There are two primary means of modeling systems using reaction networks: the deterministic and stochastic approaches. The deterministic approach treats the system’s species, i.e., the state space, as a continuous process that is governed by a set of coupled ordinary differential equations (the ‘reaction-rate equations’). In the stochastic approach, the system state is a vector of counts, where each entry corresponds to the (discrete and non-negative) number of copies of each species type. The state space of the species’ counts evolves over continuous time, but system updates occur at discrete random times, and any changes in the state space are by an integer amount. This type of system behavior is clearly a closer approximation to real world systems, which usually involve a finite number of species, or particles, interacting with each other at random times to change the system, in contrast to a deterministic ordinary differential equation (ODE) with continuous state space. The system updates in the stochastic formulation occur as integer valued changes to the coordinates of the state space. The stochastic approach also appears to be more realistic in terms of modeling randomness in interactions of the species. For example, in gene expression, random movement associated with Brownian motion affects particle interactions. In the SIR system and in the LV system, interactions between susceptible individuals and infectious individuals and between fox (predator) and rabbits (prey), respectively, are subject to noise [
4]. Analysis using the stochastic model also solves the parameter identifiability issues inherent in fitting some of their deterministic analogs [
4]. Since we are now in a stochastic setting, the object that describes the system evolution is a probability distribution function, which turns out to be the solution to a single differential equation (the “master equation”) [
5]. The master equation solution gives the probability that the system is in a particular state at a particular time and was derived for chemical reaction networks by [
6].
2.1. Deterministic Systems
An RN consists of a set of reactant species, that interact via a set of reactions, to produce products. Specifically, an RN consists of three sets (X, C, R) defined as follows:
- 1
Species X: , which represents the quantities of the d different species types in the system.
- 2
Complexes C: a linear combination of the species. These can be represented as vectors , where gives the number of each species consumed by the rth reaction and gives the number of each species produced by the rth reaction.
- 3
Reactions R: with the reaction’s stoichiometry
The classical chemical notation for the
rth reaction is then
where
and
are non-negative stoichiometric coefficients denoting the number(s) of
consumed and produced respectively for the
rth reaction. The parameter
is the rate constant for the
rth reaction. We say that the
rth reaction is of order
m if
, i.e., if it involves
m reactant molecules.
Guldberg and Waage modeled the dynamics of chemical reaction systems in a macroscopic setting using the Law of Mass Action in the 1860s [
7]. The macroscopic setting implies the system contains a sufficiently large number of molecules, so that the state space may be viewed as a concentration, by dividing the species’ numbers by an appropriate volume term,
, and considering both to be large. Let
, where
denotes the concentration of species
, and
is the system volume, so that
is a continuous quantity. In a well-mixed system, the reaction rate for reaction
r under the Law of Mass Action is given by
We call
the macroscopic rate function of the
rth reaction, and its form implies that the reaction rates are proportional to the concentrations of the reactant molecules in each reaction. The stoichiometric matrix
S is defined as
The column
describes the net change in the state space when reaction
r occurs, and
describes the net change in the number of molecules of
when reaction
r occurs. The deterministic ODE
for species
i gives a deterministic approximation of species
i’s concentration over time, which may be derived by adding up the product of the macroscopic rate functions and the net change in species
i from each reaction. This gives the system of reaction rate equations as [
8]
where
. Clearly the solution to Equation (
4) depends on the parameter values, initial condition and time, and we denote this whenever necessary as
. We suppress this dependence in some places for convenience, but
should be understood implicitly to depend on these. The determinsitic approximation is valid in well-mixed macroscopic systems, i.e., with large numbers of molecules such that
when
grows large. To give a simple concrete example of the notation, consider the following example
. Here there are three species
,
since there is only 1 reaction, and the complexes are
,
, with stoichiometry
and macroscopic reaction rate
, that is, two molecules of dihydrogen react with one molecule of dioxygen to give two molecules of water. Then the reaction rate equation describing concentration over time in a macroscopic system is
2.2. Stochastic Systems
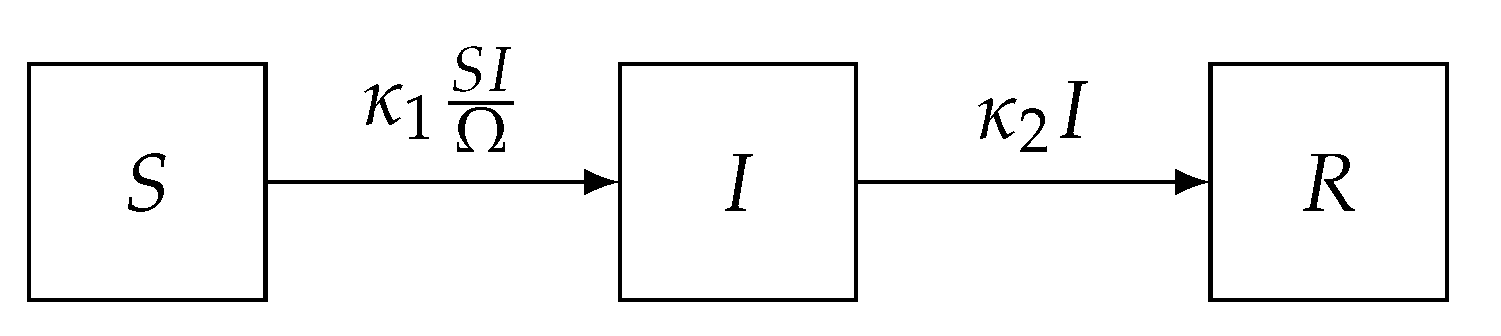
Often, systems will contain species occurring with relatively low abundance, for example in a predator–prey system when say the prey is on the verge of extinction, and hence such systems will not be completely amenable to a macroscopic analysis. In these systems, stochasticity, for instance due to Brownian motion in the chemical example, becomes an important factor affecting system evolution. While the deterministic ODE provides a sort of approximate ‘mean’ trajectory (this will only be exactly true for linear systems), a stochastic approach requires the probability distribution of different states over time, which turns out is the solution to the so-called master equation (ME).
Consider a system of d species and R reactions, , under a fixed volume () and temperature. Further, let be the probability that reaction r occurs in the interval , given system state X at time t. Then is the probability that reaction r occurs in , given the system is in state , and thus moves to state X. A mathematical representation of the master equation may be arrived at by answering the following question. How did we get to state X at time , there are two possibilities
Writing these probabilities out, dividing by
and taking the limit as
, gives the master equation
The expression in Equation (
5) is typically referred to as the chemical master equation, note that the expression above is merely the Chapman–Kolmogorov equation for a general stochastic process, not necessarily one of a chemical nature. The solution to Equation (
5) gives the probability distribution that the system is at state
X at time
t, and there is an implicit dependence on the initial state
which is suppressed for convenience. Hence, given a set of observed data from a reaction network, one would ideally perform parameter estimation and statistical inference using the solution to Equation (
5) in order to compute the likelihood function. Likelihood-based inference has strong axiomatic justification via the likelihood principle, and its computation is required for the frequentist maximum likelihood estimation and in Bayesian analysis to compute the posterior distribution. Unfortunately, it is well known that solving Equation (
5) is intractable in all but a few simple systems, for example in linear systems. This is because no analytic solutions to the ME exist in general, and in the cases where solutions exist, the computational complexity involved makes solving them impractical. For instance, when the size of the state space is finite, a solution to Equation (
5) can be computed in principle via matrix exponentiation. Even when the state space is finite, it is usually very large, large enough so that exact solutions to the ME using matrix exponentiation is infeasible. For example, in the SIR model we fit to the Eyam plague data below, the size of the state space is 62,128. An exact solution to the ME via matrix exponentiation requires the computation of the power series of a 62,128 × 62,128 matrix, and this calculation would be required each time the likelihood needs to be evaluated at a particular parameter value.
To completely specify the stochastic systems under consideration, we must determine the form of the stochastic reaction rates
. Under the assumptions that the system is well-stirred and at constant temperature, these rates mirror the deterministic mass action rates and follow the so-called stochastic law of mass action of the form
These are adjustments of the determinsitic rates that account for finite molecule numbers and the combinatorial number of ways to choose reactant molecules. A rigorous derivation of these rates was given by [
6] and McQuarrie was one of the first to review the stochastic nature of chemically reacting systems [
9]. Under the assumptions that the systems are well-mixed and thermally equilibrated, the reaction rates have the form in Equation (
6) and the master equation with these rates describe a density dependent Markov process (DDMJP). DDMJPs evolve over continuous time and make jumps at discrete time points, which update the system’s state. Further, their transition probabilities depend on the state space only through the density
, hence the name. We give two important results on the limiting properties of DDMJPs in the
Appendix A developed by Tom Kurtz [
10,
11,
12,
13], which we make use of in
Section 3 to prove results on the properties of the proposed estimation method. In the context of chemical systems, the master equation is often referred to as the chemical master equation (CME).
While exact solutions to the ME/CME are usually not available, several useful approximations have been developed which are more amenable to analysis. These often involve Taylor expanding the ME and truncating higher order terms. Consider writing the ME in terms of the step operator as follows
Here,
is the number of molecules of species
i and
is the step operator defined by
. Taylor expansion of the step operator and truncation of terms of order higher than 3 leads to a partial differential equation which gives an approximation to the ME. This partial differential equation is sometimes referred to as a Fokker–Planck equation, whose solution describes the probability distribution of a Langevin equation (CLE), or diffusion process. In general, a Fokker–Planck approximation will still not have analytic solutions, but simulating process trajectories from the CLE will be typically much more efficient than simulation from the exact process via Gillespie’s stochastic simulation algorithm (SSA). If we carry this out one step further and Taylor expand both the step operator and the rate functions
about the ODE solution Equation (
4), we arrive at yet another partial differential equation. The solution to this equation describes the probability distribution of the so-called linear noise approximation (LNA), see [
14,
15,
16] for details on these expansions. The LNA has several nice properties that make it useful for computation, one of them being that the process variances and covariances can be computed by a matrix system of ODEs. The LNA process variance at time
t, which we denote
, satisfies
where
,
and is subject to initial condition
. Note that
is the vector
, and the explicit dependence on time
t has been suppressed. Our goal was to use the ideas introduced in this section to propose a method for estimating the parameters
of a stochastic reaction network. We do this in the following section and prove results on properties of the proposed estimators.






{kind=link}
{kind=link}
{kind=link}