already pointed out that some of these assignments are inconsistent with both the theory and other ESR studies [16]; while a singlet was assigned to O– in this study, theory shows that the signal should be a triplet [43], which is what was found for many other compounds, including closely related TiO2 [44,45]. The analysis of ESR spectra is quite complicated, and there is no consensus on the assignment of some peaks; thus, there is no conclusive evidence of paramagnetic monoatomic oxygen species on SnO2.
On the other hand, multiple ESR studies show triplet signals consistent with O2− adsorbates [46,47,48,49], which was confirmed by the appearance of the hyperfine structure in 17O isotope exchange experiments [49]. This signal appears only on reduced surfaces, which have been heat-treated and cooled in UHV and exposed to O2 below 150 °C, and its intensity is increased on Pt-doped surfaces [46]. These findings are in agreement with computational models of oxygen adsorption onto SnO2, which only show adsorption on reduced surfaces [17,28,32]. However, ESR cannot provide evidence for the other computationally predicted species, the peroxide O22−, which is not paramagnetic.
The existence of such low-temperature molecular adsorbates is also confirmed by TPD studies, which show that oxygen desorption from SnO2 may be classified into three peaks, α, β and γ, as presented in Figure 4. The appearance of the α peak is correlated with low-temperature adsorption onto a surface preheated in vacuum at 700 and 550 °C (Figure 4a,b, respectively) for several hours and cooled down to below 150 °C before O2 exposure. However, the peak does not appear if the sample was exposed to O2 at temperatures above 300 °C; instead, two different desorption events occur, the β and γ peaks, which were assigned to O− and lattice oxygen, respectively. However, the assignments are made based solely on the observed desorption temperatures. In one of the studies, the identity of the α and β adsorbates was investigated by ESR [50]; while α was confirmed to be O2−, no paramagnetic signal was detected for β. Therefore, the assignment of β as O− is again doubtful, and other plausible explanations for the origin of β peak ought to be considered. It should also be pointed out that the onset and intensity of the β desorption peak depend not only on the pressure but also on the temperature at which oxygen dosing was performed [50,51,52]. Considering this and the fact that the peak coincides with the temperature range at which SnO2 is known to reduce spontaneously [53], the β peak could be explained by the desorption of Obr (surface reduction). Such assignment would also explain why the β peak does not appear after low-temperature dosing, where the chemisorbed oxygen species have insufficient energy to dissociate and heal the bridging vacancies. However, more data are necessary to make this determination conclusively.
The more recent evidence of adsorbed species comes from an operando DRIFTS study of O2 and H2 on a UHV treated sample, which is expected to have an appreciable initial density of vacancies. The spectra collected in several-minute intervals display emerging absorption bands attributable to superoxide and peroxide species [40]. The bands appear over the first few minutes and stabilise by about 15 min after gas introduction. If the atmosphere is then changed to H2, the bands are removed rapidly over the first 5 min and disappear completely after just 10 min. Reintroduction of oxygen leads to the reappearance of the bands, showing the reversible formation of these adsorbates on reduced surfaces of SnO2.
The development of near ambient pressure photoelectron analysers opened up the possibility of operando XPS analysis. Conveniently, the thermocouple lead of most NAP XPS systems can be used to measure the resistance of the sample in situ, therefore allowing analysis of a real, working sensor. The recent report of such an investigation does not show changes in the shape of O 1 s emission, which would have confirmed the presence of adsorbed species [19]. However, the spectra were collected at temperatures above 200 °C, so diatomic adsorbates (α peak in TPD) were unlikely to persist at the surface. On the other hand, a rigid shift in the binding energy (BE) scale was observed for all core levels, suggesting a change in the Fermi level of the sample, which is evidence of band bending. The peaks shift to lower BE in the O2 atmosphere relative to UHV, which the authors interpret as evidence of upward band bending caused by chemisorbed oxygen. However, another possible interpretation of the observed Fermi level change stems from a different choice of reference. The formation of vacancies in SnO2 introduces shallow electron donors, which may become thermally ionised and cause downward band bending. This would manifest itself as a shift of peaks to higher BE in photoelectron spectroscopy, which was observed in both UPS and XPS on reduced and oxidised SnO2 samples [53]. These authors conclude that the flat band condition corresponds to a perfect, stoichiometric surface on which no adsorbates can exist, and the bands bend downward from that, moving peaks in both UPS and XPS spectra to higher binding energies.
4. Interactions with Reducing Gases
4.1. Ionosorption Based Models—Reactions with Preadsorbed Oxygen
Since the foundations of oxygen ionosorption were established, the detection of reducing gases was understood in terms of the reaction with surface oxygen ions and the release of electrons trapped at the surface into the conduction band. The papers on the detection of common reducing gases, such as CO, H2 or VOC, describe the surface reactions in a manner presented in Table 1.
Similar to the core theory behind them, these mechanisms do not refer to the adsorption configurations of the species involved. As discussed earlier, computational studies suggest that O− cannot be implicitly assumed to exist, and empirical studies systematically fail to show O− on SnO2, suggesting that these reactions may be an oversimplification that defines a particular stoichiometry for the surface processes rather than defining the involvement of specific adsorbed species.
In the previous chapter, the adsorption configuration of an oxygen molecule onto a Vbr was discussed. Computational studies show that various combustible gases, such as CO and NO, can adsorb on top of an O2 molecule and subsequently react with it [17,28,30,32]. On both the (110) and (101) surfaces, one half of the dioxygen heals a Vbr, and the other reacts with the combustible molecule, as shown in Figure 5 and Figure 6.
By the example of CO, the overall process occurring at a vacancy can therefore be described as:
VO…O2− + CO → OO + CO2 + e−
Similar to the O− based detection in Table 1, this process involves releasing one electron per molecule detected; however, it is described by a species known to exist on SnO2, and the calculated transition state energies are reasonably low (0.80 eV on the (110) surface and 0.43 eV on the (101) surface) [17,32]. The products of the reactions are initially adsorbed but may either desorb or further react with the surface. The latter possibility is explored below.
While the reaction of CO and NO with preadsorbed oxygen species leads to the healing of a surface oxygen vacancy, the coadsorption of O2 and H2 is predicted to look quite different. In this case, one hydrogen atom binds to each of the adsorbed dioxygen atoms to form two distinct hydroxyl groups [63]. Hydrogenation of the oxygen atom bound to the VO results in a rooted hydroxyl ((OH)O), which occupies the bridging lattice site, while the other oxygen atom becomes a terminal hydroxyl bound to a neighbouring Sn5c site (Sn5c…OH). The two hydroxyl groups have dissimilar properties and hence affect the electronic structure of SnO2 differently [59]. The Sn5c…OH hydroxyl forms a dipole, and it does not contribute to the sensor’s resistance other than by removing the oxygen adsorbate during hydroxyl formation. On the other hand, the (OH)O is an electron donor (relative to the equivalent Obr it replaces), which can donate charge carriers to the conduction band and become positively charged; the formation of such hydroxyl surface donors is expected to have a noticeable effect on the sensor’s resistance [58,59,64].
Although these mechanisms show how reducing gases may be detected on the surfaces of SnO2, they do not provide the complete picture. If a sensor’s resistance were regulated solely by the formation and consumption of oxygen adsorbates, then the lowest attainable resistance would be in the absence of oxygen. However, it was shown experimentally that the resistance of a sensor might fall below the no-oxygen baseline during CO detection in humid synthetic air [64]. Therefore, mechanisms extending beyond the interaction with preadsorbed oxygen need to be considered.
4.2. Direct Adsorption of Reducing Gases
Humidity is virtually always present in the atmosphere in varying amounts, which is a potential problem for the reliability of gas sensors, as the adsorption of water could cause the resistance of a sensor to change. Computational studies show that the interaction between the surface and H2O is complex; many adsorption configurations are possible, including both molecular and dissociative modes, as shown in Figure 7.
On a stoichiometric surface, H2O can adsorb onto an Sn5c site [29]. The adsorption energy is relatively small compared to other possible configurations but can be further lowered by the dissociation of the H2O molecule, where one of the H atoms coordinates to a neighbouring Obr. The remaining OH coordinated to the Sn5c becomes a terminal hydroxyl (Sn5c…OH), and the reaction of Obr with H atom transforms it into a rooted hydroxyl ((OH)O); an example of such an interaction is shown in Figure 7b. In fact, the result of dissociative adsorption of H2O on a stoichiometric surface is quite similar to the reaction of H2 with preadsorbed O2, as discussed above.
The formation of an oxygen vacancy on the surface strongly influences the adsorption of H2O; the adsorption energy of H2O onto an Sn5c is more negative on a surface with a Vbr in the vicinity, both in the molecular and dissociative configurations [29]. However, the vacancy is also a new adsorption site for H2O, leading to more stable species. Although molecular adsorption onto Vbr is the least stable of all configurations, the dissociative mode on Vbr, as shown in Figure 7f, is the opposite, being the most favourable adsorption configuration of all. This configuration is also unique in that it produces two (OH)O instead of one of each type of hydroxyl. Therefore, it should affect the resistance more strongly as two electron donors are formed per molecule detected instead of one [29,65].
Although dissociative adsorption is more stable than its molecular counterpart on both adsorption sites, H2O cannot always spontaneously dissociate upon adsorption. The process was shown to be thermally activated in an operando DRIFTS study, where the appearance of bands corresponding to OH was observed only after dosing H2O at temperatures above 200 °C [66].
The preferred adsorption mode of H2O, therefore, depends strongly on the surface of SnO2, which determines the range of possible configurations based on the availability of adsorption sites, for example neighbouring Vbr and Obr, to allow dissociative adsorption. This may not necessarily be a common occurrence, as the bridging vacancies tend to group together, for example, in the proposed structure of the 2 × 1 reconstruction of the (110) surface, where every other row of bridging oxygen atoms is removed. This suggests that depending on the preparation, the surfaces should behave differently in the presence of humidity, which was shown experimentally in an H2O/D2O exchange study using DRIFTS [67]. The study also confirms that SnO2 surface hydroxylation is a reversible process in a dynamic equilibrium with the gas phase. Additional TPD data show that H2O desorbs from the surface over a wide range of temperatures, suggesting multiple adsorption configurations with different adsorption energies [51,52]. The TPD chromatograms (Figure 8) show two broad peaks, one around 100 °C, which corresponds to molecularly adsorbed water, and another above 400 °C, which results from various dissociatively adsorbed species. Since the adsorption energy decreases with increasing OH coverage, it takes progressively more energy to desorb H2O from SnO2, which is why temperatures of at least 600 °C are required for complete dehydroxylation [66].
An operando DRIFTS study with simultaneous resistance measurements reveals that H2O does not always induce a reducing response. [67]. Two differently prepared samples of SnO2 were exposed to increasingly humid synthetic air. While one of the samples shows a strong response, the effect of humidity on the other sample is negligible. However, spectra reveal that the former is accompanied by relatively small changes in the OH bands, while intense OH formation is observed for the latter. Therefore, experimental data suggest that there must be two adsorption mechanisms, one which causes a reducing type response, likely linked to the formation of (OH)O electron donors, and one which is relatively electroneutral, which could be dominated by the formation of Sn5c…OH [59].
Although ionosorption is the prevalent sensor operation mechanism discussed in the literature, it has been known for a long time that CO elicits a response on SnO2-based sensors in the absence of oxygen [56]. Since no CO2 was observed during CO adsorption in the absence of O2, the change in resistance was attributed to the direct adsorption of CO onto SnO2, which produces a surface donor state that can become ionised by donating an electron into the conduction band, increasing the sensor’s resistance. The authors support their argument with the study of Heiland et al., who studied the interactions of SnO2 with acetic acid by TPD [68]. According to the TPD results, the reaction products desorb from the surface primarily as CO instead of CO2, which Hahn interprets as a lack of reaction between CO and the surface. However, these results are at odds with the findings of Yamazoe et al., who recorded TPD chromatograms after dosing CO, rather than acetone, onto SnO2 powder. They found only CO2 desorbing from the surface and concluded that CO irreversibly adsorbs onto SnO2 and desorbs as CO2 upon heating, suggesting the reaction follows a Mars-van Krevelen mechanism [69]. Therefore, it is likely that multiple adsorption pathways for CO are possible.
It is well known that humidity has a substantial impact on the detection of CO in SnO2-based gas sensors. One of the possible contributory processes is the transformation of Obr into (OH)O when a CO molecule reacts with a terminal OH, which was found on a (101) surface [32]. The initial and final adsorbate configurations and the transition state are shown in Figure 9. Although the process forms a donor state, which will influence the sensor’s resistance, it permanently removes important adsorption sites for CO, as explained in the next section.
Computational investigation of NO adsorption onto SnO2 reveals that various geometries are possible, but the most stable ones are linked to nitrogen (N−)down configurations [30]. Contrary to H2O adsorption, the bonding interaction between NO and Sn5c is more favourable on a stoichiometric surface than on a reduced one. However, when present at the surface, the Vbr is again the preferred adsorption site. Although a different study reports no O-down geometries [18], both studies agree that the most stable adsorption configuration is N-down onto a Vbr with an adsorption energy of about 1.2 eV. Analysis of the charge density transfer reveals that, in terms of the influence on the electronic structure, NO can act not only like a reducing gas but also like an oxidising one [18]. While adsorption onto Obr results in a donor-like NO+ state, the same molecule adsorbing onto a Vbr results in electron density being withdrawn from the surface and the formation of an NO–. Effectively, NO adsorption will produce a reducing gas response on a stoichiometric surface but an oxidising gas response on a fully reduced surface. For intermediate surfaces, a mixture of the adsorbates is expected at the surface; such behaviour complicates NO detection.
The detection mechanisms of many gases involve their transformation into reaction products such as H2O (from H2), NO2 (from NO) and CO2 (from CO). Of these, the formation of H2O is usually lost in the background humidity, but the ambient concentrations of CO2 and NO2 are respectively small and negligible, which means that their interference should be taken into consideration. NO2 is considered an oxidising gas, which strongly increases the resistance of the sensor. Its adsorption is discussed in the next section.
On the other hand, CO2 seems to have little effect on the sensor’s resistance compared to other gases; considerable concentration changes produce a relatively small response. A computational study found that CO2 does not adsorb onto a stoichiometric (110) surface of SnO2, as only very weak interactions were found [70]. Moreover, on a reduced (110) surface with preadsorbed VO…O2, the CO2 does not interact with O2, as the two molecules are localised far apart from each other in their equilibrium position. These two conclusions imply that CO2 should not substantially affect the sensor’s resistance, as they eliminate two main detection mechanisms. However, a different study suggests the possibility of CO2 adsorption on a stoichiometric (101) SnO2 surface, with the molecule coordinating to an Obr and two Sn5c sites in a tridentate carbonate form, as shown in Figure 10 [32]. A charge transfer occurs from the surface, with each forming carbonate gaining 0.120 e−, determined from population analysis. Moreover, it was shown that the (110) surface could also be activated towards CO2 detection; an investigation of CO2 adsorption onto a hydroxylated surface shows a possible reaction leading to the formation of a bidentate carbonate ion adsorbed onto two Sn5c, with the remaining H atom coordinating to a neighbouring Obr to form an (OH)O. It appears that the presence of Sn5c…OH also activates neighbouring Obr to react with CO2 forming a bidentate CO32− between an Sn5c and Vbr. These findings show how humidity should enhance the sensing properties of SnO2 towards CO2, which has been demonstrated experimentally [70]. However, because of the poor performance of pure SnO2 in CO2 detection, most studies involve modification of the surface, for example, by lanthanum doping [71,72], and relatively few studies investigate the interaction of CO2 with undoped SnO2.
4.3. Reduction of the Surface
There is another reaction pathway for detecting CO, which recently has gained more recognition within the gas sensing community. As mentioned before, the direct reduction of the SnO2 surface by CO was initially discounted as no CO2 was detected in the outflowing gas [56]. However, such behaviour would vary significantly from many other oxides, on which CO oxidation proceeds via a Mars-van Krevelen (MvK) mechanism, which features a cyclic reduction/oxidation of the adsorption sites [73,74]. Indeed, DFT studies on various SnO2 surfaces have shown repeatedly that the direct reduction of the surface by CO should also be possible [17,28,32,75]. Several mechanisms were proposed, through which the CO molecule may react with Obr, for example, through a carbonate intermediary, as shown in Figure 11.
Although the formation of a carbonate species on the surface is often associated with CO detection, it is likely a secondary process resulting from the adsorption of the produced CO2. Experimentally, it was found that carbonates do not always appear during CO dosing on SnO2 and that identical features in the DRIFTS spectra may be produced by dosing CO2 instead of CO [76]. Indeed, the formation of surface carbonates seems to be related to the immediate sample pre-treatment, with the species appearing after preparation at 450 °C but not after 1000 °C [67,76].
Whether the carbonates are formed during or after the interaction of SnO2 with CO, the predicted reduction of the surface by CO was confirmed experimentally in operando UV-Vis diffuse reflectance spectroscopy [77]. The analysis revealed strong absorption in the visible region upon dosing with CO, consistent with the formation of in-gap electronic states of oxygen vacancies, making the typically transparent semiconductor opaque. Additional evidence comes from a TPD study, which shows that CO desorbs from the SnO2 surface almost exclusively as CO2, which indicates that CO reacts with the surface upon adsorption [69]. Moreover, the amount of CO2 desorbed after CO dosing depends strongly on the degree of surface reduction, and therefore the density of Obr (versus Vbr) on the surface. This indicates that surface oxygen atoms are important adsorption sites and that non-reductive adsorption of CO onto SnO2 is limited. Therefore, there is strong experimental evidence supporting this mechanism of gas–surface interactions.
It seems fortuitous that CO can reduce the surface of SnO2 directly by reacting with Obr; the Vbr are essential adsorption sites for oxygen that enable the secondary reaction of CO with the preadsorbed oxygen, reforming the Obr lost in the reduction. This cyclic process results in a dynamically changing density of Obr on the surface. The steady-state coverage, which eventually determines the electronic properties of the surface, will depend on the rates of reduction and oxidation, and therefore the concentration of CO, assuming the ambient oxygen concentration is constant. Because of the strong evidence supporting the reaction, it is more reasonable to perceive the detection mechanism of CO in terms of plausible reactions of well-established species rather than an elusive monoatomic adsorbate; such descriptions of CO detection have been proposed recently [76], which foreshadows a new era in gas-sensor modelling.
Although the reactant geometry of the reduction of SnO2 by NO is similar to CO, the reaction is less likely in the case of NO. DFT calculation determined that this process is endothermic, making it less likely to occur at noticeable rates [30]. Nevertheless, there is a possibility of a direct surface reduction by NO, which results in the formation of NO2 adsorbed onto a bridging vacancy. The production of this interfering gas is somewhat problematic, as it is an oxidising gas that causes a significant increase in the sensor’s resistance. The adsorption mechanisms of NO2 are the subject of the following section.
5. Interactions with Oxidising Gases—NO2
5.1. Adsorption onto Stoichiometric Surfaces
Even though oxygen lends its name to this category of target gases, it rarely is considered one, as it defines the baseline resistance of sensors. Therefore, oxidising gases are those that increase the sensor’s resistance relative to an ambient oxygen background, for example, NO2 or SO2. The current research focuses on the former, and there is very little research on other oxidising gases, with SO2 sometimes included as a frequent interfering gas to NO2.
Conventionally, NO2 detection is considered in terms similar to oxygen ionosorption; the adsorbates withdraw electrons from the conduction band of SnO2. However, the formed states are deeper than those of oxygen, causing stronger band bending and a more significant resistance increase [78]. Even though this description is fairly straightforward, the reactions of NO2 on the surface of SnO2 seem to be quite complex. In contrast to carbon, which only forms two oxides, nitrogen can form a variety of oxides, such as N2O, NO, NO2, N2O4 and the NO3− ion, many of which are formed during parasitic reactions on the surface of the sensor [78] as evidenced by IR and Raman spectroscopic analyses [66,79]. Long sensor recovery time, during which conductivity of the sensing layer returns to its baseline value after removal of the target gas, has been linked to the formation of the NO3− ion, which has significantly larger adsorption energy than NO2, and therefore desorbs at a slower rate [80]. In order to improve the performance of sensors, it is crucial to understand the formation mechanisms of various nitrogen oxides on the surfaces of SnO2 and to design sensitive layers that could hinder parasitic reactions.
On stoichiometric SnO2 (101) surfaces, NO2 can adsorb in a monodentate or bidentate bridging geometry with one or two neighbouring Sn5c sites [81]. The adsorption energy between the two configurations differs by only about 0.13 eV. It means that even at room temperature, the molecule should convert easily between the two states, which would result in a “random walk” along the Sn5c rows. This could eventually lead to two NO2 molecules happening upon one another at the surface, resulting in them reacting; one of the molecules abstracts an oxygen atom from the other to become an NO3− ion, while the other is reduced to NO, which may adsorb onto a neighbouring site or desorb from the surface. The occurrence of this reaction is supported by the observation of an intermediate NO2 dimer, denoted as ONONO2, using in-situ Raman spectroscopy [79].
Whereas the interaction of SnO2 with the produced NO is relatively weak, the NO3− adsorbs much more strongly than NO2. Contrary to NO2, the energy difference between the monodentate and bridging geometries of NO3− is considerable [81]; therefore, a random walk along the Sn paths is unlikely. Moreover, because of the stronger adsorption of NO3−, temperatures of up to 700 °C are necessary to remove the adsorbates altogether. In terms of their electronic effects, the Bader charge trapped by the NO3− is over twice as large as that trapped by an adsorbed NO2 molecule, which suggests it should affect the sensor’s resistance much more strongly [80]. As a result, the formation of NO3− may lead to sensor poisoning and long recovery times.
5.2. Adsorption onto Reduced Surfaces
Although a “walking” NO2 molecule is interesting to picture, the preferable adsorption site for this gas is a surface oxygen vacancy, as shown in independent computational studies [18,31]. The molecule interacts with a vacancy via the oxygen atoms. Both studies determined that NO2 adsorption onto a bridging vacancy is stronger than at an in-plane vacancy. NO2 adsorbed onto Vbr is most stable in a monodentate configuration, though bidentate geometries coordinated to a nearby Sn5c or another Vbr are also possible. In all such geometries, the molecule is immobile, which lowers its chance of reacting with another NO2 to form nitrates. Many studies highlight the importance of oxygen vacancies in the low-temperature detection of NO2 on SnO2-based gas sensors, although they sparsely comment on the atomistic origin of the superior sensitivity [82,83,84].
Adsorption onto an oxygen-deficient surface offers a reaction pathway that is unavailable on a stoichiometric one, which is the dissociation of NO2, as shown in Figure 12. We have already considered the mechanism by which reducing gases affect the sensor’s resistance; the current process can be thought of as the direct opposite. Similar to oxygen, the adsorption of NO2 at a surface vacancy can lead to the oxidation of the surface, as shown in a computational study of the (110) surface [18]. The adsorption of NO2 onto Vbr leads to the dissociation of the molecule and healing the vacancy; the authors comment on the likeness between the final structure achieved here and during the adsorption of NO onto a stoichiometric surface. Therefore, apart from the electron-withdrawing properties of NO2 adsorption, the removal of VO self-doping in SnO2 must be considered. Moreover, the molecule preferentially heals the bridging oxygen site on a surface where in-plane vacancies are also available, in agreement with the preferable adsorption onto Obr. This selectivity is inverted in the case of SO2, which preferentially adsorbs onto Vpl [31]. Therefore, in theory, one can regulate the selectivity of a sensor by selectively creating bridging vacancies.
Adsorption of NO2 onto SnO2 with various distributions of both bridging and in-plane vacancies was studied via first-principles thermodynamics, and the results show clearly that bridging vacancies are the more favourable of the two [85]. Based on these results and the Redhead equation, a simulated TPD chromatogram was computed and compared to experimental data, as shown in Figure 13. The match is reasonable, although experimental data show some high-temperature desorption of NO, which was not predicted by DFT. Moreover, the empirical TPD study that benchmarks simulated desorption shows NO desorption only at temperatures above 200 °C on a differently prepared surface [66]. This could be explained by a temperature-activated dissociation of NO2 at a surface vacancy, resulting in NO desorbing at temperatures above typical, which the DFT study did not consider. However, it should be mentioned that while the desorption is dominated by NO2 in this study, an earlier TPD study found only NO and some N2O desorbing from SnO2 powder after adsorption at room temperature [69]. This interesting difference, if confirmed, could indicate the importance of the atomistic state of the surface in determining its interactions with gas adsorbates.
6. Oxygen Vacancies versus Gas Sensing Mechanism
The previous chapters discussed the importance of oxygen vacancies as adsorption sites, their formation during the interaction with reducing gases, and their healing via the complex interactions with oxygen or oxidising gases. However, as mentioned before, oxygen vacancies are also a crucial self-doping mechanism of SnO2 and largely determine its intrinsic conductivity [24]. Although a detailed description of the involvement of VO in the transducer function of gas sensors is beyond the scope of this review, there are several points worth considering.
Even though oxygen vacancies have long been recognised as the source of conductivity in bulk SnO2, recent computational studies show these electronic states to be deep in the bandgap, thus unlikely to contribute to conductivity [23]. Moreover, the defects are now considered negative-U type, where the attractive interaction between two electrons bound at a vacancy means that the system’s energy with a singly occupied vacancy can be lowered by capturing a conduction electron at the defect [23]. By implication, singly ionised vacancies are not thermodynamically stable for any Fermi level position [86]. This computational finding contradicts the well-established evidence of paramagnetic (singly occupied) oxygen vacancies observed in ESR studies [22,46], which could hint at a difference between the nature of bulk and near-surface vacancies. The effect of vacancies on the electronic properties of SnO2 could depend on their depth below the surface, with bulk vacancies being energetically deep, negative-U and likely neutral, and surface vacancies singly or doubly ionised depending on the temperature. The singly charged vacancies, which could emerge in the substoichiometric coordination of a reduced surface, should then lead to the formation of a surface conductivity layer, while the bulk of the material remains an insulator (relative to the surface). Indeed, such surface conductivity layer models have been proposed recently for In2O3, a gas-sensitive material analogous to SnO2, whose conductivity as a thick particulate film is several times larger than bulk conductivity models can account for [87].
The existence of singly ionised vacancies is important to acknowledge, as it reveals certain parallelism between ionosorption and surface reduction. If the formed oxygen vacancy is a singly ionised donor, the conduction band has gained one electron per lattice oxygen atom lost, similar to the formation of O−. Moreover, such interpretation of the meaning of phenomenological “ionosorbed oxygen” could explain why some studies observe macroscopic changes consistent with the appearance of ionosorbed O2− at elevated temperatures, even though such species are known to be unstable outside of the stabilising influence of the Madelung potential on oxygen lattice sites.
7. Conclusions
The insight provided by recent molecular modelling studies, backed by emerging operando spectroscopy, offers a new perspective on the gas-sensitive phenomena of SnO2-based gas sensors, and therefore our understanding of the sensing mechanisms needs to be re-evaluated. Although the prevalent model of oxygen ionosorption can explain some of the observed macroscopic behaviour of the sensors, it is based on species thus far unconfirmed. The systematic lack of evidence after so many years of research indicates that new mechanisms need to be considered.
The only convincing evidence of oxygen adsorbates on the SnO2 surface is linked to diatomic species, whose effect on the sensor’s resistance is limited according to ionosorption theory, which requires O− to work. Although O− may exist under special circumstances, most interactions between the surface, O2 and target gases are mediated by the diatomic oxygen adsorbates. It is important to mention that both the dissociation of O2 and the reaction of a target gas with preadsorbed oxygen results in the healing of a surface vacancy, which should affect the equilibrium density of vacancies, providing a mechanism for resistance change.
Furthermore, direct evidence of the reduction of the SnO2 surface was finally obtained, which settles the debate of whether CO can react with the Obr, as predicted for some years based on DFT calculations. The lack of CO2 observed during the dosing of CO on SnO2 in the absence of oxygen is then somewhat puzzling but could be explained by a slow rate of reaction and the depletion of Obr, which cannot be replenished by ambient oxygen. This is supported by the strong dependence on the amount of desorbed species after CO dosing based on the degree of surface reduction.
The importance of bridging sites is further highlighted in the adsorption mechanisms of H2O. Obr is a reaction partner for the dissociation of H2O on both reduced and stoichiometric surfaces, and the dissociation on a Vbr leads to the formation of an additional electron donor, thus causing a larger resistance change. Moreover, the transformation of Obr into (OH)O is the most likely candidate for the interference in CO detection, as the essential adsorption site is removed.
Finally, the bridging sites are heavily involved in the detection of both NO and NO2, which transform into one another in a network of complex interactions with the surface. In the process, they either form or heal bridging vacancies, which should strongly affect the baseline conductivity of SnO2. However, these two gases also adsorb readily onto various other sites, which should also have a noticeable effect on the sensor’s resistance. The mutual coexistence of the absorbed species in these processes is evidenced by the results of the adsorption of one or the other, which leads to a similar final state, as noticed independently by both computational and empirical scientists.
In conclusion, the majority of evidence points to the mechanisms centred around oxygen vacancies. The surface of SnO2 is a dynamic assembly, which can support a large density of vacancies both at the surface (bridging and in-plane) and in several atomic layers below the surface. As evidenced by experimental studies, the in-plane oxygen vacancies play an important role in determining its conductivity and reactivity. Therefore, the surface of SnO2 is an exchange zone between bulk and gas-phase oxygen, and the sensors response can be seen as resulting from the change in the position of the exchange equilibrium, on top of which the effects of direct adsorption may manifest as additional resistance change.
Funding
This work was supported by the Engineering and Physical Sciences Research Council (EP/R512400/1).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rieu, M.; Camara, M.; Tournier, G.; Viricelle, J.P.; Pijolat, C.; de Rooij, N.F.; Briand, D. Fully Inkjet Printed SnO2 Gas Sensor on Plastic Substrate. Sens. Actuators B Chem. 2016, 236, 1091–1097. [Google Scholar] [CrossRef] [Green Version]
- Devabharathi, N.; Umarji, A.M.; Dasgupta, S. Fully Inkjet-Printed Mesoporous SnO2-Based Ultrasensitive Gas Sensors for Trace Amount NO2 Detection. ACS Appl. Mater. Interfaces 2020, 12, 57207–57217. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhou, Q.; Lu, Z.; Wei, Z.; Xu, L.; Gui, Y. Recent Advances of SnO2-Based Sensors for Detecting Fault Characteristic Gases Extracted from Power Transformer Oil. Front. Chem. 2018, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Huang, K.; Yuan, F.; Xie, C. Gas Sensing Properties and in Situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy Study of Acetone Adsorption and Reactions on SnO2 Films. Sens. Mater. 2014, 26, 649–663. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, Q.; Zhang, S.; He, Z. The Enhanced H2 Selectivity of SnO2 Gas Sensors with the Deposited SiO2 Filters on Surface of the Sensors. Sensors 2019, 19, 2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zhang, Q.; Lv, R.; Wu, D.; Zhang, S. Enhancing Formaldehyde Selectivity of SnO2 Gas Sensors with the ZSM-5 Modified Layers. Sensors 2021, 21, 3947. [Google Scholar] [CrossRef]
- Ding, J.; McAvoy, T.J.; Cavicchi, R.E.; Semancik, S. Surface State Trapping Models for SnO2-Based Microhotplate Sensors. Sens. Actuators B Chem. 2001, 77, 597–613. [Google Scholar] [CrossRef]
- Bârsan, N.; Hübner, M.; Weimar, U. Conduction Mechanisms in SnO2 Based Polycrystalline Thick Film Gas Sensors Exposed to CO and H2 in Different Oxygen Backgrounds. Sens. Actuators B Chem. 2011, 157, 510–517. [Google Scholar] [CrossRef]
- Barsan, N.; Weimar, U. Conduction Model of Metal Oxide Gas Sensors. J. Electroceramics 2001, 7, 143–167. [Google Scholar] [CrossRef]
- Morrison, S.R. Mechanism of Semiconductor Gas Sensor Operation. Sens. Actuators 1987, 11, 283–287. [Google Scholar] [CrossRef]
- Heiland, G. Homogeneous Semiconducting Gas Sensors. Sens. Actuators 1981, 2, 343–361. [Google Scholar] [CrossRef]
- Geistlinger, H. Electron Theory of Thin-Film Gas Sensors. Sens. Actuators B Chem. 1993, 17, 47–60. [Google Scholar] [CrossRef]
- Göpel, W.; Schierbaum, K.D. SnO2 Sensors: Current Status and Future Prospects. Sens. Actuators B Chem. 1995, 26, 1–12. [Google Scholar] [CrossRef]
- Hauffe, K. The Application of the Theory of Semiconductors to Problems of Heterogeneous Catalysis. Adv. Catal. 1955, 7, 213–257. [Google Scholar] [CrossRef]
- Pulkkinen, U.; Rantala, T.T.; Rantala, T.S.; Lantto, V. Kinetic Monte Carlo Simulation of Oxygen Exchange of SnO2 Surface. J. Mol. Catal. A Chem. 2001, 166, 15–21. [Google Scholar] [CrossRef]
- Gurlo, A. Interplay between O2 and SnO2: Oxygen Ionosorption and Spectroscopic Evidence for Adsorbed Oxygen. Chem. Phys. Chem. 2006, 7, 2041–2052. [Google Scholar] [CrossRef]
- Lu, Z.; Ma, D.; Yang, L.; Wang, X.; Xu, G.; Yang, Z. Direct CO Oxidation by Lattice Oxygen on the SnO2(110) Surface: A DFT Study. Phys. Chem. Chem. Phys. 2014, 16, 12488–12494. [Google Scholar] [CrossRef]
- Hong, S.N.; Kye, Y.H.; Yu, C.J.; Jong, U.G.; Ri, G.C.; Choe, C.S.; Kim, K.H.; Han, J.M. Ab Initio Thermodynamic Study of the SnO2 (110) Surface in an O2 and NO Environment: A Fundamental Understanding of the Gas Sensing Mechanism for NO and NO2. Phys. Chem. Chem. Phys. 2016, 18, 31566–31578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorokhta, M.; Khalakhan, I.; Vondráček, M.; Tomeček, D.; Vorokhta, M.; Marešová, E.; Nováková, J.; Vlček, J.; Fitl, P.; Novotný, M.; et al. Investigation of Gas Sensing Mechanism of SnO2 Based Chemiresistor Using near Ambient Pressure XPS. Surf. Sci. 2018, 677, 284–290. [Google Scholar] [CrossRef]
- Bolzan, A.A.; Fong, C.; Kennedy, B.J.; Howard, C.J. Structural Studies of Rutile-Type Metal Dioxides. Acta Crystallogr. Sect. B Struct. Sci. 1997, 53, 373–380. [Google Scholar] [CrossRef]
- Fröhlich, D.; Kenklies, R.; Helbig, R. Band-Gap Assignment in SnO2 by Two-Photon Spectroscopy. Phys. Rev. Lett. 1978, 41, 1750–1751. [Google Scholar] [CrossRef]
- D’Arienzo, M.; Cristofori, D.; Scotti, R.; Morazzoni, F. New Insights into the SnO2 Sensing Mechanism Based on the Properties of Shape Controlled Tin Oxide Nanoparticles. Chem. Mater. 2013, 25, 3675–3686. [Google Scholar] [CrossRef]
- Buckeridge, J.; Catlow, C.R.A.; Farrow, M.R.; Logsdail, A.J.; Scanlon, D.O.; Keal, T.W.; Sherwood, P.; Woodley, S.M.; Sokol, A.A.; Walsh, A. Deep vs Shallow Nature of Oxygen Vacancies and Consequent N-Type Carrier Concentrations in Transparent Conducting Oxides. Phys. Rev. Mater. 2018, 2, 56–59. [Google Scholar] [CrossRef] [Green Version]
- De Frésart, E.; Darville, J.; Gilles, J.M. Influence of the Surface Reconstruction on the Work Function and Surface Conductance of (110) SnO2. Appl. Surf. Sci. 1982, 11–12, 637–651. [Google Scholar] [CrossRef]
- Kamp, B.; Merkle, R.; Lauck, R.; Maier, J. Chemical Diffusion of Oxygen in Tin Dioxide: Effects of Dopants and Oxygen Partial Pressure. J. Solid State Chem. 2005, 178, 3027–3039. [Google Scholar] [CrossRef]
- Batzill, M.; Katsiev, K.; Burst, J.M.; Diebold, U.; Chaka, A.M.; Delley, B. Gas-Phase-Dependent Properties of SnO2 (110), (100), and (101) Single-Crystal Surfaces: Structure, Composition, and Electronic Properties. Phys. Rev. B-Condens. Matter Mater. Phys. 2005, 72, 1–20. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Nagasawa, Y.; Shimomura, S.; Tabata, K.; Suzuki, E. A Density Functional Theory Study of the Interaction of Oxygen with a Reduced SnO2 (110) Surface. Chem. Phys. Lett. 2000, 316, 477–482. [Google Scholar] [CrossRef]
- Wang, X.; Qin, H.; Chen, Y.; Hu, J. Sensing Mechanism of SnO2 (110) Surface to CO: Density Functional Theory Calculations. J. Phys. Chem. C 2014, 118, 28548–28561. [Google Scholar] [CrossRef]
- Eslamian, M.; Salehi, A.; Nadimi, E. The Role of Oxygen Vacancies on SnO2 Surface in Reducing Cross-Sensitivity between Ambient Humidity and CO: A First Principles Investigation. Surf. Sci. 2021, 708, 121817. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, L.; He, C.; Ma, D.; Lu, Z. Adsorption and Oxidation of NO on Various SnO2(1 1 0) Surfaces: A Density Functional Theory Study. Sens. Actuators B Chem. 2015, 221, 717–722. [Google Scholar] [CrossRef]
- Prades, J.D.; Cirera, A.; Morante, J.R.; Pruneda, J.M.; Ordejón, P. Ab Initio Study of NOx Compounds Adsorption on SnO2 Surface. Sens. Actuators B Chem. 2007, 126, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Ducéré, J.-M.; Hemeryck, A.; Estève, A.; Rouhani, M.D.; Landa, G.; Ménini, P.; Tropis, C.; Maisonnat, A.; Fau, P.; Chaudret, B. A Computational Chemist Approach to Gas Sensors: Modeling the Response of SnO2 to CO, O2, and H2O Gases. J. Comput. Chem. 2012, 33, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Abokifa, A.A.; Haddad, K.; Fortner, J.; Lo, C.S.; Biswas, P. Sensing Mechanism of Ethanol and Acetone at Room Temperature by SnO2 Nano-Columns Synthesized by Aerosol Routes: Theoretical Calculations Compared to Experimental Results. J. Mater. Chem. A 2018, 6, 2053–2066. [Google Scholar] [CrossRef]
- Oviedo, J.; Gillan, M.J. Energetics and Structure of Stoichiometric SnO2 Surfaces Studied by First-Principles Calculations. Surf. Sci. 2000, 463, 93–101. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Nagasawa, Y.; Murakami, A.; Tabata, K. Stability of Oxygen Anions and Hydrogen Abstraction from Methane on Reduced SnO2 (110) Surface. Int. J. Quantum Chem. 1998, 69, 669–678. [Google Scholar] [CrossRef]
- Cox, D.F.; Fryberger, T.B.; Semancik, S. Oxygen Vacancies and Defect Electronic States on the SnO2 (110)-1 × 1 Surface. Phys. Rev. B 1988, 38, 2072–2083. [Google Scholar] [CrossRef]
- Sopiha, K.V.; Malyi, O.I.; Persson, C.; Wu, P. Chemistry of Oxygen Ionosorption on SnO2 Surfaces. ACS Appl. Mater. Interfaces 2021, 13, 33664–33676. [Google Scholar] [CrossRef]
- Trani, F.; Causà, M.; Ninno, D.; Cantele, G.; Barone, V. Density Functional Study of Oxygen Vacancies at the SnO2 Surface and Subsurface Sites. Phys. Rev. B-Condens. Matter Mater. Phys. 2008, 77, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Liang, J.; Liu, Y.; Liu, Y.; Xu, X.; Fang, X.; Zhong, W.; Wang, X. Identifying Surface Active Sites of SnO2: Roles of Surface O2−, O22− Anions and Acidic Species Played for Toluene Deep Oxidation. Ind. Eng. Chem. Res. 2019, 58, 18569–18581. [Google Scholar] [CrossRef]
- Simion, C.E.; Schipani, F.; Papadogianni, A.; Stanoiu, A.; Budde, M.; Oprea, A.; Weimar, U.; Bierwagen, O.; Barsan, N. Conductance Model for Single-Crystalline/Compact Metal Oxide Gas-Sensing Layers in the Nondegenerate Limit: Example of Epitaxial SnO2 (101). ACS Sens. 2019, 4, 2420–2428. [Google Scholar] [CrossRef]
- Chang, S. Oxygen Chemisorption on Tin Oxide: Correlation between Electrical Conductivity and EPR Measurements. J. Vac. Sci. Technol. 1980, 17, 366–369. [Google Scholar] [CrossRef]
- Che, M.; Tench, A.J. Characterization and Reactivity of Mononuclear Oxygen Species on Oxide Surfaces. Adv. Catal. 1982, 31, 77–133. [Google Scholar] [CrossRef]
- Coronado, J.M.; Maira, A.J.; Conesa, J.C.; Yeung, K.L.; Augugliaro, V.; Soria, J. EPR Study of the Surface Characteristics of Nanostructured TiO2 under UV Irradiation. Langmuir 2001, 17, 5368–5374. [Google Scholar] [CrossRef]
- Coronado, J.M.; Soria, J. ESR Study of the Initial Stages of the Photocatalytic Oxidation of Toluene over TiO2 Powders. Catal. Today 2007, 123, 37–41. [Google Scholar] [CrossRef]
- Armelao, L.; Barreca, D.; Bontempi, E.; Canevali, C.; Depero, L.E.; Mari, C.M.; Ruffo, R.; Scotti, R.; Tondello, E.; Morazzoni, F. Can Electron Paramagnetic Resonance Measurements Predict the Electrical Sensitivity of SnO2-Based Film? Appl. Magn. Reson. 2002, 22, 89–100. [Google Scholar] [CrossRef]
- Grishina, D.A.; Mironov, A.A.; Pentegov, I.S.; Marikutsa, A.V.; Konstantinova, E.A. Electron Spin Resonance Characterization of Defects in Sensor Materials Based on Nanocrystalline Tin Dioxide. Opt. Micro-Nanometrol. IV 2012, 8430, 84300A. [Google Scholar] [CrossRef]
- Che, M.; Tench, A.J. Characterization and Reactivity of Molecular Oxygen Species on Oxide Surfaces. Adv. Catal. 1983, 32, 1–148. [Google Scholar] [CrossRef]
- Volodin, A.M.; Cherkashin, A.E. ESR Spectra of O2−on SnO2. Effect of Adsorbed CO on the Conditions of Stabilization of O2−. React. Kinet. Catal. Lett. 1981, 17, 323–327. [Google Scholar] [CrossRef]
- Yamazoe, N.; Fuchigami, J.; Kishikawa, M.; Seiyama, T. Interactions of Tin Oxide Surface with O2, H2O and H2. Surf. Sci. 1979, 86, 335–344. [Google Scholar] [CrossRef]
- Matsuura, Y.; Takahata, K.; Ihokura, K. Mechanism of Gas Sensitivity Change with Time of SnO2 Gas Sensors. Sens. Actuators 1988, 14, 223–232. [Google Scholar] [CrossRef]
- Matsuura, Y.; Takahata, K.; Matsuura, S. Temperature Programmed Desorption Study on Mechanism of Long-Term Deterioration of SnO2 Gas Sensor. Denki Kagaku Oyobi Kogyo Butsuri Kagaku 1990, 58, 1154–1161. [Google Scholar] [CrossRef]
- Cavicchi, R.; Tarlov, M.; Semancik, S. Preparation of Well-ordered, Oxygen-rich SnO2 (110) Surfaces via Oxygen Plasma Treatment. J. Vac. Sci. Technol. A Vac. Surf. Film. 1990, 8, 2347–2352. [Google Scholar] [CrossRef]
- Saukko, S.; Lassi, U.; Lantto, V.; Kroneld, M.; Novikov, S.; Kuivalainen, P.; Rantala, T.T.; Mizsei, J. Experimental Studies of O2-SnO2 Surface Interaction Using Powder, Thick Films and Monocrystalline Thin Films. Thin Solid Film. 2005, 490, 48–53. [Google Scholar] [CrossRef]
- Capone, S.; Siciliano, P.; Quaranta, F.; Rella, R.; Epifani, M.; Vasanelli, L. Moisture Influence and Geometry Effect of Au and Pt Electrodes on CO Sensing Response of SnO2 Microsensors Based on Sol-Gel Thin Film. Sens. Actuators B Chem. 2001, 77, 503–511. [Google Scholar] [CrossRef]
- Hahn, S.H.; Bârsan, N.; Weimar, U.; Ejakov, S.G.; Visser, J.H.; Soltis, R.E. CO Sensing with SnO2thick Film Sensors: Role of Oxygen and Water Vapour. Thin Solid Film. 2003, 436, 17–24. [Google Scholar] [CrossRef]
- Umar, A.; Ammar, H.Y.; Kumar, R.; Almas, T.; Ibrahim, A.A.; AlAssiri, M.S.; Abaker, M.; Baskoutas, S. Efficient H2 Gas Sensor Based on 2D SnO2 Disks: Experimental and Theoretical Studies. Int. J. Hydrog. Energy 2020, 45, 26388–26401. [Google Scholar] [CrossRef]
- Suematsu, K.; Watanabe, K.; Yuasa, M.; Kida, T.; Shimanoe, K. Effect of Ambient Oxygen Partial Pressure on the Hydrogen Response of SnO2 Semiconductor Gas Sensors. J. Electrochem. Soc. 2019, 166, B618–B622. [Google Scholar] [CrossRef]
- Grossmann, K.; Pavelko, R.G.; Barsan, N.; Weimar, U. Interplay of H2, Water Vapor and Oxygen at the Surface of SnO2 Based Gas Sensors—An Operando Investigation Utilizing Deuterated Gases. Sens. Actuators B Chem. 2012, 166–167, 787–793. [Google Scholar] [CrossRef]
- Hübner, M.; Pavelko, R.G.; Barsan, N.; Weimar, U. Influence of Oxygen Backgrounds on Hydrogen Sensing with SnO2 Nanomaterials. Sens. Actuators B Chem. 2011, 154, 264–269. [Google Scholar] [CrossRef]
- Choi, P.G.; Izu, N.; Shirahata, N.; Masuda, Y. SnO2 Nanosheets for Selective Alkene Gas Sensing. ACS Appl. Nano Mater. 2019, 2, 1820–1827. [Google Scholar] [CrossRef]
- Kooti, M.; Keshtkar, S.; Askarieh, M.; Rashidi, A. Progress toward a Novel Methane Gas Sensor Based on SnO2 Nanorods-Nanoporous Graphene Hybrid. Sens. Actuators B Chem. 2019, 281, 96–106. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Shi, C.; Li, L.; Qin, H.; Hu, J. Sensing Mechanism of SnO2 (1 1 0) Surface to H2: Density Functional Theory Calculations. Sens. Actuators B Chem. 2015, 220, 279–287. [Google Scholar] [CrossRef]
- Barsan, N.; Rebholz, J.; Weimar, U. Conduction Mechanism Switch for SnO2 Based Sensors during Operation in Application Relevant Conditions; Implications for Modeling of Sensing. Sens. Actuators B Chem. 2015, 207, 455–459. [Google Scholar] [CrossRef]
- Santarossa, G.; Hahn, K.; Baiker, A. Free Energy and Electronic Properties of Water Adsorption on the SnO2 (110) Surface. Langmuir 2013, 29, 5487–5499. [Google Scholar] [CrossRef]
- Leblanc, E.; Perier-Camby, L.; Thomas, G.; Gibert, R.; Primet, M.; Gelin, P. NOx Adsorption onto Dehydroxylated or Hydroxylated Tin Dioxide Surface. Application to SnO2-Based Sensors. Sens. Actuators B Chem. 2000, 62, 67–72. [Google Scholar] [CrossRef]
- Wicker, S.; Guiltat, M.; Weimar, U.; Hémeryck, A.; Barsan, N. Ambient Humidity Influence on CO Detection with SnO2 Gas Sensing Materials. A Combined DRIFTS/DFT Investigation. J. Phys. Chem. C 2017, 121, 25064–25073. [Google Scholar] [CrossRef] [Green Version]
- Thoren, W.; Kohl, D.; Heiland, G. Kinetic Studies of the Decomposition of CH3COOH and CH3COOD on SnO2 Single Crystals. Surf. Sci. 1985, 162, 402–410. [Google Scholar] [CrossRef]
- Tamaki, J.; Nagaishi, M.; Teraoka, Y.; Miura, N.; Yamazoe, N.; Moriya, K.; Nakamura, Y. Adsorption Behavior of CO and Interfering Gases on SnO2. Surf. Sci. 1989, 221, 183–196. [Google Scholar] [CrossRef]
- Wang, D.; Chen, Y.; Liu, Z.; Li, L.; Shi, C.; Qin, H.; Hu, J. CO2-Sensing Properties and Mechanism of Nano-SnO2 Thick-Film Sensor. Sens. Actuators B Chem. 2016, 227, 73–84. [Google Scholar] [CrossRef]
- Kim, D.H.; Yoon, J.Y.; Park, H.C.; Kim, K.H. CO2-Sensing Characteristics of SnO2 Thick Film by Coating Lanthanum Oxide. Sens. Actuators B Chem. 2000, 62, 61–66. [Google Scholar] [CrossRef]
- Hsu, K.C.; Fang, T.H.; Hsiao, Y.J.; Chan, C.A. Highly Response CO2 Gas Sensor Based on Au-La2O3 Doped SnO2 Nanofibers. Mater. Lett. 2020, 261, 127144. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, H.M.; Pala, R.G.S.; Shapovalov, V.; Metiu, H. CO Oxidation by Rutile TiO2 (110) Doped with V, W, Cr, Mo, and Mn. J. Phys. Chem. C 2008, 112, 12398–12408. [Google Scholar] [CrossRef]
- Ramesh, K.; Chen, L.; Chen, F.; Liu, Y.; Wang, Z.; Han, Y.F. Re-Investigating the CO Oxidation Mechanism over Unsupported MnO, Mn2O3 and MnO2 Catalysts. Catal. Today 2008, 131, 477–482. [Google Scholar] [CrossRef]
- Zakaryan, H.A.; Aroutiounian, V.M. Ab Initio Investigation of CO Gas Sensing Mechanism on SnO2 Surfaces. Allsensor 2017, 27, 50–55. [Google Scholar]
- Degler, D.; Wicker, S.; Weimar, U.; Barsan, N. Identifying the Active Oxygen Species in SnO2 Based Gas Sensing Materials: An Operando IR Spectrsocopy Study. J. Phys. Chem. C 2015, 119, 11792–11799. [Google Scholar] [CrossRef]
- Degler, D.; Barz, N.; Dettinger, U.; Peisert, H.; Chassé, T.; Weimar, U.; Barsan, N. Extending the Toolbox for Gas Sensor Research: Operando UV/Vis Diffuse Reflectance Spectroscopy on SnO2-Based Gas Sensors. Sens. Actuators B Chem. 2016, 224, 256–259. [Google Scholar] [CrossRef]
- Ruhland, B.; Becker, T.; Müller, G. Gas-Kinetic Interactions of Nitrous Oxides with SnO2surfaces. Sens. Actuators B Chem. 1998, 50, 85–94. [Google Scholar] [CrossRef]
- Sergent, N.; Epifani, M.; Pagnier, T. In Situ Raman Spectroscopy Study of NO2 Adsorption onto Nanocrystalline Tin (IV) Oxide. J. Raman Spectrosc. 2006, 37, 1272–1277. [Google Scholar] [CrossRef]
- Maeng, S.; Kim, S.W.; Lee, D.H.; Moon, S.E.; Kim, K.C.; Maiti, A. SnO2 Nanoslab as NO2 Sensor: Identification of the NO2 Sensing Mechanism on a SnO2 Surface. ACS Appl. Mater. Interfaces 2014, 6, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Rodriguez, J.A.; Law, M.; Kung, P.; McKinney, J.R.; Yang, P. SnO2 Nanoribbons as NO2 Sensors: Insights from First Principles Calculations. Nano Lett. 2003, 3, 1025–1028. [Google Scholar] [CrossRef]
- Epifani, M.; Prades, J.D.; Comini, E.; Pellicer, E.; Avella, M.; Faglia, G.; Cirera, A.; Scotti, R.; Morazzoni, F.; Morante, J.R. The Role of Surface Oxygen Vacancies in the NO2 Sensing Properties of SnO2 Nanocrystals. J. Phys. Chem. C 2008, 112, 19540–19546. [Google Scholar] [CrossRef]
- Wei, Y.; Chen, C.; Yuan, G.; Gao, S. SnO2nanocrystals with Abundant Oxygen Vacancies: Preparation and Room Temperature NO2sensing. J. Alloys Compd. 2016, 681, 43–49. [Google Scholar] [CrossRef]
- Zhong, Y.; Li, W.; Zhao, X.; Jiang, X.; Lin, S.; Zhen, Z.; Chen, W.; Xie, D.; Zhu, H. High-Response Room-Temperature NO2 Sensor and Ultrafast Humidity Sensor Based on SnO2 with Rich Oxygen Vacancy. ACS Appl. Mater. Interfaces 2019, 11, 13441–13449. [Google Scholar] [CrossRef] [PubMed]
- Prades, J.D.; Cirera, A.; Morante, J.R. First-Principles Study of NOx and SO2 Adsorption onto SnO2 (110). J. Electrochem. Soc. 2007, 154, H675. [Google Scholar] [CrossRef]
- Watkins, G.D. Negative-U Properties for Defects in Solids. In Advances in Solid State Physics; Springer: Berlin/Heidelberg, Germany, 1984; Volume 24, pp. 163–189. [Google Scholar] [CrossRef]
- Lany, S.; Zakutayev, A.; Mason, T.O.; Wager, J.F.; Poeppelmeier, K.R.; Perkins, J.D.; Berry, J.J.; Ginley, D.S.; Zunger, A. Surface Origin of High Conductivities in Undoped In2O3 Thin Films. Phys. Rev. Lett. 2012, 108, 2–6. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Progressing reduction of the (110,101) surfaces of SnO2. Image prepared in VESTA [35].
Figure 1.
Progressing reduction of the (110,101) surfaces of SnO2. Image prepared in VESTA [35].
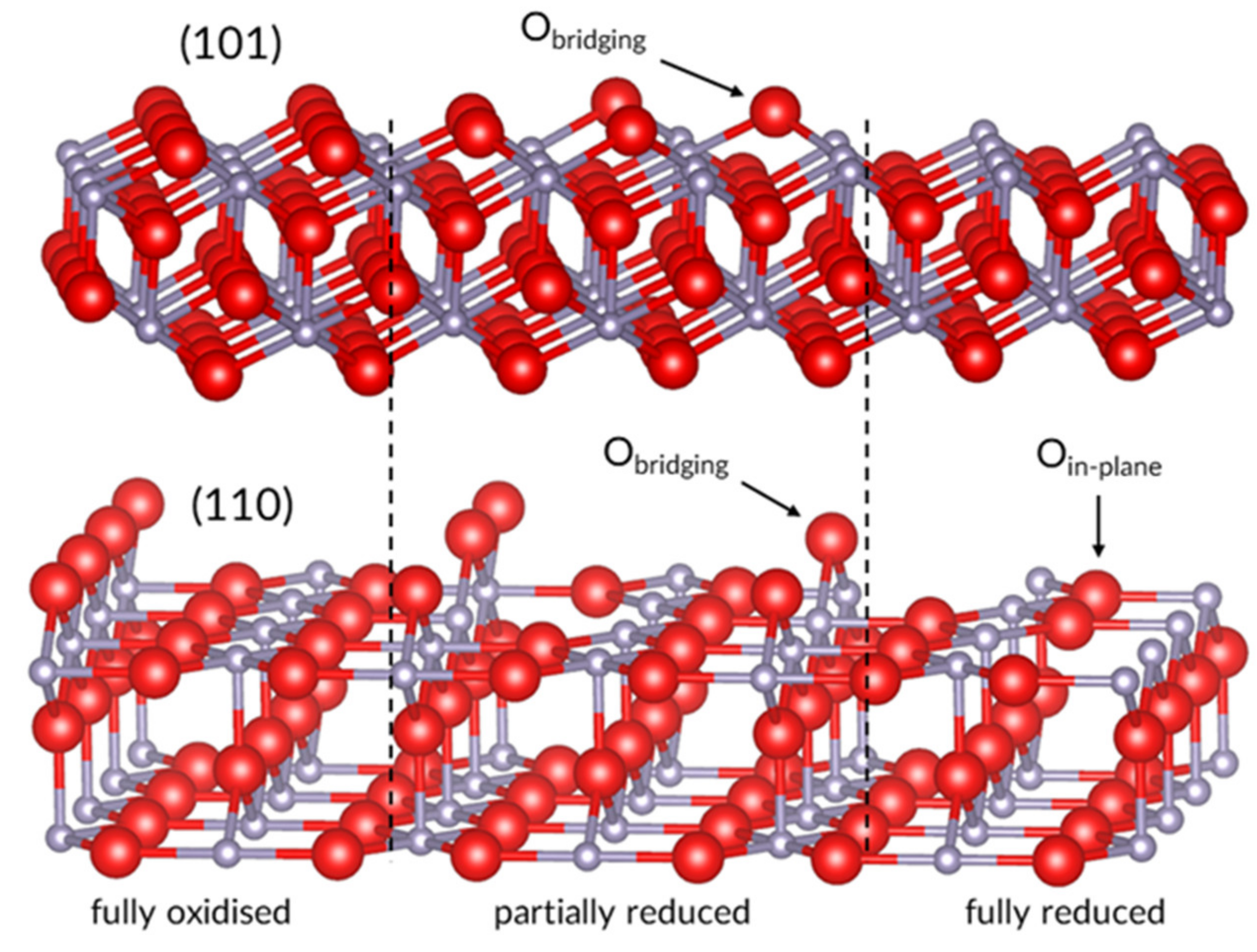
Figure 2.
Adsorption of O2 onto a (110) surface of SnO2 with an isolated vacancy. From left: a (110) surface with a single (periodic) bridging vacancy (blue), “O-stand” superoxide molecular adsorbate in the end-on configuration, “O-lie” peroxide molecular adsorbate in the side-on configuration and “O-cleavage” monoatomic O– adsorbate adjacent to a healed vacancy—now Obr. Reproduced with permission from Reference [17].
Figure 2.
Adsorption of O2 onto a (110) surface of SnO2 with an isolated vacancy. From left: a (110) surface with a single (periodic) bridging vacancy (blue), “O-stand” superoxide molecular adsorbate in the end-on configuration, “O-lie” peroxide molecular adsorbate in the side-on configuration and “O-cleavage” monoatomic O– adsorbate adjacent to a healed vacancy—now Obr. Reproduced with permission from Reference [17].
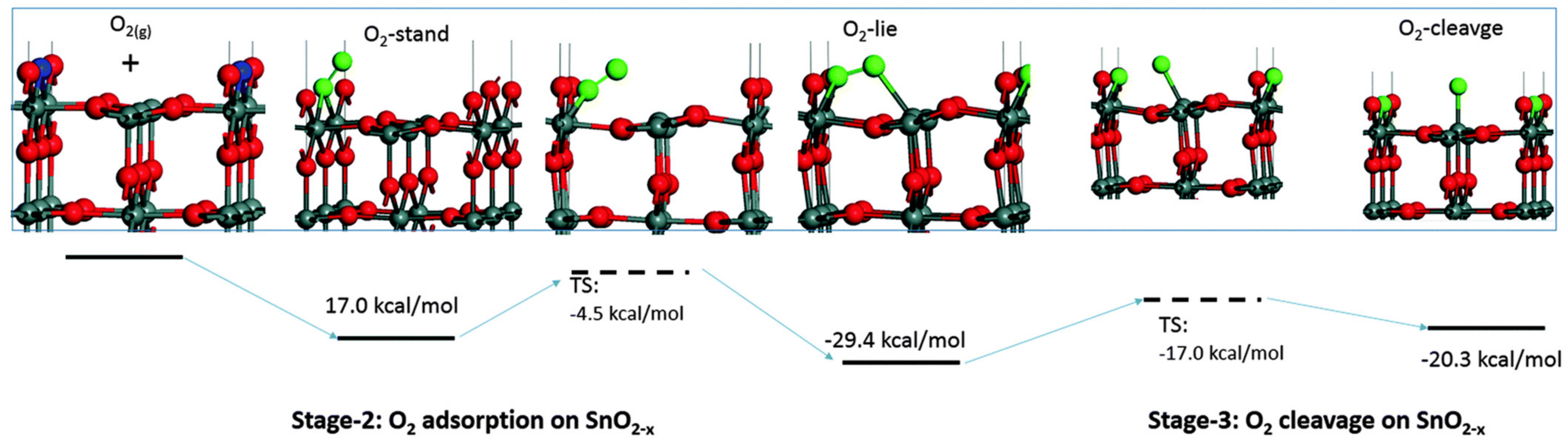
Figure 3.
ESR spectra of SnO2 in UHV. The sample was calcined in UHV at 550 °C for 4 h. (a) Sample at 20 °C. (b) The sample was warmed up to 100 °C. (c) The sample was warmed up to 150 °C. Reproduced with permission from Reference [42].
Figure 3.
ESR spectra of SnO2 in UHV. The sample was calcined in UHV at 550 °C for 4 h. (a) Sample at 20 °C. (b) The sample was warmed up to 100 °C. (c) The sample was warmed up to 150 °C. Reproduced with permission from Reference [42].
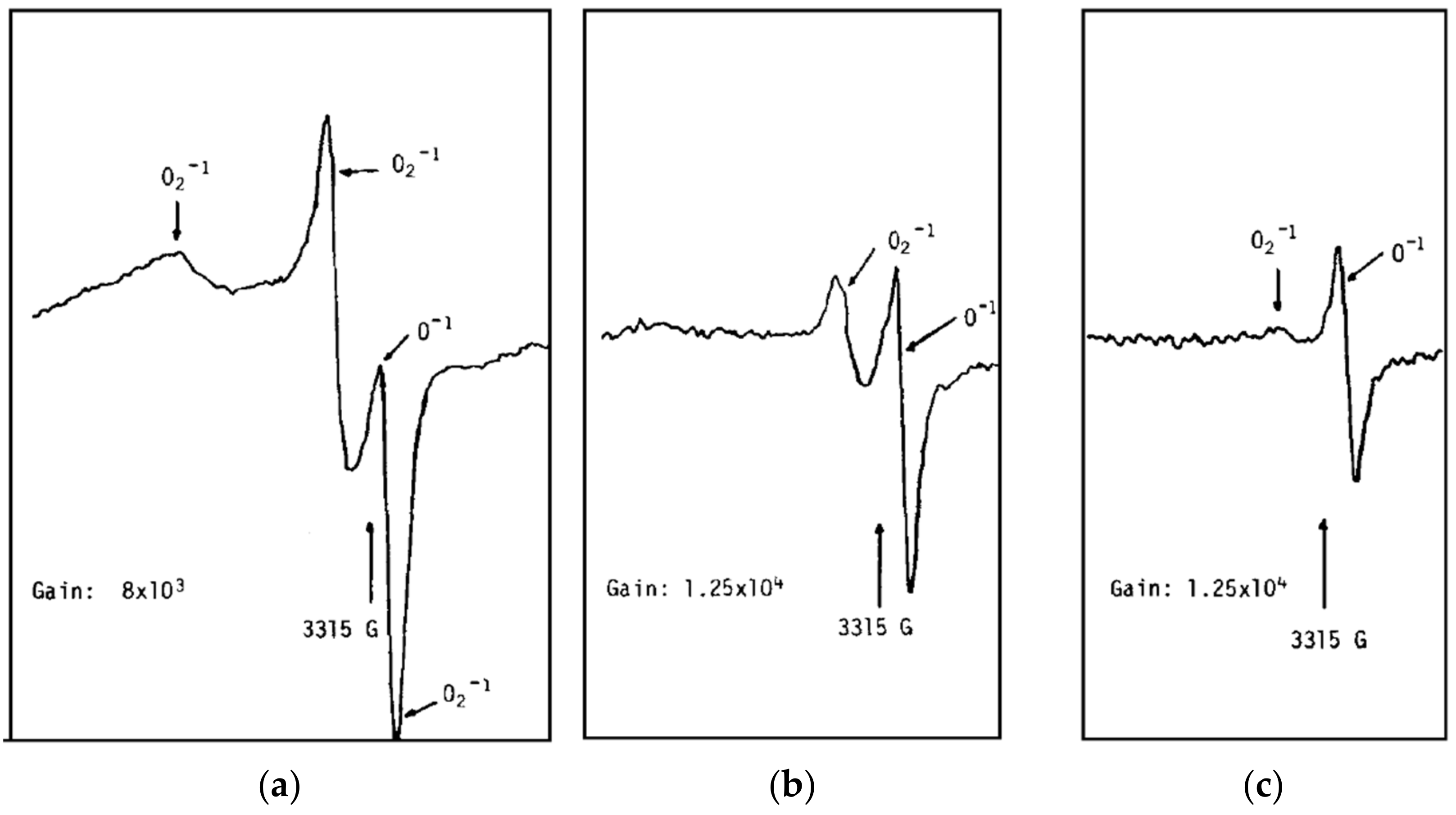
Figure 4.
TPD chromatograms of O2 desorption from SnO2. (a) Traces 1–2 correspond to the low-temperature adsorption following the evacuation at 700 °C. Traces 3–8 correspond to oxygen dosing at 400 °C and pressures of 99, 48, 16, 6, 3 and 2 Torr, respectively. Reproduced with permission from Reference [50]. (b) TPD of SnO2 after exposure to O2. Trace α denotes sample cooled in UHV, while β denotes cooling in O2 background. Reproduced with permission from Reference [54].
Figure 4.
TPD chromatograms of O2 desorption from SnO2. (a) Traces 1–2 correspond to the low-temperature adsorption following the evacuation at 700 °C. Traces 3–8 correspond to oxygen dosing at 400 °C and pressures of 99, 48, 16, 6, 3 and 2 Torr, respectively. Reproduced with permission from Reference [50]. (b) TPD of SnO2 after exposure to O2. Trace α denotes sample cooled in UHV, while β denotes cooling in O2 background. Reproduced with permission from Reference [54].
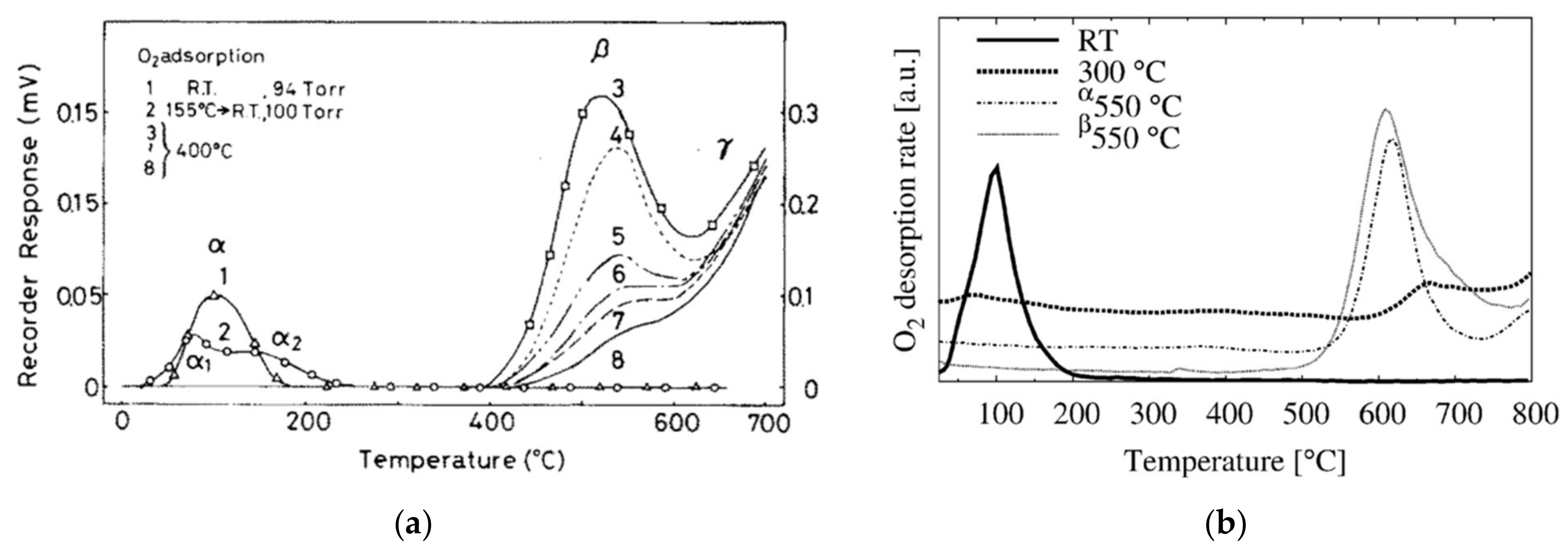
Figure 5.
Coadsorption of NO and O2 onto a reduced (110) surface of SnO2. Reproduced with permission from Reference [30].
Figure 5.
Coadsorption of NO and O2 onto a reduced (110) surface of SnO2. Reproduced with permission from Reference [30].
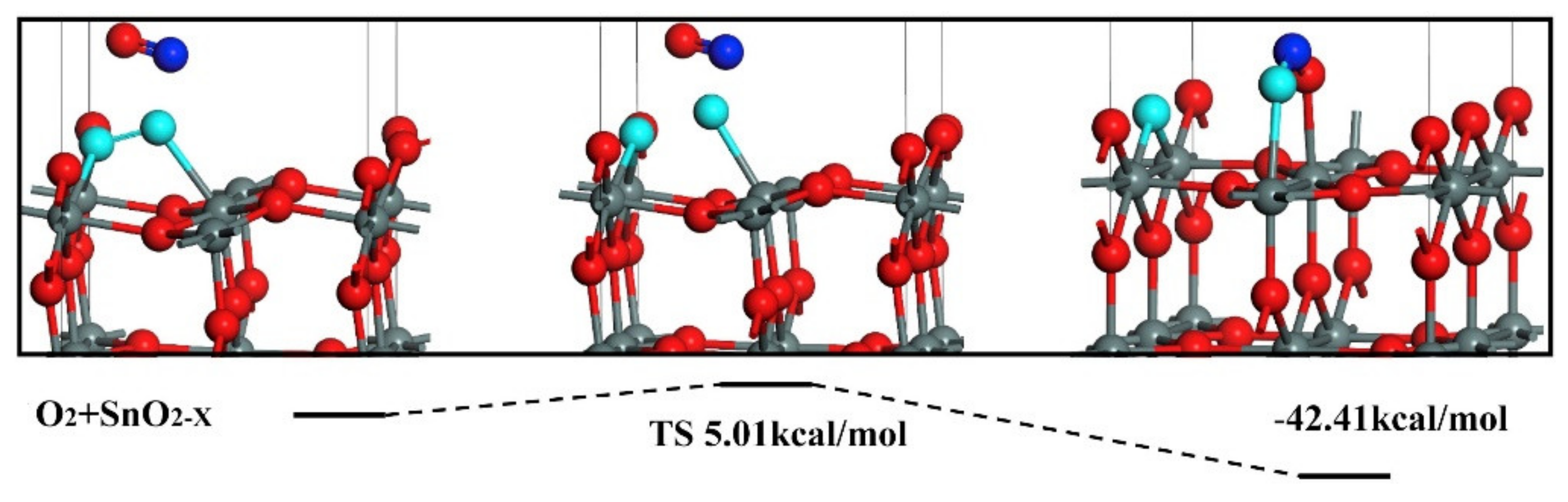
Figure 6.
Coadsorption of CO and O2 onto a reduced (101) surface of SnO2. CO reacts with O2 preadsorbed onto a VO. Reproduced with permission from reference [32].
Figure 6.
Coadsorption of CO and O2 onto a reduced (101) surface of SnO2. CO reacts with O2 preadsorbed onto a VO. Reproduced with permission from reference [32].
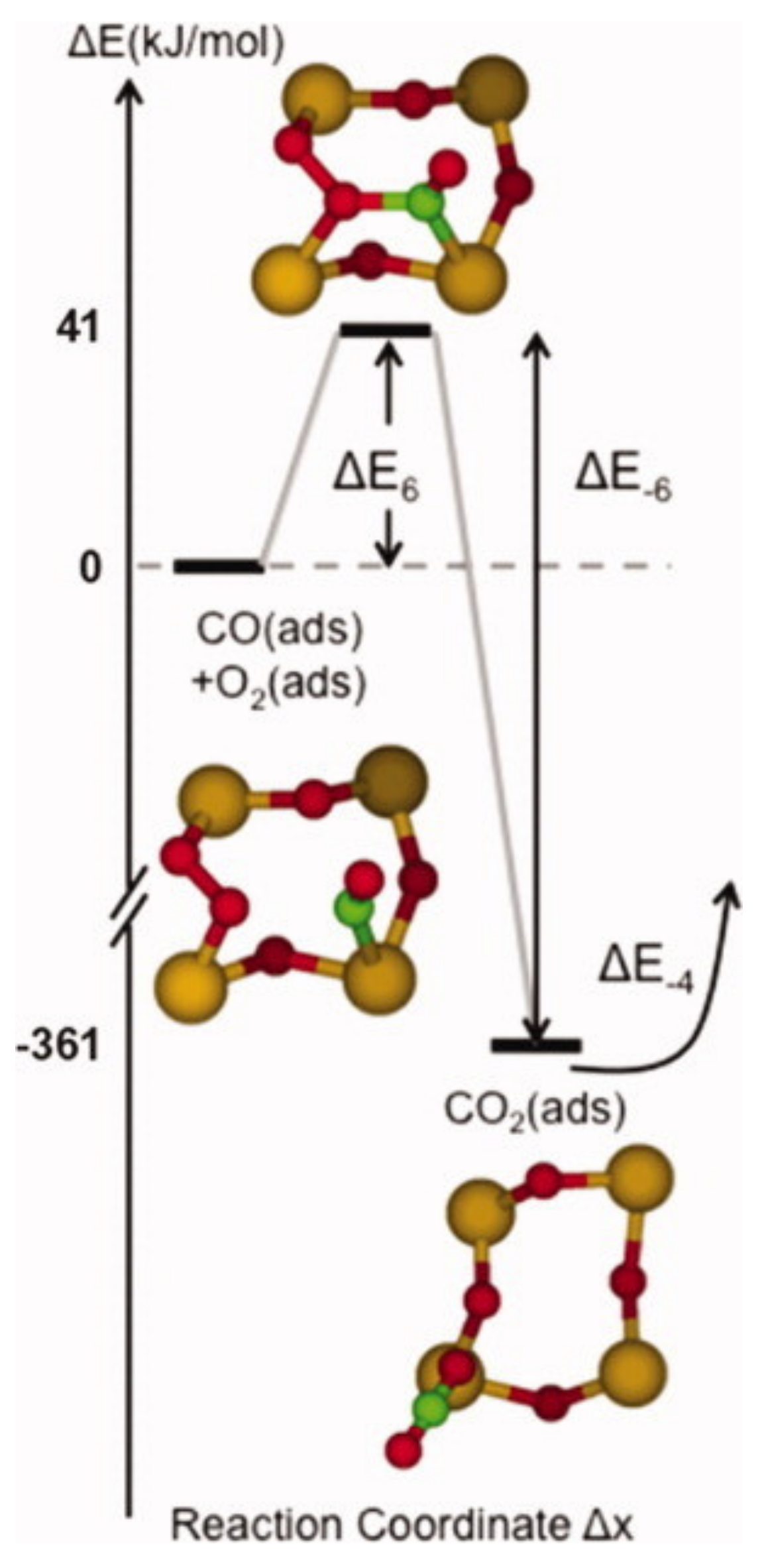
Figure 7.
Adsorption of H2O onto (a,b) stoichiometric and (c–f) reduced (110) surfaces of SnO2. Adsorption energies are given for each configuration. (a) Molecular adsorption onto Sn5c, −1.29 eV. (b) Dissociative adsorption on Sn5c, −1.85 eV. (c) Molecular adsorption onto Vbr, −0.81 eV. (d) Molecular adsorption onto Sn5c with a neighbouring Vbr, −1.32 eV. (e) Disscheme 5. c with a neighbouring Vbr, −1.92 eV. (f) Dissociative adsorption on Vbr, −2.48 eV. Reproduced with permission from Reference [29].
Figure 7.
Adsorption of H2O onto (a,b) stoichiometric and (c–f) reduced (110) surfaces of SnO2. Adsorption energies are given for each configuration. (a) Molecular adsorption onto Sn5c, −1.29 eV. (b) Dissociative adsorption on Sn5c, −1.85 eV. (c) Molecular adsorption onto Vbr, −0.81 eV. (d) Molecular adsorption onto Sn5c with a neighbouring Vbr, −1.32 eV. (e) Disscheme 5. c with a neighbouring Vbr, −1.92 eV. (f) Dissociative adsorption on Vbr, −2.48 eV. Reproduced with permission from Reference [29].
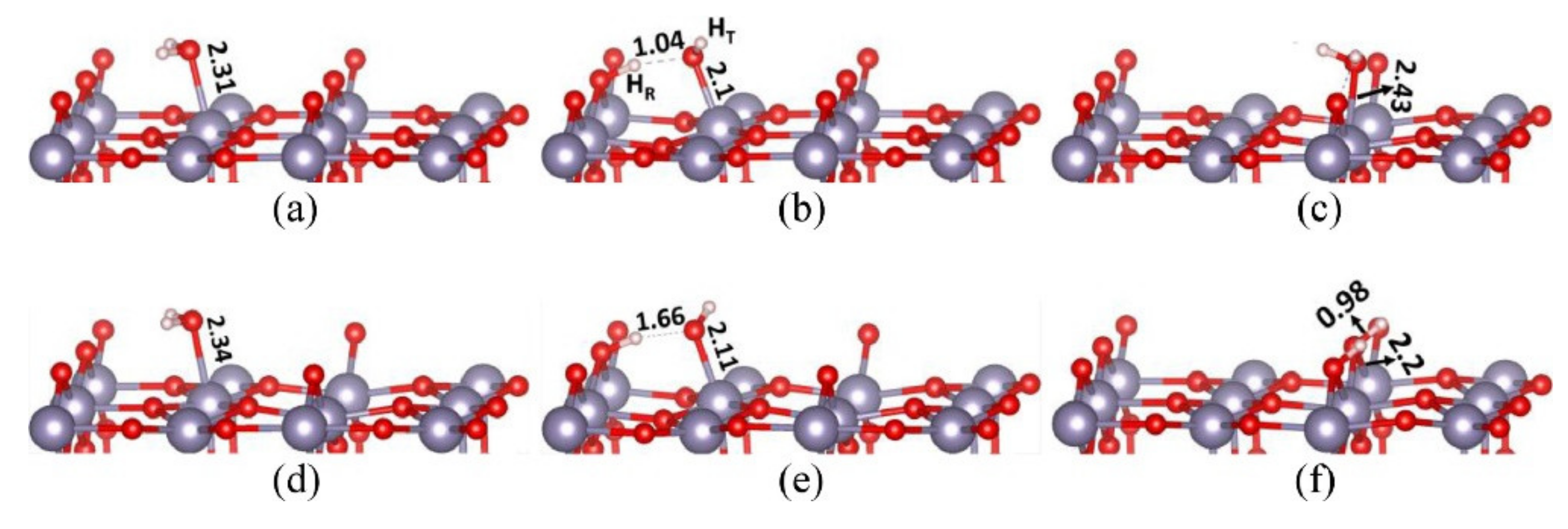
Figure 8.
TPD chromatograms of H2O and O2 desorption from SnO2. Reproduced with permission from Reference [51].
Figure 8.
TPD chromatograms of H2O and O2 desorption from SnO2. Reproduced with permission from Reference [51].

Figure 9.
Adsorption of CO onto a (101) surface of SnO2 with a terminal hydroxyl on an adjacent Sn5c site. Reproduced with permission from Reference [32].
Figure 9.
Adsorption of CO onto a (101) surface of SnO2 with a terminal hydroxyl on an adjacent Sn5c site. Reproduced with permission from Reference [32].
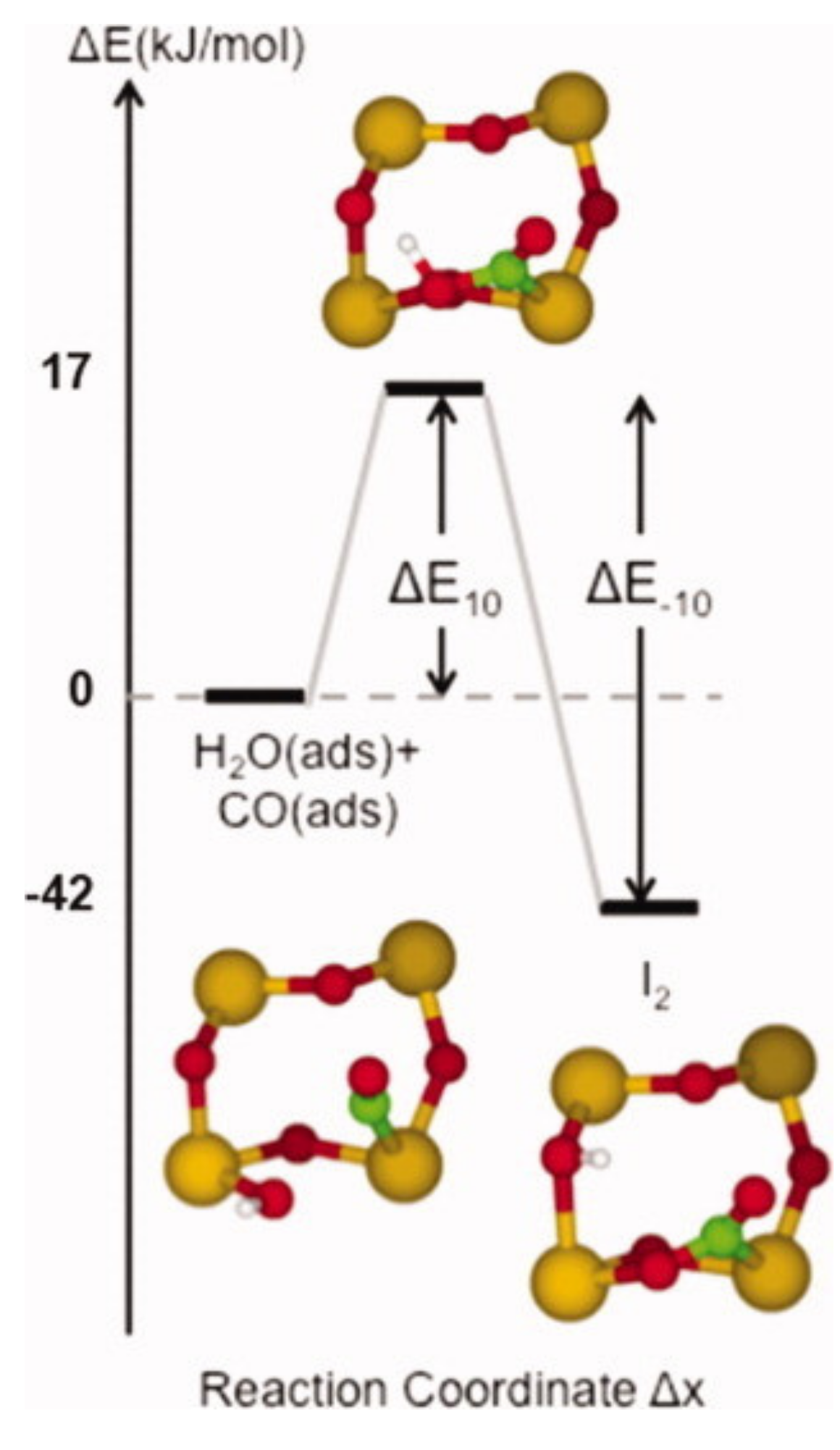
Figure 10.
Adsorption of CO2 onto a stoichiometric (101) surface of SnO2. The formed adsorbate is a tridentate carbonate-like species. Reproduced with permission from Reference [32].
Figure 10.
Adsorption of CO2 onto a stoichiometric (101) surface of SnO2. The formed adsorbate is a tridentate carbonate-like species. Reproduced with permission from Reference [32].

Figure 11.
Direct reduction of the (110) surface of SnO2 during CO adsorption. Reproduced with permission from Reference [17].
Figure 11.
Direct reduction of the (110) surface of SnO2 during CO adsorption. Reproduced with permission from Reference [17].
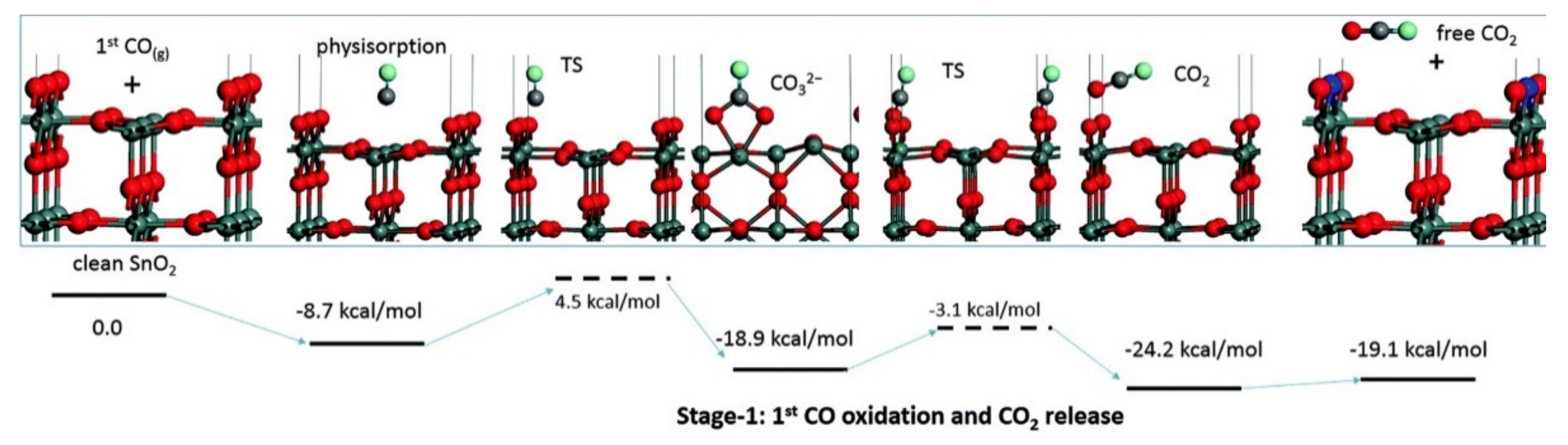
Figure 12.
Adsorption of NO2 onto (110) surface of SnO2. (a) Stoichiometric surface, monodentate adsorption onto Sn5c. (b) Reduced surface, dissociative adsorption onto a Vbr, leading to the healing of the vacancy and an adsorbed NO molecule. Reproduced with permission from Reference [18].
Figure 12.
Adsorption of NO2 onto (110) surface of SnO2. (a) Stoichiometric surface, monodentate adsorption onto Sn5c. (b) Reduced surface, dissociative adsorption onto a Vbr, leading to the healing of the vacancy and an adsorbed NO molecule. Reproduced with permission from Reference [18].
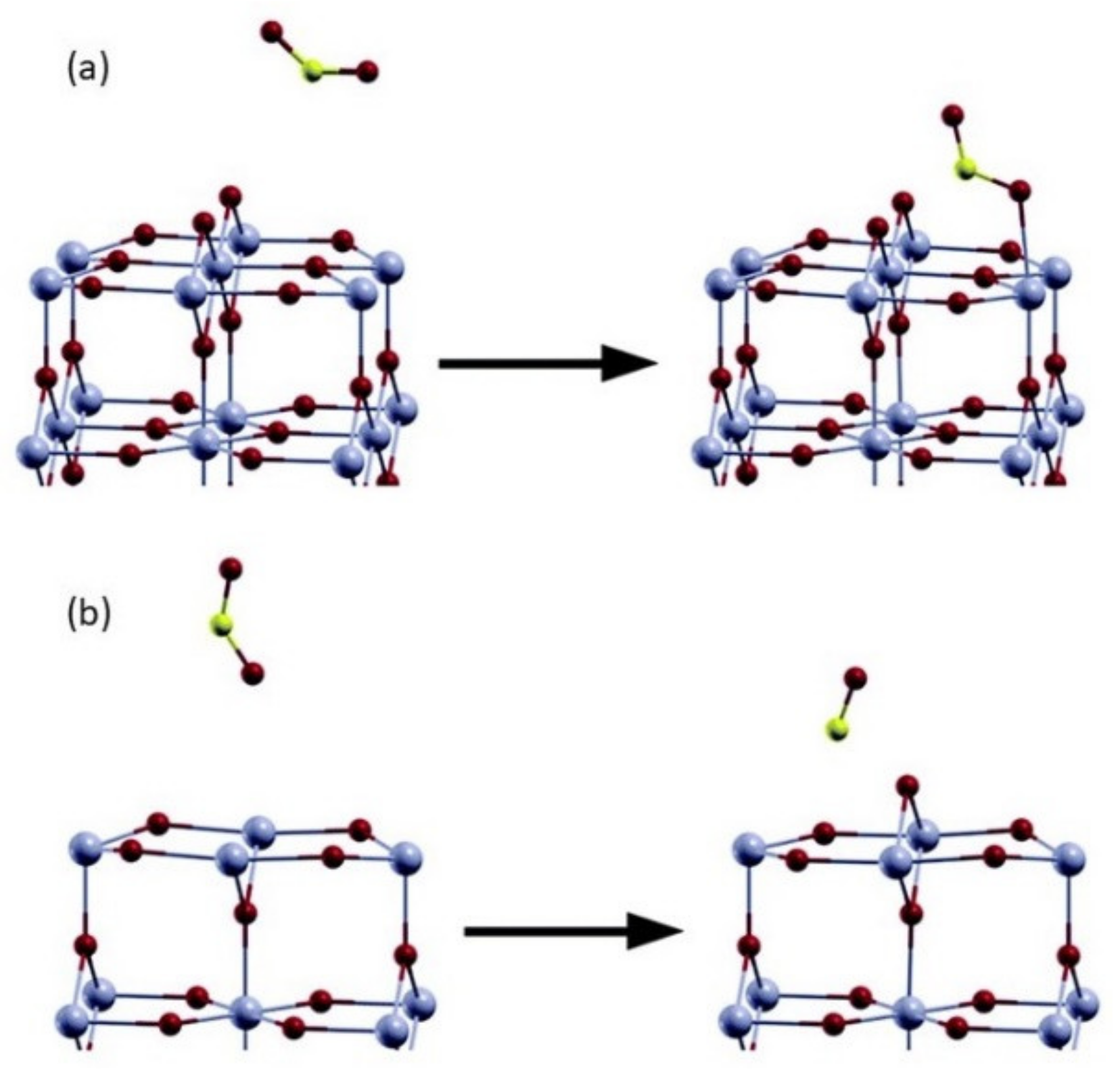
Figure 13.
Comparison of empirical and simulated NOx desorption in TPD of SnO2 after exposure to NO2. Reproduced with permission from Reference [85].
Figure 13.
Comparison of empirical and simulated NOx desorption in TPD of SnO2 after exposure to NO2. Reproduced with permission from Reference [85].
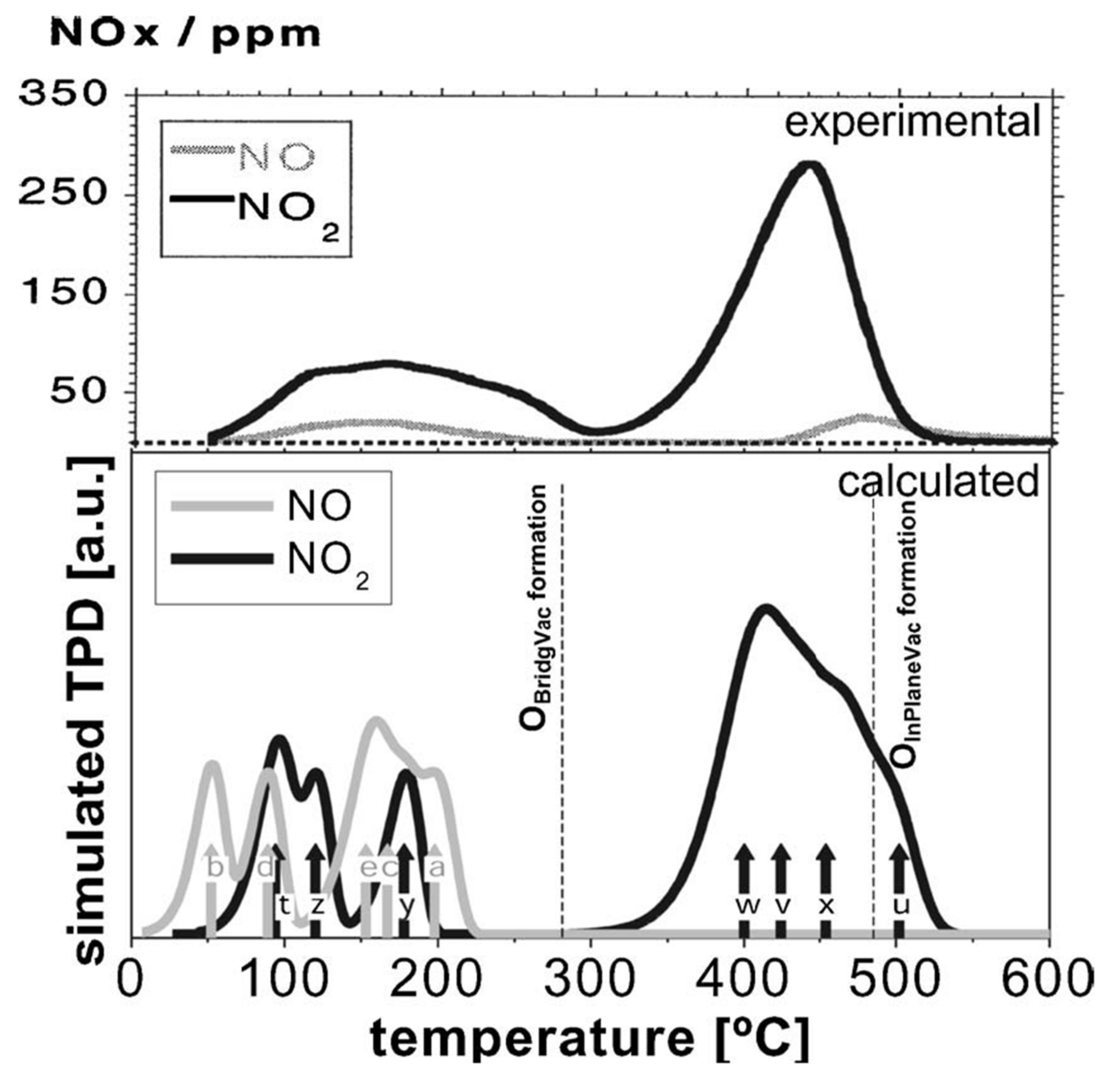

{kind=link}
{kind=link}
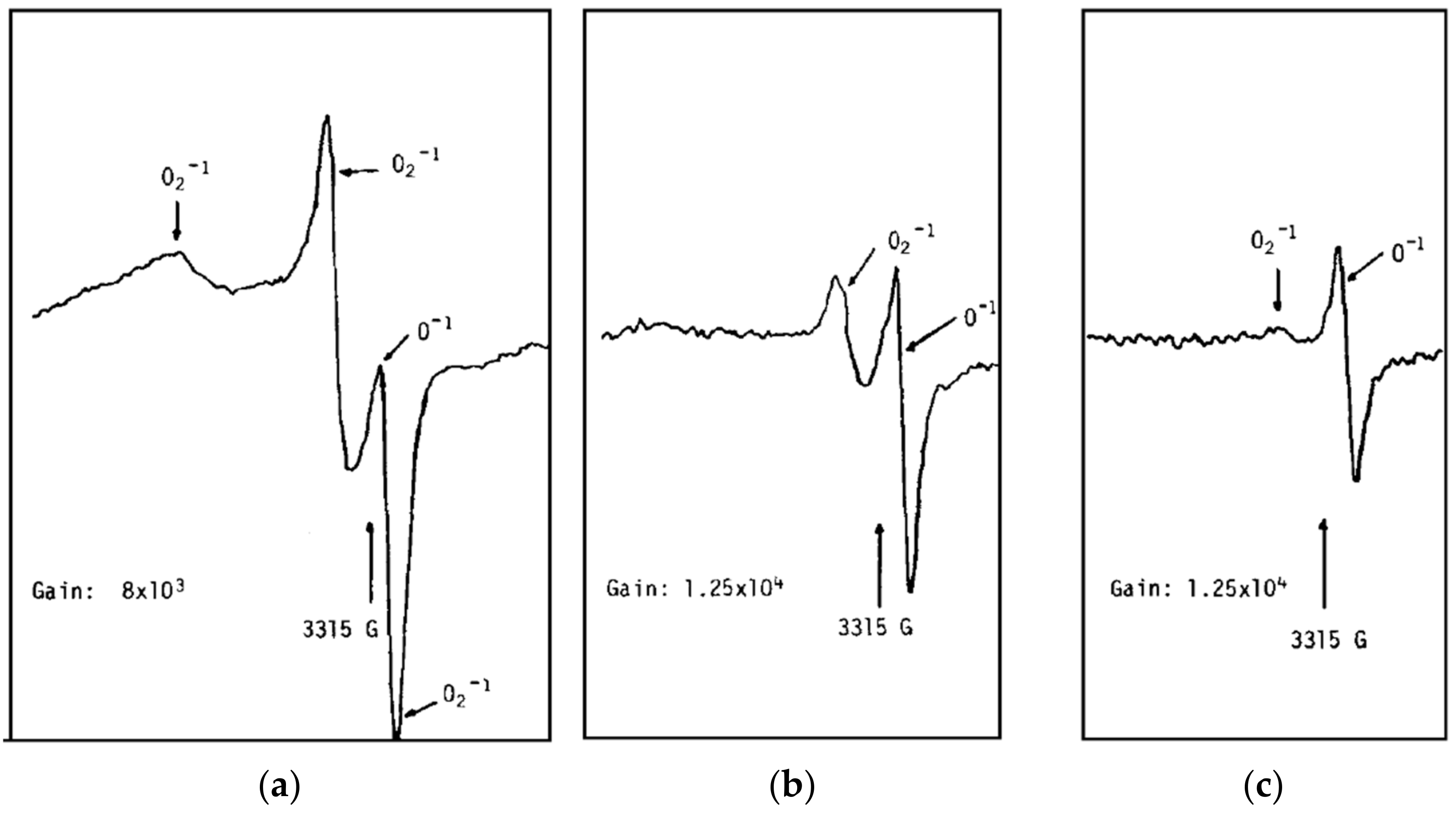{kind=link}
{kind=link}
{kind=link}
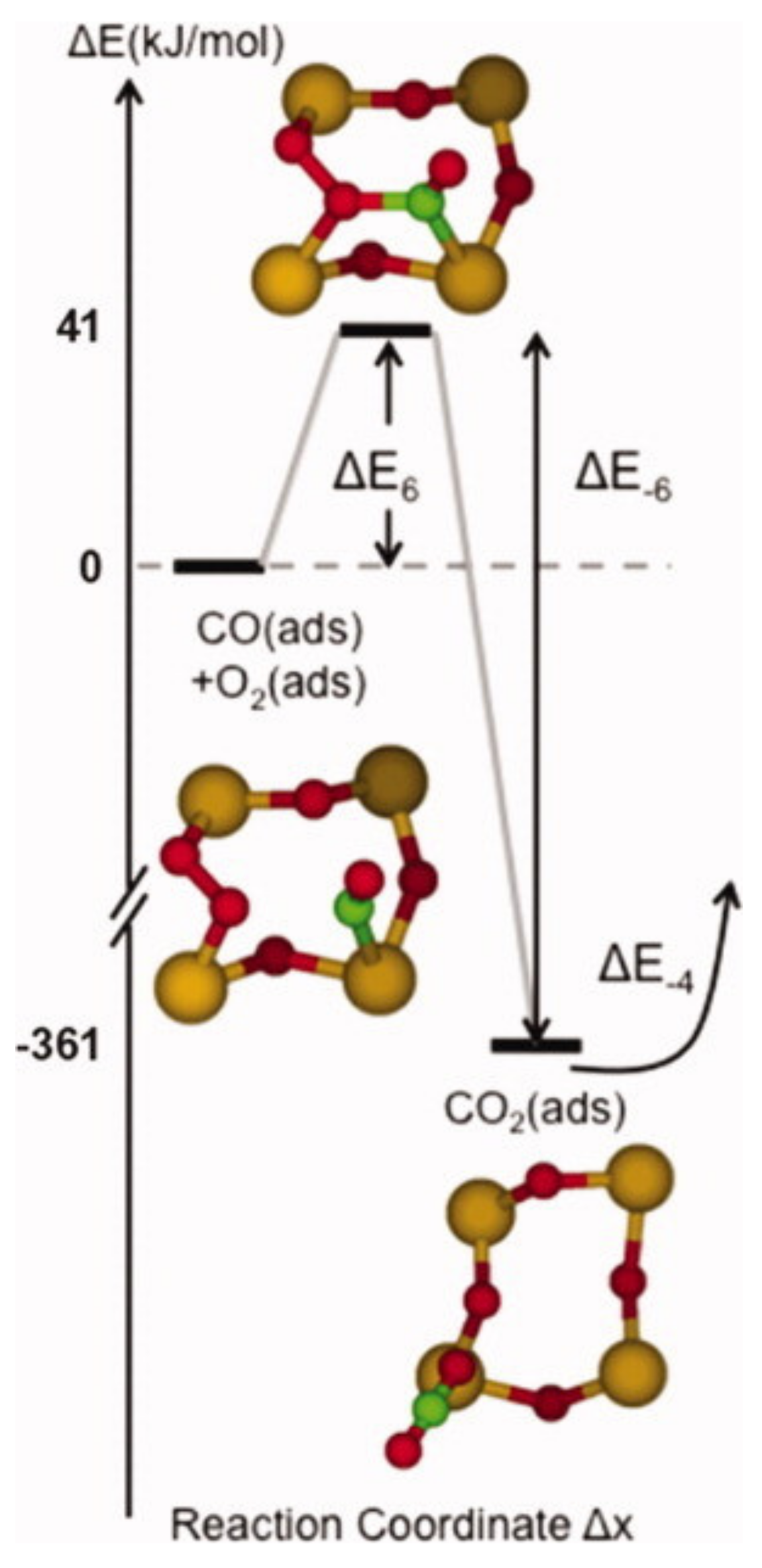{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kucharski, S.; Blackman, C. Atomistic Descriptions of Gas-Surface Interactions on Tin Dioxide. Chemosensors 2021, 9, 270. https://doi.org/10.3390/chemosensors9090270
AMA Style
Kucharski S, Blackman C. Atomistic Descriptions of Gas-Surface Interactions on Tin Dioxide. Chemosensors. 2021; 9(9):270. https://doi.org/10.3390/chemosensors9090270
Chicago/Turabian StyleKucharski, Stefan, and Chris Blackman. 2021. "Atomistic Descriptions of Gas-Surface Interactions on Tin Dioxide" Chemosensors 9, no. 9: 270. https://doi.org/10.3390/chemosensors9090270
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.