
Expediting Disulfiram Assays through a Systematic Analytical Quality by Design Approach

 ,
,  ,
, 
 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
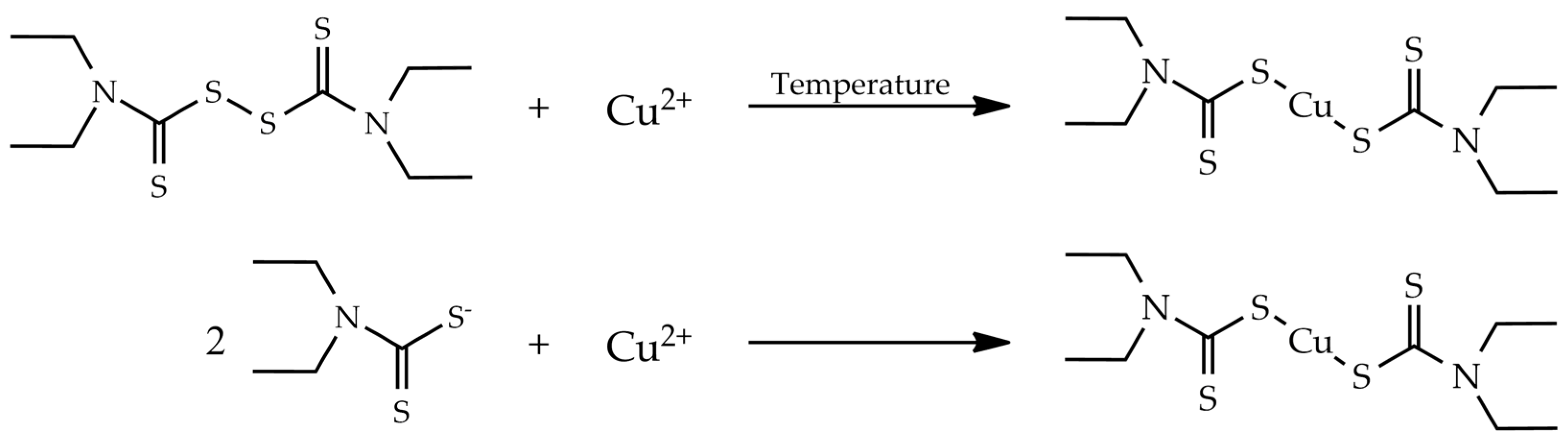
2.2. Copper (II) Diethyldithiocarbamate Synthesis
2.3. Chromatographic Instrument and Conditions
2.4. Calibration Standards and Quality Control Solutions
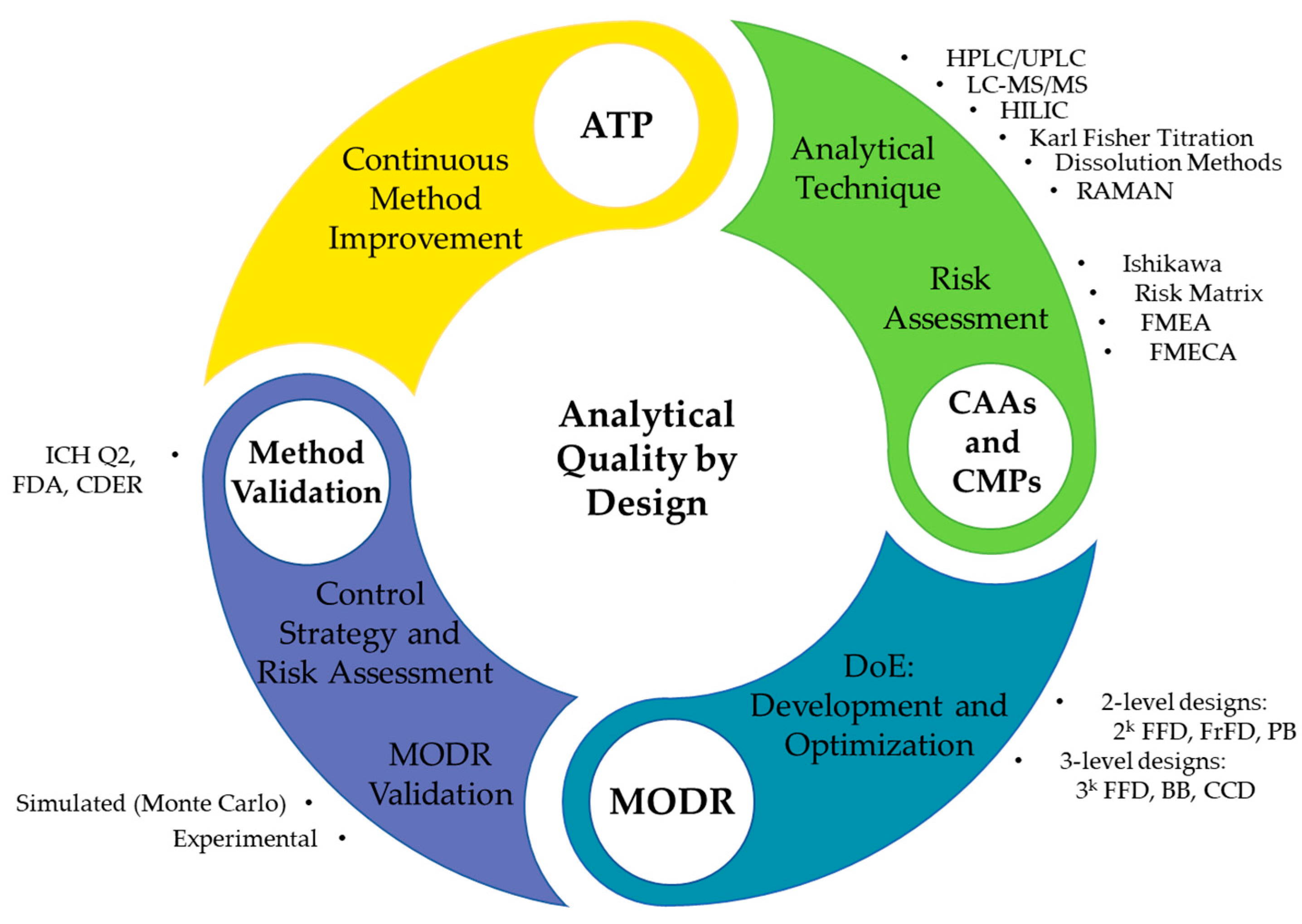
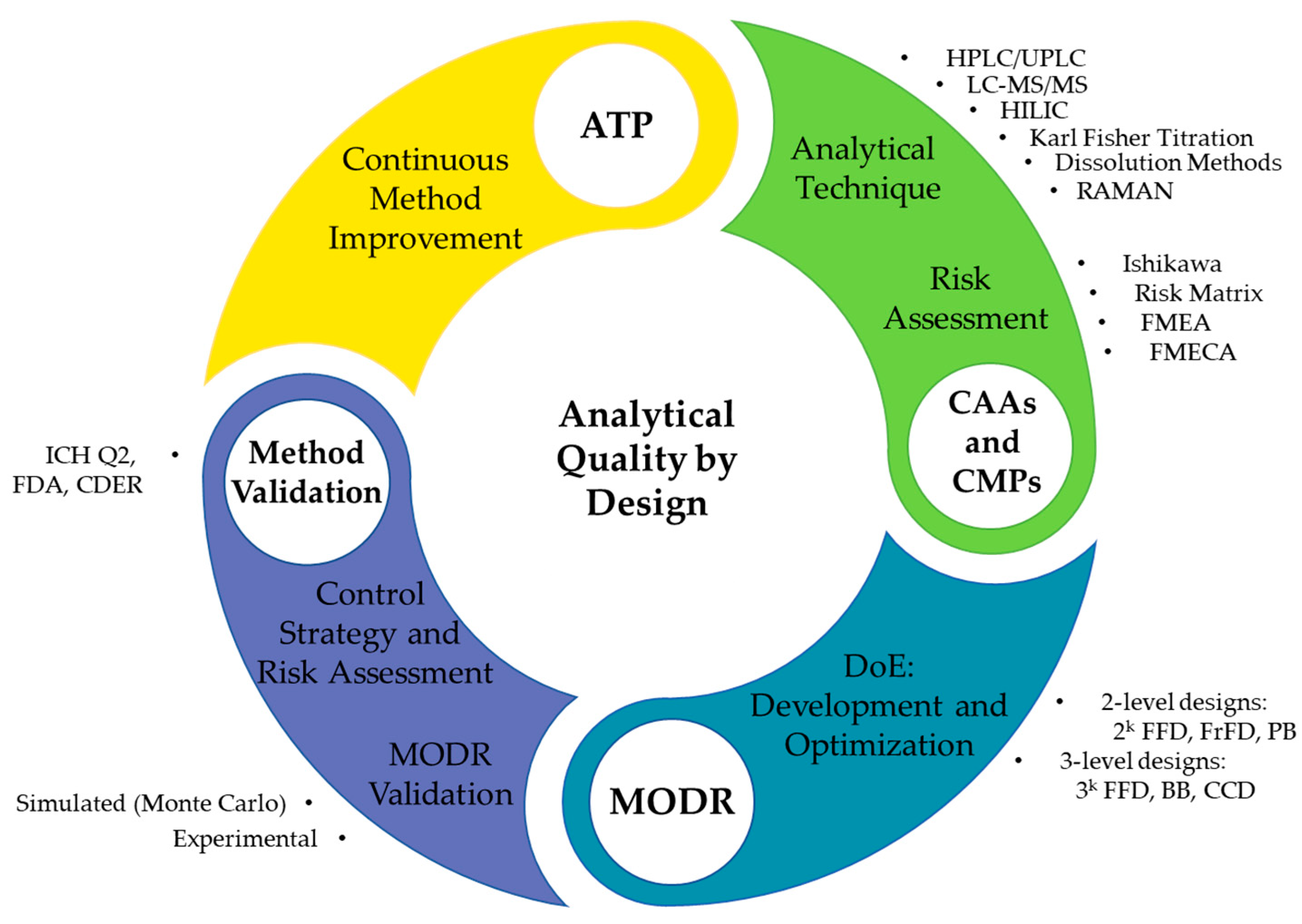
2.5. AQbD HPLC Method Development
2.5.1. Analytical Target Profile Definition
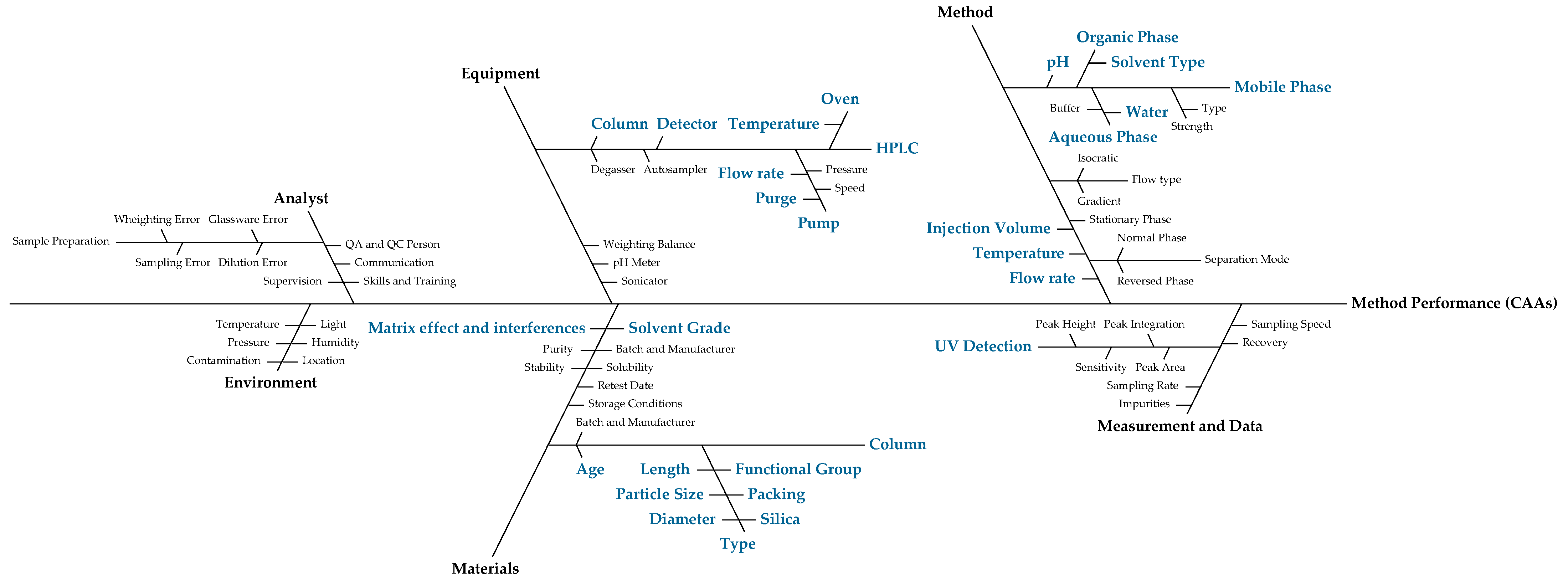
2.5.2. Risk Assessment
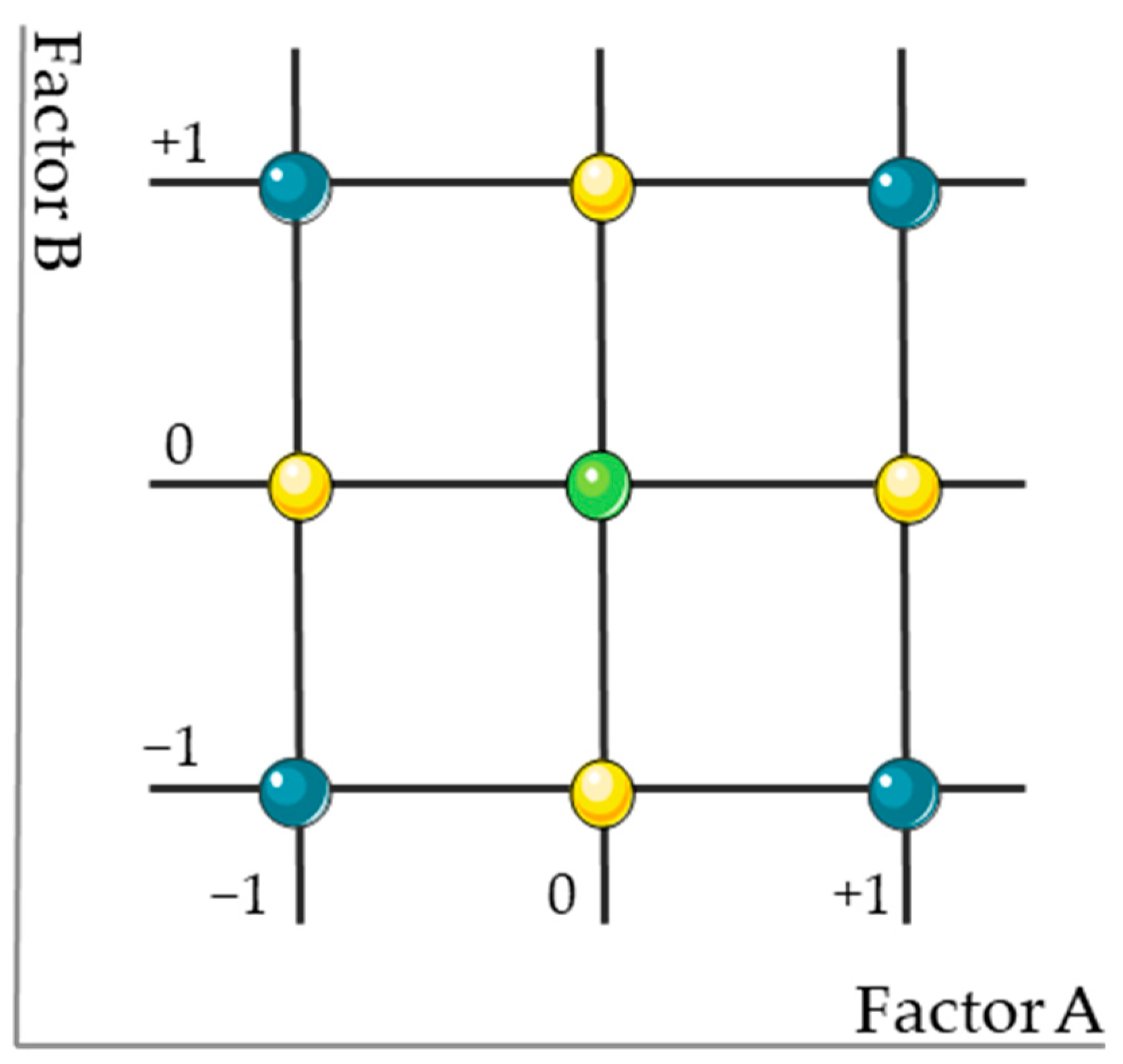
2.5.3. Method Development and Optimization
2.5.4. MODR Design and Validation
2.6. Method Validation
2.6.1. System Suitability
2.6.2. Detection and Quantification Limits
2.6.3. Linearity
2.6.4. Accuracy and Precision
2.6.5. Robustness and Ruggedness
2.6.6. Specificity
2.6.7. Stability
2.7. Method Applicability: Nanostructured Lipid Carriers
2.7.1. NLC Production
2.7.2. Entrapment Efficiency and Drug Loading Determination
3. Results and Discussion
3.1. Risk Assessment
3.2. Method Development and Optimization: Critical Analytical Attributes
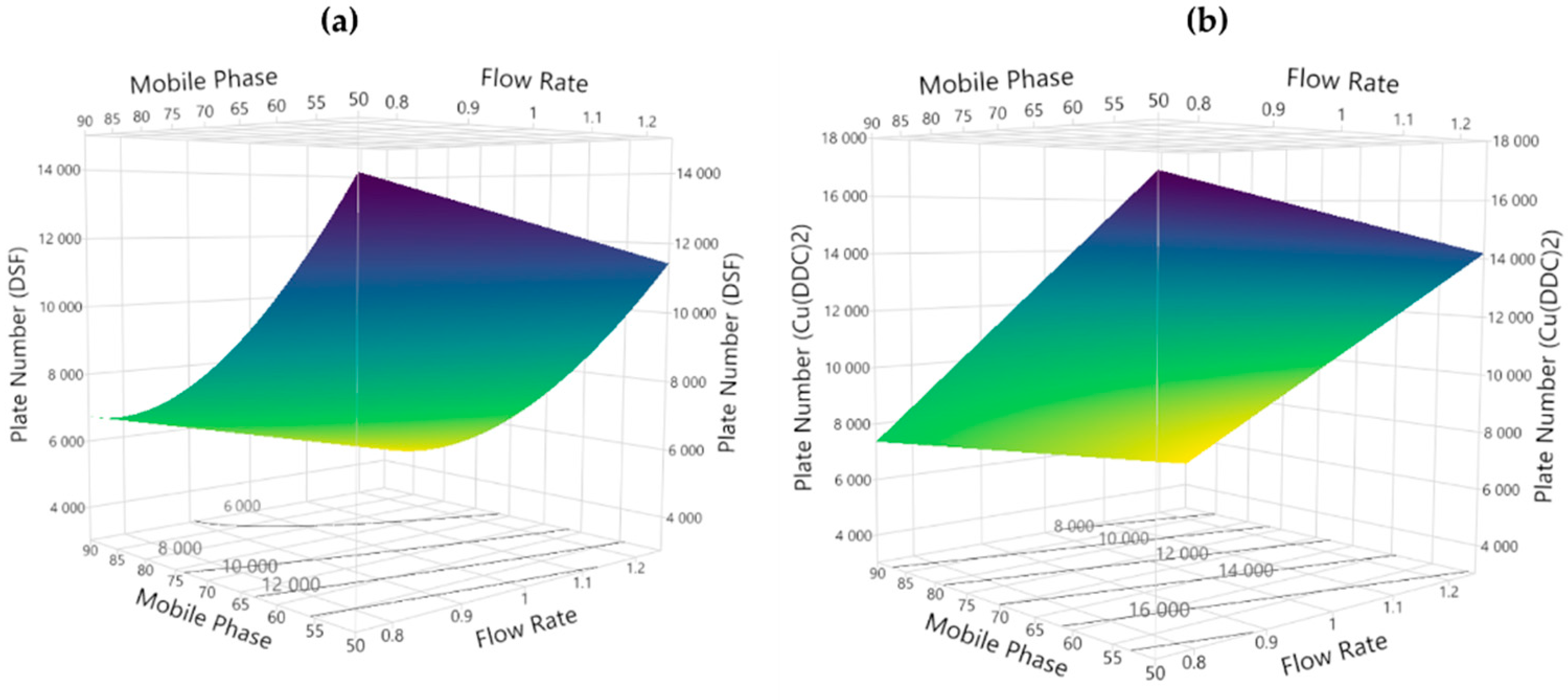
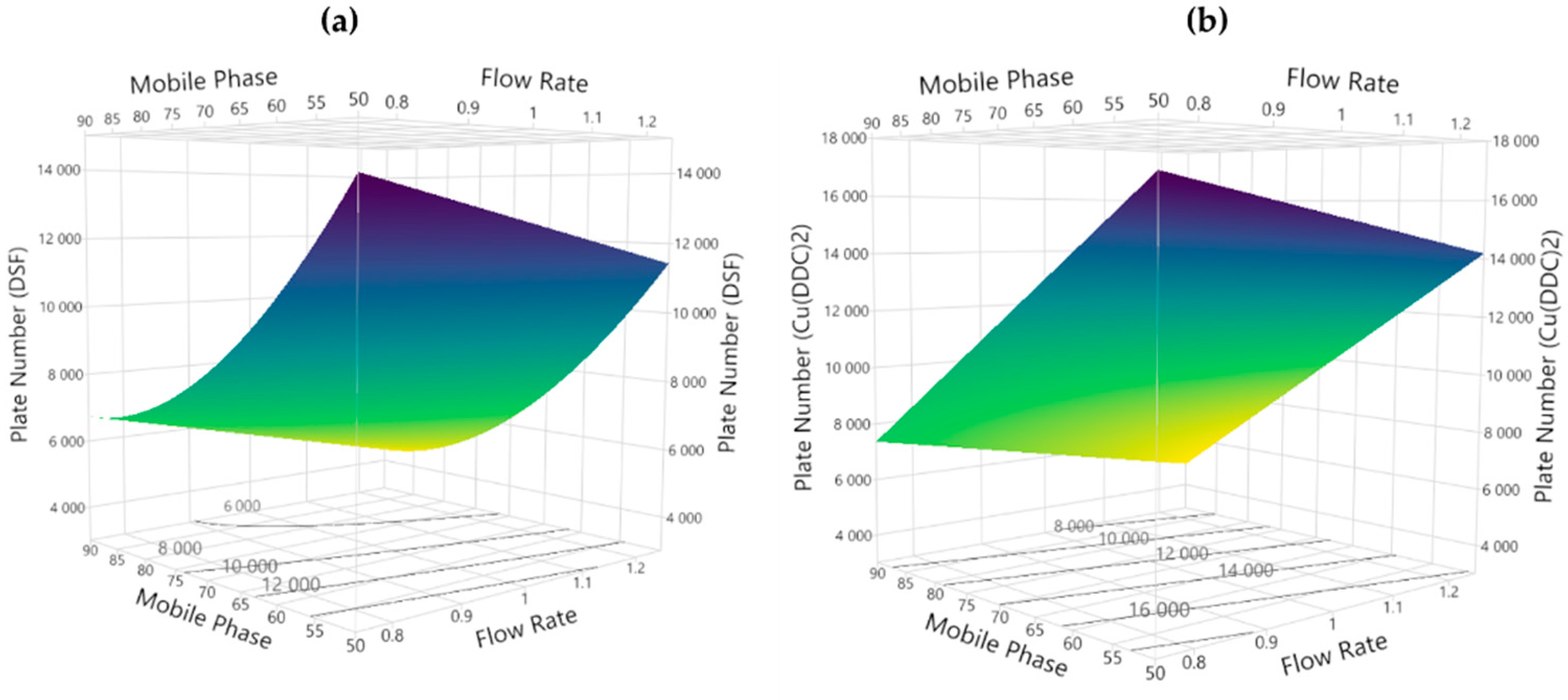
3.2.1. Theoretical Plates
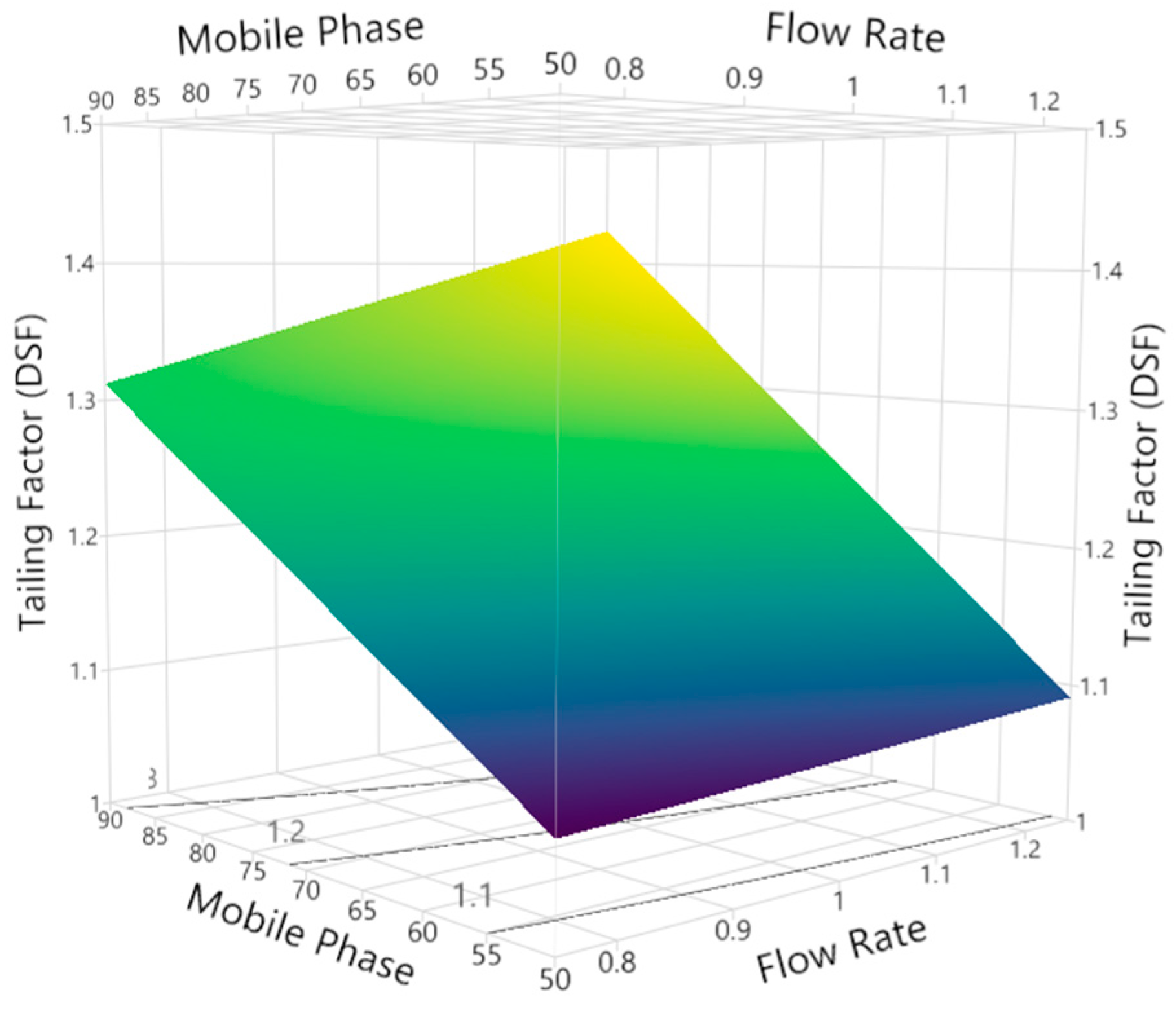
3.2.2. Tailing Factor
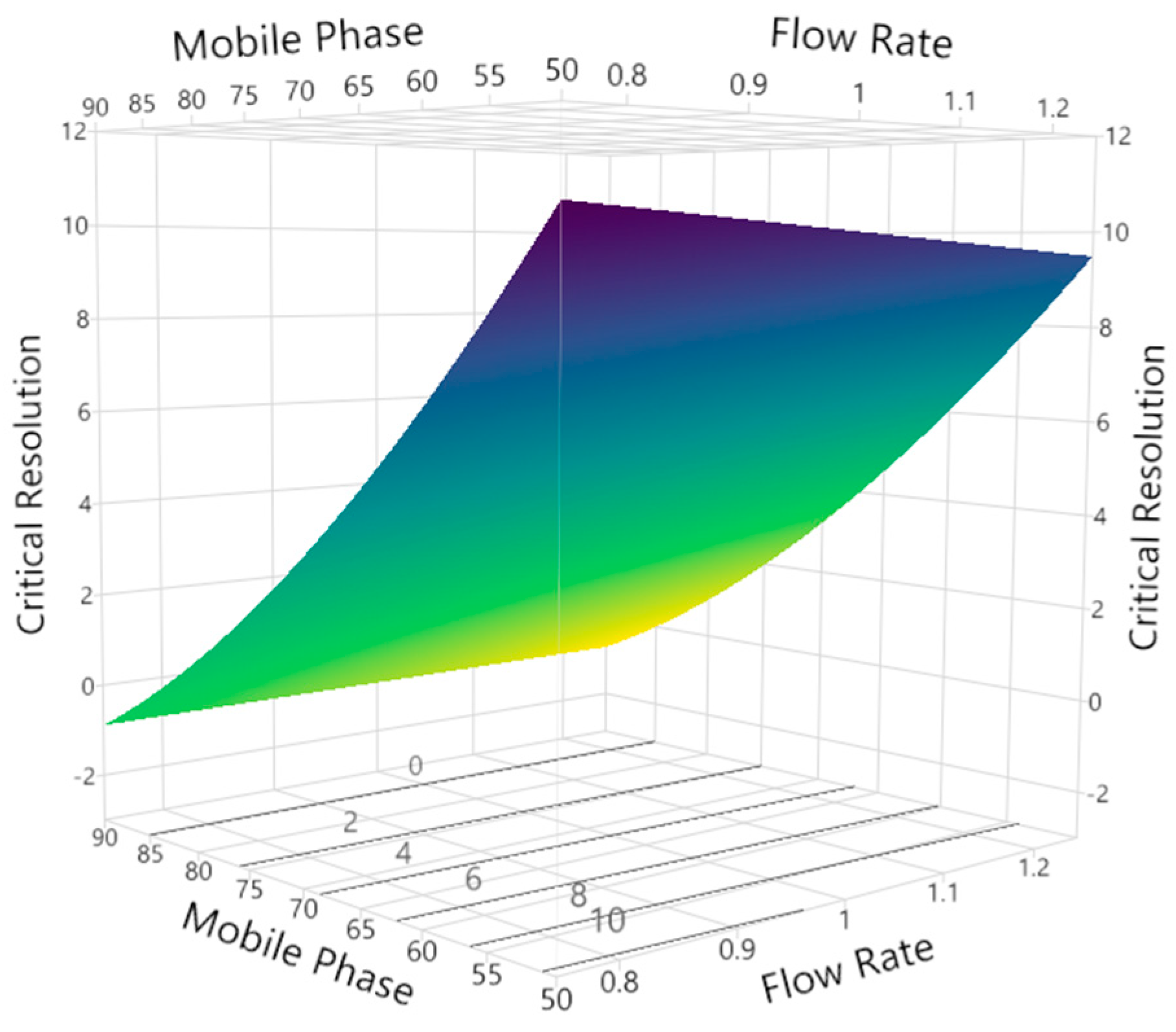
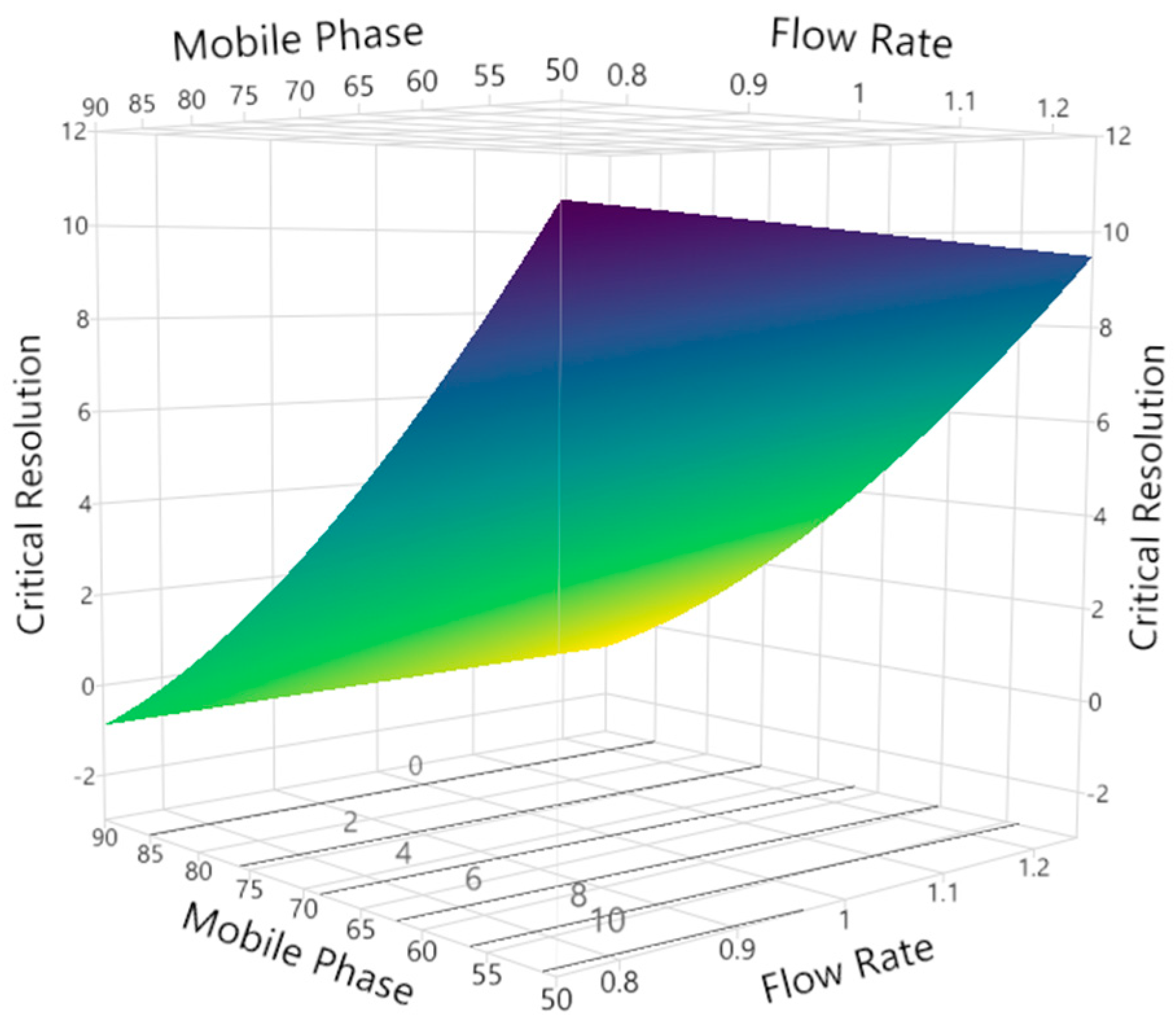
3.2.3. Critical Peak Resolution
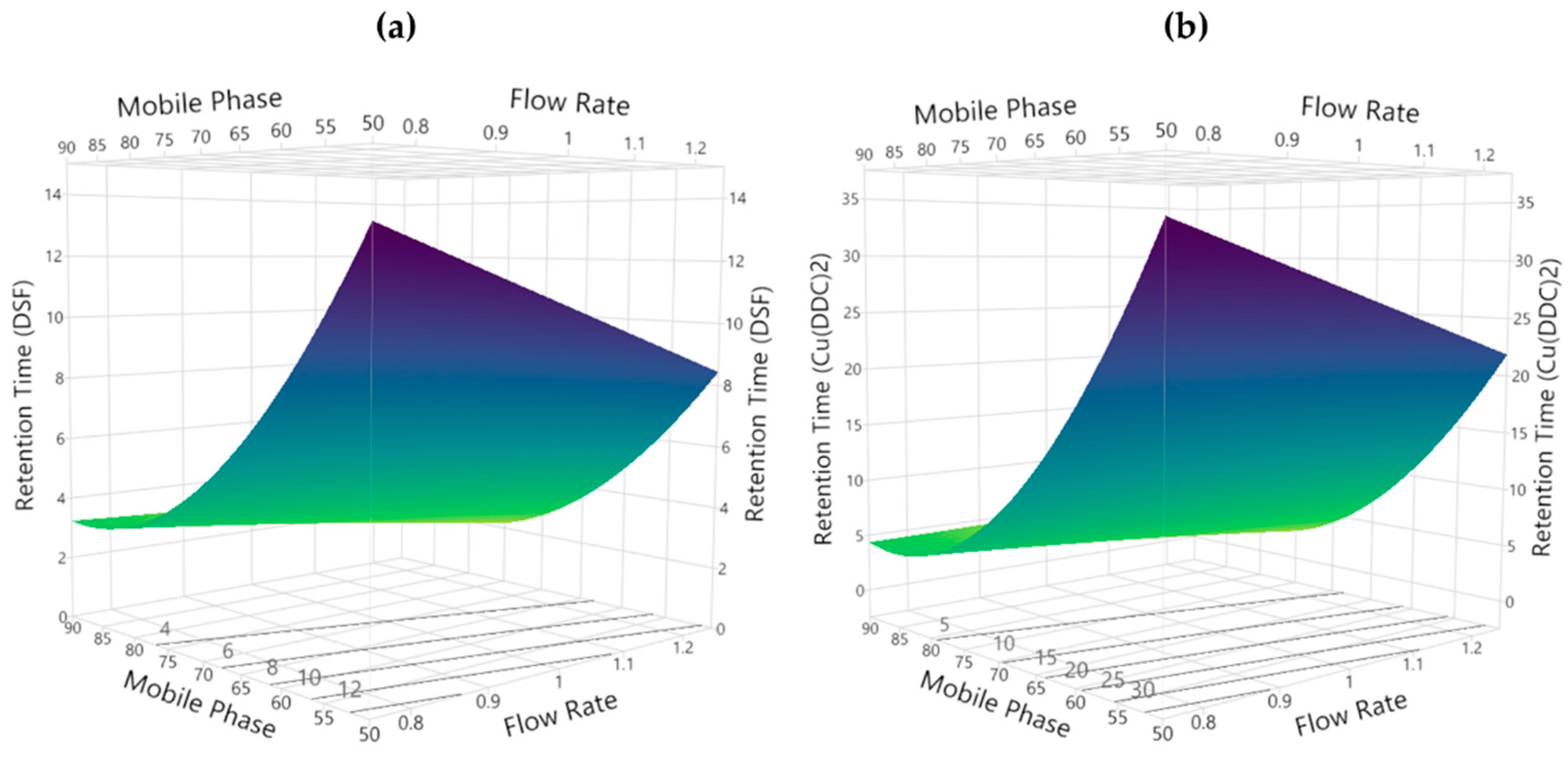
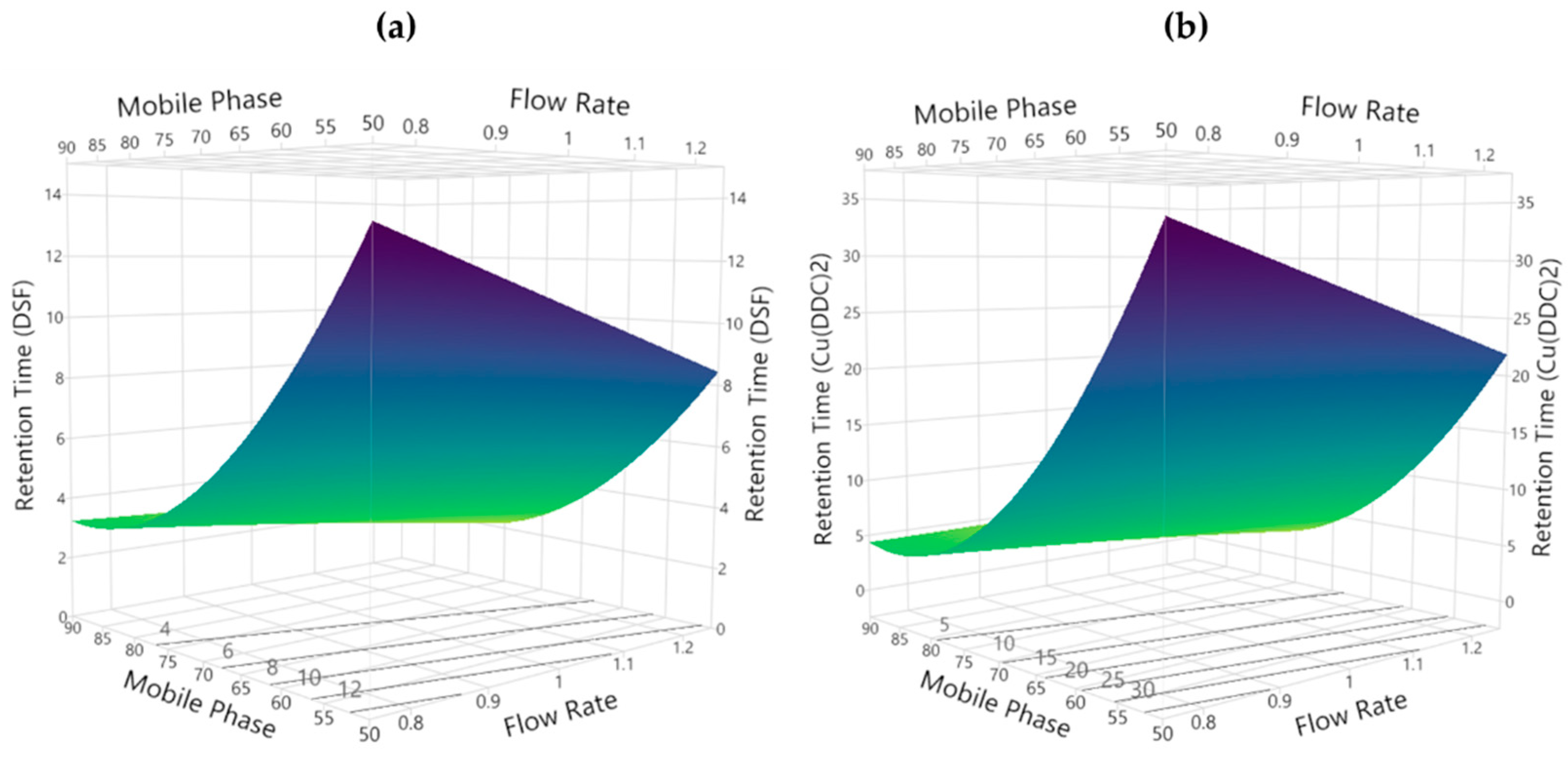
3.2.4. Retention Time
3.2.5. Capacity Factor
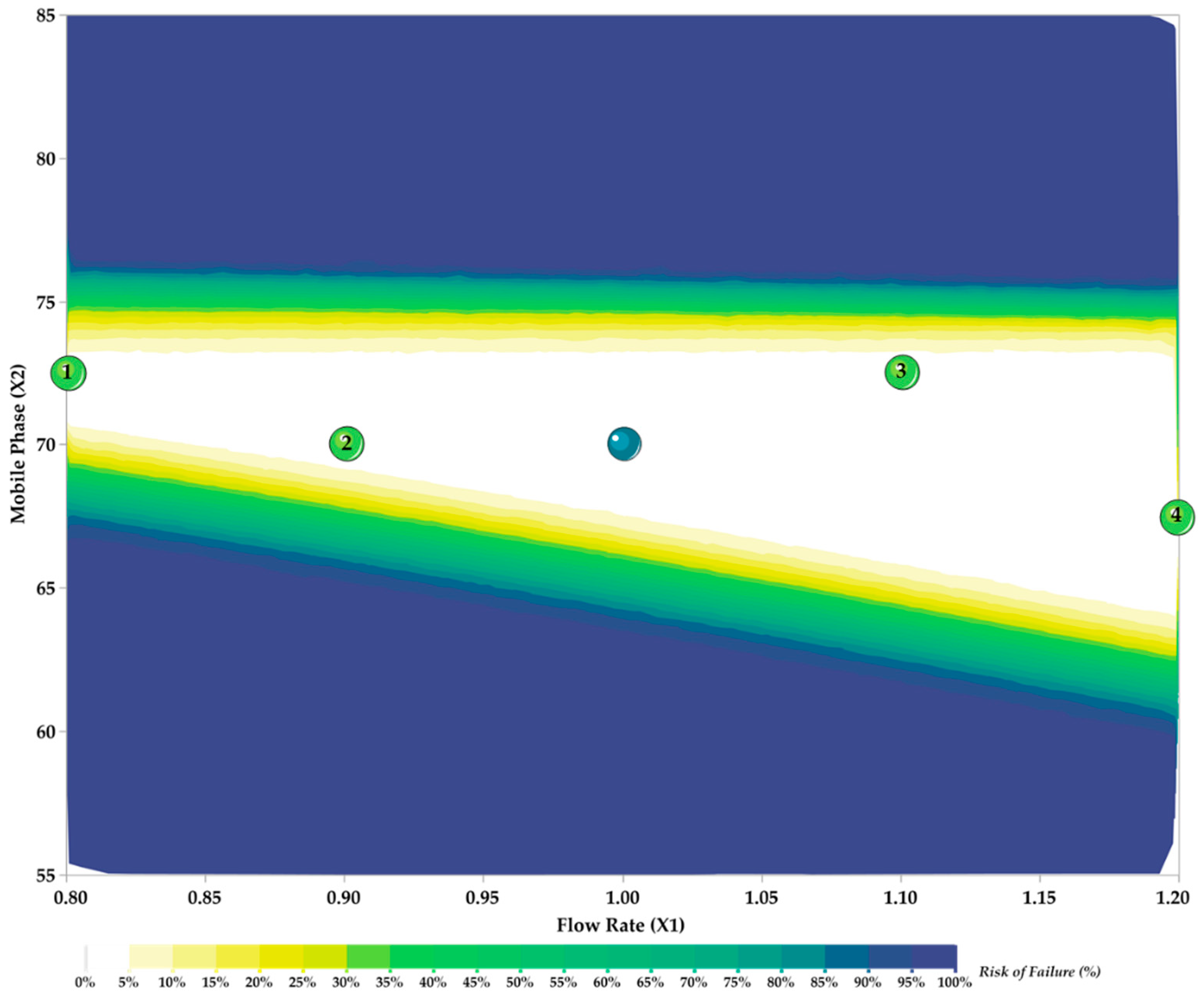
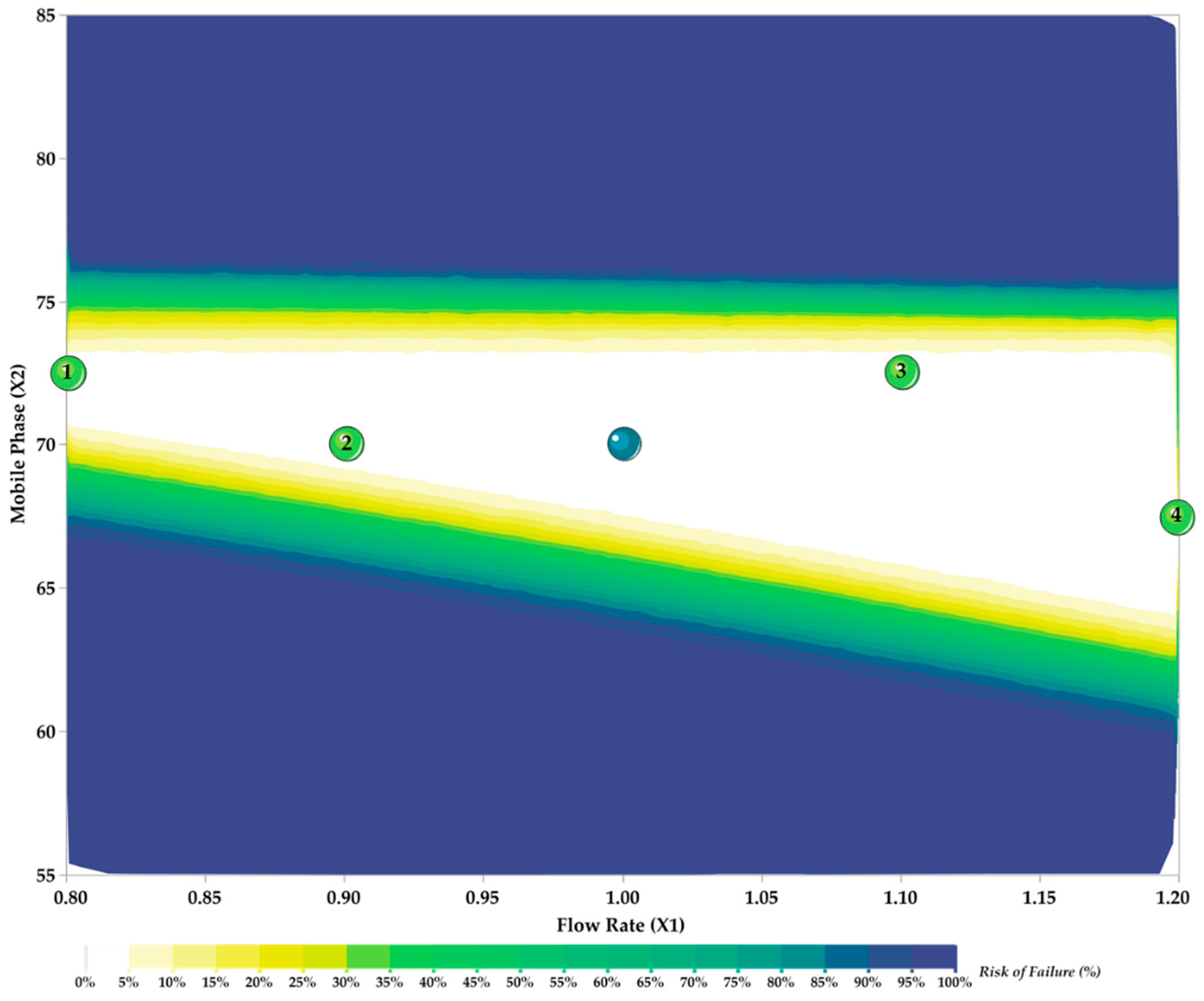
3.3. Method Development and Optimization: MODR
3.4. Method Validation
3.4.1. System Suitability
3.4.2. Detection and Quantification Limits
3.4.3. Linearity
3.4.4. Accuracy and Precision
3.4.5. Robustness and Ruggedness
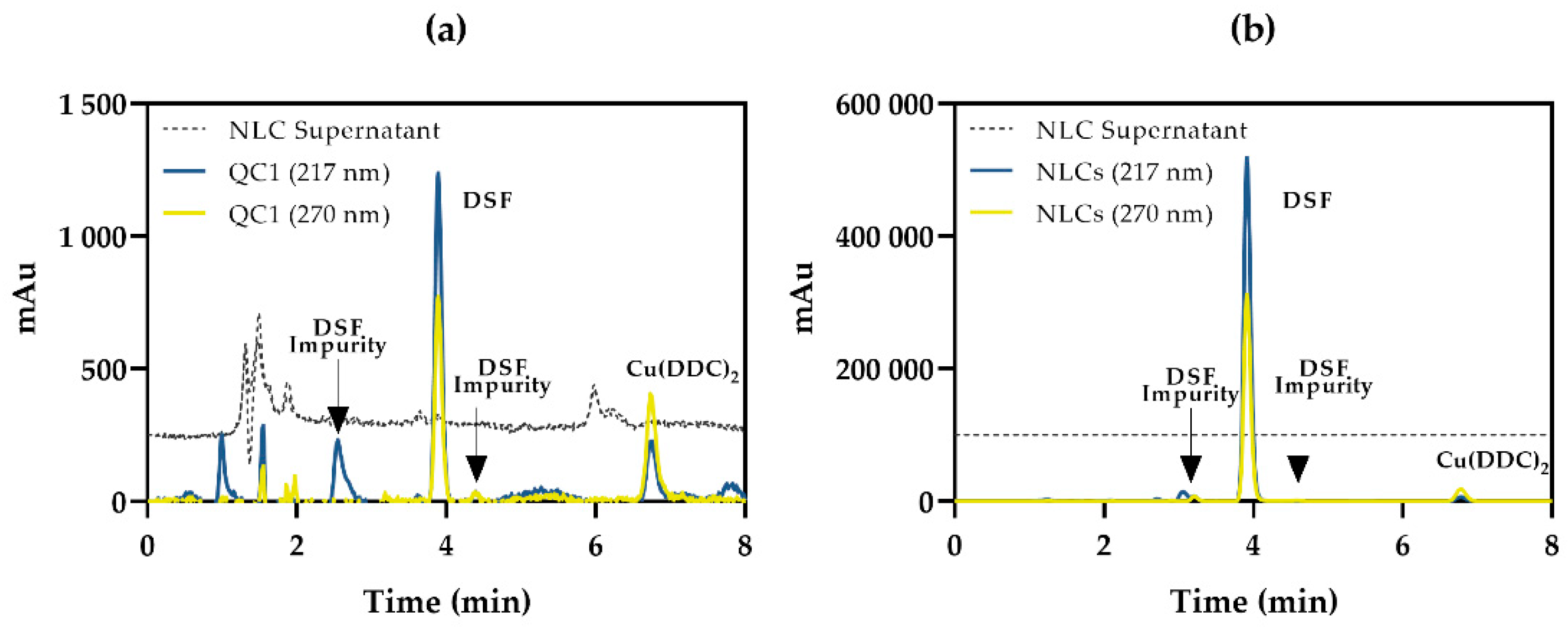
3.4.6. Specificity
3.4.7. Stability
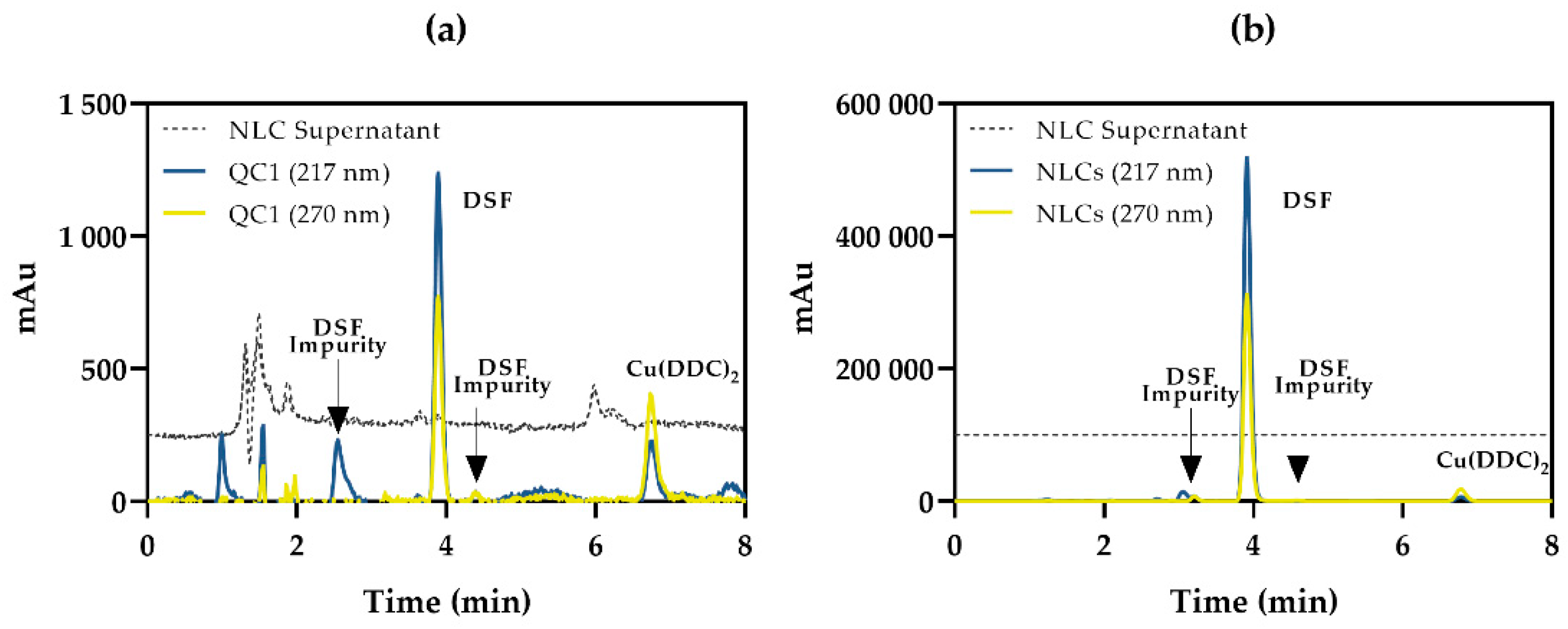
3.5. Method Applicability: Nanostructured Lipid Carriers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Mihic, S.J.; Koob, G.F.; Mayfield, J.; Harris, R.A. Chapter 23: Ethanol. In Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, 13 ed.; Brunton, L.L., Hilal-Dandan, R., Knollmann, B.C., Eds.; McGraw-Hill Education: New York, NY, USA, 2018. [Google Scholar]
- Basso, J.; Mendes, M.; Fortuna, A.; Vitorino, R.; Sousa, J.; Pais, A.; Vitorino, C. Nanotechnological approaches in cancer: The role of celecoxib and disulfiram. In Drug Repurposing in Cancer Therapy; Elsevier: Amsterdam, The Netherlands, 2020; pp. 353–393. [Google Scholar] [CrossRef]
- Cen, D.; Brayton, D.; Shahandeh, B.; Meyskens, F.L.; Farmer, P.J. Disulfiram Facilitates Intracellular Cu Uptake and Induces Apoptosis in Human Melanoma Cells. J. Med. Chem. 2004, 47, 6914–6920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehbe, M.; Anantha, M.; Backstrom, I.; Leung, A.; Chen, K.; Malhotra, A.; Edwards, K.; Bally, M.B. Nanoscale reaction vessels designed for synthesis of copper-drug complexes suitable for preclinical development. PLoS ONE 2016, 11, e0153416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said Suliman, A.; Khoder, M.; Tolaymat, I.; Webster, M.; Alany, R.G.; Wang, W.; Elhissi, A.; Najlah, M. Cyclodextrin Diethyldithiocarbamate Copper II Inclusion Complexes: A Promising Chemotherapeutic Delivery System against Chemoresistant Triple Negative Breast Cancer Cell Lines. Pharmaceutics 2021, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Irth, H.; De Jong, G.; Brinkman, U.T.; Frei, R. Determination of disulfiram and two of its metabolites in urine by reversed-phase liquid chromatography and spectrophotometric detection after post-column complexation. J. Chromatogr. B 1988, 424, 95–102. [Google Scholar] [CrossRef]
- Houson, I. Process Understanding: For Scale-up and Manufacture of Active Ingredients; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Tome, T.; Zígart, N.; Cásar, Z.; Obreza, A. Development and optimization of liquid chromatography analytical methods by using AQbD principles: Overview and recent advances. Org. Process Res. Dev. 2019, 23, 1784–1802. [Google Scholar] [CrossRef] [Green Version]
- Rozet, E.; Ziemons, E.; Marini, R.; Boulanger, B.; Hubert, P. Quality by design compliant analytical method validation. Anal. Chem 2012, 84, 106–112. [Google Scholar] [CrossRef] [PubMed]
- International Conference on Harmonization. Quality Risk Management Q9; ICH: London, UK, 2005; pp. 1–23. [Google Scholar]
- Sahu, P.K.; Ramisetti, N.R.; Cecchi, T.; Swain, S.; Patro, C.S.; Panda, J. An overview of experimental designs in HPLC method development and validation. J. Pharm. Biomed. Anal. 2018, 147, 590–611. [Google Scholar] [CrossRef] [PubMed]
- International Conference on Harmonization. Pharmaceutical Development Q8 R2; ICH: London, UK, 2009; pp. 1–28. [Google Scholar]
- Rozet, E.; Lebrun, P.; Hubert, P.; Debrus, B.; Boulanger, B. Design spaces for analytical methods. Trends Analyt Chem 2013, 42, 157–167. [Google Scholar] [CrossRef]
- Peraman, R.; Bhadraya, K.; Padmanabha Reddy, Y. Analytical quality by design: A tool for regulatory flexibility and robust analytics. Int. J. Anal. Chem. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Duan, H.; Wei, D.; Cui, B.; Wang, X. Stability of diethyl dithiocarbamate chelates with Cu (II), Zn (II) and Mn (II). J. Mol. Struct. 2019, 1184, 375–381. [Google Scholar] [CrossRef]
- Panda, S.S.; Ravi Kumar Bera, V.V.; Beg, S.; Mandal, O. Analytical quality by design (AQbD)-oriented RP-UFLC method for quantification of lansoprazole with superior method robustness. J Liq. Chrom. Relat. Tech. 2017, 40, 479–485. [Google Scholar] [CrossRef]
- Singh, B.; Kumar, R.; Ahuja, N. Optimizing drug delivery systems using systematic “design of experiments.” Part I: Fundamental aspects. Crit. Rev. Ther. Drug Carrier Syst. 2005, 22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, C. Application of quality by design concept to develop a dual gradient elution stability-indicating method for cloxacillin forced degradation studies using combined mixture-process variable models. J. Chromatogr. A 2017, 1514, 44–53. [Google Scholar] [CrossRef] [PubMed]
- International Conference on Harmonization. Validation of Analytical Procedures: Text and Methodology Q2(R1); ICH: London, UK, 2005; pp. 1–17. [Google Scholar]
- Food and Drug Administration. Analytical Procedures and Methods Validation for Drugs and Biologics; FDA: Silver Spring, MD, USA, 2015; pp. 1–18. [Google Scholar]
- Center for Drug Evaluation and Research. Validation of Chromatographic Methods; FDA: Silver Spring, MD, USA, 1994; pp. 1–33. [Google Scholar]
- Food and Drug Administration. Bioanalytical Method Validation; FDA: Silver Spring, MD, USA, 2018; pp. 1–44. [Google Scholar]
- Almeida, A.M.D.; Castel-Branco, M.M.; Falcao, A. Linear regression for calibration lines revisited: Weighting schemes for bioanalytical methods. J. Chromatogr. B 2002, 774, 215–222. [Google Scholar] [CrossRef]
- USP Validation of Compendial Methods. In United States Pharmacopeia and National Formulary; US Pharmcopeia: Rockville, MD, USA, 2005; pp. 2748–2751.
- Basso, J.; Mendes, M.; Cova, T.F.; Sousa, J.J.; Pais, A.A.; Vitorino, C. Analytical Quality by Design (AQbD) as a multiaddressable platform for co-encapsulating drug assays. Anal. Methods 2018, 10, 5659–5671. [Google Scholar] [CrossRef]
- Mendes, M.; Basso, J.; Silva, J.; Cova, T.; Sousa, J.; Pais, A.; Vitorino, C. Biomimeting ultra-small lipid nanoconstructs for glioblastoma treatment: A computationally guided experimental approach. Int. J. Pharm. 2020, 587, 119661. [Google Scholar] [CrossRef] [PubMed]
- Stamatis, D.H. Failure Mode and Effect Analysis: FMEA from Theory to Execution; American Society for Quality, Quality Press: Seattle, WA, USA, 2003. [Google Scholar]
- Dong, M.W. HPLC and UHPLC for Practicing Scientists; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- González, O.; Blanco, M.E.; Iriarte, G.; Bartolomé, L.; Maguregui, M.I.; Alonso, R.M. Bioanalytical chromatographic method validation according to current regulations, with a special focus on the non-well defined parameters limit of quantification, robustness and matrix effect. J. Chromatogr. A 2014, 1353, 10–27. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ATP Element | Target | Justification |
|---|---|---|
| Analyte | DSF, Cu(DDC)2 | Development of an analytical method for the estimation of the analytes in solution or co-encapsulated in nanosystems for routine and stability analysis. |
| Sample | Liquid | Analytes must be suitably dissolved, with complete solvent miscibility. |
| Analyticaltechnique | RP-HPLC | Highly lipophilic analytes are better separated and eluted in RP-HPLC methods due to the increased retention with the non-polar C18 stationary phase. Apart from the increased resolution, RP-HPLC provides a fast analysis with small sample volume and organic solvent consumption. |
| Instrument | HPLC with a quaternary pump, autosampler, and PDA detector | A quaternary pump allows an improved mixing of the mobile phase solvents and higher resolution, whereas the PDA detector allows the detection of several compounds at their λmax, thus maximizing sensitivity (DSF, λmax = 217 nm, Cu(DDC)2, λmax = 270 nm). |
| Method | Specific, accurate, precise, linear, reproducible, robust, cost-effective, and simple | The method should comply with formal validation criteria, presenting a short run time and reduced use of organic solvents. |
| Application | Assay | Method should be able to determine DSF and Cu(DDC)2 in solution or co-encapsulated in nanosystems for routine and stability analysis. |
| CAAs | Number of theoretical plates, retention time, tailing factor, critical peak resolution, capacity factor | Chromatographic parameters that allow a robust and reliable determination of the analytes. Should meet their formal and commonly accepted quality criteria. |
| S and O Score | Criteria |
| 1 | Negligible risk that does not require attention |
| 2–3 | Minor effect that can be easily corrected inline |
| 4–5 | Moderately severe effect that requires attention |
| 6–7 | Highly severe effect that requires particular attention |
| D Score | Criteria |
| 1 | Easily detectable, negligible risk that does not require attention |
| 2–3 | Detectable, with a minor effect that can be easily corrected inline |
| 4–5 | Not easily detectable, presents a moderate risk that requires immediate attention |
| 6–7 | Difficult to detect, presents a severe effect that requires the utmost attention |
| Failure Mode | Effect | S | O | D | RPN | Strategy |
|---|---|---|---|---|---|---|
| Mobile Phase Composition | Multiple | 7 | 5 | 7 | 245 | Evaluate through RSM |
| Column age | Non-selectivity | 6 | 3 | 3 | 54 | Monitor column use |
| Column equilibration | Extraneous peaks | 3 | 3 | 3 | 27 | Guarantee equilibration prior to analysis |
| Column type | Retention variation | 5 | 5 | 2 | 50 | Guarantee ATP compliance |
| Flow Rate | Multiple effects | 6 | 5 | 6 | 180 | Evaluate through RSM |
| Injection Volume | Sensitivity | 4 | 2 | 3 | 24 | Guarantee method compliance |
| Matrix effect | Extraneous peaks | 3 | 3 | 2 | 18 | Guarantee ATP compliance |
| Mobile Phase pH | Retention variation | 2 | 1 | 6 | 12 | Low risk, no actions taken |
| Oven Temperature | Column pressure | 3 | 2 | 2 | 12 | Low risk, no actions taken |
| Purge | Multiple | 3 | 2 | 4 | 24 | Guarantee purge prior to analysis |
| Solvent Grade | Extraneous peaks | 3 | 3 | 2 | 18 | Guarantee ATP compliance |
| UV Detection | Sensitivity | 4 | 2 | 1 | 8 | Guarantee ATP compliance |
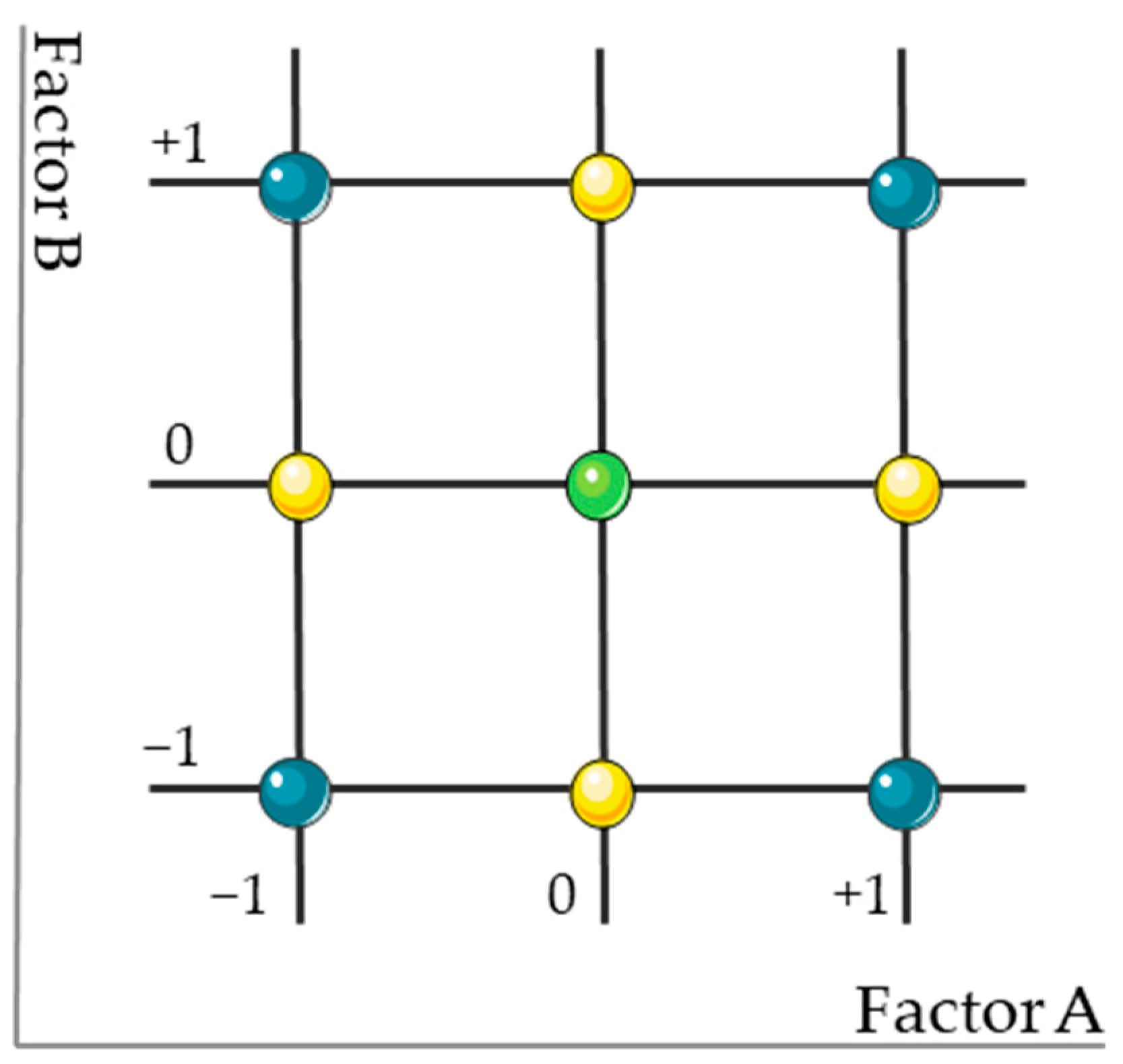
| Run | Code | X1 (Flow Rate, mL/min) | X2 (Mobile Phase, % ACN) | |
|---|---|---|---|---|
| 1 | Factorial Design Points | − − | 0.8 | 55 |
| 2 | − + | 0.8 | 85 | |
| 3 | + − | 1.2 | 55 | |
| 4 | + + | 1.2 | 85 | |
| 5 | Central Point | 0 0 | 1.0 | 70 |
| 6 | Axial Points | −α 0 | 0.8 | 70 |
| 7 | 0 −α | 1.0 | 55 | |
| 8 | +α 0 | 1.2 | 70 | |
| 9 | 0 +α | 1.0 | 85 |
| CAA | N DSF | N Cu(DDC)2 | Tf DSF | Tf Cu(DDC)2 | Res | Rt DSF | Rt Cu(DDC)2 | k’ DSF | k’ Cu(DDC)2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Specification Conditions | >2000 | >2000 | <2.0 | <2.0 | >2.0 | <6.0 | <10.0 | >2.0 | >2.0 | |
| Predicted | 1 | 7916 | 11,173 | 1.21 | 1.21 | 2.83 | 4.52 | 7.50 | 2.40 | 4.25 |
| 2 | 7860 | 11,260 | 1.21 | 1.19 | 3.44 | 4.55 | 8.01 | 2.82 | 5.46 | |
| 3 | 6507 | 9694 | 1.26 | 1.21 | 2.65 | 3.12 | 4.75 | 2.40 | 4.25 | |
| 4 | 7436 | 10,943 | 1.21 | 1.15 | 4.59 | 3.94 | 7.66 | 3.89 | 8.67 | |
| Confidence Interval (CI, 95%) | 1 | [7251, 8580] | [10,323, 12,023] | [1.21, 1.22] | [1.18, 1.24] | [2.80, 2.89] | [4.32, 4.72] | [6.92, 8.08] | [2.26, 2.53] | [3.89, 4.60] |
| 2 | [7276, 8443] | [10,635, 11,886] | [1.21, 1.22] | [1.16, 1.22] | [3.41, 3.48] | [4.37, 4.72] | [7.51, 8.52] | [2.68, 2.95] | [5.10, 5.83] | |
| 3 | [5935, 7080] | [9060, 10,329] | [1.25, 1.26] | [1.18, 1.24] | [2.62, 2.68] | [2.94, 3.29] | [4.25, 5.24] | [2.26, 2.53] | [3.89, 4.60] | |
| 4 | [6797, 8075] | [10,073, 11,814] | [1.21, 1.22] | [1.12, 1.18] | [4.55, 4.62] | [3.74, 4.14] | [7.09, 8.22] | [3.77, 4.02] | [8.33, 9.00] | |
| Experimental | 1 | 8051 | 10,440 | 1.22 | 1.22 | 2.86 | 4.45 | 7.32 | 2.35 | 4.17 |
| 2 | 7356 | 11,721 | 1.22 | 1.20 | 3.49 | 4.42 | 7.46 | 2.77 | 5.47 | |
| 3 | 6837 | 8996 | 1.26 | 1.19 | 2.62 | 3.24 | 5.03 | 2.36 | 4.14 | |
| 4 | 7205 | 11,545 | 1.22 | 1.15 | 4.56 | 3.99 | 7.63 | 3.79 | 8.42 | |
| Analyte | Conc. (µg/mL) | Rt (min) | Peak Area | N | Res | Tf | k’ | ||
|---|---|---|---|---|---|---|---|---|---|
| Mean | RSD (%) | Mean | RSD (%) | ||||||
| DSF | 0.2 | 3.89 | 0.08 | 8334 | 1 | 6336 | 5.99 | 1.23 | 2.80 |
| 20 | 3.9 | 0.1 | 716,585 | 0.07 | 6319 | 3.35 | 1.24 | 2.66 | |
| 90 | 3.89 | 0.08 | 3,673,964 | 0.02 | 6273 | 3.29 | 1.24 | 2.72 | |
| Cu(DDC)2 | 0.2 | 6.75 | 0.07 | 8497 | 0.8 | 10,838 | 12.5 | 1.17 | 5.46 |
| 20 | 6.71 | 0.08 | 1,086,184 | 0.03 | 10,151 | 8.28 | 1.15 | 5.19 | |
| 90 | 6.71 | 0.09 | 4,359,938 | 0.7 | 9957 | 8.35 | 1.13 | 5.37 | |
| Acceptance Criteria | - | ≤2.0% | - | ≤2.0% | >2000 | >2.0 | ≤2.0 | >2.0 | |
| Analyte | Conc. (µg/mL) | Intraday (n = 6) | Interday (n = 18) | ||||
|---|---|---|---|---|---|---|---|
| Measured Conc. (µg/mL) | Accuracy (%) | Precision (%) | Measured Conc. (µg/mL) | Accuracy (%) | Precision (%) | ||
| DSF | 0.2 | 0.201 ± 0.001 | −0.3 | 0.7 | 0.196 ± 0.004 | 1.9 | 2.0 |
| 20 | 20 ± 1 | 0.9 | 5.5 | 21 ± 2 | −7.0 | 7.3 | |
| 90 | 90.8 ± 0.5 | −0.9 | 0.6 | 90 ± 1 | −0.2 | 1.1 | |
| Cu(DDC)2 | 0.2 | 0.21 ± 0.01 | −2.8 | 5.3 | 0.22 ± 0.02 | −8.0 | 11.4 |
| 20 | 21 ± 1 | −3.1 | 2.9 | 19 ± 1 | 0.9 | 5.7 | |
| 90 | 91 ± 2 | −1.1 | 2. | 87 ± 2 | 3.8 | 2.8 | |
| Analyte | Conc. (µg/mL) | Rt (min) | Peak Area | N | Res | Tf | k’ | ||
|---|---|---|---|---|---|---|---|---|---|
| Mean | RSD (%) | Mean | RSD (%) | ||||||
| DSF | 0.2 | 3.48 | 0.06 | 8631 | 0.9 | 8041 | 4.45 | 1.18 | 2.24 |
| 20 | 3.46 | 0.06 | 791 979 | 0.04 | 8083 | 4.40 | 1.15 | 2.23 | |
| 90 | 3.46 | 0.04 | 3,808,929 | 0.04 | 7850 | 4.36 | 1.15 | 2.21 | |
| Cu(DDC)2 | 0.2 | 6.300 | 0.004 | 7038 | 1 | 12,755 | 14.87 | 1.24 | 4.84 |
| 20 | 6.224 | 0.002 | 948,910 | 0.2 | 11,711 | 10.28 | 1.03 | 4.17 | |
| 90 | 6.25 | 0.01 | 3,977,465 | 0.4 | 11,388 | 10.18 | 1.02 | 4.75 | |
| Acceptance Criteria | - | ≤2.0% | - | ≤2.0% | >2000 | >2.0 | ≤2.0 | >2.0 | |
| Analyte | Conc. (µg/mL) | Intraday (n = 6) | ||
|---|---|---|---|---|
| Measured Conc. (µg/mL) | Accuracy (%) | Precision (%) | ||
| DSF | 0.2 | 0.21 ± 0.01 | −7.5 | 5.3 |
| 20 | 20 ± 2 | −1.6 | 7.9 | |
| 90 | 92.0 ± 0.3 | −2.2 | 0.3 | |
| Cu(DDC)2 | 0.2 | 0.176 ± 0.002 | 12.2 | 1.4 |
| 20 | 20.6 ± 0.7 | −3.1 | 3.5 | |
| 90 | 88 ± 3 | 2.4 | 2.9 | |
| Analyte | Conc. (µg/mL) | Freeze-Thaw | 7 Days, −20 °C | ||||
| Measured Conc. (µg/mL) | Accuracy (%) | Precision (%) | Measured Conc. (µg/mL) | Accuracy (%) | Precision (%) | ||
| DSF | 0.2 | 0.196 ± 0.007 | 2.0 | 3.7 | 0.198 ± 0.008 | 0.9 | 3.9 |
| 20 | 19.9 ± 0.2 | 0.2 | 0.9 | 20.0 ± 0.4 | 0.2 | 1.8 | |
| 90 | 88.3 ± 0.8 | 1.9 | 0.8 | ±2 | 0.5 | 2.4 | |
| Cu(DDC)2 | 0.2 | 0.22 ± 0.01 | −7.6 | 4.6 | 0.212 ± 0.003 | −6.2 | 1.6 |
| 20 | 18.5 ± 0.3 | 7.4 | 1.7 | 19.6 ± 0.5 | 2.2 | 2.7 | |
| 90 | 93 ± 2 | −3.7 | 2.3 | 95. ± 2 | −6.0 | 2.1 | |
| Analyte | Conc. (µg/mL) | Autosampler (24 h) | Short term (72 h, 4 °C) | ||||
| Measured Conc. (µg/mL) | Accuracy (%) | Precision (%) | Measured Conc. (µg/mL) | Accuracy (%) | Precision (%) | ||
| DSF | 0.2 | 0.197 ± 0.006 | 1.3 | 2.8 | 0.197 ± 0.008 | 1.4 | 4.1 |
| 20 | 20.9 ± 0.9 | −4.5 | 4.2 | 23 ± 2 | −16.5 | 7.3 | |
| 90 | 93 ± 3 | −3.2 | 3.1 | 91 ± 2 | −1.5 | 1.7 | |
| Cu(DDC)2 | 0.2 | 0.197 ± 0.005 | 1.6 | 2.7 | 0.19 ± 0.01 | 4.9 | 6.7 |
| 20 | 19.0 ± 0.5 | 4.8 | 2.7 | 18.4 ± 0.7 | 7.9 | 4.0 | |
| 90 | 91 ± 2 | −1.6 | 2.2 | 92 ± 4 | −2.4 | 4.6 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basso, J.; Ramos, M.L.; Pais, A.; Vitorino, R.; Fortuna, A.; Vitorino, C. Expediting Disulfiram Assays through a Systematic Analytical Quality by Design Approach. Chemosensors 2021, 9, 172. https://doi.org/10.3390/chemosensors9070172
Basso J, Ramos ML, Pais A, Vitorino R, Fortuna A, Vitorino C. Expediting Disulfiram Assays through a Systematic Analytical Quality by Design Approach. Chemosensors. 2021; 9(7):172. https://doi.org/10.3390/chemosensors9070172
Chicago/Turabian StyleBasso, João, Maria Luísa Ramos, Alberto Pais, Rui Vitorino, Ana Fortuna, and Carla Vitorino. 2021. "Expediting Disulfiram Assays through a Systematic Analytical Quality by Design Approach" Chemosensors 9, no. 7: 172. https://doi.org/10.3390/chemosensors9070172
APA StyleBasso, J., Ramos, M. L., Pais, A., Vitorino, R., Fortuna, A., & Vitorino, C. (2021). Expediting Disulfiram Assays through a Systematic Analytical Quality by Design Approach. Chemosensors, 9(7), 172. https://doi.org/10.3390/chemosensors9070172