Abstract
Oxygen and nitrogen free radicals (RONS) form an exceptionally reactive molecular assembly within eukaryote cells. This perspective article gives a combined overview of different facets of research covering molecular reactivity, resultant tissue damage and final tissue outcomes as they relate to major disease. There is an emphasis on cardiovascular disease, as the damage processes are best liked to the pathology. The overriding importance of inflammation in driving damage across all tissues is highlighted. Brief coverage is also provided of measurement approaches, respectively for antioxidant status, using potentiometry, and voltammetry for selected target species. Whilst damage due to RONS is a common focus, the fundamental importance of RONS to biological signalling is also covered here as an indispensable basis for life. The article thus provides a global overview of this topic for anyone wishing to understand the current status across multiple fronts.
Keywords:
free radicals; ROS; nitric oxide; superoxide; cell signaling; mitochondria; antioxidants; hormesis 1. Introduction
The aim of this perspective article is to provide a clinical context for the series of sensors papers in the journal’s Special Issue “Electrochemical Sensors for Antioxidant/Oxidant Activity Monitoring”. Hard-wired into our life on earth as oxygen dependent eukaryote organisms is the need to deal with the consequences of generating energy from conversion of the oxygen diradical to water. The cellular mitochondrial machine that achieves this is an exceptional immobilised phase redox system, that takes a series of intermediate energetic steps to final oxygen reduction in its inner surface. However, through this, it sets up a continuous outward flow of reactive oxygen species (ROS), which emanate from the nanopores of its outer membrane. Our appreciation of free radical chemistry is partly derivative of radiation biology, and so naturally emphasises ROS as a damage causing agency. Cellular co-existence with ROS from the inception of eukaryotic life has also meant not only that biology has exceptional countermeasures to mitigate free radical damage, the antioxidant system, but more remarkably, it has harnessed ROS for signalling purposes [1,2]. As such, these reactive species have become fully integrated into the biochemical information sharing systems of the cell, and without them, life would not be possible. Their special utility is that they are highly reactive at low concentrations, broad in their molecular interactions, require no catalytic facility and have very short half-lives. These intrinsic properties are, however, conditioned by the specific biological matrix [3,4,5]. Thus, whilst slower reacting H2O2 has a half-life of 10−3 s, it drops to 10−8 s in the presence of tissue catalase. For the more reactive O2.− free radical, the already short half-life of 10−9 s drops to a remarkable 10−15 s in the presence of tissue superoxide dismutase (SOD). The non-radical H2O2 still has a sufficiently fast reaction kinetics and is able to operate as an efficient signalling molecule; this it does mostly through oxidative reactions with protein thiol residues at rate constants that show a varying repertoire from 100–107 M−1 s−1.
The true RONS free radicals have a much faster, and less discriminating, reactivity that extends across all cell constituents, lipids, proteins and nucleic acids. Though this is rapid, it leaves a trail of sustained molecular damage, a “memory” effect, characterised by products such as oxidized lipid, fragmented protein and DNA strand breaks; rate constants here are 105–109 M−1 s−1. One indicator of the reactive tendency of these molecules is the differences in steady state concentrations, for example, 10−8 M/L for H2O2 and 10−11 M/L for superoxide. As such, levels are important to control and within this containment scenario, feedback can be exquisitely controlled, and yet pervasive influence exerted across all tissues and organs allowing a universality of action [6]. It is necessary to balance our consideration with the damaging aspects of ROS, with an in-depth biochemical understanding of their role in disease and health is to be understood sufficiently for meaningful intervention. There is no doubt of the capacity of ROS, especially if unregulated, to wreak indiscriminate structural havoc across the cell’s structures, from nucleic acids, proteins and lipids through to organelles [7].
This article provides an accessible overview of what we have learned so far about free radicals in disease, and our attempts at tracking these metastable entities, made difficult by their unique presence in biological fluids as hyper-reactive species. Headline biological observations are highlighted to enable the analytical scientist to place their research in context, and to help define priority measurement needs.
2. The Cell Context
2.1. The Key ROS/NOS Players
The starting point for this is the need to define the rampant species that are summarised as causing “oxidative stress”, not necessarily an informative term. Firstly, they encompass nitrogen as well as oxygen species, and are not all free radicals, nor are they all super-hyperreactive [8]. The most dominant force in the ROS armoury is the superoxide ion (O2.−) which par excellence combines the above duality of signalling and damage, followed by the non-radical and much slower acting H2O2, though produced by the same system. They are relevant to both our understanding of normal biochemistry and of multiple diseases. Paradoxically, less informative or biologically relevant is the extreme high reactivity OH. free radical, typically formed in the secondary process of the metal accelerated Fenton reaction, and operating essentially as an isolated agent, causing damage unless specially harnessed as a bactericide. At the opposite extreme, the NO. free radical has minimal damage potential, but is a critical signalling molecule. Its further importance, however, is through conversion to the slower reacting peroxynitrite ion (ONOO−) following reaction with O2.− (Figure 1). This is an important agent for the nitrosation and nitration, respectively, of cysteine and tyrosine residues on proteins and thereby achieves an influential role in functional properties of a wide range of strategically important enzymes and proteins [9].
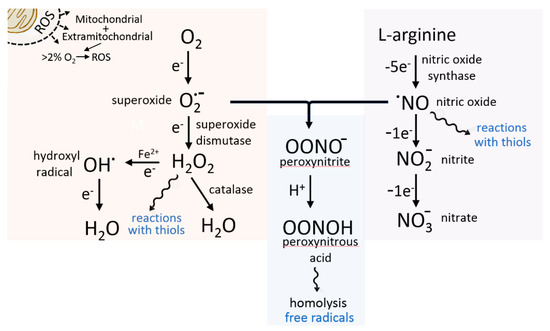
Figure 1.
Schematic showing connectivity between mitochondrially generated superoxide and enzymically produced (NO synthase) nitric oxide. Peroxynitrous acid can decompose spontaneously to create further free radicals. The hydrogen peroxide reactions with protein thiols are a part of its regulatory role as biochemical signal molecule. S-nitrosylation reactions have complex signalling through to reservoir and NO transport functions.
The combined force of reactive oxygen and nitrogen species is referred to as RONS. Measurement access to these would put them on par with other diagnostic markers for medicine, but unfortunately their very short lives in biological fluids, 10−6–10−9 s, renders biologically meaningful measurement challenging. Moreover, they are at the limit and beyond of the measurement sensitivity of many analytical techniques, unless present in excess for some local or disease reason. Their instability also does not allow for a standard mode of sample storage and transport. Despite their dynamic orchestration, we are left mostly with ‘single shot’ measurement of the more enduring stable end products of their reactions. These provide a diverse offering, including the products of lipid peroxidation, such as malondialdehyde, lipid–protein adducts, oxidised nucleic acid bases and modified amino acid residues [10]. The latter may extend to allowing monitoring of circulating proteins such as albumin, so facilitating access. In the case of the functionally critical phospholipids, adverse effects on cell function may be more acutely apparent. Crossover reactions between ROS and NO. also offer a monitoring opportunity. With the formation of ONOO− and the attachment of this group to a protein, the assessment of oxidative stress is possible simply by measuring nitrogen content [11]. The degree of phospholipid oxidation may determine the whole direction of a tissue towards inflammatory change vs. suppressing inflammation. The diverse markers at our disposal creates an informative tapestry, but as a rather misleading static representation that, moreover, compresses sequential time points into one. In this way, acute, short lived events may be missed, but chronic disease associations will be more likely to be picked up providing the first window into damage mechanisms.
2.2. Signalling
The fast signalling interrelationships of the free radicals are complicated and protean, and so not as readily summarised as are the standard biochemical cascades. Mapping of multi-molecule redox state and associated redox (electro-) chemistry are a good fit to the interpretation challenge, however. When signaling is involved, rapid reversibility is imperative, and this does appear to be the general case. It is when there are uncoordinated, excess interactions that tissue damage results. This is not just through a direct molecule to molecule interactive event, but via the triggering of biological non-radical tissue mediators such as growth factors and cytokines which then orchestrate wholesale tissue change, but not necessarily in a beneficial way, and so an expression of disease. What is observed in say, diabetes, neurodegenerative change and cardiovascular disease is really, thus, sustained by this biological aftermath and not the initial free radical chemistry. The pre-eminent normal state in which cumulative damage is well evident is the ageing process. Here, in particular, particles previously thought to be non-functional debris, the lipofuscin granules [12], turn out to be protein–lipid aggregates containing iron with intrinsic oxidative properties, themselves representative of the chronological drift in the cell’s redox state. None of us are immune to this oxidative stress chronicity, but we are still at a stage where we do not understand it fully, still face it as refractory to manipulation, notwithstanding the well-meaning efforts of boost antioxidant intake, long term, using vitamins C, E and other supplements, such as the ever-popular green tea, as a source of polyphenols.
2.3. The Disease Connection
Ultimately, the RONS mechanistic link to disease is as complex as that to the normal biochemistry, but we do now understand that it is the sustained dysregulation of RONS output and interactions, coupled with a limited intrinsic antioxidant capacity that is the cause. A useful descriptor of the RONS driven oxidative stress and antioxidant balance paradigm is provided by the nature of a hormetic response to a damaging agent (Figure 2). Whilst this shows the later, obvious, dose response curve of increased cell damage with higher RONS, it critically highlights the fact that where there is a low pre-existing background of free radical production in a tissue, rather than leading to damage, it primes a more effective antioxidant upregulation in the event of a free radical surge [13]. This primed antioxidant activity thereby increases tissue survival as compared with when no free radicals had existed there in the first place. This not only shows how background RONS is a vehicle for signalling inside a tissue, but it raises the question of the wisdom of unrestricted antioxidant use which attempts to abolish the influence of RONS. The concept of protective antioxidant priming based on low level stimulation by RONS has been added to by a new concept invoking a distinctly positive benefit due to such priming RONS levels. This centres on their added signalling value in normal physiology, and defined as the oxidative eustress state; the damage counterpart at high RONS levels has been termed oxidative distress [14].
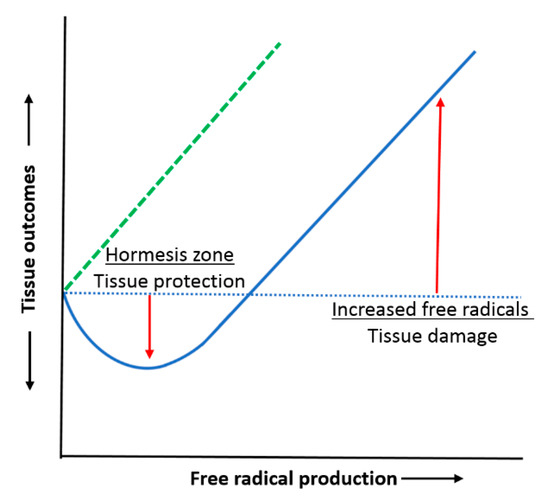
Figure 2.
Hormesis curve showing enhanced tissue protection by antioxidants through their priming by low level free radical production: protection from free radical surge; hormesis zone (blue curve). Lack of added protection (hormesis) with free radical surge when free radical exposure is already pre-reduced using antioxidants, with subsequent greater damage at higher free radical levels: damage curve shifted to the left (dashed green line).
3. Genesis and Wider Interactions
3.1. NADPH Oxidases and ROS Activity
Central to the production of ROS by the mitochondrion are the membrane outward facing NADPH (reduced nicotinamide adenine dinucleotide phosphate) oxidases, which have the sole purpose of creating either O2.− or H2O2 [15]. Therefore, an excess of cell mitochondria might well make more high energy ATP (adenosine triphosphate) as the cell’s “battery”, but the danger to the cell also escalates, and indeed is manifested in many diseases. The NADPH oxidase isoforms in the blood vessel lining cells, the endothelial cells, exemplify the control that that may be exerted on them, e.g., as exerted by hormonal and thrombin effectors in the circulation. The system, more generally, has a self-amplifying capability with, for example, the capacity for oxidative conversion of xanthine dehydrogenase to an O2.− generating xanthine oxidase, stimulating yet more production of NADPH oxidase. The H2O2 product also has a key role in effecting oxidative folding of protein inside the cell’s endoplasmic reticulum (ER) prior to entry into the Golgi apparatus and final export. However, with excess, ER oxidative stress results, and then this has a major disruptive effect on the protein genesis pathway, sufficient indeed for total cell failure and cell death [16].
The best understood routes to signalling by ROS are the oxidation of protein cysteines, metal oxidation and direct molecular attachment. Reversibility of cysteine/cystine switching provides a ready basis for redox switches [17]. If the coordination is lost, then oxidative drift will cause cumulative damage. Mechanistic detail has yet to be worked out, but where the molecular changes occur, lie the clues to future drug design. There is also a cooperativity of ROS effects on cell phenotypic expression through modulated interaction with the DNA’s transcription machinery, a basis for epigenetic change, along with direct structural changes to the DNA [18]. With respect to the drugs pipeline, these known loci offer points of precision therapeutic leverage for disease (Figure 3). What is much less certain, despite our decades of research, is why a blanket use of dietary supplement antioxidants does not have the disease mitigating impact that their chemical reactivity and redox potential would suggest [19]. More analytical tools will certainly help here, including Western blotting and ELISA for the global characterisation of protein population shifts under oxidative stress and, of course, recovery. Alternatively, high throughput proteomics, bioinformatics and focussed attention to the new horizon of protein redox states will be of substantial future value [6].
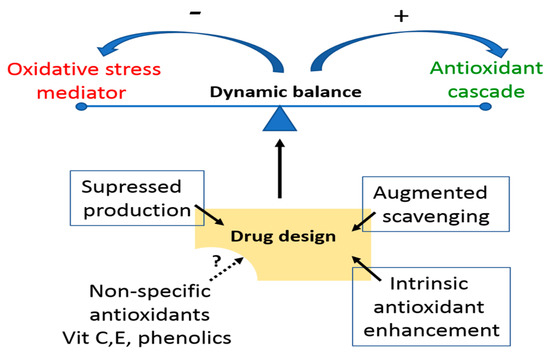
Figure 3.
Options for targeted therapies to shift the prevailing oxidant status of a tissue through the balance of supply and loss. Low specificity dietary and other exogenous antioxidants have uncertain role in disease management beyond population level health benefits.
3.2. Extracellular Influence
The influence of ROS outside of the cell is also becoming a feature of attention. Under oxidative stress duress, cells release microparticles from their plasma membranes [20]. These are not fixed organelles, but are variable aggregates of insoluble cell components, and follow on from lipid peroxidation. Importantly, here, they have their own ROS activity and, moreover, recruit stress responses in nearby cells, essentially infecting them. Non-coding, short sequence, microRNAs (miRNAs) are another released entity that enters the circulation and causing redox pathway shifts in other cells [21]. These special single stranded loop RNAs regulate gene expression, and by redox shifts, for example, can move the response of an inflamed or injured tissue between a pathological fibrotic reaction to one that is non-fibrotic. This section is divided by subheadings. It provides a concise and precise description of the experimental results, their interpretation, as well as experimental conclusions.
4. Measurement
Electron paramagnetic resonance (EPR), with spin trapping [22] and fluorescence probes [6], have been the mainstay of measurement approaches as investigative techniques, but are not applicable to routine medical use, and do not necessarily provide rapid dynamic tracking capability or fine-tuned speciation that we need. The nearest to dynamic tracking at low levels we have is sensors and probes for H2O2 and NO.. Alternatively, higher level more trackable antioxidant measurement can give us information about the net balance of what is being produced and what has been consumed within the oxidative stress process. Electrochemistry is one tool that is well suited to this, providing mapping and quantitation grounded in Nernstian principles [23], pivotal to the capacity for electron donation by the body’s antioxidant repertoire, the polarisation voltages at which it occurs, its thermodynamic link and the rate at which this occurs, i.e., the kinetics of biological effectiveness.
There is no ideal or universal, indicator of for measuring sample antioxidant capacity (antioxidant activity, AOA), as in a laboratory single species redox titration; biological fluids have far too complex a makeup to allow for this. However, as an empirical assessment, indicator species in the formal potential range 0–+0.62 V vs. NHE would seem to be able to cover the reactions of most of the constituent antioxidant species. This has led to the use of tailored reporters, e.g., the Fe3+/Fe2+ couple in haem-like configurations such as with 2,4,6-tripyridyls-triazine, 1,10-phenanthrolin and cyanide ion. This particular stratagem is codified in the term FRAP (ferric reducing/antioxidant power). The equilibrium balance of Fe3+/Fe2+ is then readily determined by potentiometry using a Pt reporter electrode.
Antioxidant status assessed with such summarizing reactions is semi-empirical and does not generate mechanistic information. However, it does offer an accessible biological index of AOA that may be correlated subsequently with nutritional and clinical state. If such an assay can be rigidly standardized, as is the case for all clinical assays, it could enable a reproducible measure of AOA that can be cross-referenced across laboratories and studies. Here, also, EPR is a useful adjunct, because it can track electrochemically silent free radicals during the measurement process.
Speciation of RONS is possible through electrochemical sensors [24] amongst other transducer techniques. Voltammetry is especially appropriate given the redox properties of these species. Numerous innovative designs have been reported, summarized in Figure 4, but biological assay challenges have not receded. In particular, critical appraisal is needed if the intended use is clinical. Despite the simple redox interconversions, the extremely short lifetimes impose a unique measurement challenge. Speciation may well be reliably demonstrated, but accurate quantitation is quite a different matter. The demonstration of concentration trends in biological models or of recovery data is sometimes mistakenly used to confirm analytical fidelity. This cannot be the case any more than it would be for a glucose sensor. There is room for a different level of evaluation in line with standard practice in laboratory medicine.
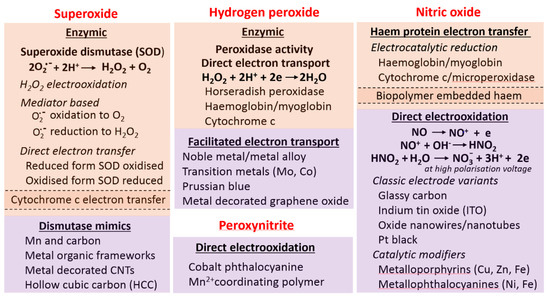
Figure 4.
Summary of electrode chemistries and electrode surface modifications used for electrochemical RONS measurement. Associated barrier membranes and permselective layers are not included here but are an indispensable element of any practical device. The different shaded blocks identify the different generic approaches: biological, catalytic/inorganic. Few peroxyinitrite sensors have been attempted; selective barrier films are typically needed; an alternate, less specific anion affinity micro-architectured surface is not included here.
Of all the candidate species, the more stable, and higher concentration, H2O2 is the most likely to be used clinically. It can be variously accessed through inert metal and carbon electrodes (Figure 4), along with nanostructured modifications for greater sensitivity. The most used enzymic system is that relying on horseradish peroxidase (HRP) or allied enzymes, the former possessing a high turnover number. Parallel approaches for O2.−, variously with superoxide dismutase (SOD) and enzyme mimics (Figure 4), including the use of porous materials and positively charged surfaces to promote binding of the anion.
The practical concern in all these situations is the absence of a reference method against which an assay can be benchmarked. Therefore, any measurement outcome needs to be considered at best as being semi-quantitative and at worst semi-intuitive based on a post hoc understanding of the biochemistry. Yet, there needs to be a credible reference on which to base our measurements; without this, our mining of new data on RONS activity and what is recruited to protect from its damaging aspects must remain limited, as indeed does any quantitative interpretation of disease progression.
5. Disease Profiles
5.1. General Considerations
Supraphysiological levels of ROS, whether diffused or localized, have the capacity to do substantial harm. We have a good understanding of the reactive chemistry with specific functional groups, but bridging this to macro-structural pathological change has proven to be difficult, and in some cases controversial.
The tissue surveillance system recognizes the injury signal due to ROS damage through release of cell constituents from damaged cells. It then sets up an inflammatory cascade, itself a source of free radicals, which then either leads to healing or progression to chronic inflammation stage. The biochemical players in this include a host of pro-inflammatory cytokines and mediators derived from arachidonic acid. The most important focal effect is due to the recruited inflammatory cells which then operate as self-sustaining ROS producers [25]. Whilst the resulting pathological features at any individual tissue may be unique to its biology, the same injury induces processes in operation (Figure 5).
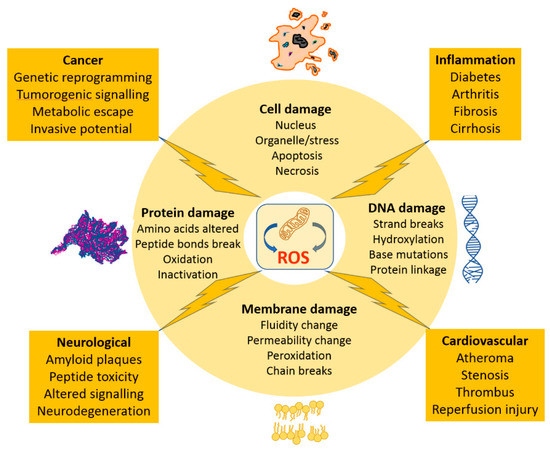
Figure 5.
Schematic of the tissue outcomes of excess mitochondrial generated ROS, describing the generic molecular damage accruing to lipid membranes, proteins, DNA and ultimately the single cell. The subsequent whole tissue outcomes are provided for selected tissue types and with respect to inflammatory disease and cancer.
A late-stage accumulation of fibrous tissue is one common feature which can be equally attributed to ROS, such as in liver cirrhosis or in chronic lung and inflammatory disease. The controversy lies in the relative importance to attach to ROS in any give tissue change. An added feature depleting functional cell mass is the specific ROS induction of programmed cell death, apoptosis. ROS outcomes are, perhaps, better understood and so much of the commentary below is provided in relation to this group.
5.2. Cardiovascular Disease
The most investigated of the systems diseases is the cardiovascular system. From earlier work on NO. signalling in the vascular endothelium and its regulation/relaxation and surface protection from blood coagulation, we have learnt more about the interaction with ROS. Firstly, even H2O2 has beneficial ‘housekeeping’ effects on the endothelial lining. However, O2.− increase in oxidative stress leads to an uncoupling of the enzymic NO. synthesis process, needed for continuous NO. to be effective, with the result that not only is vascular damage instituted, but the endothelial protective role is compromised [26]. The focal expression of extreme NO. dysregulation in this way is the vascular atheromatous plaque (atheroma). Again, driven by O2.−, a harbinger of dramatic wall damage and inflammatory change, it is programmed to self-generate through the amplified recruitment of monocytes from the circulation; these are basically mobile O2.− factories [27]. In the reaction with NO. now, the spill over to ONOO− product now makes for potent levels of cell toxicity. In the right place, of course, monocytes and other white cells have immense value in internalising and neutralising microbial attack. This they couple with initiating inflammatory change as basis for tissue protection, but in the wrong place, such pro-inflammatory action, most notably the coronary artery, has given us one of humanity’s biggest killers. Here, no amount of antioxidant data mining is going to be predictive of coronary artery disease or blockage, and nor is it the case that raising antioxidant levels by nutritional intervention is going to have a curative effect. In particular, the intensity of local change such as this, however damaging and dramatic locally, will have little impact at the remote systemic level, including in blood. For therapy, we will need targeted drugs (Figure 3), and it would be somewhat facile to expect the general administration of antioxidants to turn the fate of an individual, whatever the statistically significant health benefits at population level.
The post-blockage resumption of blood flow through a tissue presents us with another ROS dependent basis for cellular damage. The result is accelerated, “catchup” oxygen consumption and a burst release of ROS; reperfusion injury. Here, especially if there has been a background low level exposure to RONS, then primed antioxidant activity will be in place, and with it, the hormetic outcome profile (Figure 2). Therefore, in a state such as myocardial infarction recovery, decisions about the generic institution of antioxidant-based population health become quite sensitive ones.
With regard to the myocardium itself, its cells are replete with 30% filling by mitochondria, necessary for high ATP production, but this may also mean that it takes only minor disequilibrium in activity, content or turnover for serious oxidative damage to occur. This is evident it some cardiomyopathies and though these are long term conditions, the free radical effect on cell contractile properties is immediate [28].
Vascular tissues have the capacity to regenerate blood vessels, both normally and in disease. ROS signalling plays a role here also, with promotion of the biochemical cascades that result in micro-vessel endothelial budding and vessel connectivity: angiogenesis. This is proposed as being the consequence of oxidative action on proteins, together with protein adduct formation. This latter proves to be important in stabilising the most important of all the angiogenesis factors: hypoxia-inducible factor 1α (HIF-1α) [29].
5.3. Other Tissue Scenarios
The brain is a uniquely configured organ which variously uses up high amounts of oxygen, is predominantly lipid in composition and has low reserves of antioxidant. The tendency for hypoxic, peroxidative damage is therefore high, the ability to recruit an antioxidant response more limited and the vulnerability of the lipids to oxidation quite special. On a long-term basis, we are beginning to understand that background ROS is associated with neurodegeneration, but yet again, there is a contradictory biology here; in the developing brain, ROS are indispensable to neuronal development and connectivity [6]. In Alzheimer’s, the depletion of neurons as dementia progresses has been linked to indirect ROS damage, via stimulated deposition of ß-amyloid, a peptide complex with toxic neuronal activity.
Tissue fibrosis is a frequent end stage of inflammation, because it can reinforce tissue mechanical strength and bridging, but it can also have a disruptive effect on the subtle architecture of major internal organs such as the kidney, liver and lungs, where tissue distortion through scarring may be as damaging as any loss of functioning cells. In the case of inflammatory lung disease, at the chronic stage, the permissive effects of ROS with their promotion of particular transcription factors, proteins controlling the transfer of genetic information from DNA, leads to promoted inflammation. In the case of rheumatic joint disease, ROS stimulates the production of pro-inflammatory prostaglandins. Right across the spectrum of renal diseases, from glomerular to tubular, the damage seen can be attributed to ROS; this can even be the case where the causal agent is a toxin. In the ocular lens, light transparency is critically dependent on protein crystallinity, and if its intricate organization is disrupted through ROS damage, crosslinks form and the lens becomes light scattering, i.e., cataract formation. ROS have been specifically linked to the liver fibrosis seen after viral hepatitis [30], here further exacerbated by a diminished antioxidant reserve.
The β cell of the pancreas is a tablespoon quantity of cells that can be dramatically destroyed in type I diabetes through inflammatory change and are, moreover, highly sensitive to the oxidative stress brought about by the damaging synergy of glucose and lipid [31]. There are also peripheral effects with ROS on a lowered insulin effectiveness. Whilst we know some of these basic facts, we need to accumulate quantitative links between disease state, its genesis and the amount of ROS loading.
Cancer cells, finally, provide an object lesson in innate biological control and compensation. This regressed cell system is functionally uncoordinated in many ways, one of which is in mitochondrial organisation, whereby high rates of ROS production occur. This is so greatly linked to cancer cell survival that the cells also self-protect by upregulating their antioxidant systems [32]. Cancer, ultimately, can be attributed to DNA damage and resulting aberrant genetic messaging which remains corrected. A prime basis for the damage is oxidative through ROS, and associated DNA hydroxylation, but other processes are involved (Figure 5).
6. Antioxidant Perspectives in Disease
No parameters, be it vitamin C, E, urate, flavonoid or other antioxidants, when individually measured, can gauge whole body antioxidant status. Nor can a single parameter, beyond identifying a dietary deficiency, give a guide to nutritional needs for antioxidants for long term health. Part of the reason for poor linkage to outcomes is that there are submerged body antioxidant systems we have yet to understand. With the best current redox indicators of AOA assessment, we cannot yet access all the complex intermolecular and inter-compartmental redox species necessary for health. An option might be to use multiparameter biochemical processing across redox species, from metabolites to proteins, with informatics tools for cluster analysis in disease and health. If disease ‘fingerprints’ arising from this can be made diagnostically robust, they may lead us to the better stratification of normal, pre-disease and disease. A starting point here might be just a focus on the thiol couple, then extended to other redox pairs. Analytical development would need to run in parallel with workup of the biological disease process. It is clear that the AOA-disease gap in understanding will need to be made up somehow, in view of the disappointing results with antioxidant nutritional advice: there has been no “magic bullet”. A further benefit would be in informing the development of drug therapies to mitigate RONS effects (Figure 3).
7. Conclusions
The work of free radicals in biology and disease has expanded considerably in recent years and shows, increasingly, how “hardwired” RONS are in normal biochemistry as messengers. The combined description in this article of the uniquely fast kinetics, the breadth of the tissue damage and the outcomes for whole tissues has shown the full context in which RONS need to be studied for a better understanding. The one aspect that it highlights as important is the presence of inflammatory change across all tissue processes. Therefore, once we better understand the free radical environment here, a better universal understanding of RONS can result in both healthy and diseased tissues. The measurement developments are given in an outline, but their limitations are also indicated in order to highlight the need for practical consideration, if advances are not to be consigned to the research laboratory. The fact that little has been stated about endogenous antioxidant systems attests to the fact that we know so little about them, and so the article indirectly points to a serious gap in our understanding that will need to be filled. The measurement of individual RONS can only take us so far in what is essentially a biological research need.
The analytical science relating to free radical biochemistry cannot operate in isolation; there has to be a linked approach with biology and the questions that it is attempting to answer. The challenges are greater than with other biological molecular players, because the RONS pathways are integrated into innumerable signalling networks. There is a precedent, in that the burgeoning complexity of immune pathways has been accommodated into a rational understanding. As with immune system analysis, the ultimate prize will be better drug therapies able to cover the wide spectrum, where we know that RONS have either active or permissive roles.
Funding
This research received no external funding.
Institutional Review Board Statement
All descriptions of biomedical research is from the literature already published.
Informed Consent Statement
No new human studies were included other than those already reported in the literature.
Data Availability Statement
No new data was obtained for the writing of this perspective.
Conflicts of Interest
The author declares no conflict of interest.
References
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Gutsche, M.; Sobotta, M.C.; Wabnitz, G.H.; Ballikaya, S.; Meyer, A.J.; Samstag, Y.; Dick, T.P. Proximity-based Protein Thiol Oxidation by H2O2-scavenging Peroxidases. J. Biol. Chem. 2009, 284, 31532–31540. [Google Scholar] [CrossRef]
- Ma, J.Z.; Zhou, H.X.; Yan, S.W.; Song, W.H. Kinetics studies and mechanistic considerations on the reactions of superoxide radical ions with dissolved organic matter. Water Res. 2019, 149, 56–64. [Google Scholar] [CrossRef]
- Taverne, Y.; Bogers, A.; Duncker, D.J.; Merkus, D. Reactive Oxygen Species and the Cardiovascular System. Oxid. Med. Cell. Longev. 2013, 862423. [Google Scholar] [CrossRef]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Gorlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Zhou, T.; Pannell, B.K.; Ziegler, A.C.; Best, T.M. Biological and physiological role of reactive oxygen species—The good, the bad and the ugly. Acta Physiol. 2015, 214, 329–348. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Ferrer-Sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballa, S.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of peroxynitrite and protein tyrosine nitration. Chem. Rev. 2018, 118, 1338–1408. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: How should you do it and what do the results mean? Brit. J. Pharmacol. 2004, 142, 231–255. [Google Scholar] [CrossRef]
- Sitar, M.E.; Aydin, S.; Cakatay, U. Human serum albumin and its relation with oxidative stress. Clin. Lab. 2013, 59, 945–952. [Google Scholar] [CrossRef]
- Hohn, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin-bound iron is a major intracellular source of oxidants: Role in senescent cells. Free Radic. Biol. Med. 2010, 48, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Barcena, C.; Mayoral, P.; Quiros, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Hillion, M.; Antelmann, H. Thiol-Based redox switches in prokaryotes. Biol. Chem. 2015, 396, 415–444. [Google Scholar] [CrossRef]
- Wang, Y.S. Bulky DNA lesions induced by reactive oxygen species. Chem. Res. Toxicol. 2008, 21, 276–281. [Google Scholar] [CrossRef]
- Hertog, M.G.L.; Feskens, E.J.M.; Hollman, P.C.H.; Katan, M.B.; Kromhout, D. Dietary antioxidant flavonoids and risk of coronary heart disease—The Zutphen elderly study. Lancet 1993, 342, 1007–1011. [Google Scholar] [CrossRef]
- Burger, D.; Turner, M.; Munkonda, M.N.; Touyz, R.M. Endothelial microparticle-derived reactive oxygen species: Role in endothelial signaling and vascular function. Ox. Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Torma, F.; Gombos, Z.; Jokai, M.; Berkes, I.; Takeda, M.; Mimura, T.; Radak, Z.; Gyori, F. The roles of microRNA in redox metabolism and exercise-mediated adaptation. J. Sport Health Sci. 2020, 9, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Abbas, K.; Babic, N.; Peyrot, F. Use of spin traps to detect superoxide production in living cells by electron paramagnetic resonance (EPR) spectroscopy. Methods 2016, 109, 31–43. [Google Scholar] [CrossRef]
- Ivanova, A.V.; Gerasimova, E.L.; Brainina, K.Z. Potentiometric study of antioxidant activity: Development and prospects. Crit. Rev. Anal. Chem. 2015, 45, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Calas-Blanchard, C.; Catanante, G.; Noguer, T. Electrochemical Sensor and Biosensor Strategies for ROS/RNS Detection in Biological Systems. Electroanalysis 2014, 26, 1277–1286. [Google Scholar] [CrossRef]
- Juranek, I.; Bezek, S. Controversy of free radical hypothesis: Reactive oxygen species—Cause or consequence of tissue injury? Gen. Physiol. Biophys. 2005, 24, 263–278. [Google Scholar] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Nowak, W.N.; Deng, J.C.; Ruan, X.Z.; Xu, Q.B. Reactive oxygen species generation and atherosclerosis. Arterioscl. Throm. Vas. 2017, 37, E41–E52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.H.; Li, Y.F.; Heims-Waldron, D.; Bezzerides, V.; Guatimosim, S.; Guo, Y.X.; Gu, F.; Zhou, P.Z.; Lin, Z.Q.; Ma, Q.; et al. Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ. Res. 2018, 122, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Gorlach, A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin. Cell Dev. Biol. 2005, 16, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Fabregat, I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox. Biol. 2015, 6, 106–111. [Google Scholar] [CrossRef]
- Gerber, P.A.; Rutter, G.A. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid. Redox. Sign. 2017, 26, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).