

Design a Clinical Research Protocol: Influence of Real-World Setting
Abstract
1. Introduction
2. Methodology
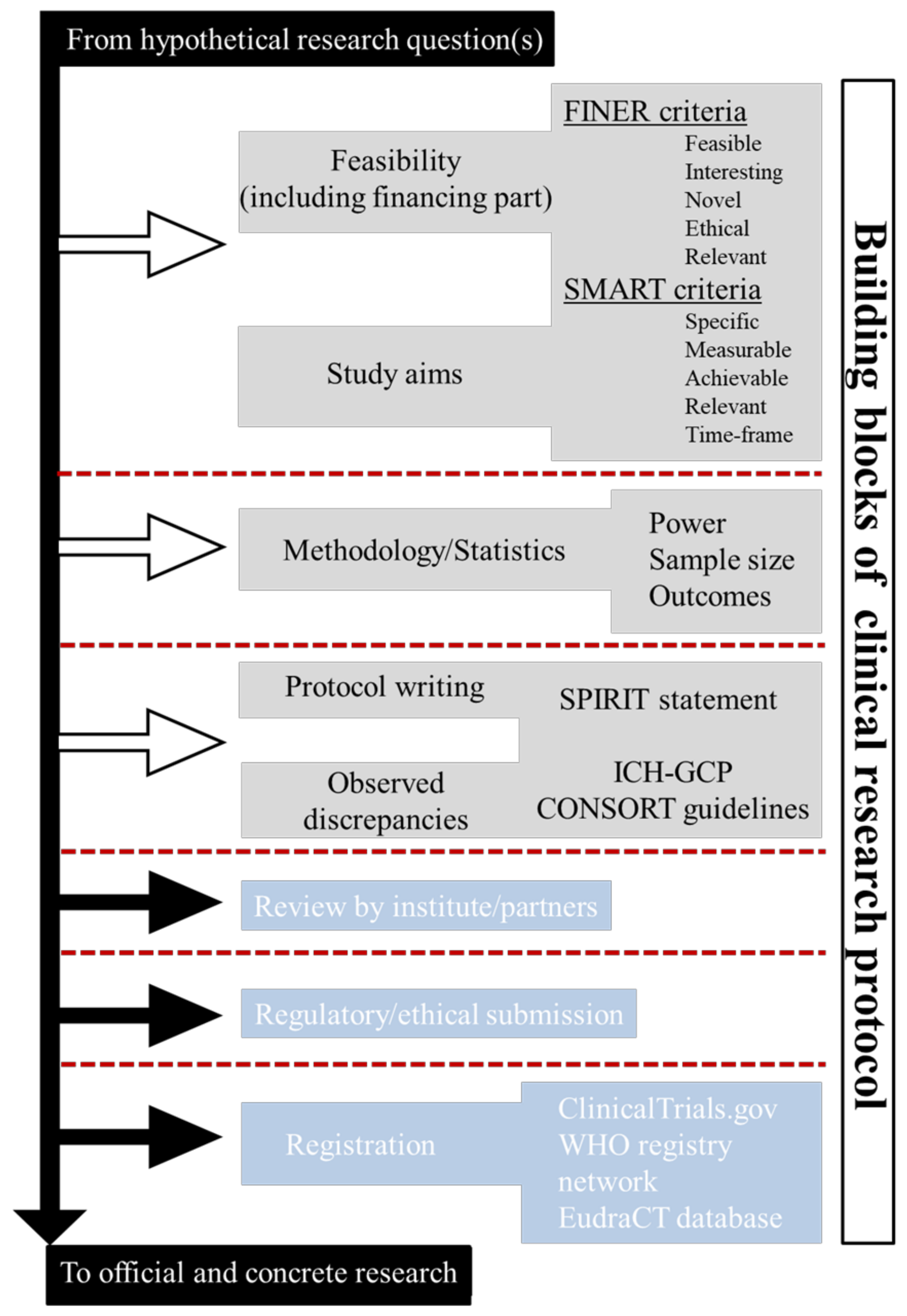
2.1. Building Blocks of Clinical-Research Protocol
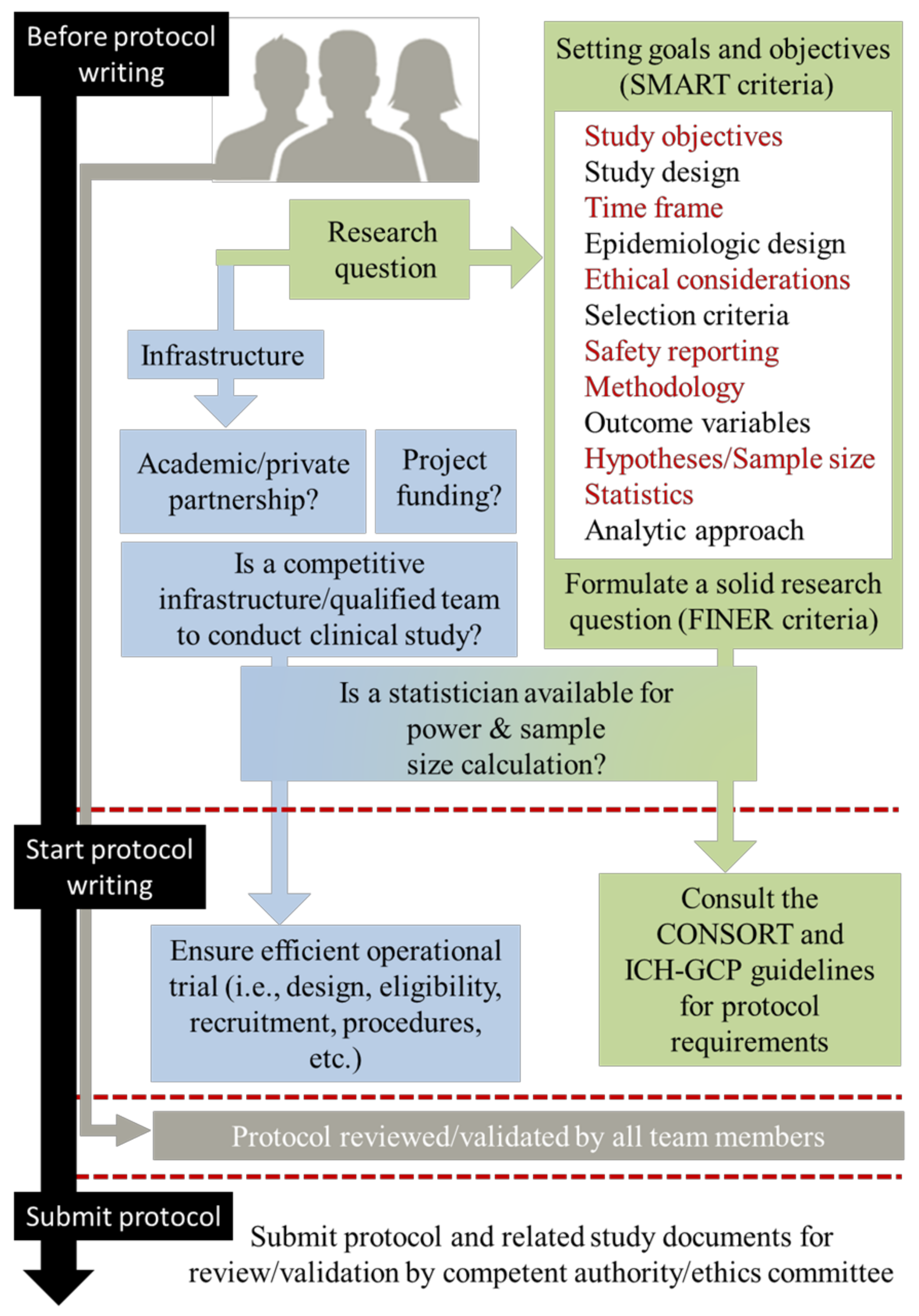
2.2. Before Writing Phase: Feasibility, Study Aims, and Methodology
- “What are the aims of the study?”, This encompasses the SMART (specific, measurable, achievable, relevant, and time-frame) criteria.
- “Why, Where and How the study should be conducted?”. This encompasses the FINER (feasible, interesting, novel, ethical, and relevant) criteria.
- (i)
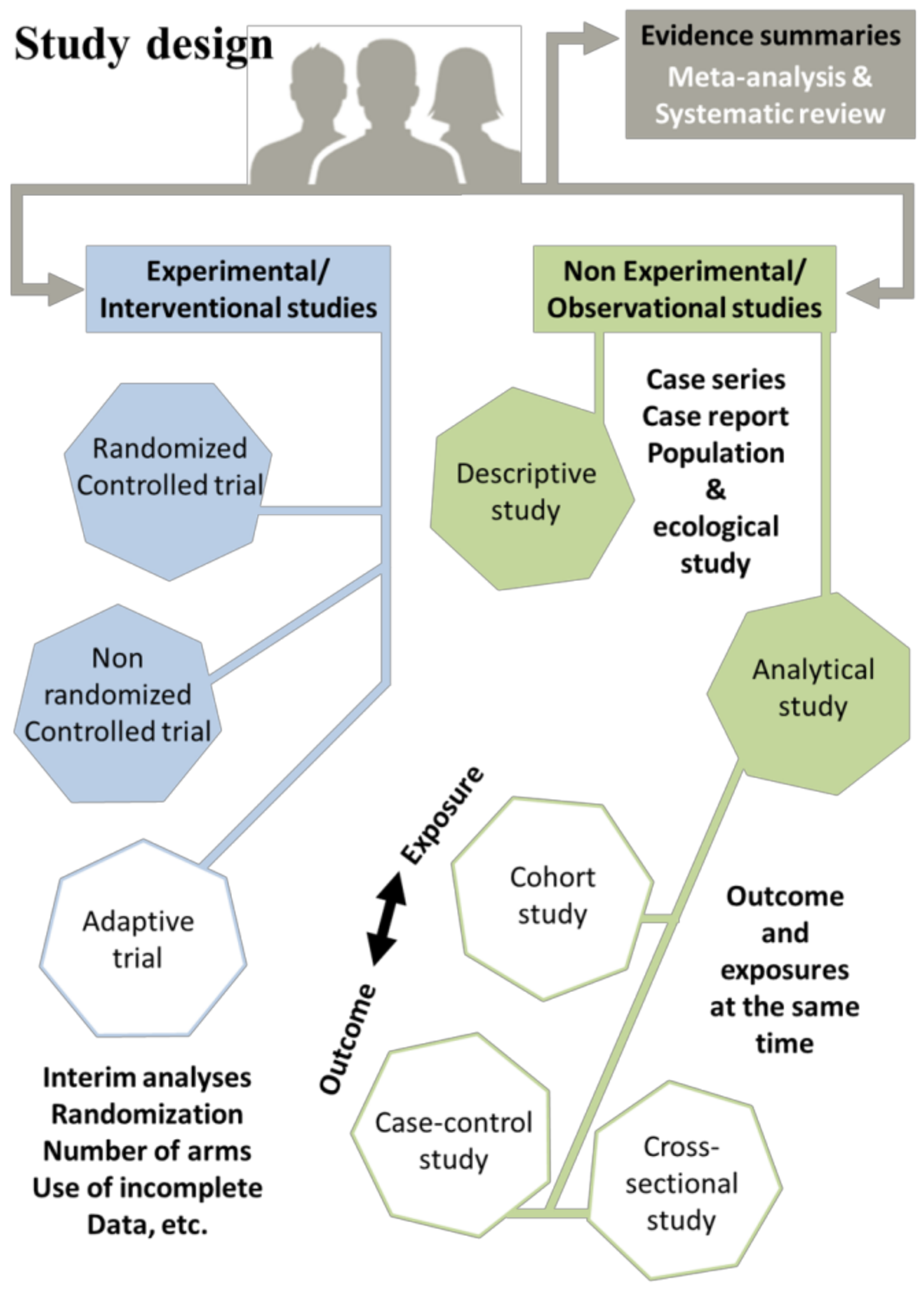
- Case–control studies, which are conducted retrospectively and are relatively straightforward to execute. They involve comparison between two distinct groups: a group with a disease (cases) and a group without this disease (controls). For instance, researchers might examine the likelihood of individuals being diabetic among obese patients compared to non-obese individuals. To accomplish this, a cohort of patients diagnosed with diabetes is selected, along with a control group exhibiting normal blood-sugar levels. Their weight histories are then examined to identify any potential correlations [54,55].
- (ii)
- Cohort studies employ a longitudinal design, which can be retrospective, using medical records, or prospective, by following participants over time. These studies involve comparisons of two samples from a population, one with a specific risk factor and the other without this risk factor, by measuring the relative risk (RR). For example, researchers might investigate the RR of developing non-small-cell lung cancer (NSCLC) among people who smoke. A sample is selected, consisting of both smokers and non-smokers, and the number of individuals with NSCLC is calculated within each group [56].
- (iii)
- Cross-sectional studies (translational design, easy to conduct) are observational research designs that aim to collect data on a particular population at a specific point in time. Unlike longitudinal studies, which follow a group of individuals over an extended period, cross-sectional studies provide a snapshot of the population’s characteristics, behaviors, or attitudes at a given moment. This type of study can be conducted using surveys, interviews, or physical examinations to gather information on various variables of interest. Cross-sectional studies are often used to explore the prevalence of certain diseases or risk factors in a population, as well as to identify associations between variables. However, it is important to note that cross-sectional studies do not establish causality and are limited by their inability to capture changes over time. As an example, the examination of the prevalence of smoking among teenagers in a particular city can be considered. The researcher selects a sample of teenagers from different schools in the city and administers a survey that asks them about their smoking habits. The survey also collects demographic information, such as age, gender, and socioeconomic status. The researcher then analyzes the data to determine the proportion of teenagers who smoke, as well as any patterns or associations between smoking and demographic factors [57,58].
2.3. Writing Phase: Guidelines and Observed Discrepancies
- (i)
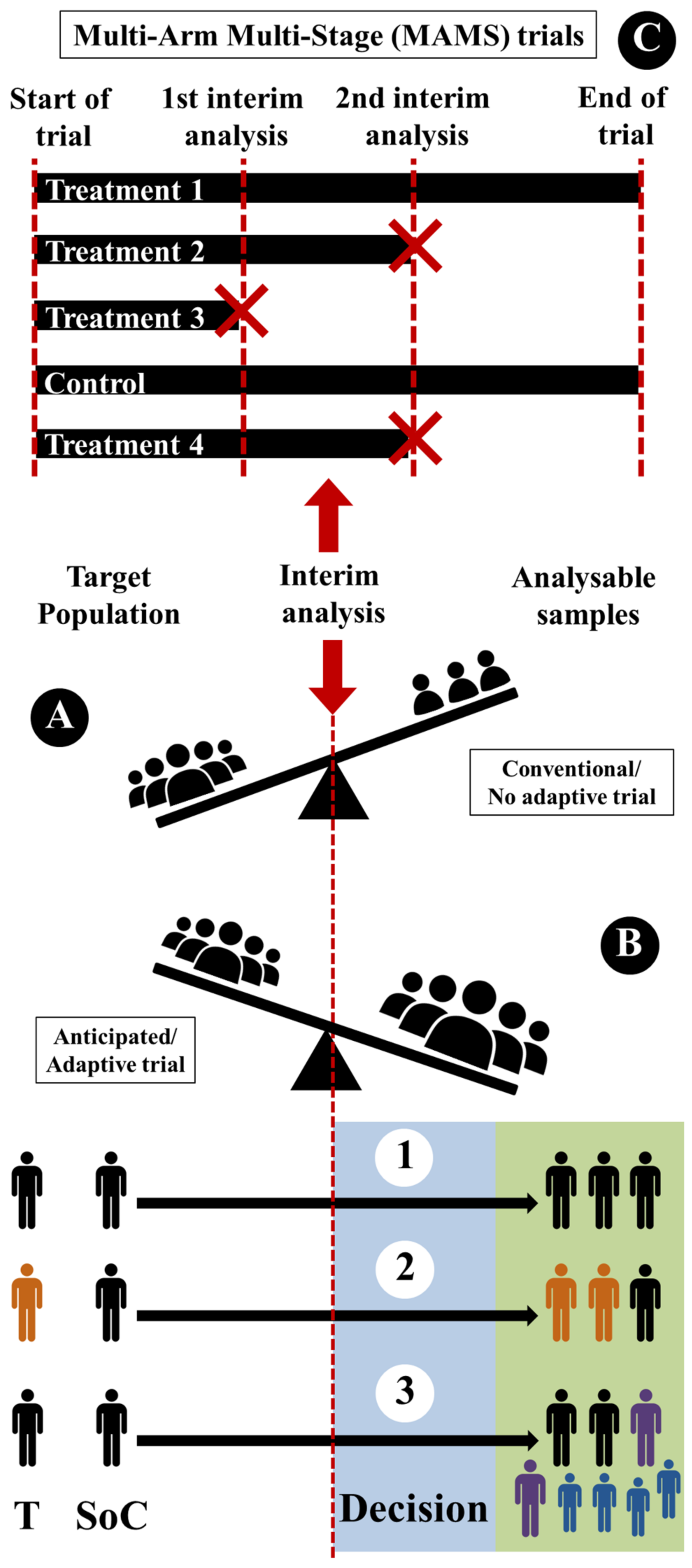
- Sample-size reassessment (Figure 5(B1)): sample size can be reassessed (after interim analysis) and increased to ensure that the trial is adequately powered.
- (ii)
- Response adaptive randomization (Figure 5(B2)): treatment-allocation ratio can be modified (after interim analysis) to favor enrolment in specific treatment.
- (iii)
- Adaptive enrichment (Figure 5(B3)): study-eligibility criteria can be modified with a sample-size reassessment (after interim analysis) to investigate the efficacy of the intervention in the related subgroup.
2.4. Internal Review
2.5. Regulatory and Ethics Validations
- A.
- Submitting a clinical-trial application to the European Medicines Agency (EMA) and obtaining a favorable opinion from the Member State Ethics Committee before initiating a trial.
- B.
- Designing clinical trials in accordance with good clinical practice (GCP) guidelines and ensuring that studies are scientifically valid, feasible, and ethical.
- C.
- Ensuring that all the study staff involved in a trial are appropriately qualified and trained to carry out their duties.
- D.
- Establishing procedures for the monitoring, recording, and reporting of adverse events and serious adverse events that occur during trials.
- E.
- Ensuring that informed consent is obtained from all of a study’s participants, and that they are provided with clear and accurate information about the trial.
- F.
- Ensuring that trials are registered on a publicly accessible database, and that the results of trials are publicly disclosed.
- G.
- Ensuring that trials are conducted in compliance with applicable data-protection and privacy laws.
2.6. Registration of the Protocol
- The selection of which trial to register on the publicly available database;
- Requests for users to set up accounts (PI organizations, like hospitals, universities, medical centers, etc.);
- The creation ClinicalTrials.gov research contents;
- The validation of the accuracy of entered content;
- Checking “Record Status” on ClinicalTrials.gov;
- The validation of notification emails from ClinicalTrials.gov within 15 (registration) and 25 days (results);
- Maintaining records entered on ClinicalTrials.gov in line with policy lines;
- Contacting ClinicalTrials.gov in case of expected/unexpected PI changes within 15 to 30 days.
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohnstamm, N.M. Approaching Judgment Day: The Influence of Brexit on the EU Pharmaceutical Framework. Leg. Issues Econ. Integr. 2019, 46, 161–179. [Google Scholar]
- Vijayananthan, A.; Nawawi, O. The importance of Good Clinical Practice guidelines and its role in clinical trials. Biomed. Imaging Interv. J. 2008, 4, e5. [Google Scholar] [CrossRef] [PubMed]
- Downing, N.S.; Aminawung, J.A.; Shah, N.D.; Krumholz, H.M.; Ross, J.S. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005–2012. JAMA 2014, 311, 368–377. [Google Scholar] [PubMed]
- Rubin, E.H.; Gilliland, D.G. Drug development and clinical trials—The path to an approved cancer drug. Nat. Rev. Clin. Oncol. 2012, 9, 215–222. [Google Scholar] [PubMed]
- Sahragardjoonegani, B.; Beall, R.F.; Kesselheim, A.S.; Hollis, A. Repurposing existing drugs for new uses: A cohort study of the frequency of FDA-granted new indication exclusivities since 1997. J. Pharm. Policy Pract. 2021, 14, 3. [Google Scholar] [CrossRef]
- Sanghera, S.; Coast, J.; Martin, R.M.; Donovan, J.L.; Mohiuddin, S. Cost-effectiveness of prostate cancer screening: A systematic review of decision-analytical models. BMC Cancer 2018, 18, 84. [Google Scholar]
- Eichler, H.G.; Sweeney, F. The evolution of clinical trials: Can we address the challenges of the future? Clin. Trials 2018, 15 (Suppl. S1), 27–32. [Google Scholar]
- Nass, S.J.; Rothenberg, M.L.; Pentz, R.; Hricak, H.; Abernethy, A.; Anderson, K.; Gee, A.W.; Harvey, R.D.; Piantadosi, S.; Bertagnolli, M.M.; et al. Accelerating anticancer drug development—Opportunities and trade-offs. Nat. Rev. Clin. Oncol. 2018, 15, 777–786. [Google Scholar]
- Sertkaya, A.; Wong, H.H.; Jessup, A.; Beleche, T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials 2016, 13, 117–126. [Google Scholar]
- Lanzerath, D. Research Ethics and Research Ethics Committees in Europe. In Medical Research Ethics: Challenges in the 21st Century; Springer International Publishing: Cham, Switzerland, 2023; pp. 423–439. [Google Scholar]
- Navone, N.M.; van Weerden, W.M.; Vessella, R.L.; Williams, E.D.; Wang, Y.; Isaacs, J.T.; Nguyen, H.M.; Culig, Z.; Van Der Pluijm, G.; Rentsch, C.A.; et al. Movember GAP1 PDX project: An international collection of serially transplantable prostate cancer patient-derived xenograft (PDX) models. Prostat 2018, 78, 1262–1282. [Google Scholar]
- Harrer, S.; Shah, P.; Antony, B.; Hu, J. Artificial intelligence for clinical trial design. Trends Pharmacol. Sci. 2019, 40, 577–591. [Google Scholar] [PubMed]
- Barr, L.H.; Crofton, J.; Lin, Y.H. A community hospital clinical trials program: Infrastructure for growth. Surg. Oncol. Clin. N. Am. 2011, 20, 447–453. [Google Scholar] [PubMed]
- Gamble, C.; Krishan, A.; Stocken, D.; Lewis, S.; Juszczak, E.; Doré, C.; Williamson, P.R.; Altman, D.G.; Montgomery, A.; Lim, P.; et al. Guidelines for the content of statistical analysis plans in clinical trials. JAMA 2017, 318, 2337–2343. [Google Scholar]
- Yuan, I.; Topjian, A.A.; Kurth, C.D.; Kirschen, M.P.; Ward, C.G.; Zhang, B.; Mensinger, J.L. Guide to the statistical analysis plan. Pediatr. Anesth. 2019, 29, 237–242. [Google Scholar]
- Thomas, L.; Peterson, E.D. The value of statistical analysis plans in observational research: Defining high-quality research from the start. JAMA 2012, 308, 773–774. [Google Scholar]
- Al-Jundi, A.; SakkA, S. Protocol writing in clinical research. J. Clin. Diagn. Res. JCDR 2016, 10, ZE10. [Google Scholar]
- Fisher, W.W.; Fuhrman, A.M.; Greer, B.D.; Ibañez, V.F.; Peterson, K.M.; Piazza, C.C. Ethical considerations with balancing clinical effectiveness with research design. In Research Ethics in Behavior Analysis; Academic Press: Cambridge, MA, USA, 2023; pp. 149–168. [Google Scholar]
- Corneli, A.; Pierre, C.; Hinkley, T.; Lin, L.; Fordyce, C.B.; Hamre, G.; Roe, M.T. One and done: Reasons principal investigators conduct only one FDA-regulated drug trial. Contemp. Clin. Trials Commun. 2017, 6, 31–38. [Google Scholar] [PubMed]
- Abzug, M.J.; Esterl, E.A. Establishment of a clinical trials office at a children’s hospital. Pediatrics 2001, 108, 1129–1134. [Google Scholar] [PubMed]
- Croghan, I.T.; Viker, S.D.; Limper, A.H.; Evans, T.K.; Cornell, A.R.; Ebbert, J.O.; Gertz, M.A. Developing a clinical trial unit to advance research in an academic institution. Contemp. Clin. Trials 2015, 45, 270–276. [Google Scholar]
- Simera, I.; Moher, D.; Hoey, J.; Schulz, K.F.; Altman, D.G. A catalogue of reporting guidelines for health research. Eur. J. Clin. Investig. 2010, 40, 35–53. [Google Scholar]
- Farrugia, P.; Petrisor, B.A.; Farrokhyar, F.; Bhandari, M. Research questions, hypotheses and objectives. Can. J. Surg. 2010, 53, 278. [Google Scholar] [PubMed]
- Demotes-Mainard, J.; Cornu, C.; Guerin, A.; Bertoye, P.H.; Boidin, R.; Bureau, S.; Chrétien, J.M.; Delval, C.; Deplanque, D.; Dubray, C.; et al. How the new European data protection regulation affects clinical research and recommendations? Therapies 2019, 74, 31–42. [Google Scholar]
- Timmers, M.; Van Veen, E.B.; Maas, A.I.; Kompanje, E.J. Will the Eu data protection regulation 2016/679 inhibit critical care research? Med. Law Rev. 2019, 27, 59–78. [Google Scholar] [PubMed]
- Casali, P.G.; Vyas, M. Data protection and research in the European Union: A major step forward, with a step back. Ann. Oncol. 2021, 32, 15–19. [Google Scholar] [PubMed]
- Chico, V. The impact of the general data protection regulation on health research. Br. Med. Bull. 2018, 128, 109–118. [Google Scholar] [CrossRef]
- Minssen, T.; Rajam, N.; Bogers, M. Clinical trial data transparency and GDPR compliance: Implications for data sharing and open innovation. Sci. Public Policy 2020, 47, 616–626. [Google Scholar]
- Gloy, V.; Speich, B.; Griessbach, A.; Taji Heravi, A.; Schulz, A.; Fabbro, T.; Magnus, C.P.; McLennan, S.; Bertram, W.; Briel, M. Scoping review and characteristics of publicly available checklists for assessing clinical trial feasibility. BMC Med. Res. Methodol. 2022, 22, 142. [Google Scholar]
- Abbott, J.H. The distinction between randomized clinical trials (RCTs) and preliminary feasibility and pilot studies: What they are and are not. J. Orthop. Sports Phys. Ther. 2014, 44, 555–558. [Google Scholar]
- Fiteni, F.; Westeel, V.; Pivot, X.; Borg, C.; Vernerey, D.; Bonnetain, F. Endpoints in cancer clinical trials. J. Visc. Surg. 2014, 151, 17–22. [Google Scholar] [CrossRef]
- De Gruttola, V.G.; Clax, P.; DeMets, D.L.; Downing, G.J.; Ellenberg, S.S.; Friedman, L.; Gail, M.H.; Prentice, R.; Wittes, J.; Zeger, S.L. Considerations in the evaluation of surrogate endpoints in clinical trials: Summary of a National Institutes of Health workshop. Control. Clin. Trials 2001, 22, 485–502. [Google Scholar]
- Pazdur, R. Endpoints for assessing drug activity in clinical trials. Oncologist 2008, 13, 19–21. [Google Scholar] [PubMed]
- Mallinckrod, C.H.; Lane, P.W.; Schnell, D.; Peng, Y.; Mancuso, J.P. Recommendations for the primary analysis of continuous endpoints in longitudinal clinical trials. Drug Inf. J. 2008, 42, 303–319. [Google Scholar]
- Fleming, T.R.; Powers, J.H. Biomarkers and surrogate endpoints in clinical trials. Stat. Med. 2012, 31, 2973–2984. [Google Scholar] [PubMed]
- Walton, M.K.; Powers, I.I.I.J.H.; Hobart, J.; Patrick, D.; Marquis, P.; Vamvakas, S.; Isaac, M.; Molsen, E.; Cano, S.; Burke, L.B. Clinical outcome assessments: Conceptual foundation—Report of the ISPOR clinical outcomes assessment–emerging good practices for outcomes research task force. Value Health 2015, 18, 741–752. [Google Scholar] [PubMed]
- Elliott, M.R. Surrogate Endpoints in Clinical Trials. Annu. Rev. Stat. Its Appl. 2023, 10, 75–96. [Google Scholar]
- Walia, A.; Haslam, A.; Prasad, V. FDA validation of surrogate endpoints in oncology: 2005–2022. J. Cancer Policy 2022, 34, 100364. [Google Scholar]
- Buyse, M.; Molenberghs, G.; Paoletti, X.; Oba, K.; Alonso, A.; Van der Elst, W.; Burzykowski, T. Statistical evaluation of surrogate endpoints with examples from cancer clinical trials. Biom. J. 2016, 58, 104–132. [Google Scholar]
- Molenberghs, G.; Buyse, M.; Geys, H.; Renard, D.; Burzykowski, T.; Alonso, A. Statistical challenges in the evaluation of surrogate endpoints in randomized trials. Control. Clin. Trials 2002, 23, 607–625. [Google Scholar]
- Buyse, M.; Molenberghs, G. The Evaluation of Surrogate Endpoints; Burzykowski, T., Ed.; Springer: New York, NY, USA, 2005; Volume 427. [Google Scholar]
- Gotay, C.C.; Kawamoto, C.T.; Bottomley, A.; Efficace, F. The prognostic significance of patient-reported outcomes in cancer clinical trials. J. Clin. Oncol. 2008, 26, 1355–1363. [Google Scholar]
- Mercieca-Bebber, R.; King, M.T.; Calvert, M.J.; Stockler, M.R.; Friedlander, M. The importance of patient-reported outcomes in clinical trials and strategies for future optimization. Patient Relat. Outcome Meas. 2018, 9, 353–367. [Google Scholar]
- Deshpande, P.R.; Rajan, S.; Sudeepthi, B.L.; Nazir, C.A. Patient-reported outcomes: A new era in clinical research. Perspect. Clin. Res. 2011, 2, 137. [Google Scholar]
- Brundage, M.; Blazeby, J.; Revicki, D.; Bass, B.; De Vet, H.; Duffy, H.; Efficace, F.; King, M.; Lam, C.L.; Moher, D.; et al. Patient-reported outcomes in randomized clinical trials: Development of ISOQOL reporting standards. Qual. Life Res. 2013, 22, 1161–1175. [Google Scholar] [PubMed]
- Buyse, M.; Michiels, S.; Sargent, D.J.; Grothey, A.; Matheson, A.; De Gramont, A. Integrating biomarkers in clinical trials. Expert Rev. Mol. Diagn. 2011, 11, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Freidlin, B.; McShane, L.M.; Korn, E.L. Randomized clinical trials with biomarkers: Design issues. J. Natl. Cancer Inst. 2010, 102, 152–160. [Google Scholar] [PubMed]
- Dancey, J.E.; Dobbin, K.K.; Groshen, S.; Jessup, J.M.; Hruszkewycz, A.H.; Koehler, M.; Parchment, R.; Ratain, M.J.; Shankar, L.K.; Stadler, W.M.; et al. Guidelines for the development and incorporation of biomarker studies in early clinical trials of novel agents. Clin. Cancer Res. 2010, 16, 1745–1755. [Google Scholar]
- Van Schaeybroeck, S.; Allen, W.L.; Turkington, R.C.; Johnston, P.G. Implementing prognostic and predictive biomarkers in CRC clinical trials. Nat. Rev. Clin. Oncol. 2011, 8, 222–232. [Google Scholar]
- Lipkovich, I.; Dmitrienko, A. Strategies for identifying predictive biomarkers and subgroups with enhanced treatment effect in clinical trials using SIDES. J. Biopharm. Stat. 2014, 24, 130–153. [Google Scholar] [CrossRef]
- Holm, K.E.; Casaburi, R.; Cerreta, S.; Gussin, H.A.; Husbands, J.; Porszasz, J.; Prieto-Centurion, V.; Sandhaus, R.A.; Sullivan, J.L.; Walsh, L.J.; et al. Patient involvement in the design of a patient-centered clinical trial to promote adherence to supplemental oxygen therapy in COPD. Patient-Patient-Centered Outcomes Res. 2016, 9, 271–279. [Google Scholar] [CrossRef]
- González-Bueno, J.; Sevilla-Sánchez, D.; Puigoriol-Juvanteny, E.; Molist-Brunet, N.; Codina-Jané, C.; Espaulella-Panicot, J. Improving medication adherence and effective prescribing through a patient-centered prescription model in patients with multimorbidity. Eur. J. Clin. Pharmacol. 2022, 78, 127–137. [Google Scholar] [CrossRef]
- Hulley, S.B. (Ed.) Designing Clinical Research; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Setia, M.S. Methodology series module 2: Case-control studies. Indian J. Dermatol. 2016, 61, 146. [Google Scholar]
- Schulz, K.F.; Grimes, D.A. Case-control studies: Research in reverse. Lancet 2002, 359, 431–434. [Google Scholar] [PubMed]
- Camargo, L.M.A.; Silva, R.P.M.; de Oliveira Meneguetti, D.U. Research methodology topics: Cohort studies or prospective and retrospective cohort studies. J. Hum. Growth Dev. 2019, 29, 433–436. [Google Scholar]
- Wang, X.; Cheng, Z. Cross-sectional studies: Strengths, weaknesses, and recommendations. Chest 2020, 158, S65–S71. [Google Scholar]
- Kesmodel, U.S. Cross-sectional studies—What are they good for? Acta Obstet. Gynecol. Scand. 2018, 97, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Hariton, E.; Locascio, J.J. Randomised controlled trials—The gold standard for effectiveness research. BJOG Int. J. Obstet. Gynaecol. 2018, 125, 1716. [Google Scholar] [CrossRef]
- Kaptchuk, T.J. The double-blind, randomized, placebo-controlled trial: Gold standard or golden calf? J. Clin. Epidemiol. 2001, 54, 541–549. [Google Scholar]
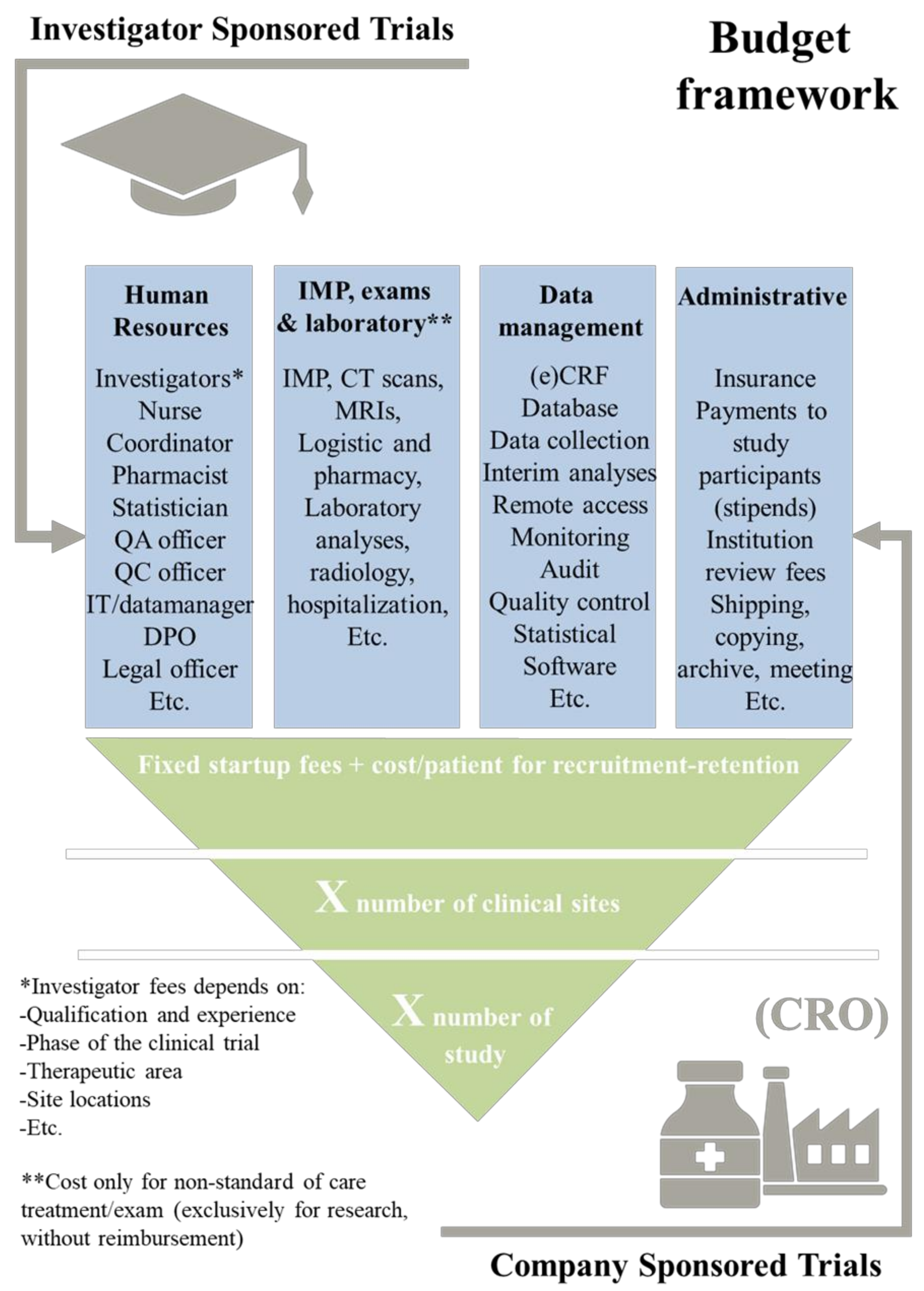
- Cimino, J.; Braun, C. Building a competitive infrastructure to support clinical research in healthcare institution. Eur. J. Clin. Investig. 2021, 51, e13641. [Google Scholar] [CrossRef]
- Hwang, T.J.; Carpenter, D.; Lauffenburger, J.C.; Wang, B.; Franklin, J.M.; Kesselheim, A.S. Failure of investigational drugs in late-stage clinical development and publication of trial results. JAMA Intern. Med. 2016, 176, 1826–1833. [Google Scholar]
- Vernon, J.A.; Golec, J.H.; DiMasi, J.A. Drug development costs when financial risk is measured using the Fama–French three-factor model. Health Econ. 2010, 19, 1002–1005. [Google Scholar]
- Sheffet, A.J.; Flaxman, L.; Tom, M.; Hughes, S.E.; Longbottom, M.E.; Howard, V.J.; Marler, J.R.; Brott, T.G. CREST Investigators. Financial management of a large multisite randomized clinical trial. Int. J. Stroke 2014, 9, 811–813. [Google Scholar]
- Martin, L.; Hutchens, M.; Hawkins, C.; Radnov, A. How much do clinical trials cost. Nat. Rev. Drug Discov. 2017, 16, 381–382. [Google Scholar] [CrossRef]
- Makris, U.E.; Ferrante, L.E.; Mody, L. Leadership Lessons: Building and Leading a High-Performing Clinical Research Team. J. Am. Geriatr. Soc. 2018, 66, 1258. [Google Scholar]
- Chan, A.W.; Tetzlaff, J.M.; Altman, D.G.; Laupacis, A.; Gøtzsche, P.C.; Krleža-Jerić, K.; Hróbjartsson, A.; Mann, H.; Dickersin, K.; Berlin, J.A.; et al. SPIRIT 2013 statement: Defining standard protocol items for clinical trials. Ann. Intern. Med. 2013, 158, 200–207. [Google Scholar]
- MacPherson, H.; Altman, D.G.; Hammerschlag, R.; Youping, L.; Taixiang, W.; White, A.; Moher, D. Revised standards for reporting interventions in clinical trials of acupuncture (STRICTA): Extending the CONSORT statement. J. Altern. Complement. Med. 2010, 16, ST-1. [Google Scholar]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Mehta, C. Adaptive designs for clinical trials. N. Engl. J. Med. 2016, 375, 65–74. [Google Scholar] [CrossRef]
- Pallmann, P.; Bedding, A.W.; Choodari-Oskooei, B.; Dimairo, M.; Flight, L.; Hampson, L.V.; Holmes, J.; Mander, A.P.; Odondi, L.O.; Sydes, M.R.; et al. Adaptive designs in clinical trials: Why use them, and how to run and report them. BMC Med. 2018, 16, 29. [Google Scholar]
- Angus, D.C.; Berry, S.; Lewis, R.J.; Al-Beidh, F.; Arabi, Y.; van Bentum-Puijk, W.; Bhimani, Z.; Bonten, M.; Broglio, K.; Brunkhorst, F.; et al. The REMAP-CAP (randomized embedded multifactorial adaptive platform for community-acquired pneumonia) study. Rationale and design. Ann. Am. Thorac. Soc. 2020, 17, 879–891. [Google Scholar]
- Barker, A.D.; Sigman, C.C.; Kelloff, G.J.; Hylton, N.M.; Berry, D.A.; Esserman, L. I-SPY 2: An adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin. Pharmacol. Ther. 2009, 86, 97–100. [Google Scholar]
- Saleh, M.; Naik, G. So you want to be a principal investigator. J. Oncol. Pract. 2018, 14, e384–e392. [Google Scholar] [CrossRef]
- Rowinsky, E.K. Erosion of the principal investigator role in a climate of industry dominance. Eur. J. Cancer 2005, 41, 2206–2209. [Google Scholar] [CrossRef] [PubMed]
- Cingi, C.; Bayar Muluk, N. Quick Guide to Good Clinical Practice; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Verma, K. Base of a research: Good clinical practice in clinical trials. J. Clin. Trials 2013, 3, 100–128. [Google Scholar]
- Tenti, E.; Simonetti, G.; Bochicchio, M.T.; Martinelli, G. Main changes in European clinical trials regulation (no 536/2014). Contemp. Clin. Trials Commun. 2018, 11, 99–101. [Google Scholar] [CrossRef]
- Califf, R.M.; Zarin, D.A.; Kramer, J.M.; Sherman, R.E.; Aberle, L.H.; Tasneem, A. Characteristics of clinical trials registered in ClinicalTrials. gov, 2007–2010. JAMA 2012, 307, 1838–1847. [Google Scholar] [CrossRef]
- Chang, S.M.; Slutsky, J. Debunking myths of protocol registration. Syst. Rev. 2012, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Henon, C.; Lissa, D.; Paoletti, X.; Thibault, C.; Le Tourneau, C.; Lanoy, E.; Hollebecque, A.; Massard, C.; Soria, J.C.; Postel-Vinay, S. Patient-reported tolerability of adverse events in phase 1 trials. ESMO Open 2017, 2, e000148. [Google Scholar] [CrossRef]
- Pitrou, I.; Boutron, I.; Ahmad, N.; Ravaud, P. Reporting of safety results in published reports of randomized controlled trials. Arch. Intern. Med. 2009, 169, 1756–1761. [Google Scholar]
- Levenson, M.; He, W.; Chen, J.; Fang, Y.; Faries, D.; Goldstein, B.A.; Ho, M.; Lee, K.; Mishra-Kalyani, P.; Rockhold, F.; et al. Biostatistical considerations when using RWD and RWE in clinical studies for regulatory purposes: A landscape assessment. Stat. Biopharm. Res. 2023, 15, 3–13. [Google Scholar]
- Seeger, J.D.; Nunes, A.; Loughlin, A.M. Using RWE research to extend clinical trials in diabetes: An example with implications for the future. Diabetes Obes. Metab. 2020, 22, 35–44. [Google Scholar]
- Uecke, O.; Reszka, R.; Linke, J.; Steul, M.; Posselt, T. Clinical trials: Considerations for researchers and hospital administrators. Health Care Manag. Rev. 2008, 33, 103–112. [Google Scholar]
- García-Romero, A.; Escribano, Á.; Tribó, J.A. The impact of health research on length of stay in Spanish public hospitals. Res. Policy 2017, 46, 591–604. [Google Scholar]
- Demotes-Mainard, J.; Ohmann, C. European Clinical Research Infrastructures Network: Promoting harmonisation and quality in European clinical research. Lancet 2005, 365, 107–108. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Role in Clinical Research | Responsibility |
|---|---|
| Principal investigator (PI) | Responsible for the entire study (funding, design, data, staffing, conduct, and reporting data and outcomes through publications. |
| Additional investigator (co-PI) | Shares responsibility with the PI for ensuring that study is conducted in compliance with applicable laws and regulations and institutional policy. |
| Project manager | Management of all study activities. |
| Research assistant (nurse or clinical research associate) | In contact with the patients, ensures that the recruitment (according to the eligibility criteria) is smooth and that the planned visits are carried out. |
| Quality-control coordinator | Oversees quality control and verifies that all staff are following standard operating procedures (SOPs). |
| IT manager | Coordinates the organization, implementation, and management of IT processes. |
| Data manager | Implements the data-entry system and management. |
| Programmer/analyst | Data-quality analyses. |
| Statistician | Vital role in the design (including sample size), planning, and analysis of clinical trials (including safety-monitoring guidelines). |
| Administrative assistant | Ensures the smooth operation and organization of various administrative tasks throughout the trial process. |
| Legal officer | Coordinates legal contracts with collaborators and sponsors. |
| Data-protection officer | Verifies that personal data used in trials are in accordance with local data-protection laws. |
| Financial manager | Verifies specific research budget and manages invoices. |
| Human-resources manager | Handles the human resources necessary to conduct research. |
| Contents | Specificity | Topics |
|---|---|---|
| Title, identifying number (EudraCT if applicable), version, and date | Must be modified in case of amendment (version/date) | A |
| Sponsor details | Monitor (if different from the sponsor for, e.g., CRO) | |
| Contact details of the person(s) authorized by the sponsor to validate/sign the protocol | Even way for amendments | |
| Details of the on-site PI who is responsible for conducting the study | Include the full contact details for the trial site(s) | |
| Details of all medical departments/institutions involved in the trial | Include subcontracted laboratories/platform | |
| Full description of the investigational medical product (IMP) | Include drug register | B |
| A summary of previous results from clinical and non-clinical studies that are relevant for the trial | References to relevant research and data | |
| A summary of the potential risks and benefits to human subjects (if any) | Include all previous preclinical studies | |
| Scientific justification of dosage, route of administration, and treatment period(s) of IMP | ||
| Legal statement that the trial will be conducted in compliance with the GCP, validated protocol version, and the applicable data-protection requirement(s) | Include GDPR requirement(s) | |
| Description of the population evaluated | ||
| Description of the purpose and objectives of research | C | |
| Description of the primary endpoints and the secondary endpoints | Include sample-size calculation | |
| A description of the type/design and procedures | (e.g., placebo-controlled, double-blind, crossover design, etc.) | |
| Explanation of the methodology followed to minimize risk and avoid bias | (e.g., blinding and randomization processes) | |
| Description of the management of IMP | Include description of the dosage form, packaging, and labeling | D |
| Expected duration of subject participation | Duration and number of follow-ups | |
| Description of the study-discontinuation criteria | ||
| IMP-accountability procedures | ||
| Description of IMP-randomization codes | (e.g., breaking codes in case of emergency) | |
| Definition of source data to be recorded on the CRFs | (electronic or paper records of data) | |
| Subject-inclusion/exclusion criteria | ||
| Subject-withdrawal criteria (when and how) | Follow-up and how withdrawn subjects will be replaced | E |
| The description of IMP used in the trial | Investigator’s brochure included | F |
| Medication(s) permitted/not permitted before and/or during the trial | Include rescue medication | |
| Monitoring of subject compliance | ||
| Efficacy-parameter evaluation | Assessment, recording, and analysis of efficacy parameters | G |
| Safety-parameter evaluation | Assessment, recording, and analysis of safety parameters | |
| Procedures for recording and reporting adverse events | Follow-up of adverse events. | H |
| Description of the statistical methods to be employed | Planned interim analysis | |
| The estimated number of participants to be enrolled (total target number) | Sample size, including power calculations (should be specified in case of multicenter trials) | |
| Trial-termination criteria | ||
| Accounting procedure for unused and missing clinical data. | ||
| Deviation(s)-reporting procedure | From the original statistical plan, should be justified in protocol | I |
| Subject-selection criteria | (e.g., eligibility, dosage, and randomization criteria) | |
| Direct access to source data/document statement | Sponsor and IRB/IEC/regulatory monitoring | J |
| The quality plan | Describes quality-assurance practices and processes | K |
| Description of ethical and regulatory considerations relating to the trial | L | |
| Description of the following trial-related procedures: data handling, data verification, statistical analyses, and preparation of trial reports | M | |
| Financing of the project and insurance for participant details | Can be covered in a separate document | N |
| Publication policy between the partners | Can be covered in a separate document | O |
| Supplements | P |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cimino, J.; Braun, C. Design a Clinical Research Protocol: Influence of Real-World Setting. Healthcare 2023, 11, 2254. https://doi.org/10.3390/healthcare11162254
Cimino J, Braun C. Design a Clinical Research Protocol: Influence of Real-World Setting. Healthcare. 2023; 11(16):2254. https://doi.org/10.3390/healthcare11162254
Chicago/Turabian StyleCimino, Jonathan, and Claude Braun. 2023. "Design a Clinical Research Protocol: Influence of Real-World Setting" Healthcare 11, no. 16: 2254. https://doi.org/10.3390/healthcare11162254
APA StyleCimino, J., & Braun, C. (2023). Design a Clinical Research Protocol: Influence of Real-World Setting. Healthcare, 11(16), 2254. https://doi.org/10.3390/healthcare11162254