
Mathematical Modelling of Glioblastomas Invasion within the Brain: A 3D Multi-Scale Moving-Boundary Approach

 ,
,  and
and 
Abstract
1. Introduction
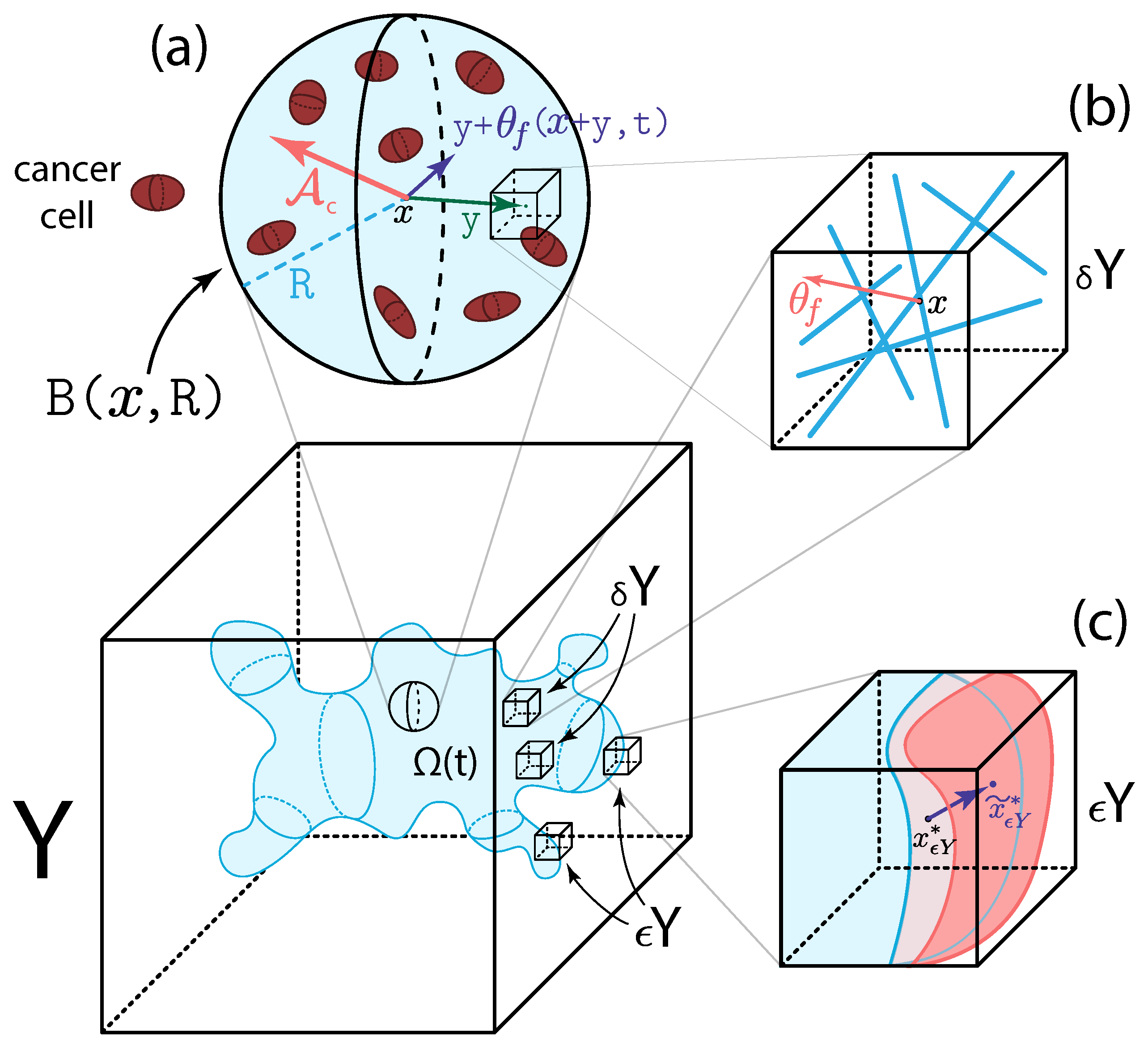
2. Multi-Scale Modelling of the Tumour Dynamics
2.1. Macro-Scale Dynamics
2.1.1. Cancer Cell Population Dynamics
2.1.2. Two-Phase ECM Macro-Scale Dynamics
2.1.3. The Complete Macro-Dynamics
2.2. Micro-Scale Processes and the Double Feedback Loop
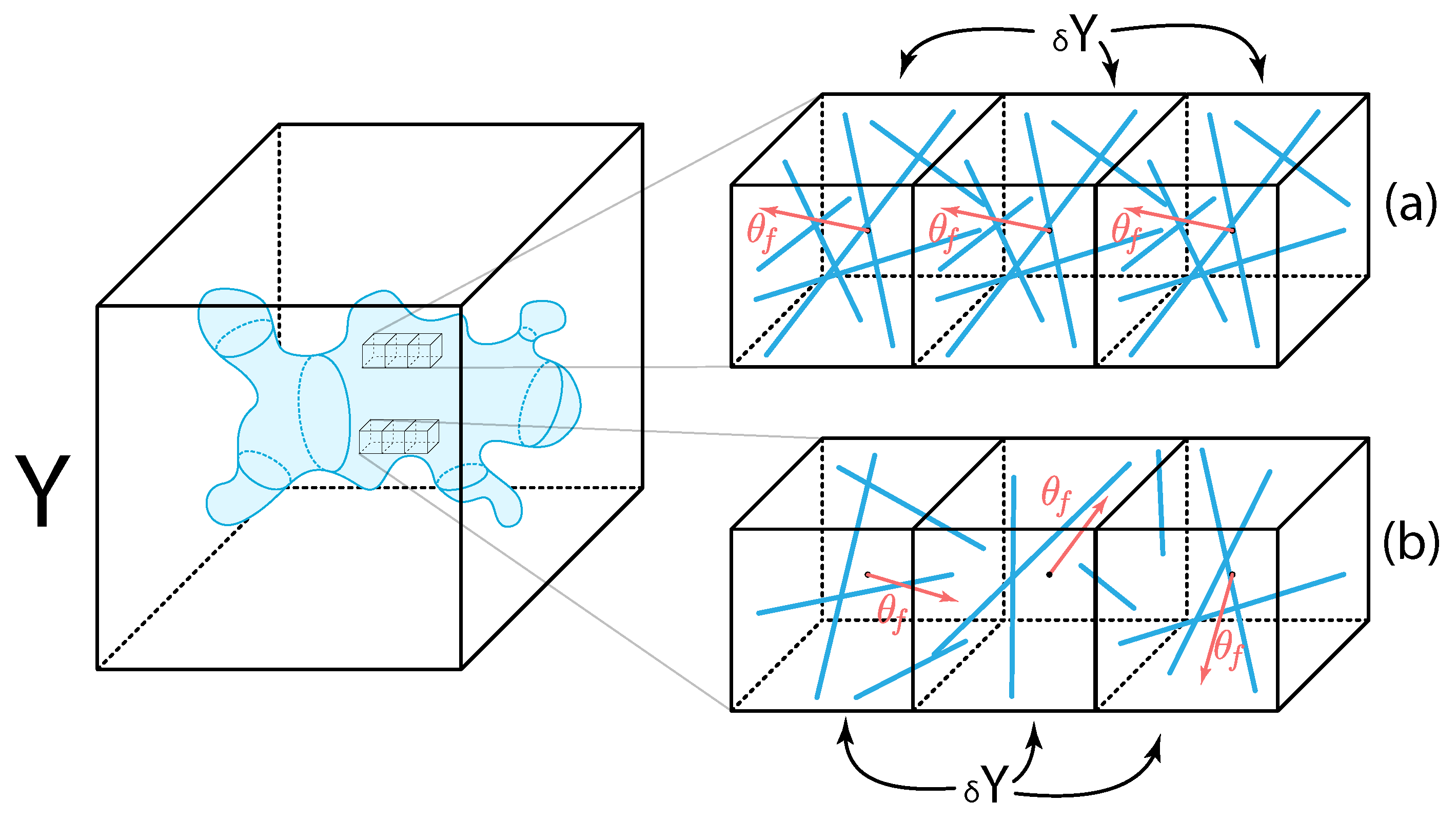
2.2.1. Two-Scale Representation and Dynamics of Fibres
2.2.2. MDE Micro-Dynamics and Its Links
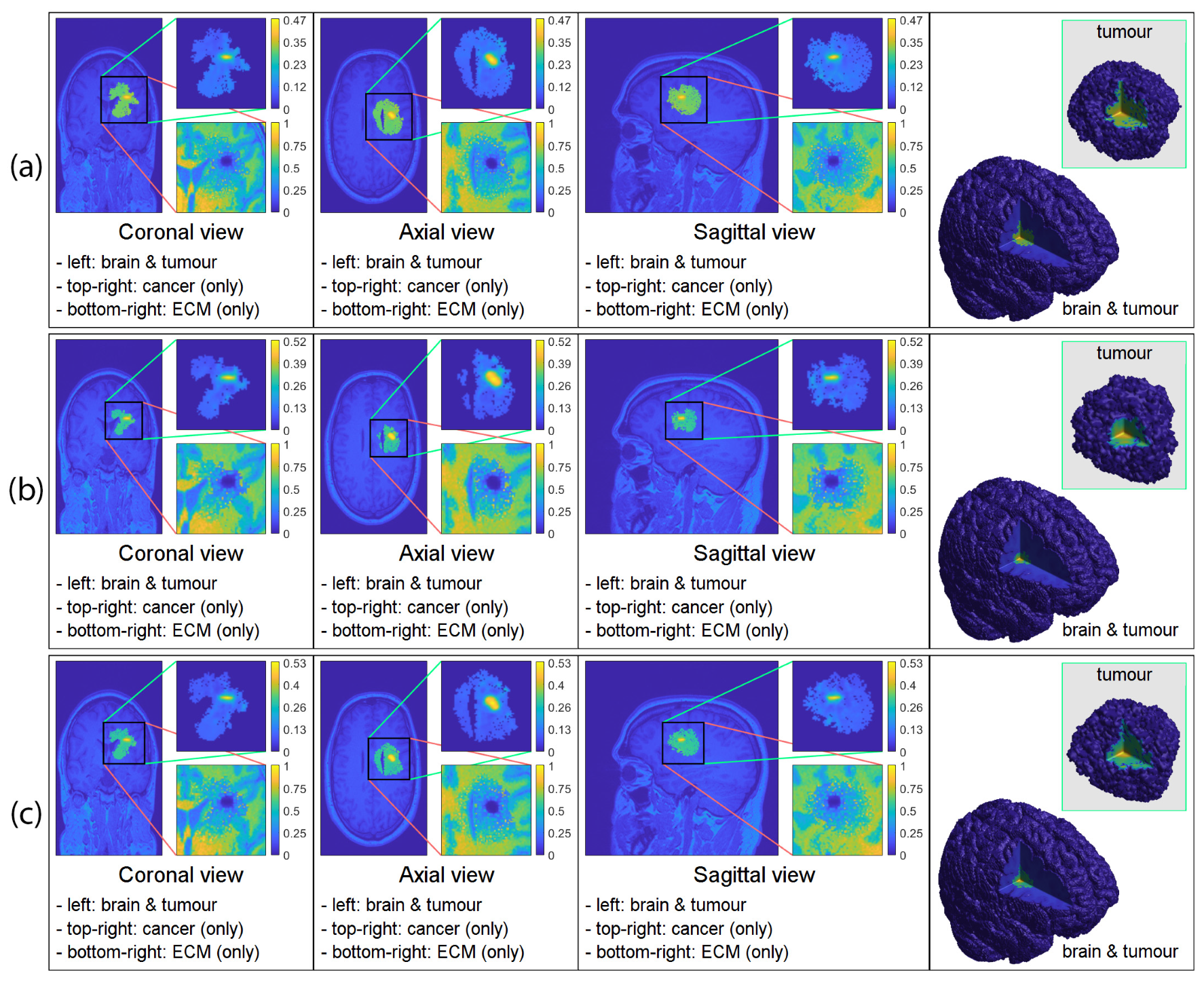
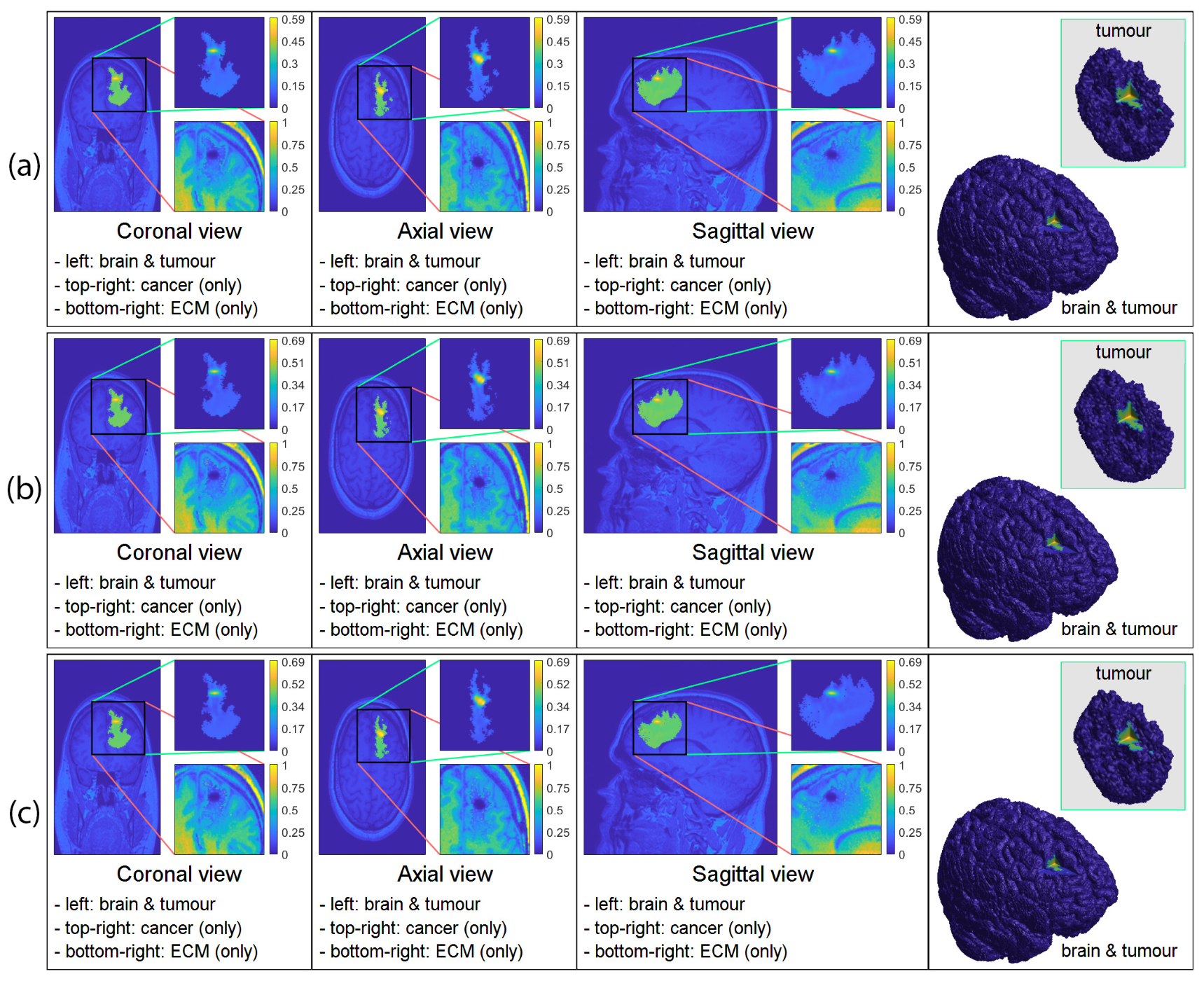
3. Computational Results: Numerical Simulations in 3D
3.1. Initial Conditions
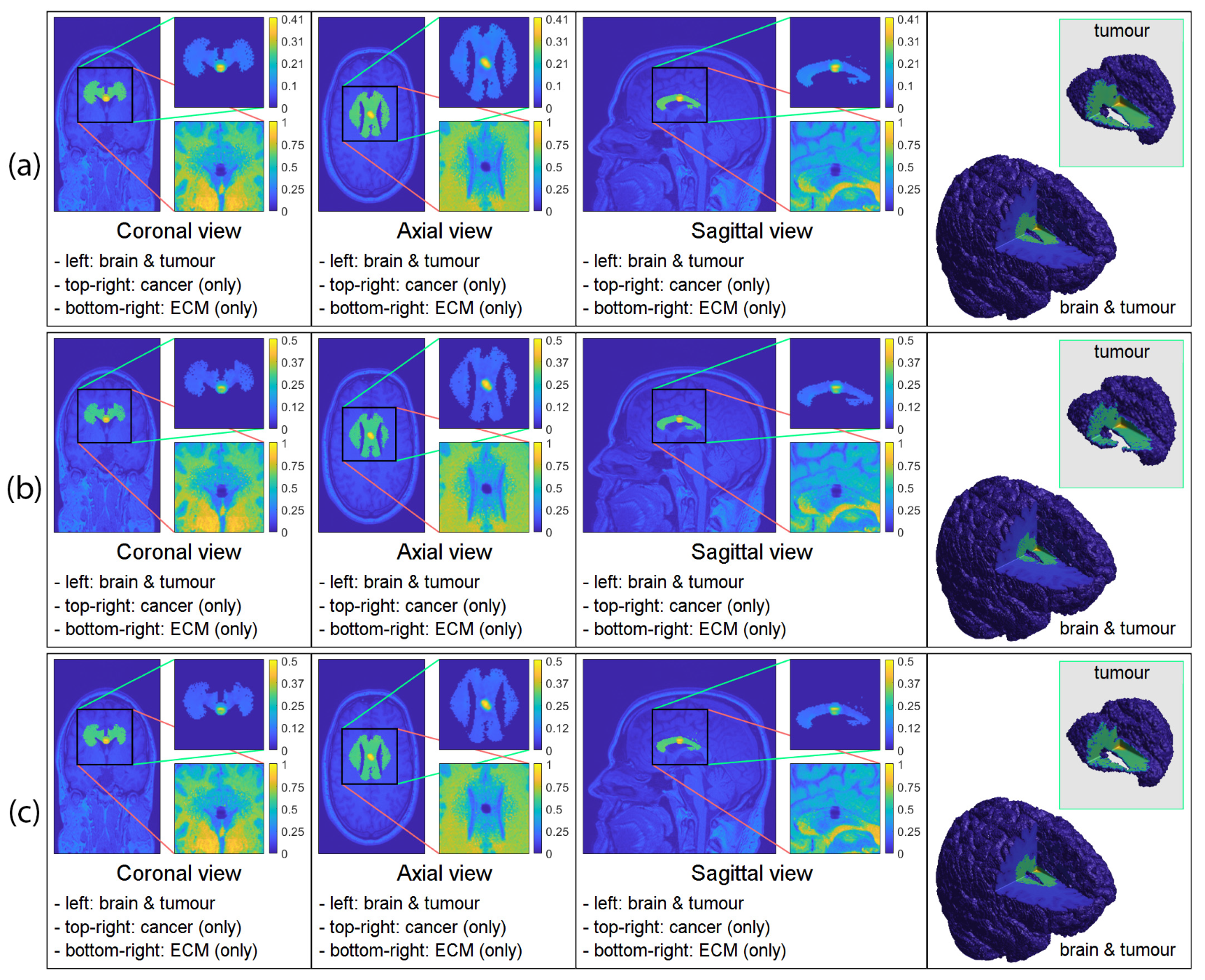
3.2. Numerical Simulations in 3D
4. Discussion and Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| MRI | Magnetic resonance imaging |
| DTI | Diffusion tensor imaging |
| ECM | Extracellular matrix |
| MDE | Matrix degrading enzymes |
| PDE | Partial differential equation |
Appendix A. Parameter Values

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Value | Description | Reference |
|---|---|---|---|
| Diffusion coeff. for the cancer cell population | [37] | ||
| Grey matter regulator coefficient | Estimated | ||
| r | Degree of randomised turning | [37] | |
| a | 0 | Model switching parameter | Estimated |
| 100 | Cell’s sensitivity to the directional information | [37] | |
| Cell–cell adhesion coeff. | [26] | ||
| Minimum level of cell–cell adhesion | [29] | ||
| Cell–non-fibre adhesion coeff. | [26] | ||
| Cell–fibre adhesion coeff. | [19] | ||
| Proliferation coeff. for cancer cell population | [19] | ||
| Degradation coeff. of the fibre ECM | [30] | ||
| Degradation coeff. of the non-fibre ECM | [30] | ||
| Optimal tissue environment controller | [20] | ||
| R | Sensing radius | [26] | |
| Maximum of micro-fibre density at any point | [26] | ||
| Macro-scale spatial step-size | [20] | ||
| Size of a boundary micro-domain | [20] | ||
| Size of a fibre micro-domain | [26] | ||
| 450 | Number of random points used for the | Estimated | |
| approximation of the adhesion integral |
Appendix B. Further Details on the Micro-Fibre Rearrangement Process
Appendix C. Further Details on the MDE Micro-Scale
References
- Burri, S.H.; Gondi, V.; Brown, P.D.; Mehta, M.P. The Evolving Role of Tumor Treating Fields in Managing Glioblastoma. Am. J. Clin. Oncol. 2018, 41, 191–196. [Google Scholar] [CrossRef]
- Davis, M. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef]
- Klopfenstein, Q.; Truntzer, C.; Vincent, J.; Ghiringhelli, F. Cell lines and immune classification of glioblastoma define patient’s prognosis. Br. J. Cancer 2019, 120, 806–814. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef]
- Meneceur, S.; Linge, A.; Meinhardt, M.; Hering, S.; Löck, S.; Bütof, R.; Krex, D.; Schackert, G.; Temme, A.; Baumann, M.; et al. Establishment and Characterisation of Heterotopic Patient-Derived Xenografts for Glioblastoma. Cancers 2020, 12, 871. [Google Scholar] [CrossRef]
- Preusser, M.; de Ribaupierre, S.; Wöhrer, A.; Erridge, S.C.; Hegi, M.; Weller, M.; Stupp, R. Current concepts and management of glioblastoma. Ann. Neurol. 2011, 70, 9–21. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Brodbelt, A.; Greenberg, D.; Winters, T.; Williams, M.; Vernon, S.; Collins, V.P. Glioblastoma in England: 2007–2011. Eur. J. Cancer 2015, 51, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.; Chaplain, M.; Newman, E.; Steele, R.; Thompson, A. Mathematical modelling of tumour invasion and metastasis. J. Theorl. Medic. 2000, 2, 129–154. [Google Scholar] [CrossRef]
- Anderson, A.R.; Hassanein, M.; Branch, K.M.; Lu, J.; Lobdell, N.A.; Maier, J.; Basanta, D.; Weidow, B.; Narasanna, A.; Arteaga, C.L.; et al. Microenvironmental Independence Associated with Tumor Progression. Cancer Res. 2009, 69, 8797–8806. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.R.A. A hybrid mathematical model of solid tumour invasion: The importance of cell adhesion. Math. Medic. Biol. 2005, 22, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Basanta, D.; Simon, M.; Hatzikirou, H.; Deutsch, A. Evolutionary game theory elucidates the role of glycolysis in glioma progression and invasion. Cell Prolif. 2008, 41, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Basanta, D.; Scott, J.G.; Rockne, R.; Swanson, K.R.; Anderson, A.R.A. The role of IDH1 mutated tumour cells in secondary glioblastomas: An evolutionary game theoretical view. Phys. Biol. 2011, 8, 015016. [Google Scholar] [CrossRef] [PubMed]
- Böttger, K.; Hatzikirou, H.; Chauviere, A.; Deutsch, A. Investigation of the Migration/Proliferation Dichotomy and its Impact on Avascular Glioma Invasion. Math. Model. Nat. Phenom. 2012, 7, 105–135. [Google Scholar] [CrossRef][Green Version]
- Chaplain, M.; Lolas, G. Mathematical modelling of cancer cell invasion of tissue: The role of the urokinase plasminogen activation system. Math. Model. Meth. Appl. Sci. 2005, 15, 1685–1734. [Google Scholar] [CrossRef]
- Chaplain, M.A.J.; Lolas, G. Mathematical modelling of cancer invasion of tissue: Dynamic heterogeneity. Netw Heterog Media 2006, 1, 399–439. [Google Scholar] [CrossRef]
- Deakin, N.E.; Chaplain, M.A.J. Mathematical modelling of cancer cell invasion: The role of membrane-bound matrix metalloproteinases. Front. Oncol. 2013, 3, 1–9. [Google Scholar] [CrossRef]
- Deisboeck, T.S.; Wang, Z.; Macklin, P.; Cristini, V. Multiscale Cancer Modeling. Annu. Rev. Biomed. Eng. 2011, 13, 127–155. [Google Scholar] [CrossRef]
- Domschke, P.; Trucu, D.; Gerisch, A.; Chaplain, M. Mathematical modelling of cancer invasion: Implications of cell adhesion variability for tumour infiltrative growth patterns. J. Theor. Biol. 2014, 361, 41–60. [Google Scholar] [CrossRef]
- Trucu, D.; Lin, P.; Chaplain, M.A.J.; Wang, Y. A Multiscale Moving Boundary Model Arising In Cancer Invasion. Multiscale Model. Simul. 2013, 11, 309–335. [Google Scholar] [CrossRef]
- Hatzikirou, H.; Brusch, L.; Schaller, C.; Simon, M.; Deutsch, A. Prediction of traveling front behavior in a lattice-gas cellular automaton model for tumor invasion. Comput. Math. Appl. 2010, 59, 2326–2339. [Google Scholar] [CrossRef]
- Kiran, K.L.; Jayachandran, D.; Lakshminarayanan, S. Mathematical modelling of avascular tumour growth based on diffusion of nutrients and its validation. Can. J. Chem. Eng. 2009, 87, 732–740. [Google Scholar] [CrossRef]
- Knútsdóttir, H.; Pálsson, E.; Edelstein-Keshet, L. Mathematical model of macrophage-facilitated breast cancer cells invasion. J. Theor. Biol. 2014, 357. [Google Scholar] [CrossRef] [PubMed]
- Macklin, P.; McDougall, S.; Anderson, A.R.A.; Chaplain, M.A.J.; Cristini, V.; Lowengrub, J. Multiscale modelling and nonlinear simulation of vascular tumour growth. J. Math. Biol. 2009, 58, 765–798. [Google Scholar] [CrossRef]
- Mahlbacher, G.; Curtis, L.; Lowengrub, J.; Frieboes, H. Mathematical modelling of tumour-associated macrophage interactions with the cancer microenvironment. J. Immunother. Cancer 2018, 6, 10. [Google Scholar] [CrossRef]
- Shuttleworth, R.; Trucu, D. Multiscale Modelling of Fibres Dynamics and Cell Adhesion within Moving Boundary Cancer Invasion. Bull. Math. Biol. 2019, 81, 2176–2219. [Google Scholar] [CrossRef]
- Shuttleworth, R.; Trucu, D. Multiscale dynamics of a heterotypic cancer cell population within a fibrous extracellular matrix. J. Theor. Biol. 2020, 486, 110040. [Google Scholar] [CrossRef]
- Shuttleworth, R.; Trucu, D. Cell-Scale Degradation of Peritumoural Extracellular Matrix Fibre Network and Its Role Within Tissue-Scale Cancer Invasion. Bull. Math. Biol. 2020, 82, 65. [Google Scholar] [CrossRef]
- Suveges, S.; Eftimie, R.; Trucu, D. Directionality of Macrophages Movement in Tumour Invasion: A Multiscale Moving-Boundary Approach. Bull. Math. Biol. 2020, 82, 148. [Google Scholar] [CrossRef]
- Suveges, S.; Eftimie, R.; Trucu, D. Re-polarisation of macrophages within a multi-scale moving boundary tumour invasion model. arXiv 2021, arXiv:2103.03384. [Google Scholar]
- Szymańska, Z.; Morales-Rodrigo, C.; Lachowicz, M.; Chaplain, M.A.J. Mathematical modelling of cancer invasion of tissue: The role and effect of nonlocal interactions. Math. Model. Methods Appl. Sci. 2009, 19, 257–281. [Google Scholar] [CrossRef]
- Tektonidis, M.; Hatzikirou, H.; Chauvière, A.; Simon, M.; Schaller, K.; Deutsch, A. Identification of intrinsic in vitro cellular mechanisms for glioma invasion. J. Theor. Biol. 2011, 287, 131–147. [Google Scholar] [CrossRef]
- Xu, J.; Vilanova, G.; Gomez, H. A Mathematical Model Coupling Tumor Growth and Angiogenesis. PLoS ONE 2016, 11, e0149422. [Google Scholar] [CrossRef] [PubMed]
- Alfonso, J.C.L.; Köhn-Luque, A.; Stylianopoulos, T.; Feuerhake, F.; Deutsch, A.; Hatzikirou, H. Why one-size-fits-all vaso-modulatory interventions fail to control glioma invasion: In silico insights. Sci. Rep. 2016, 6, 37283. [Google Scholar] [CrossRef] [PubMed]
- Engwer, C.; Hillen, T.; Knappitsch, M.; Surulescu, C. Glioma follow white matter tracts: A multiscale DTI-based model. J. Math. Biol. 2014, 71, 551–582. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.; Surulescu, C. A Multiscale Modeling Approach to Glioma Invasion with Therapy. Vietnam. J. Math. 2016, 45, 221–240. [Google Scholar] [CrossRef]
- Painter, K.; Hillen, T. Mathematical modelling of glioma growth: The use of Diffusion Tensor Imaging (DTI) data to predict the anisotropic pathways of cancer invasion. J. Theor. Biol. 2013, 323, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Scribner, E.; Saut, O.; Province, P.; Bag, A.; Colin, T.; Fathallah-Shaykh, H.M. Effects of Anti-Angiogenesis on Glioblastoma Growth and Migration: Model to Clinical Predictions. PLoS ONE 2014, 9, e115018. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.R.; Alvord, E.C.; Murray, J.D. A quantitative model for differential motility of gliomas in grey and white matter. Cell Prolif. 2000, 33, 317–329. [Google Scholar] [CrossRef]
- Swanson, K.R.; Rostomily, R.C.; Alvord, E.C. A mathematical modelling tool for predicting survival of individual patients following resection of glioblastoma: A proof of principle. Br. J. Cancer 2007, 98, 113–119. [Google Scholar] [CrossRef]
- Swanson, K.R.; Rockne, R.C.; Claridge, J.; Chaplain, M.A.; Alvord, E.C.; Anderson, A.R. Quantifying the Role of Angiogenesis in Malignant Progression of Gliomas: In Silico Modeling Integrates Imaging and Histology. Cancer Res. 2011, 71, 7366–7375. [Google Scholar] [CrossRef]
- Syková, E.; Nicholson, C. Diffusion in Brain Extracellular Space. Physiol. Rev. 2008, 88, 1277–1340. [Google Scholar] [CrossRef]
- Clatz, O.; Sermesant, M.; Bondiau, P.Y.; Delingette, H.; Warfield, S.; Malandain, G.; Ayache, N. Realistic simulation of the 3-D growth of brain tumors in MR images coupling diffusion with biomechanical deformation. IEEE Trans. Med. Imaging 2005, 24, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Cobzas, D.; Mosayebi, P.; Murtha, A.; Jagersand, M. Tumor Invasion Margin on the Riemannian Space of Brain Fibers. In Medical Image Computing and Computer-Assisted Intervention— MICCAI 2009; Springer: Berlin/Heidelberg, Germany, 2009; pp. 531–539. [Google Scholar] [CrossRef]
- Jbabdi, S.; Mandonnet, E.; Duffau, H.; Capelle, L.; Swanson, K.R.; Pélégrini-Issac, M.; Guillevin, R.; Benali, H. Simulation of anisotropic growth of low-grade gliomas using diffusion tensor imaging. Magn. Reson. Med. 2005, 54, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Konukoglu, E.; Clatz, O.; Bondiau, P.Y.; Delingette, H.; Ayache, N. Extrapolating glioma invasion margin in brain magnetic resonance images: Suggesting new irradiation margins. Med. Image Anal. 2010, 14, 111–125. [Google Scholar] [CrossRef]
- Suarez, C.; Maglietti, F.; Colonna, M.; Breitburd, K.; Marshall, G. Mathematical Modeling of Human Glioma Growth Based on Brain Topological Structures: Study of Two Clinical Cases. PLoS ONE 2012, 7, e39616. [Google Scholar] [CrossRef]
- Yan, H.; Romero-López, M.; Benitez, L.I.; Di, K.; Frieboes, H.B.; Hughes, C.C.; Bota, D.A.; Lowengrub, J.S. 3D Mathematical Modeling of Glioblastoma Suggests That Transdifferentiated Vascular Endothelial Cells Mediate Resistance to Current Standard-of-Care Therapy. Cancer Res. 2017, 77, 4171–4184. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Trucu, D.; Lin, P.; Thompson, A.; Chaplain, M.A.J. A multiscale mathematical model of tumour invasive growth. Bull. Math. Biol. 2017, 79, 389–429. [Google Scholar] [CrossRef]
- Laird, A.K. Dynamics of Tumour Growth. Br. J. Cancer 1964, 13, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Laird, A.K. Dynamics of Tumour Growth: Comparison of Growth Rates and Extrapolation of Growth Curve to One Cell. Br. J. Cancer 1965, 19, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Tjorve, K.M.C.; Tjorve, E. The use of Gompertz models in growth analyses, and new Gompertz-model approach: An addition to the Unified-Richards family. PLoS ONE 2017, 12, e0178691. [Google Scholar] [CrossRef]
- IXI Dataset—Information eXtraction from Images. Available online: http://brain-development.org/ixi-dataset (accessed on 7 September 2021).
- Chen, Q.; Zhang, X.H.F.; Massagué, J. Macrophage Binding to Receptor VCAM-1 Transmits Survival Signals in Breast Cancer Cells that Invade the Lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate Partners for Tumor Cell Migration, Invasion, and Metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Huda, S.; Weigelin, B.; Wolf, K.; Tretiakov, K.V.; Polev, K.; Wilk, G.; Iwasa, M.; Emami, F.S.; Narojczyk, J.W. Lévy-like movement patterns of metastatic cancer cells revealed in microfabricated systems and implicated in vivo. Nat. Commun. 2018, 9, 4539. [Google Scholar] [CrossRef] [PubMed]
- Petrie, R.J.; Doyle, A.D.; Yamada, K.M. Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 538–549. [Google Scholar] [CrossRef]
- Weiger, M.C.; Vedham, V.; Stuelten, C.H.; Shou, K.; Herrera, M.; Sato, M.; Losert, W.; Parent, C.A. Real-Time Motion Analysis Reveals Cell Directionality as an Indicator of Breast Cancer Progression. PLoS ONE 2013, 8, e0058859. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.H.; Giri, A.; Sun, S.X.; Wirtz, D. Three-dimensional cell migration does not follow a random walk. Proc. Natl. Acad. Sci. USA 2014, 111, 3949–3954. [Google Scholar] [CrossRef]
- Basser, P.; Mattiello, J.; LeBihan, D. Diagonal and off-diagonal components of the self-diffusion tensor:their relation to and estimation from the NMR spin-echo signal. In Proceedings of the 11th Society of Magnetic Resonance in Medicine Meeting, Berlin, Germany, 8–14 August 1992. [Google Scholar]
- Basser, P.; Mattiello, J.; Robert, T.; LeBihan, D. Diffusion tensor echo-planar imaging of human brain. In Proceedings of the SMRM, New York, NY, USA, 14–20 August 1993; Volume 584. [Google Scholar]
- Basser, P.; Mattiello, J.; Lebihan, D. Estimation of the Effective Self-Diffusion Tensor from the NMR Spin Echo. J. Magn. Reson. Ser. B 1994, 103, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Basser, P.; Mattiello, J.; LeBihan, D. MR diffusion tensor spectroscopy and imaging. Biophys. J. 1994, 66, 259–267. [Google Scholar] [CrossRef]
- O’Donnell, L.J.; Westin, C.F. An Introduction to Diffusion Tensor Image Analysis. Neurosurg. Clin. 2011, 22, 185–196. [Google Scholar] [CrossRef]
- Hillen, T.; Painter, K.J.; Swan, A.C.; Murtha, A.D. Moments of von mises and fisher distributions and applications. Math. Biosci. Eng. 2017, 14, 673–694. [Google Scholar] [CrossRef]
- Mardia, K.V. Directional Statistics; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
- Hagmann, P.; Jonasson, L.; Maeder, P.; Thiran, J.P.; Wedeen, V.J.; Meuli, R. Understanding Diffusion MR Imaging Techniques: From Scalar Diffusion-weighted Imaging to Diffusion Tensor Imaging and Beyond. RadioGraphics 2006, 26, S205–S223. [Google Scholar] [CrossRef]
- Chicoine, M.R.; Silbergeld, D.L. Assessment of brain tumor cell motility in vivo and in vitro. J. Neurosurg. 1995, 82, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.J.; Hunt, C. The limited value of cytoreductive surgery in elderly patients with malignant gliomas. Neurosurgery 1994, 34, 62–66; discussion 66–67. [Google Scholar] [PubMed]
- Silbergeld, D.L.; Chicoine, M.R. Isolation and characterization of human malignant glioma cells from histologically normal brain. J. Neurosurg. 1997, 86, 525–531. [Google Scholar] [CrossRef]
- Damelin, S.B.; Miller, W.J. The Mathematics of Signal Processing; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar] [CrossRef]
- Gondi, C.S.; Lakka, S.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Tung, C.H.; Weissleder, R.; Rao, J.S. Adenovirus-Mediated Expression of Antisense Urokinase Plasminogen Activator Receptor and Antisense Cathepsin B Inhibits Tumor Growth, Invasion, and Angiogenesis in Gliomas. Cancer Res. 2004, 64, 4069–4077. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, I.; Braghetta, P.; Bonaldo, P.; Cescon, M. Collagen VI in healthy and diseased nervous system. Dis. Model. Mech. 2018, 11, dmm032946. [Google Scholar] [CrossRef]
- Kalinin, V. Cell – extracellular matrix interaction in glioma growth. In silico model. J. Integr. Bioinform. 2020, 17. [Google Scholar] [CrossRef]
- Mohanam, S. Biological significance of the expression of urokinase-type plasminogen activator receptors (uPARs) in brain tumors. Front. Biosci. 1990, 4, d178. [Google Scholar] [CrossRef]
- Persson, M.; Nedergaard, M.K.; Brandt-Larsen, M.; Skovgaard, D.; Jorgensen, J.T.; Michaelsen, S.R.; Madsen, J.; Lassen, U.; Poulsen, H.S.; Kjaer, A. Urokinase-Type Plasminogen Activator Receptor as a Potential PET Biomarker in Glioblastoma. J. Nucl. Med. 2015, 57, 272–278. [Google Scholar] [CrossRef]
- Pointer, K.B.; Clark, P.A.; Schroeder, A.B.; Salamat, M.S.; Eliceiri, K.W.; Kuo, J.S. Association of collagen architecture with glioblastoma patient survival. J. Neurosurg. 2016, 126, 1812–1821. [Google Scholar] [CrossRef]
- Pullen, N.; Pickford, A.; Perry, M.; Jaworski, D.; Loveson, K.; Arthur, D.; Holliday, J.; Meter, T.V.; Peckham, R.; Younas, W.; et al. Current insights into matrix metalloproteinases and glioma progression: Transcending the degradation boundary. Met. Med. 2018, 5, 13–30. [Google Scholar] [CrossRef]
- Ramachandran, R.K.; Sørensen, M.D.; Aaberg-Jessen, C.; Hermansen, S.K.; Kristensen, B.W. Expression and prognostic impact of matrix metalloproteinase-2 (MMP-2) in astrocytomas. PLoS ONE 2017, 12, e0172234. [Google Scholar] [CrossRef] [PubMed]
- Veeravalli, K.K.; Rao, J.S. MMP-9 and uPAR regulated glioma cell migration. Cell Adhes. Migr. 2012, 6, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Veeravalli, K.K.; Ponnala, S.; Chetty, C.; Tsung, A.J.; Gujrati, M.; Rao, J.S. Integrin α9β1-mediated cell migration in glioblastoma via SSAT and Kir4.2 potassium channel pathway. Cell. Signal. 2012, 24, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Young, N.; Pearl, D.K.; Brocklyn, J.R.V. Sphingosine-1-Phosphate Regulates Glioblastoma Cell Invasiveness through the Urokinase Plasminogen Activator System and CCN1/Cyr61. Mol. Cancer Res. 2009, 7, 23–32. [Google Scholar] [CrossRef]
- Armstrong, N.J.; Painter, K.J.; Sherratt, J.A. A continuum approach to modelling cell–cell adhesion. J. Theor. Biol. 2006, 243, 98–113. [Google Scholar] [CrossRef]
- Gerisch, A.; Chaplain, M. Mathematical modelling of cancer cell invasion of tissue: Local and non-local models and the effect of adhesion. J. Theor. Biol. 2008, 250, 684–704. [Google Scholar] [CrossRef]
- Ghosh, S.; Salot, S.; Sengupta, S.; Navalkar, A.; Ghosh, D.; Jacob, R.; Das, S.; Kumar, R.; Jha, N.N.; Sahay, S.; et al. p53 amyloid formation leading to its loss of function: Implications in cancer pathogenesis. Cell Death Differ. 2017, 24, 1784–1798. [Google Scholar] [CrossRef]
- Gras, S.L. Chapter 6- Surface- and Solution-Based Assembly of Amyloid Fibrils for Biomedical and Nanotechnology Applications. In Engineering Aspects of Self-Organizing Materials; Koopmans, R.J., Ed.; Academic Press: Cambridge, MA, USA, 2009; Volume 35, pp. 161–209. [Google Scholar] [CrossRef]
- Gras, S.L.; Tickler, A.K.; Squires, A.M.; Devlin, G.L.; Horton, M.A.; Dobson, C.M.; MacPhee, C.E. Functionalised amyloid fibrils for roles in cell adhesion. Biomaterials 2008, 29, 1553–1562. [Google Scholar] [CrossRef]
- Jacob, R.S.; George, E.; Singh, P.K.; Salot, S.; Anoop, A.; Jha, N.N.; Sen, S.; Maji, S.K. Cell Adhesion on Amyloid Fibrils Lacking Integrin Recognition Motif. J. Biol. Chem. 2016, 291, 5278–5298. [Google Scholar] [CrossRef]
- Wolf, K.; Alexander, S.; Schacht, V.; Coussens, L.; Andrian, U.; Rheenen, J.; Deryugina, E.; Friedl, P. Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev. Biol. 2009, 20, 931–941. [Google Scholar] [CrossRef]
- Wolf, K.; Friedl, P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Tren. Cel. Biol. 2011, 21, 736–744. [Google Scholar] [CrossRef]
- Gu, Z.; Liu, F.; Tonkova, E.A.; Lee, S.Y.; Tschumperlin, D.J.; Brenner, M.B.; Ginsberg, M.H. Soft matrix is a natural stimulator for cellular invasiveness. Mol. Biol. Cell 2014, 25, 457–469. [Google Scholar] [CrossRef]
- Hofer, A.M.; Curci, S.; Doble, M.A.; Brown, E.M.; Soybel, D.I. Intercellular communication mediated by the extracellular calcium-sensing receptor. Nat. Cell Biol. 2000, 2, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The Biology of Cancer; Garland Science: New York, NY, USA, 2006. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.L.; Watson, S.A.; Brown, P.D.; Collins, H.M.; Steele, R.J. Matrix metalloproteinases. Brit. J. Surg. 1997, 84, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- van Es, B.; Koren, B.; de Blank, H.J. Finite-difference schemes for anisotropic diffusion. J. Comput. Phys. 2014, 272, 526–549. [Google Scholar] [CrossRef]
- Günter, S.; Yu, Q.; Krüger, J.; Lackner, K. Modelling of heat transport in magnetised plasmas using non-aligned coordinates. J. Comput. Phys. 2005, 209, 354–370. [Google Scholar] [CrossRef]
- Raffelt, D.A.; Tournier, J.D.; Smith, R.E.; Vaughan, D.N.; Jackson, G.; Ridgway, G.R.; Connelly, A. Investigating white matter fibre density and morphology using fixel-based analysis. NeuroImage 2017, 144, 58–73. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suveges, S.; Hossain-Ibrahim, K.; Steele, J.D.; Eftimie, R.; Trucu, D. Mathematical Modelling of Glioblastomas Invasion within the Brain: A 3D Multi-Scale Moving-Boundary Approach. Mathematics 2021, 9, 2214. https://doi.org/10.3390/math9182214
Suveges S, Hossain-Ibrahim K, Steele JD, Eftimie R, Trucu D. Mathematical Modelling of Glioblastomas Invasion within the Brain: A 3D Multi-Scale Moving-Boundary Approach. Mathematics. 2021; 9(18):2214. https://doi.org/10.3390/math9182214
Chicago/Turabian StyleSuveges, Szabolcs, Kismet Hossain-Ibrahim, J. Douglas Steele, Raluca Eftimie, and Dumitru Trucu. 2021. "Mathematical Modelling of Glioblastomas Invasion within the Brain: A 3D Multi-Scale Moving-Boundary Approach" Mathematics 9, no. 18: 2214. https://doi.org/10.3390/math9182214
APA StyleSuveges, S., Hossain-Ibrahim, K., Steele, J. D., Eftimie, R., & Trucu, D. (2021). Mathematical Modelling of Glioblastomas Invasion within the Brain: A 3D Multi-Scale Moving-Boundary Approach. Mathematics, 9(18), 2214. https://doi.org/10.3390/math9182214