
Markov-Chain Perturbation and Approximation Bounds in Stochastic Biochemical Kinetics


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Continuous-Time Markov Chains, Approximations, and Perturbation Bounds
2.1. Approximation Approaches for the Chemical Master Equation
2.2. Continuous-Time Markov Chains and the Chemical Master Equation
2.3. Markov Chain Convergence to Steady State
2.4. Perturbation Bounds for Continuous-Time Markov Chains
3. Approximations and Perturbations for Individual Biochemical Reactions
3.1. An Illustrative Example: The Formation and Dissociation of Binary Complexes
3.2. Partial Thermodynamic Limit
3.3. Irreversible-Reaction Limit
3.4. Parametric Uncertainty Analysis
3.5. Other Approximations, Individual Reactions, and Reaction Networks
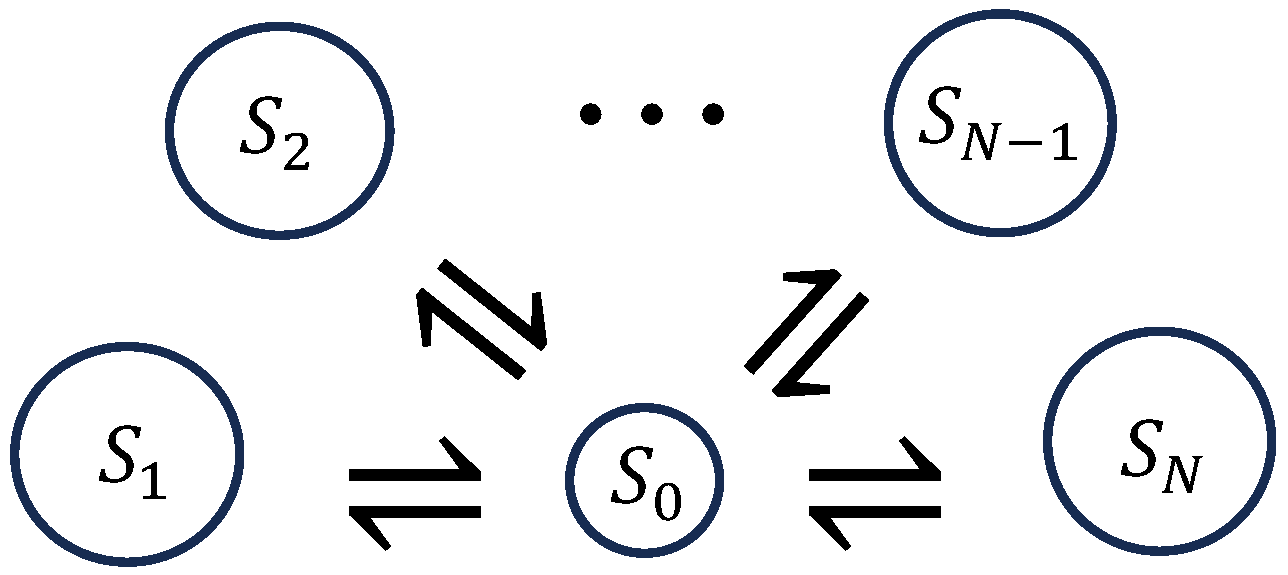
4. Linear Reaction Networks
4.1. Networks of Unimolecular Reactions
4.2. Perturbation Analysis for Unimolecular Reaction Networks
4.3. A Biological Example: Competitive Transcription-Factor Binding to DNA
5. Generalizations and Related Topics
5.1. Extensions to General Reaction Networks
5.2. The Need for Time-Inhomogeneous Markov Models
5.3. Tightness of the Perturbation Bounds
6. Conclusions: Future Work
Funding
Data Availability Statement
Conflicts of Interest
References
- Anderson, D.F.; Kurtz, T.G. Stochastic Analysis of Biochemical Systems; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Briat, C.; Khammash, M. Noise in biomolecular systems: Modeling, analysis, and control implications. Annu. Rev. Contr. Robot. 2023, 6, 283–311. [Google Scholar] [CrossRef]
- Holehouse, J.; Cao, Z.X.; Grima, R. Stochastic modeling of autoregulatory genetic feedback loops: A review and comparative study. Biophys. J. 2020, 118, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Teimouri, H.; Kolomeisky, A.B. Power of stochastic kinetic models: From biological signaling and antibiotic activities to T cell activation and cancer initiation dynamics. WIREs Comput. Mol. Sci. 2022, 12, e1612. [Google Scholar] [CrossRef]
- Wilkinson, D.J. Stochastic modelling for quantitative description of heterogeneous biological systems. Nat. Rev. Genet. 2009, 10, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.F.; Cappeletti, D.; Fan, W.T.L.; Kim, J. A new path method for exponential ergodicity of Markov processes on Zd, with applications to stochastic reaction networks. SIAM J. Appl. Dynam. Syst. 2025, 24, 1668–1710. [Google Scholar] [CrossRef]
- Anderson, D.F.; Howells, A.S. Stochastic reaction networks within interacting compartments. Bull. Math. Biol. 2023, 85, 87. [Google Scholar] [CrossRef]
- Anderson, D.F.; Kim, J. Mixing times for two classes of stochastically modeled reaction networks. Math. Biosci. Eng. 2023, 20, 4690–4713. [Google Scholar] [CrossRef]
- Mangold, K.E.; Wang, W.; Johnson, E.K.; Bhagavan, D.; Moreno, J.D.; Nerbonne, J.M.; Silva, J.R. Identification of structures for ion channel kinetic models. PLoS Comput. Biol. 2021, 17, e1008932. [Google Scholar] [CrossRef]
- Menon, V.; Spruston, N.; Kath, W.L. A state-mutating genetic algorithm to design ion-channel models. Proc. Natl. Acad. Sci. USA 2009, 106, 16829–16834. [Google Scholar] [CrossRef]
- Teed, Z.R.; Silva, J.R. A computationally efficient algorithm for fitting ion channel parameters. MethodsX 2016, 3, 577–588. [Google Scholar] [CrossRef]
- Fink, M.; Noble, D. Markov models for ion channels: Versatility versus identifiability and speed. Philos. Tranc. R. Soc. A 2009, 367, 2161–2179. [Google Scholar] [CrossRef]
- Mahdavi, S.D.; Salmon, G.L.; Daghlian, P.; Garcia, H.G.; Phillips, R. Flexibility and sensitivity in gene regulation out of equilibrium. Proc. Natl. Acad. Sci. USA 2024, 121, e2411395121. [Google Scholar] [CrossRef]
- Wong, F.; Gunawardena, J. Gene regulation in and out of equilibrium. Annu. Rev. Biophys. 2020, 49, 199–226. [Google Scholar] [CrossRef]
- Grah, R.; Zoller, B.; Tkacik, G. Nonequilibrium models of optimal enhancer function. Proc. Natl. Acad. Sci. USA 2020, 117, 31614–31622. [Google Scholar] [CrossRef]
- Hoffecker, I.T.; Shaw, A.; Sorokina, V.; Smyrlaki, I.; Högberg, B. Stochastic modeling of antibody binding predicts programmable migration on antigen patterns. Nat. Comput. Sci. 2022, 2, 179–192. [Google Scholar] [CrossRef]
- Shaw, A.; Hoffecker, I.T.; Smyrlaki, I.; Rosa, J.; Grevys, A.; Bratlie, D.; Sandlie, I.; Michaelsen, T.E.; Andersen, J.T.; Högberg, B. Binding to nanopatterned antigens is dominated by the spatial tolerance of antibodies. Nat. Nanotechnol. 2019, 14, 184–190. [Google Scholar] [CrossRef]
- Kalinkin, A.V. Markov branching processes with interaction. Russ. Math. Surv. 2002, 57, 241–304. [Google Scholar] [CrossRef]
- Vassiliou, P.-C.G. Non-Homogeneous Markov Chains and Systems: Theory and Applications; Chapman and Hall/CRC: New York, NY, USA, 2022. [Google Scholar]
- Constantino, P.H.; Vlysidis, M.; Smadbeck, P.; Kaznessis, Y.N. Modeling stochasticity in biochemical reaction networks. J. Phys. D Appl. Phys. 2016, 49, 093001. [Google Scholar] [CrossRef]
- Goutsias, J.; Jenkinson, G. Markovian dynamics on complex reaction networks. Phys. Rep. 2013, 529, 199–264. [Google Scholar] [CrossRef]
- Cao, Y.S.; Liang, S.L. Stochastic thermodynamics for biological functions. Quant. Biol. 2025, 13, e75. [Google Scholar] [CrossRef]
- Van Kampen, N.G. Stochastic Processes in Physics and Chemistry, 3rd ed.; North Holland: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Érdi, P.; Tóth, J. Mathematical Models of Chemical Reactions: Theory and Applications of Deterministic and Stochastic Models; Princeton University Press: Princeton, NJ, USA, 1989. [Google Scholar]
- Khudabukhsh, W.R.; Rempala, G.A. How to fit an SIR model to data from an SEIR model? Math. Biosci. 2024, 375, 109265. [Google Scholar] [CrossRef]
- Black, A.J.; McKane, A.J. Stochastic formulation of ecological models and their applications. Trends Ecol. Evol. 2012, 27, 337–345. [Google Scholar] [CrossRef]
- Anisimova, M.; Kosiol, C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol. Biol. Evol. 2009, 26, 255–271. [Google Scholar] [CrossRef]
- Öcal, K.; Sanguinetti, G.; Grima, R. Model reduction for the Chemical Master Equation: An information-theoretic approach. J. Chem. Phys. 2023, 158, 114113. [Google Scholar] [CrossRef]
- Schnoerr, D.; Sanguinetti, G.; Grima, R. Approximation and inference methods for stochastic biochemical kinetics-a tutorial review. J. Phys. A Math. Theor. 2017, 50, 093001. [Google Scholar] [CrossRef]
- Sharpe, D.J.; Wales, D.J. Nearly reducible finite Markov chains: Theory and algorithms. J. Chem. Phys. 2021, 155, 140901. [Google Scholar] [CrossRef]
- Woods, E.J.; Kannan, D.; Sharpe, D.J.; Swinburne, T.D.; Wales, D.J. Analysing ill-conditioned Markov chains. Phil. Trans. R. Soc. A 2023, 381, 20220245. [Google Scholar] [CrossRef]
- Gillespie, D.T. Stochastic simulation of chemical kinetics. Annu. Rev. Phys. Chem. 2007, 58, 35–55. [Google Scholar] [CrossRef]
- Gupta, A.; Mikelson, J.; Khammash, M. A finite state projection algorithm for the stationary solution of the chemical master equation. J. Chem. Phys. 2017, 147, 154101. [Google Scholar] [CrossRef]
- Margaliot, M.; Grüne, L.; Kriecherbauer, T. Entrainment in the master equation. Roy. Soc. Open Sci. 2018, 5, 172157. [Google Scholar] [CrossRef]
- Munsky, B.; Khammash, M. The finite state projection algorithm for the solution of the chemical master equation. J. Chem. Phys. 2006, 124, 044101. [Google Scholar] [CrossRef]
- Thorsley, D.; Klavins, E. Approximating stochastic biochemical processes with Wasserstein pseudometrics. IET Syst. Biol. 2010, 4, 193–211. [Google Scholar] [CrossRef]
- Kurtz, T.G. The relationship between stochastic and deterministic models for chemical reactions. J. Chem. Phys. 1972, 57, 2976–2978. [Google Scholar] [CrossRef]
- Pollett, P.K.; Vassallo, A. Diffusion approximations for some simple chemical-reaction schemes. Adv. Appl. Probab. 1992, 24, 875–893. [Google Scholar] [CrossRef]
- Horowitz, J.M. Diffusion approximations to the chemical master equation only have a consistent stochastic thermodynamics at chemical equilibrium. J. Chem. Phys. 2015, 143, 044111. [Google Scholar] [CrossRef]
- Haseltine, E.L.; Rawlings, J.B. Approximate simulation of coupled fast and slow reactions for stochastic chemical kinetics. J. Chem. Phys. 2002, 117, 6959–6969. [Google Scholar] [CrossRef]
- Salis, H.; Kaznessis, Y. Accurate hybrid stochastic simulation of a system of coupled chemical or biochemical reactions. J. Chem. Phys. 2005, 122, 054103. [Google Scholar] [CrossRef]
- Jahnke, T.; Kreim, M. Error bound for piecewise deterministic processes modeling stochastic reaction systems. Multiscale Model. Sim. 2012, 10, 1119–1147. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y. Stochastic Markov models for the process of binary complex formation and dissociation. Mat. Model. 2001, 13, 101–109. Available online: https://www.mathnet.ru/eng/mm/v13/i9/p101 (accessed on 18 April 2025).
- Kuntz, J.; Thomas, P.; Stan, G.B.; Barahona, M. Stationary distributions of continuous-time Markov chains: A review of theory and truncation-based approximations. SIAM Rev. 2021, 63, 3–64. [Google Scholar] [CrossRef]
- Zeifman, A.I.; Korotysheva, A.V.; Korolev, V.Y.; Satin, Y.A. Truncation bounds for approximations of inhomogeneous continuous-time Markov chains. Theor. Probab. Appl. 2017, 61, 513–520. [Google Scholar] [CrossRef]
- Cao, Y.F.; Terebus, A.; Liang, J. State space truncation with quantified errors for accurate solutions to discrete chemical master equation. Bull. Math. Biol. 2016, 78, 617–661. [Google Scholar] [CrossRef]
- Michel, F.; Siegle, M. Formal error bounds for the state space reduction of Markov chains. Perform. Eval. 2025, 167, 102464. [Google Scholar] [CrossRef]
- Andrieux, D. Bounding the coarse graining error in hidden Markov dynamics. Appl. Math. Lett. 2012, 25, 1734–1739. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y. The arsenal of perturbation bounds for finite continuous-time Markov chains: A perspective. Mathematics 2024, 12, 1608. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y. Ergodicity coefficient and perturbation bounds for continuous-time Markov chains. Math. Inequal. Appl. 2005, 8, 159–168. [Google Scholar] [CrossRef]
- Ho, L.S.T.; Xu, J.; Crawford, F.W.; Minin, V.N.; Suchard, M.A. Birth/birth-death processes and their computable transition probabilities with biological applications. J. Math. Biol. 2018, 76, 911–944. [Google Scholar] [CrossRef]
- Novozhilov, A.S.; Karev, G.P.; Koonin, E.V. Biological applications of the theory of birth-and-death processes. Brief. Bioinform. 2006, 7, 70–85. [Google Scholar] [CrossRef]
- Colquhoun, D.; Dowsland, K.A.; Beato, M.; Plested, A.J.R. How to impose microscopic reversibility in complex reaction mechanisms. Biophys. J. 2004, 86, 3510–3518. [Google Scholar] [CrossRef]
- Jia, C.; Jiang, D.Q.; Li, Y.M. Detailed balance, local detailed balance, and global potential for stochastic chemical reaction networks. Adv. Appl. Probab. 2021, 53, 886–922. [Google Scholar] [CrossRef]
- Malyshev, V.A.; Pirogov, S.A. Reversibility and irreversibility in stochastic chemical kinetics. Russ. Math. Surv. 2008, 63, 1–34. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y. Reversible Markov chains and spanning trees. Math. Scientist 2004, 29, 107–114. Available online: https://drive.google.com/file/d/1FkP-ODFN9wwPQeMw2dqk08Uz3uYslkv9/view (accessed on 16 June 2025).
- Mitrophanov, A.Y. Stability and exponential convergence of continuous-time Markov chains. J. Appl. Probab. 2003, 40, 970–979. [Google Scholar] [CrossRef]
- Szehr, O.; Reeb, D.; Wolf, M.M. Spectral convergence bounds for classical and quantum Markov processes. Commun. Math. Phys. 2015, 333, 565–595. [Google Scholar] [CrossRef]
- Horn, R.A.; Johnson, C.R. Matrix Analysis, 2nd ed.; Cambridge University Press: New York, NY, USA, 2013. [Google Scholar]
- Mitrophanov, A.Y. Note on Zeifman’s bounds on the rate of convergence for birth–death processes. J. Appl. Probab. 2004, 41, 593–596. [Google Scholar] [CrossRef]
- Zeifman, A.; Satin, Y.; Kryukova, A.; Razumchik, R.; Kiseleva, K.; Shilova, G. On three methods for bounding the rate of convergence for some continuous-time Markov chains. Int. J. Appl. Math. Comput. Sci. 2020, 30, 251–266. [Google Scholar] [CrossRef]
- Zeifman, A.I. Upper and lower bounds on the rate of convergence for nonhomogeneous birth and death processes. Stoch. Proc. Appl. 1995, 59, 157–173. [Google Scholar] [CrossRef]
- Dean, J.; Ganesh, A. Noise dissipation in gene regulatory networks via second order statistics of networks of infinite server queues. J. Math. Biol. 2022, 85, 14. [Google Scholar] [CrossRef]
- Szavits-Nossan, J.; Grima, R. Solving stochastic gene-expression models using queueing theory: A tutorial review. Biophys. J. 2024, 123, 1034–1057. [Google Scholar] [CrossRef] [PubMed]
- Zeifman, A.; Korolev, V.; Satin, Y. Two approaches to the construction of perturbation bounds for continuous-time Markov chains. Mathematics 2020, 8, 253. [Google Scholar] [CrossRef]
- Ipsen, I.C.F.; Selee, T.M. Ergodicity coefficients defined by vector norms. SIAM J. Matrix Anal. Appl. 2011, 32, 153–200. [Google Scholar] [CrossRef]
- Nicolis, G.; Turner, J.W. Effect of fluctuations on bifurcation phenomena. Ann. N. Y. Acad. Sci. 1979, 316, 251–262. [Google Scholar] [CrossRef]
- Remlein, B.; Seifert, U. Nonequilibrium fluctuations of chemical reaction networks at criticality: The Schlögl model as paradigmatic case. J. Chem. Phys. 2024, 160, 134103. [Google Scholar] [CrossRef]
- Beard, D.A.; Qian, H. Chemical Biophysics: Quantitative Analysis of Cellular Systems; Cambridge University Press: New York, NY, USA, 2008. [Google Scholar]
- Lauffenburger, D.A.; Lindermann, J.J. Receptors: Models for Binding, Trafficking, and Signaling; Oxford University Press: New York, NY, USA, 1993. [Google Scholar]
- Zhang, Y.N.; Ho, T.D.; Buchler, N.E.; Gordân, R. Competition for DNA binding between paralogous transcription factors determines their genomic occupancy and regulatory functions. Genome Res. 2021, 31, 1216–1229. [Google Scholar] [CrossRef]
- Laurenzi, I.J. An analytical solution of the stochastic master equation for reversible bimolecular reaction kinetics. J. Chem. Phys. 2000, 113, 3315–3322. [Google Scholar] [CrossRef]
- Zheng, Q. Note on the non-homogeneous Prendiville process. Math. Biosci. 1998, 148, 1–5. [Google Scholar] [CrossRef]
- Seneta, E. Markov chains as models in statistical mechanics. Stat. Sci. 2016, 31, 399–414. [Google Scholar] [CrossRef]
- Nicolis, G.; Prigogine, I. Fluctuations in nonequilibrium systems. Proc. Natl. Acad. Sci. USA 1971, 68, 2102–2107. [Google Scholar] [CrossRef]
- Roehner, B.; Valent, G. Solving the birth and death processes with quadratic asymptotically symmetric transition rates. SIAM J. Appl. Math. 1982, 42, 1020–1046. [Google Scholar] [CrossRef]
- Usov, I.; Satin, Y.; Zeifman, A.; Korolev, V. Ergodicity bounds and limiting characteristics for a modified Prendiville model. Mathematics 2022, 10, 4401. [Google Scholar] [CrossRef]
- Gadgil, C.; Lee, C.H.; Othmer, H.G. A stochastic analysis of first-order reaction networks. Bull. Math. Biol. 2005, 67, 901–946. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, T.; Huisinga, W. Solving the chemical master equation for monomolecular reaction systems analytically. J. Math. Biol. 2007, 54, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Darvey, I.G.; Staff, P.J. Stochastic approach to first-order chemical reaction kinetics. J. Chem. Phys. 1966, 44, 990–997. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y. The spectral gap and perturbation bounds for reversible continuous-time Markov chains. J. Appl. Probab. 2004, 41, 1219–1222. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y. Stability estimates for finite homogeneous continuous-time Markov chains. Theory Probab. Appl. 2006, 50, 319–326. [Google Scholar] [CrossRef]
- Voit, E.O. A First Course in Systems Biology; Garland Science: New York, NY, USA, 2013. [Google Scholar]
- Zhang, H.X.; Dempsey, W.P.; Goutsias, J. Probabilistic sensitivity analysis of biochemical reaction systems. J. Chem. Phys. 2009, 131, 094101. [Google Scholar] [CrossRef]
- Gunawan, R.; Cao, Y.; Petzold, L.; Doyle, F.J. Sensitivity analysis of discrete stochastic systems. Biophys. J. 2005, 88, 2530–2540. [Google Scholar] [CrossRef]
- Anderson, D.F.; Howells, A.S. Parametric sensitivity analysis for models of reaction networks within interacting compartments. J. Chem. Phys. 2025, 162, 154105. [Google Scholar] [CrossRef]
- Anisimov, V.V. Estimates for the deviations of the transition characteristics of nonhomogeneous Markov processes. Ukr. Math. J. 1988, 40, 588–592. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y.; Rosendaal, F.R.; Reifman, J. Computational analysis of the effects of reduced temperature on thrombin generation: The contributions of hypothermia to coagulopathy. Anesth. Analg. 2013, 117, 565–574. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y.; Churchward, G.; Borodovsky, M. Control of Streptococcus pyogenes virulence: Modeling of the CovR/S signal transduction system. J. Theor. Biol. 2007, 246, 113–128. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y.; Groisman, E.A. Response acceleration in post-translationally regulated genetic circuits. J. Mol. Biol. 2010, 396, 1398–1409. [Google Scholar] [CrossRef]
- Tomaiuolo, M.; Kottke, M.; Matheny, R.W.; Reifman, J.; Mitrophanov, A.Y. Computational identification and analysis of signaling subnetworks with distinct functional roles in the regulation of TNF production. Mol. BioSyst. 2016, 12, 826–838. [Google Scholar] [CrossRef]
- Nagaraja, S.; Reifman, J.; Mitrophanov, A.Y. Computational identification of mechanistic factors that determine the timing and intensity of the inflammatory response. PLoS Comput. Biol. 2015, 11, e1004460. [Google Scholar] [CrossRef]
- Satin, Y.; Razumchik, R.; Usov, I.; Zeifman, A. Numerical computation of distributions in finite-state inhomogeneous continuous time Markov chains, based on ergodicity bounds and piecewise constant approximation. Mathematics 2023, 11, 4265. [Google Scholar] [CrossRef]
- Esquível, M.L.; Krasii, N.P.; Guerreiro, G.R. Estimation–calibration of continuous-time non-homogeneous Markov chains with finite state space. Mathematics 2024, 12, 668. [Google Scholar] [CrossRef]
- Delebecque, F. A reduction process for perturbed Markov chains. SIAM J. Appl. Math. 1983, 43, 325–350. [Google Scholar] [CrossRef]
- Heidergott, B.; Leahu, H.; Löpker, A.; Pflug, G. Perturbation analysis of inhomogeneous finite Markov chains. Adv. Appl. Probab. 2016, 48, 255–273. [Google Scholar] [CrossRef]
- Aslyamov, T.; Esposito, M. General theory of static response for Markov jump processes. Phys. Rev. Lett. 2024, 133, 107103. [Google Scholar] [CrossRef]
- Aslyamov, T.; Ptaszynski, K.; Esposito, M. Nonequilibrium fluctuation-response relations: From identities to bounds. Phys. Rev. Lett. 2025, 134, 157101. [Google Scholar] [CrossRef]
- Kolchinsky, A.; Ohga, N.; Ito, S. Thermodynamic bound on spectral perturbations, with applications to oscillations and relaxation dynamics. Phys. Rev. Res. 2024, 6, 013082. [Google Scholar] [CrossRef]
- Gondolf, P.; Möbus, T.; Rouzé, C. Energy preserving evolutions over Bosonic systems. Quantum 2024, 8, 1551. [Google Scholar] [CrossRef]
- Blom, K.; Thiele, U.; Godec, A. Local order controls the onset of oscillations in the nonreciprocal Ising model. Phys. Rev. E 2025, 111, 024207. [Google Scholar] [CrossRef]
- Esquível, M.L.; Krasii, N.P. Statistics for continuous time Markov chains, a short review. Axioms 2025, 14, 283. [Google Scholar] [CrossRef]
- Vestring, Y.; Tavakoli, J. Confidence regions for steady-state probabilities and additive functionals based on a single sample path of an ergodic Markov chain. Mathematics 2024, 12, 3641. [Google Scholar] [CrossRef]
- Sherlock, C. Direct statistical inference for finite Markov jump processes via the matrix exponential. Comput. Stat. 2021, 36, 2863–2887. [Google Scholar] [CrossRef]
- Venkataramanan, L.; Sigworth, F.J. Applying hidden Markov models to the analysis of single ion channel activity. Biophys. J. 2002, 82, 1930–1942. [Google Scholar] [CrossRef]
- Mitrophanov, A.Y.; Lomsadze, A.; Borodovsky, M. Sensitivity of hidden Markov models. J. Appl. Probab. 2005, 42, 632–642. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitrophanov, A.Y. Markov-Chain Perturbation and Approximation Bounds in Stochastic Biochemical Kinetics. Mathematics 2025, 13, 2059. https://doi.org/10.3390/math13132059
Mitrophanov AY. Markov-Chain Perturbation and Approximation Bounds in Stochastic Biochemical Kinetics. Mathematics. 2025; 13(13):2059. https://doi.org/10.3390/math13132059
Chicago/Turabian StyleMitrophanov, Alexander Y. 2025. "Markov-Chain Perturbation and Approximation Bounds in Stochastic Biochemical Kinetics" Mathematics 13, no. 13: 2059. https://doi.org/10.3390/math13132059
APA StyleMitrophanov, A. Y. (2025). Markov-Chain Perturbation and Approximation Bounds in Stochastic Biochemical Kinetics. Mathematics, 13(13), 2059. https://doi.org/10.3390/math13132059