2. Materials and Methods
2.1. Specimen Collection
Dermatological specimens taken from lesions showing MD pathology were collected from 14 North American subjects who met the case definition for MD, as determined by a health care practitioner. The case definition used in this study required the presence of spontaneously developing skin lesions containing embedded or projecting red, white, blue or black filaments. Testing for LD or other pathogens prior to volunteering was not required for participation.
The MD skin specimens collected for study consisted of thickened callus material removed from lesions exhibiting embedded or projecting filaments. For comparative studies, normal healthy callused skin samples were collected from the feet of three healthy subjects, two subjects with LD but no MD, and one MD subject. Normal commercially available human skin (BioChain Institute, Newark, CA, USA) was included as a negative control for comparison purposes. MD dermatological specimens and foot callus specimens were stored as dried flakes, then forwarded to the University of New Haven Lyme Disease Research Laboratory in a blinded manner.
The study was conducted in accordance with the Declaration of Helsinki. Informed consent for study participation was obtained from MD subjects and control subjects in accordance with the specimen collection protocol approved by the Western Institutional Review Board (WIRB), Puyallup, WA, and consent to publish the study results was obtained from all subjects. The study was also approved by the University of New Haven IRB Committee as exempt under 45 CFR 46.101(b)(4). Subject identification, health status and demographic information were not provided to the research laboratory.
2.2. Culture of Bb and Hp
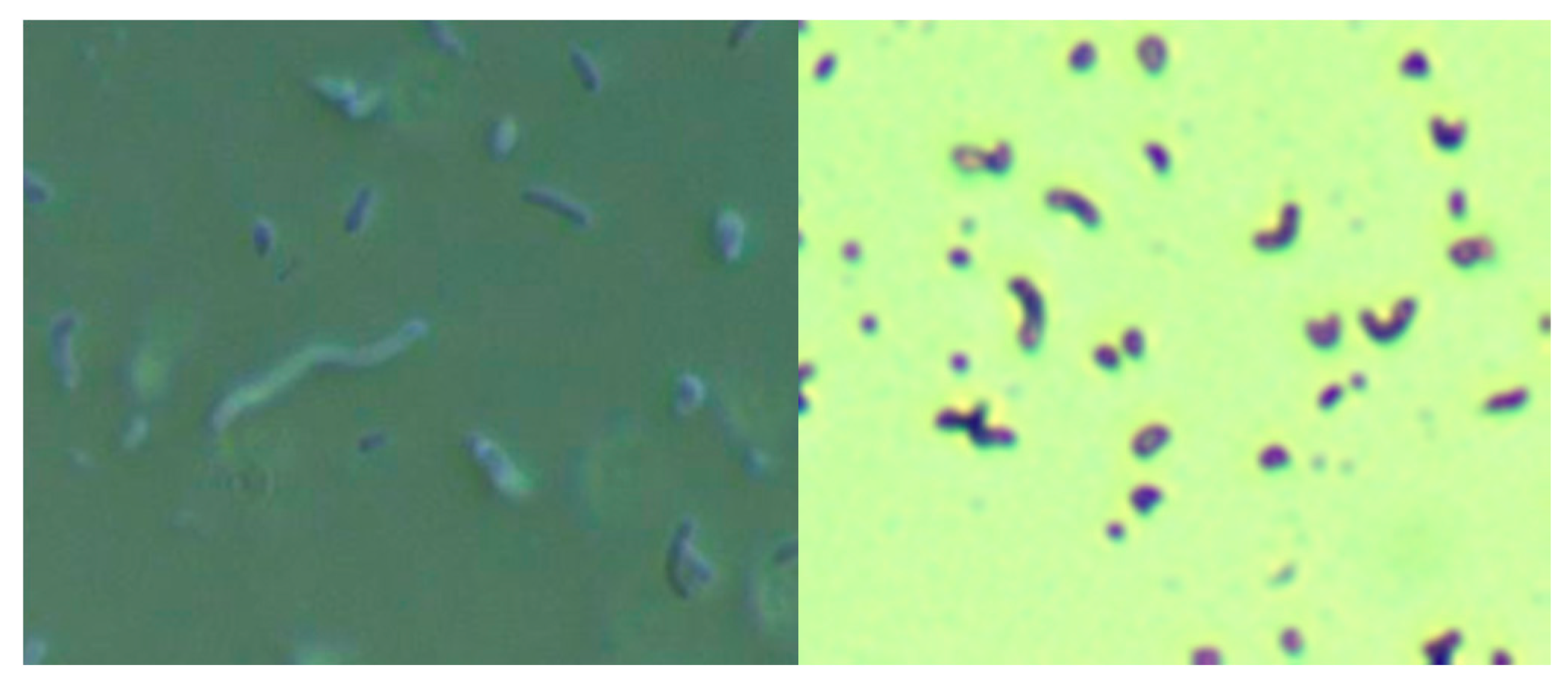
Skin specimens from MD subjects were cultured for Bb and Hp using a modification of the selective Hp culture system [
23]. In brief, 400 mL of sterile water was mixed with 19 g of Columbia blood agar base (CM0331, Thermo Scientific, Waltham, MA, USA) and then autoclaved. After cooling to 50 °C, 35 mL of laked horse blood (SR0048, Thermo Scientific), the Hp antibiotic supplement (vancomycin 1%, trimethoprim 0.5%, cefsulodin 0.5% and amphotericin B 0.5%: SR0147, Thermo Scientific) and 100 mL of BSK-H complete medium with 6% rabbit serum (Sigma Aldrich, #B8291, St. Louis, MO, USA) were added and mixed well before pouring into sterile Petri dishes. The dishes were inoculated with scabs and incubated. Culture fluid was examined by darkfield microscopy for the presence of microorganisms. Culture smears were stained with either a standard Gram stain kit (Daylynn Biologicals, Calgary, AB, USA) to determine Gram reactivity [
23], or with crystal violet (Daylynn Biologicals, Calgary, AB, USA) to visualize morphological features of the bacteria.
2.3. DNA Extraction
Dermatological specimens were homogenized by freezing in liquid nitrogen and grinding with a mortar and pestle. Specimens were resuspended in 180 µL of Buffer ATL (Qiagen Sciences, Germantown, MD, USA) with 20 µL of Proteinase K (Qiagen, Hilden, Germany), then incubated overnight at 56 °C with shaking. DNA was purified by standard phenol/chloroform extraction and alcohol precipitation the following day. Purified DNA was resuspended in 50–100 μL of 1× TE buffer at pH 8.0 (10 mM Tris, pH 8.0, and 1 mM EDTA).
2.4. Nested PCR
Nested PCR was performed to detect the presence of Bb and Hp DNA targets from purified DNA extracted from blinded MD and control specimens using “outer” primers for the first reaction and “inner” primers for the nested reaction. A volume of 10 µL of template DNA was used for the first reaction and 1 μL of PCR product from the first reaction was used as template for the second. Positive and negative controls were conducted alongside test specimens for each assay. The “No DNA” template was used as a negative control in all PCR reactions. As additional negative controls, the same PCR experiments were carried out on normal human skin samples. The positive control experiments used DNA extracted from low passage of Bb strain B-31 (ATCC #35210) and from frozen vials of Hp (ATCC #BAA-945). Primers used for the amplification of Bb and Hp were chosen from those used previously in the literature and they represented a mixture of conserved and polymorphic genes in order to define species and genotypes. PCR primers for Bb were:
pyrG,
uvrA,
fla and
OspC [
24,
25,
26,
27]. A summary of Bb PCR primers is shown in
Table 1. PCR primers for Hp were: 16S rRNA,
hsp60,
urea, and 23S rRNA [
28,
29,
30,
31]. A summary of Hp PCR primers is shown in
Table 2.
PCR reactions were performed using a final volume of 50 µL and final concentrations of 1× Buffer B (Promega), 1.5 mM MgCl2, 200 µM dNTP mix, 0.2 µM of each primer, and 2.5 U Taq polymerase (Invitrogen, Carlsbad, CA, USA). Cycling parameters were: 95 °C for 5 min followed by 35 cycles of denaturation at 95 °C for 1 min, annealing for 1 min (temperature based on the primer set used), and extension at 72 °C for 1 min, with a final extension step at 72 °C for 5 min. PCR products were visualized on 1–1.5% agarose gels stained with ethidium bromide.
Sanger sequencing was used to confirm that the target DNA detected was correct. PCR products were purified using the QIAquick Gel Extraction kit (Qiagen) according to the manufacturer’s instructions. The eluates were sequenced in both directions by Eurofins MWG Operon DNA sequencing services (Eurofins Scientific, Louisville, KY, USA). The sequences were compared to those in the National Center for Biotechnology Information (NCBI, Bethesda, MD, USA) GenBank database using Basic Local Alignment Search Tool (BLAST) analysis. Only PCR amplicons confirmed as correct by BLAST analysis were interpreted as positive.
2.5. Preparation for Histochemical Staining
MD dermatological specimens and normal human skin controls were formalin-fixed, paraffin-embedded and sectioned. Sections were deparaffinized, rehydrated, blocked with a 1:100 dilution of goat serum (Thermo Scientific) in 1× PBS phosphate buffered saline (PBS, Sigma, St. Louis, MO, USA) for 30 min at room temperature in a humidified chamber, then washed twice in 1× PBS (Sigma) for 5 min followed by distilled water for 5 min.
2.6. Dual Bb and Alginate Immunohistochemical (IHC) Staining
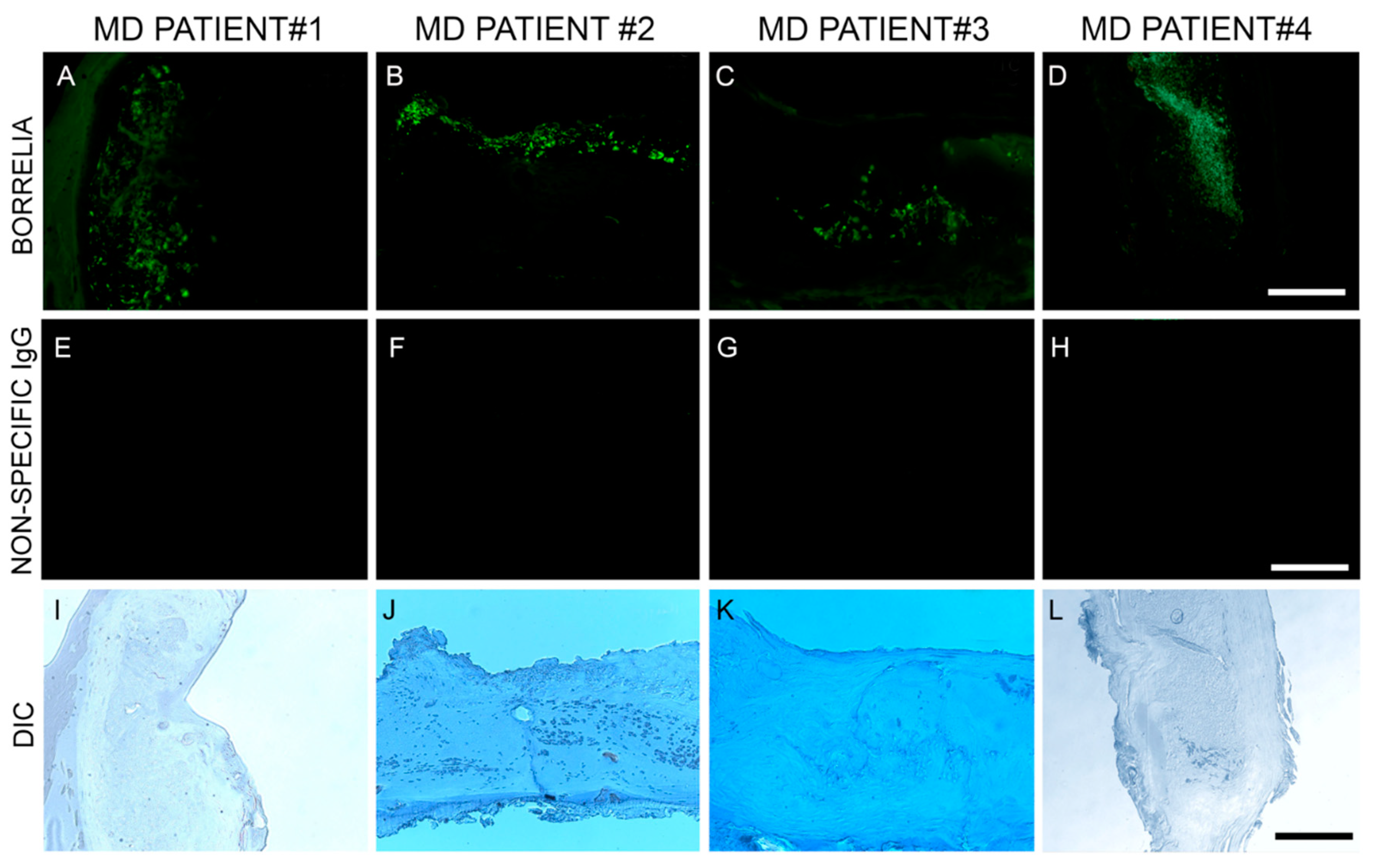
Blocked, washed sections were reacted with a 1:500 dilution (1× PBS/1%BSA) of primary anti-alginate antibody (kindly provided by Dr. Gerald Pier, Harvard Medical School, Boston, MA, USA), placed in a humidified chamber, incubated overnight (minimum of 16 h) at 4 °C, followed by washing in PBS as described in the preparation above. The sections were then reacted with 1:200 dilution of a secondary anti-rabbit antibody with a fluorescent red tag (594 nm, Thermo Scientific) for 1 h in a humidified chamber at room temperature, then washed in PBS as described above. Sections were then stained with a 1:10 dilution of mouse monoclonal antibody specifically targeting Bb sensu stricto strains (MA-1-7006, Thermo Scientific), incubated overnight (minimum of 16 h) at 4 °C, then washed as described above and further incubated with a 1:200 dilution (1× PBS/1%BSA) of goat anti-mouse secondary antibody labeled with DyLight 488 fluorescent dye in a humidified chamber at room temperature. The sections were washed again as described above and counterstained with Sudan Black (Sigma) for 20 min, then stained with 4’,6-diamidino-2-phenylindole (DAPI) for 90 s. Sections were washed, allowed to dry, then mounted with PermaFluor (Thermo Scientific). Images were captured using a Leica DM2500 fluorescent microscope at 200× and 400× magnification. As negative controls, commercially available human foreskin tissue sections and healthy human skin sections (Biomax, HuFPT136) were stained following the same procedure described above. Additional negative control experiments omitting the primary antibody and using a non-specific mouse IgG1 isotype (MA1-10406, Invitrogen) were also performed to confirm the specificity of the antibodies.
2.7. Hp IHC Staining
Consecutive sections of Bb and alginate positive blocks were further analyzed for the presence of Hp. Deparaffinized and washed sections were blocked with goat serum as described above and then incubated for 1 h with a 1:50 dilution of a fluorescently-labeled polyclonal rabbit anti-Helicobacter pylori antibody targeting ATCC 43504 whole Hp cells (PA1-73161, Thermo Scientific) for 1 h in a humidified chamber at room temperature. Sections were then washed, counterstained with Sudan Black, mounted and imaged as described above.
2.8. Dual β-Amyloid or Phospho-Tau IHC Staining and Bb or Hp IHC Staining
Staining for β-amyloid proteins and phosphorylated tau was performed on consecutive sections from blocks that were positive for Bb and Hp [
32]. Sections were processed and washed as described above, followed by incubation with a 1:200 dilution of either anti-β-amyloid antibody-1 (ABA-1) (MA1-34553, Thermo Fisher Scientific), or anti-β-amyloid antibody-2 (ABA-2) (M 0872, Daco Scientific Ltd., Reading, UK), following previously published IHC protocols [
32]. Alternatively, sections positive for Bb and Hp were incubated with 1:200 dilutions of anti-phosho-tau antibody (MN1020, Thermo Scientific). As a negative IHC control, a non-specific mouse IgG1 isotype (MA1-10406, Invitrogen) was used in place of the primary antibodies to confirm the antibody specificity of this assay.
2.9. Thioflavin Histochemical Staining
Deparaffinized, rehydrated, formalin-fixed, sections were stained with 1% Thioflavin S (Santa Cruz Biotech, Santa Cruz, CA, USA) solution in 80% ethanol and incubated for 15 min at room temperature. Sections were washed in an 80% ethanol bath, then a 70% ethanol bath for 1 min each, followed by washing in double distilled water twice for 5 min each time. Sections were then mounted and imaged as described above.
2.10. FISH Testing
FISH was adapted from previously validated probes for both Bb and Hp [
22,
33,
34,
35]. The sequences of the Bb and Hp DNA probes are listed in
Table 3. Slides of paraffin-embedded tissue sections were first deparaffinized by heating on a slide warmer for 40 min at 45 °C, then immersed in 100% xylene for 5 min three times. The tissue sections were rehydrated in series of graded alcohols (100%, 90% and 70%) and washed in PBS for 5 min followed by washing with distilled water for 15 min. The sections were then treated with 4% sodium borohydride (Sigma Aldrich) for 20 min on ice. The tissues were then digested with pre-warmed proteinase K solution (20 µg/mL in 50mM Tris) for 10 min at 37 °C and re-fixed with 4% paraformaldehyde for 10 min at room temperature. The slides were then denatured using pre-heated denaturing buffer (70% v/v formamide, 2× SSC and 0.1 mM EDTA, pH 7, Thermo Fisher, Scientific) at 70 °C followed by prehybridization for 4 h in hybridization buffer (50% v/v formamide (Sigma), 10% w/v dextran sulfate (Sigma), 1% v/v Triton X-100 (Sigma), 2× SSC, (Sigma) pH 7.0, and 2 ng of salmon sperm DNA (Sigma) in an incubator at 48 °C.
Hybridization was achieved by reacting the slides with the probe at 48 °C for 18 h in the dark. Post-hybridization, slides were washed three times with 2× SSC for 3 min each at room temperature followed by 5 washes each for 20 min in 0.1× SSC at 42 °C and a final wash in 2× SSC at room temperature. Finally, slides were blocked with freshly made blocking solution (3% w/v BSA (Fisher) in 4× SSC, 0.1% v/v Triton X-100) for 30 min and washed with wash solution (4× SSC, 0.1 % v/v Triton X-100) for 3 min at room temperature. All steps were repeated with several controls including (1) 100 ng of negative control random oligonucleotide, (2) 200 ng of unlabeled competing oligonucleotide present during the hybridization and (3) following a DNase treatment of the sections before the hybridization step to digest all genomic DNA (100 µg/mL for 60 min at 37 °C). All slides were then analyzed by fluorescent microscopy and images captured as described previously.
2.11. Confocal Microscopy
Dual IHC staining for Bb and Hp in MD skin sections was performed as described above. Deparaffinized and washed sections were stained with mouse monoclonal antibody specifically targeting Bb sensu stricto strains (MA-1-7006, Thermo Scientific) followed by staining with goat anti-mouse secondary antibody labeled with DyLight 488 fluorescent dye as described above. The sections were then washed, blocked with goat serum and incubated with fluorescently labeled polyclonal rabbit antibody targeting whole Hp cells (PA1-73161, Thermo Scientific) in a humidified chamber at room temperature. Sections were then washed and counterstained with Sudan Black. Confocal microscopy with a Leica DMI6000 was performed in order to increase optical resolution and contrast so that spatial distribution of mixed biofilm structures could be observed in 3 dimensions. Z-stacks were generated using Image J software (Wayne Rasband, NIH, Bethesda, MD, USA).
4. Discussion
In this study, Bb/Hp dual infection was detected in MD skin sections using tissue culture, PCR technology, IHC staining, FISH testing and confocal microscopy. The PCR data from this study revealed that callus material from 14 MD subjects tested positive for at least one sequence from either Bb or Hp, and eight subjects tested positive for at least one sequence from both bacteria. This finding verified that Bb and Hp are pathogens that can be present in MD skin sections. Given the fact that both Bb and Hp infections can cause illness in humans, including dermopathy, the presence of Bb and Hp in skin demonstrating MD pathology strongly suggests that these organisms could jointly contribute to MD evolution. IHC staining and FISH testing for Bb/Hp overlapped on consecutive sections, revealing that these pathogenic organisms co-localize in aggregates consistent with biofilms.
Species of
Borrelia from both the LD and RFB groups have been repeatedly detected in MD tissues, suggesting a causal relationship [
10,
12,
16]. The genus
Borrelia comprises at least 52 species from the LD group (Bbss and Bbsl) and the RFB group—members of these species being the causative agents of Lyme disease and relapsing fever, respectively [
37,
38,
39,
40,
41]. Lyme disease is a systemic infection involving multiple organs including joints, the heart, and the central nervous system [
42]. RFB produces systemic infection with cyclical fevers, flu-like symptoms and possible central nervous system involvement, especially in immunocompromised patients [
43]. Although dermatological manifestations are not well described for RFB, various skin ailments are associated with LD. There are three skin conditions commonly attributed to Bb: erythema migrans, lymphocytoma, and acrodermatitis chronica atrophicans. [
44]. In addition, other dermatological conditions have been attributed to Bb infection, including morphea, lichenoid genital lesions, B-cell lymphoma, dermatomyositis and cutaneous lymphoid hyperplasia [
44,
45,
46,
47].
Hp has been detected less frequently in MD skin sections [
10,
12,
18,
19]. Hp is a bacterium commonly encountered in humans, with as many as 90% of the population estimated to be colonized [
48,
49]. It is primarily associated with gastrointestinal inflammatory and ulcerative diseases, and it is the primary etiologic agent in chronic active gastritis, peptic ulcer disease, gastric adenocarcinoma, and mucosa-associated lymphoid malignancy [
49]. Studies have linked Hp infection to gastric cancer in humans, and infection is classified as carcinogenic [
50]. Hp infection may also be associated with extra-intestinal pathology including endocrine and metabolic disorders, immune dysfunction, rheumatological conditions and autoimmune diseases [
49,
51,
52,
53]. Hp may also play an etiological role in various skin disorders including rosacea, psoriasis, chronic urticaria, Schönlein–Henoch purpura, and alopecia areata [
51,
53,
54].
Biofilms are bacterial communities formed by many bacterial species in response to environments that are adversarial, including factors such as high pH, suboptimal oxygen levels, host immune attack or antibiotics [
55]. The formation of biofilm is a collective bacterial behavior. Bacteria use quorum sensing to regulate the gene expression required for collective behaviors, and these processes require a group-level response to extracellular signaling molecules called autoinducers that are produced and secreted by the bacteria [
56]. Biofilms can form on abiotic and biotic surfaces, including mucosal surfaces and on indwelling medical devices [
55]. Bacterial biofilms can form in vivo in human beings, resulting from infections associated with chronic conditions such as bacterial endocarditis, cystic fibrosis, human gastric mucosal lesions and struvite nephrolithiasis [
57,
58]. Biofilms along with resultant immune responses are responsible for the development of chronic recalcitrant slowly-healing wound conditions such as venous leg ulcers and diabetic foot ulcers and are acknowledged contributors to antibiotic resistance [
59,
60,
61,
62]. Like other chronic skin ulcerations, MD lesions are slow to heal and the establishment of Bb/Hp mixed biofilms in MD cutaneous tissue would help to explain the chronicity and recalcitrance of MD lesions.
Bb and Hp can independently form biofilms both in vitro and in vivo [
21,
22,
58,
60,
62,
63]. The formation of biofilms in vitro enabled the establishment of antibiotic resistant Bb cultures [
60,
64]. Although chronic LD is a controversial topic, persistent Bb infection despite antibiotics was reported in a cohort of patients with Lyme disease that included MD subjects, and viable Bb spirochetes were recovered by culture from body fluids and tissue specimens from these subjects [
65]. Antimicrobial resistance of Hp is a major concern, with 30% of clinical isolates estimated to be clarithromycin resistant [
49]. Although the exact mechanism of resistance has not been elucidated, biofilms may contribute to the establishment of recalcitrant infections caused by both Bb and Hp [
49,
60,
64].
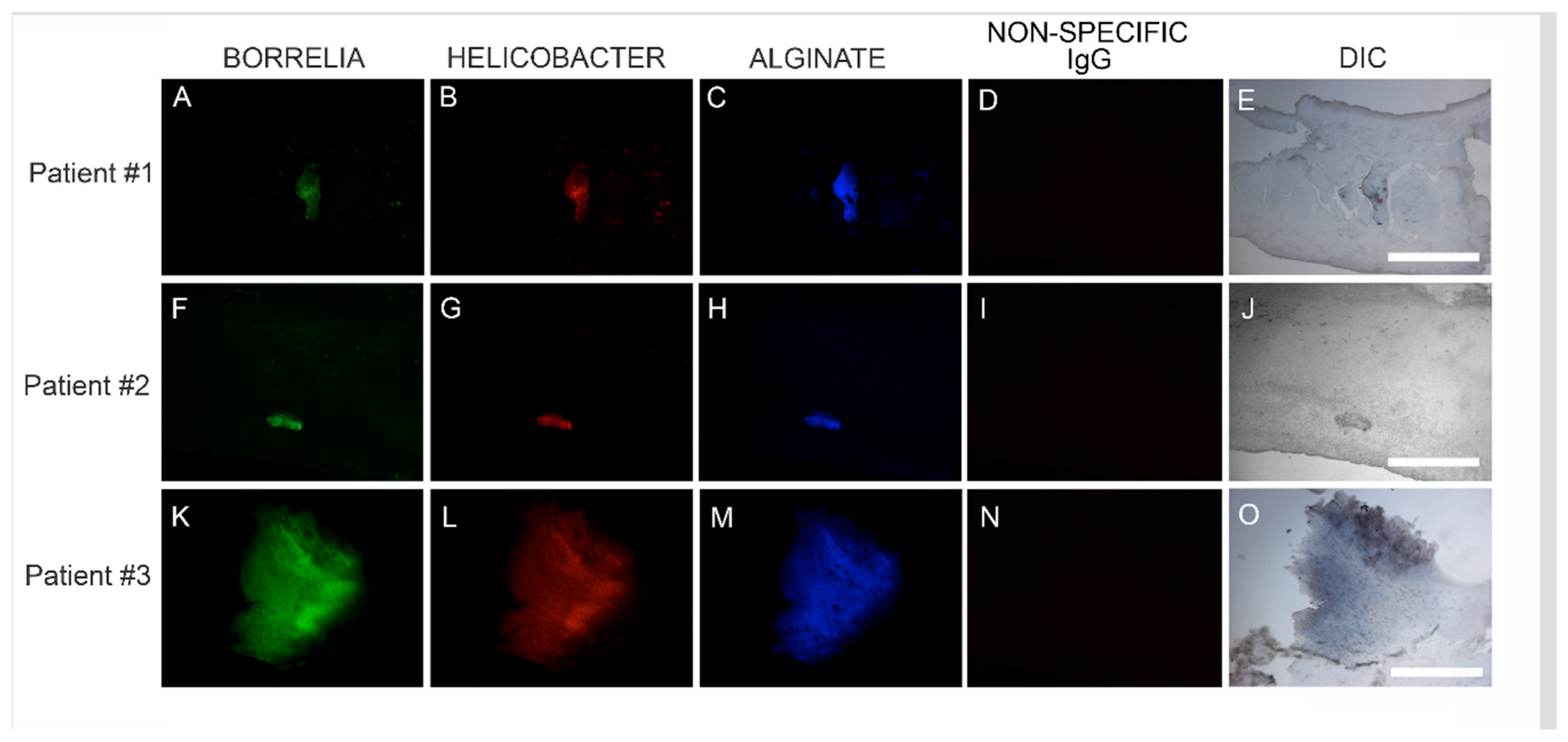
Callus sections from the six subjects with aggregates positively staining for Bb and Hp also stained positive for alginate. The presence of alginate in aggregate structures implies that mixed Bb/Hp biofilms contribute to MD pathology. Biofilm microbial communities reside in a complex self-produced exopolymeric matrix (EPM) comprised of lipids, amyloid-like proteins, exopolysaccharides, environmental DNA, and other substances [
55,
66]. The EPM serves numerous functions: structural support, protection from the external environment, gene regulation and nutrient absorption [
36,
55,
59,
60,
66]. The mucopolysaccharide component of the EPM is a protective barrier against hostile external factors, and alginate is a key mucopolysaccharide component of many bacterial biofilms [
22,
67,
68,
69]. Alginate expression occurs in
Leptospira and
Borrelia spirochetal biofilms, as well as in the biofilms formed by other bacterial species, such as those produced by
Psuedomonas aeruginosa in the lungs of cystic fibrosis patients [
22,
58,
67,
68,
69,
70].
In vitro, Bb biofilms express alginate in the EPM and exhibit elaborate structures with channels and protrusions [
22,
60,
71]. The Bb biofilms found in borrelial lymphocytomas also feature channels and protrusions that are comparable to those of Bb biofilms formed in vitro [
22]. In borrelial lymphocytoma tissue sections, alginate was associated with Bb aggregates but not isolated spirochetes, a finding that is consistent with biofilm formation. Although the exact mechanism of alginate production by
Borrelia spp. is not yet understood, an alternate method of biosynthesis involving enzymes of the Entner–Doudoroff pathway has been proposed [
60]. Alternate pathways were not tested or confirmed in the current study. Hypothetically, Hp biofilms could also express alginate as a component of their EPM [
49]. All MD calluses that demonstrated Bb/Hp aggregates were stained for alginate. Positive staining overlapped with Bb/Hp aggregates, providing convincing evidence that mixed biofilm formation occurred in vivo within calluses demonstrating MD pathology. Although alginate containing Bb/Hp aggregates were detected in MD tissue, further research is needed to determine whether Bb alone, Hp alone, or both Bb and Hp contributed towards generation of the alginate layer. Regardless of the specific origin of alginate, the presence of alginate co-localizing with Bb/Hp aggregates on MD skin tissue sections is indicative of mixed bacterial biofilm.
Confocal microscopy of MD callus tissue revealed that Bb and Hp distribution within Bb/Hp biofilm structures was species specific. Hp cells localized more superficially than Bb cells, while Bb cells localized mostly in the interior. Specific distribution suggests that these bacteria have specific functional roles within the structure that may be synergistic. Mixed bacterial infections have been reported to act in a synergistic manner, and mixed species bacterial biofilms are found in human oral cavities and have been shown to have a high degree of resistance to antibiotic treatment [
72,
73]. Simultaneous cutaneous experimental infection of laboratory animals with streptococci and
Staphylococcus aureus induced a necrotizing condition that did not occur after experimental infection with either organism alone [
72]. Synergistic interactions between bacteria in mixtures—
Acinetobacter baumanni combined with
Streptophomonas maltophila, and
Pseudomonas aeruginosa with
S. maltophila—increased biofilm formation from 20-fold to as much as 102-fold when compared to biofilm formation in single-species cultures. The synergistic augmentation of biofilm formation was noted even when one of the organisms in the mixture inherently lacked the ability to form biofilms [
74].
In addition to enhanced biofilm formation, other synergistic effects between mixed bacteria occur. For example,
Haemophilus influenzae expresses its virulence factor type IV pili in a mixed-species biofilm with
Streptococcus pneumoniae [
74]. Synergistic enhancement of biofilm production could also involve a variety of mechanisms: one species of bacteria may provide surface attachment for another species; or production of secreted factors such as quorum-sensing molecules may be increased; or production of other bacterial cellular products such as those that influence gene expression, metabolic interaction, physical contact, and synthesis of antimicrobial secretory exoproducts may be increased [
74]. Synergy in some mixed bacterial biofilms is dependent upon cell–cell contact rather than the effects of soluble bacterial products [
75]. As other pathogens such as
Treponema denticola,
Bartonella henselae, and
Rickettsia spp. have been detected in MD skin sections, they too could be co-involved in mixed biofilm formation [
10,
12,
17,
18,
19,
20]. It is possible that these and other yet undetected pathogens in MD skin could be present in mixed MD biofilms, and the constellation of pathogens contributing to MD evolution may differ from patient to patient. The synergistic effects of Bb and Hp in MD biofilms, their species-specific contributions towards establishing the biofilm structure and their joint role in MD evolution remain to be elucidated.
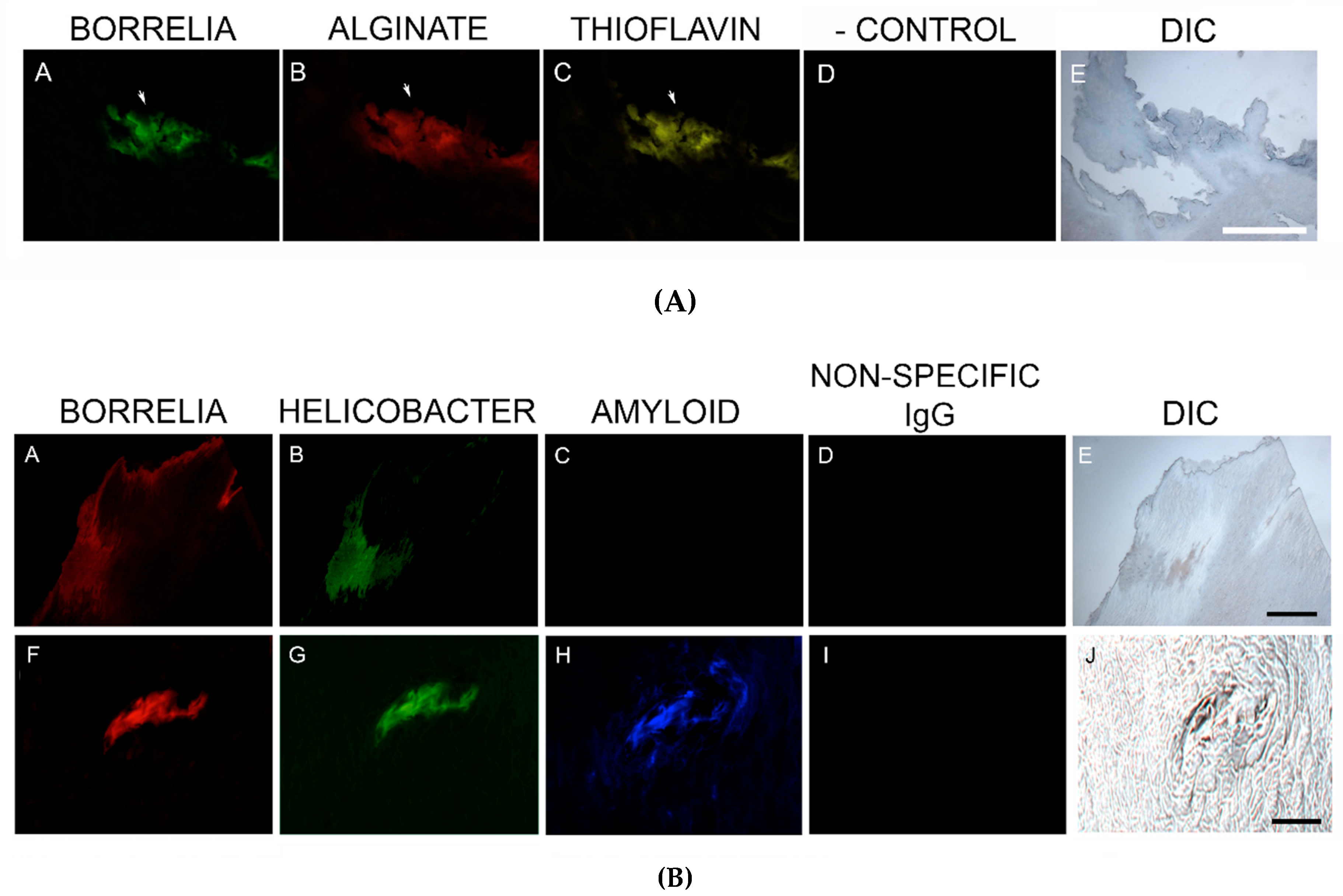
MD callus tissue sections were screened for the presence of amyloid deposits by staining with a general amyloid stain, Thioflavin S, that enhances the fluorescence of laminated β-sheets [
76], followed by more targeted detection using IHC staining with antibodies specific for amyloid-β. Amyloids are abnormal fibrous, extracellular, insoluble, proteinaceous deposits, predominantly composed of β-sheet structures in a characteristic cross-β conformation [
77]. There are 20 different plasma proteins that can form amyloid aggregates, and despite differences in their amino acid sequences, all have precursor molecules capable of forming aggregates and all have a β-strand polypeptide backbone capable of assembly into β-sheets [
77].
Degenerative diseases such as Alzheimer’s disease (AD), prion-associated spongiform encephalopathies, type II diabetes, transthyretin amyloidosis, and Parkinson’s disease are associated with accumulation of insoluble misfolded protein aggregates, such as amyloid and tau aggregates, in afflicted tissues [
77,
78,
79,
80]. AD brains are distinguished by the formation of senile plaques composed of extracellular β-amyloid protein (β-A4) deposits and/or filamentous curli fiber aggregates and the formation of neurofibrillary tangles that are composed of paired helical filaments of insoluble highly phosphorylated tau proteins and associated lipids [
32,
81]. The AD amyloid β-A4 is a 4-KDa protein resulting from the cleavage of β-amyloid precursor protein (βAPP) [
81,
82]. Under normal circumstances, βAPP is a transmembrane protein associated with neuronal development, neurite outgrowth, and axonal transport [
81].
Pathological argyrophillic filaments, histochemically consistent with amyloids, accumulate in AD brain lesions, but may also accumulate in other diseased organs such as pancreas, liver, ovaries, testes and thyroid [
32]. Amyloid deposition resulting from abnormal protein folding and assembly can occur in other tissues including skin [
83,
84,
85]. In primary cutaneous amyloidosis, the amyloid deposits are restricted to skin, while in secondary cutaneous amyloidosis (associated with systemic amyloidosis), the amyloid deposits are located in skin and other organs [
85,
86].
Amyloid fibrils are associated with both Hp and Bb infections, serving as structural components of biofilms [
83,
84,
87]. Pathology like that of AD can result from spirochetal infection. Furthermore, protein accumulation resembling that of AD occurs in patients with tertiary syphilitic dementia paralytica. [
82] Bb expresses peptides with sequences capable of forming polymers in a β-sheet structure analogous to amyloid fibrils associated with human degenerative diseases [
88]. Aggregate deposits resembling those seen in AD form in mammalian glial and neuronal cell cultures after exposure to Bb spirochetes. In addition, βAPP and phospho-tau levels in cell cultures increased after the introduction of spirochetes or borrelial lipopolysaccharides [
82]. Our study established that MD specimens stained positively for the presence of β-amyloid and positive staining overlapped with Bb/Hp aggregates. Amyloids may therefore have a functional role in establishing and maintaining mixed Bb/Hp biofilm structures in MD skin and consequently in the evolution of MD.
Although amyloid fiber formation is largely considered to be due to aberrant protein folding, functional amyloids have been identified that provide organisms with beneficial properties [
77,
89,
90]. These functional amyloids encompass a wide range of sources including a mammalian protein constituent of melanosomes called Pmel17, bacterial curli fibers and fungal hydrophobins [
77,
89,
90]. Curli are functional amyloid fibers produced by Gram-negative bacteria such as
Escherichia coli that serve as protein scaffolds within the EPM of biofilms [
91]. Curli fimbriae are biophysically prototypical amyloid fibers. They are ordered protease-resistant β-sheet-rich fibers that bind with amyloid-specific dyes such as Congo Red or Thioflavin T [
90].
It is hypothesized that bacteria-generated amyloids along with other bacterial secretory products may contribute to the pathology and evolution of amyloidogenic progressive immunological and neurological disorders [
90]. Bacterial amyloids could activate neuropathogenic signals promoting human-generated amyloid aggregation and inflammatory degeneration characteristic of degenerative neurological diseases such as AD [
90]. Bacteria- and host-derived amyloid-β are components of senile plaques and βAPP and/or βAPP-like amyloidogenic proteins are associated with spirochetes [
92]. Hypothetically, abnormal human cutaneous amyloid deposits could be functionally beneficial to bacteria, perhaps providing a scaffold for biofilm formation or providing a protective barrier for bacteria in a hostile environment. The MD specimens in this study stained positively with only one of the two anti β-amyloid antibodies. The antibodies that we used were anti-human β-amyloid antibodies but targeted to different epitopes. The crossreactivity of the anti-human β-amyloid antibodies with Bb and/or Hp-generated amyloid is not known. It is possible that the amyloidosis we observed is bacteria-generated, rather than human-generated, or possibly a combination of bacteria- and human-generated amyloid folded protein, providing a possible explanation as to why MD Bb/Hp aggregates were reactive to only one antibody.
MD callus tissue sections also stained specifically for the presence of tau protein using IHC staining. Although the “amyloid hypothesis” that β-amyloid aggregation is the primary cause of AD was the predominant theory among AD researchers, new evidence suggests that AD arises by impairment of βAPP metabolism via tau pathology [
81]. Tau protein is a microtubule-associated protein serving a functional role in promoting microtubule polymerization and stabilization, controlled by kinase regulated phosphorylation [
81,
93,
94,
95]. Six tau isoforms resulting from mRNA alternative splicing are expressed in adult human brains [
81]. Phospho-tau protein is often present in tissue from different organs demonstrating amyloid pathology, including breast, heart, kidney, lung, muscle, testis, pancreas, skin, and fibroblasts [
95,
96,
97].
Tau associated with amyloid deposits differs from normal tau in that it is hyperphosphorylated. This tau alteration decreases its capacity to bind to microtubules, compromises microtubule stabilization and axonal transport, and increases tau self-assembly and aggregate formation [
81,
98]. Phosphorylated tau, or phospo-tau, accompanies amyloid aggregate formation in many degenerative human conditions including cutaneous amyloidosis [
85,
86,
95,
96,
99], and is expressed in organs along with curli fibers and neuropil threads [
32]. In AD brains, hyperphosphorylated tau accumulates in pathological inclusions [
80], and twisted fibrils present as either paired helical filaments or related straight filaments [
81]. Neurofibrillary tangles are formed in neuronal cell bodies, while threads are formed in dendrites or axons [
81]. Phosphorylation of tau protein has been shown to trigger tau protein aggregation into filamentous structures [
100]. Tau proteins convert from an inert shape to a misfolded shape that can “seed” the growth of fibers. The misfolded “seed” triggers self-assembly of fibers that then stick together into insoluble aggregates [
80].
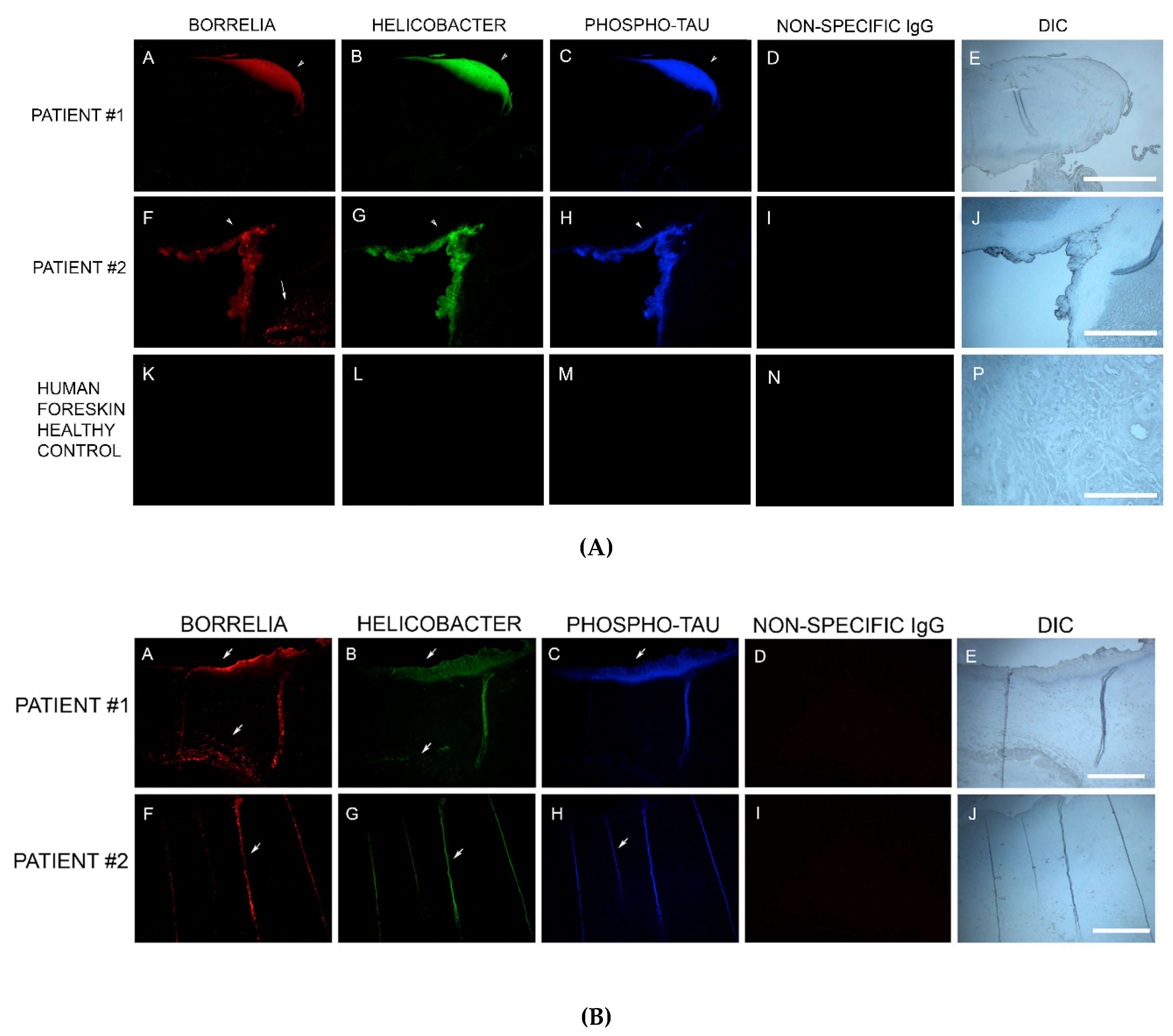
Bb in vivo biofilms may be related to tau pathology. Exposure of primary neuronal and glial cells and brain cells to Bb induces tau phosphorylation in vitro [
83]. In our study, MD skin sections were tested for the presence of phosho-tau, and all MD callus specimens stained positively, co-localizing with Bb/Hp biofilm structures and amyloid deposits. In addition, two specimens containing filamentous inclusions characteristic of MD stained positively for phospho-tau, Bb and Hp. Although MD fibers can contain collagen, keratin or both proteins, phospho-tau may be a component of some MD fibers. We hypothesize that tau protein may play a role in MD fiber formation by triggering protein aggregation into filamentous structures within MD skin. Bb/Hp positive aggregates stained positively for Thioflavin S, anti-β-amyloid protein and anti-phospho-tau, providing supportive evidence for the presence of amyloid structures within the mixed biofilms.
Although we have demonstrated biofilms containing Bb and Hp organisms in MD lesions, it is unclear whether these biofilms are responsible for the dermopathy found in MD. The fact that biofilms are associated with chronic bacterial infections and that a similar infectious dermopathy is seen in cattle and dogs argues that the biofilms containing Bb and Hp have a primary role in the skin pathology. Taken as a whole, our culture, histological and molecular findings are consistent with the modified Koch’s postulates that support a biofilm-related infectious etiology of MD lesions [
101,
102]. Additional pathogens may also be involved in formation of these biofilms [
103]. Further studies in experimental animal models are needed to confirm the role of biofilms in the pathology of MD.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}