
HSICCR: A Lightweight Scoring Criterion Based on Measuring the Degree of Causality for the Detection of SNP Interactions
 ,
,
Abstract
1. Introduction
- We propose a high-accuracy scoring criterion based on measuring the degree of causality that integrates a widely used method of measuring statistical dependencies (HSIC);
- We put forward an efficient algorithm of computing HSIC on two variables with a small sample in time, thus enabling us to compute HSIC in time in practice.
2. Related Works
3. Methodology
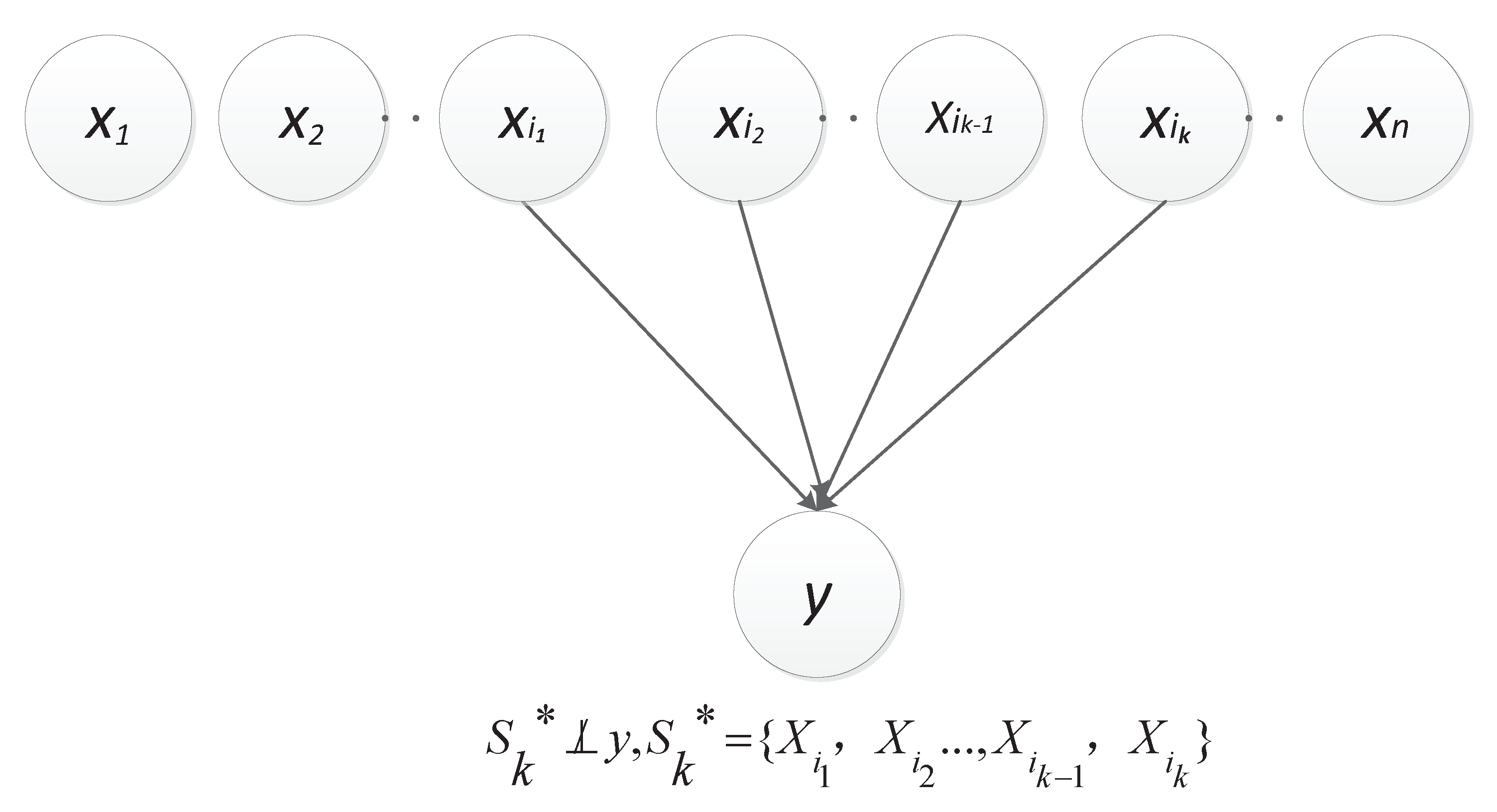
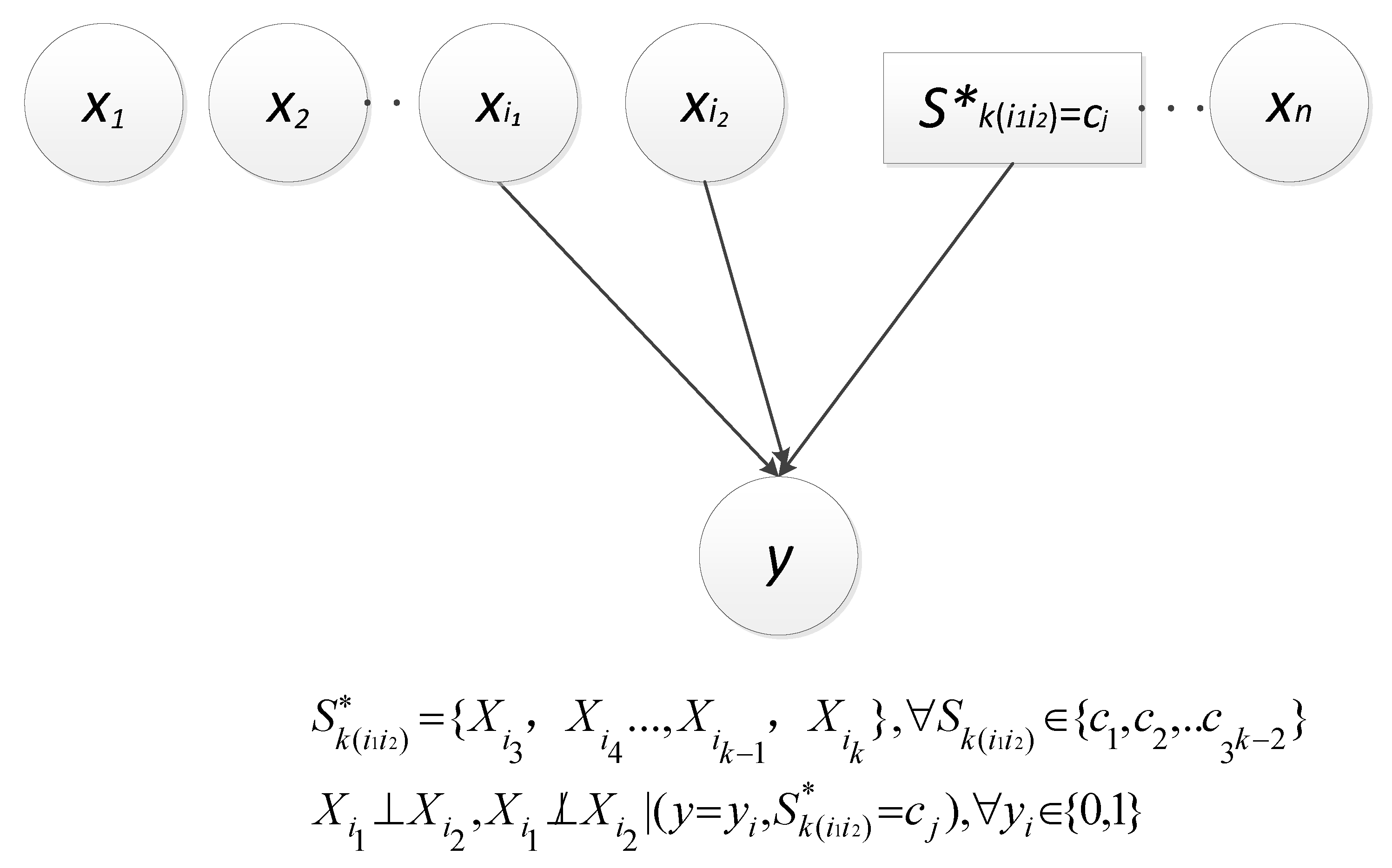
3.1. Concepts and Terms
3.2. Causal Relationship
3.3. Scoring Criterion
- 1.
- The value of is a linear weighted sum of and based on respective sample sizes;
- 2.
- For all , the value of is a linear weighted sum of all since there are v-structures given ;
- 3.
- The value of is a linear weighted sum of and based on sample size of and average sample size of all , which is a component and basis of ;
- 4.
- In particular, as is when ;
- 5.
- For robustness purposes, let = 0 if and only if the denominator of the weighted factor term is 0, like ;
- 6.
- The effort to calculate scoring criterion is reduced to calculate once, and is reduced up to times to calculate the type of problem (fortunately, in practice).
3.4. Method for Measuring Statistical Dependence
3.4.1. HSIC
3.4.2. Efficient Computation
- (X, Y) is m observations of a tuple of x and y with p and q states, respectively;
- includes two kernel functions used to calculate and , the parameters of which are and , respectively;
- (see Algorithm 2) is used to calculate , and ;
- For all , (see Algorithm 3) is used to calculate ;
- (see Algorithm 4) is used to calculate ;
- (see Algorithm 5) is used to calculate and .
| Algorithm 1 Calculate |
|
| Algorithm 2 Calculate = |
|
| Algorithm 3 Calculate |
|
| Algorithm 4 Calculate |
|
| Algorithm 5 Calculate |
|
4. Experiments
4.1. Evaluation Criterion
4.2. Simulated Datasets
4.2.1. Disease Models with k = 2
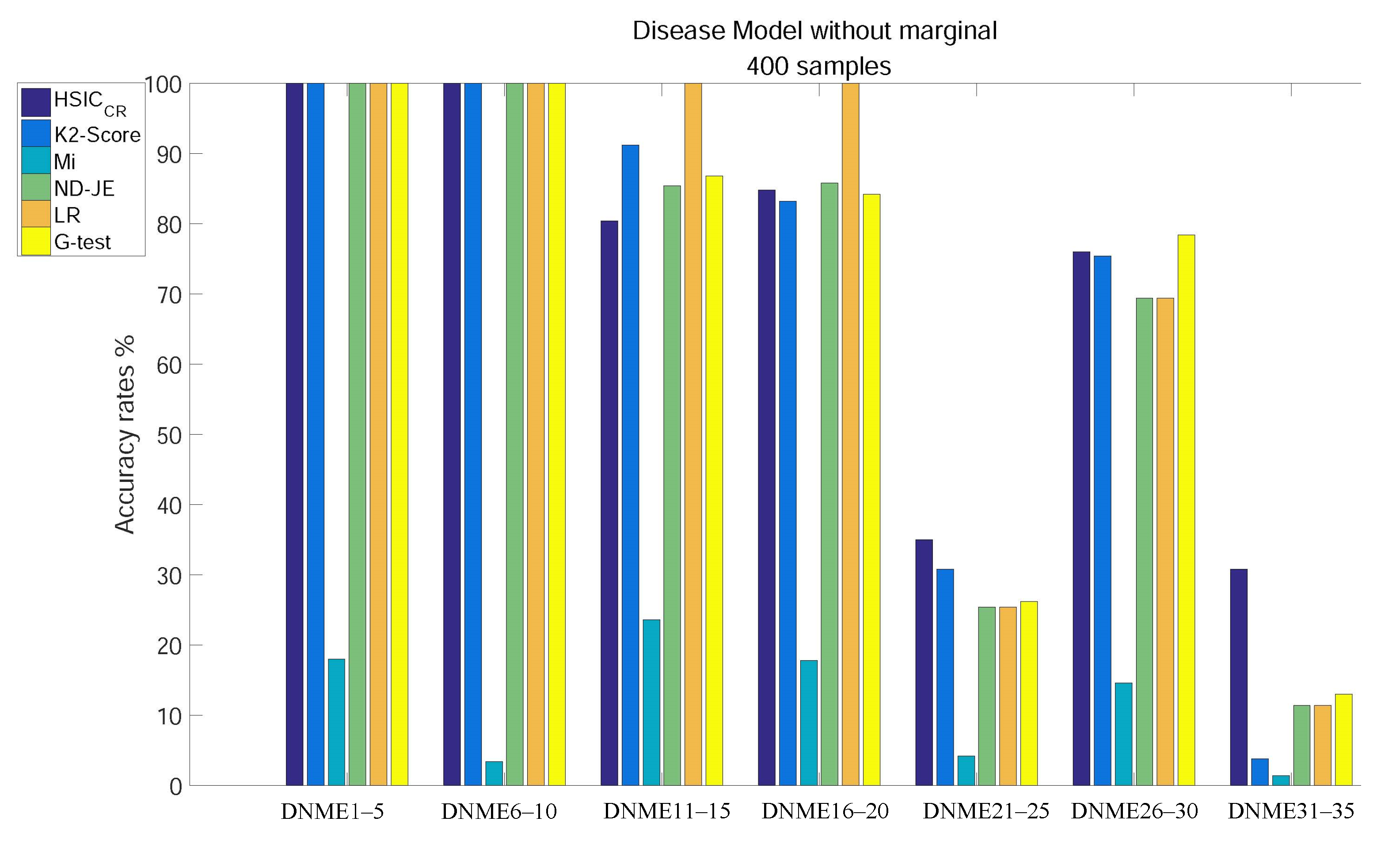
Disease Models without Marginal Effects
- Except for Mi, using tests on DNME1–10, the accuracy of all scoring criteria is close to 100%;
- All criteria are not very accurate using tests on DNME21–25 and DNME31–35;
- Mi has an extremely poor accuracy on all subgroup tests;
- LR has the highest accuracy using tests on DNME11–15 and DNME16–20, close to 100%, but is only a little more accurate than Mi on DNME21–25, DNME26–30 and DNME31–35 tests;
- The accuracy rates of both ND-JE and G-test rank in the middle overall, but G-test has the highest accuracy on the DNME26–30 test;
- The accuracy rate of K2-Score ranks second on DNME11–15 and DNME21–25 tests, third on the DNME26–30 test and slightly worse than ND-JE, HS and G-test on the DNME16–20 test, but is only a little more accurate than Mi on the most difficult model (DNME31–35) test;
- HSIC has the highest accuracy on the two most difficult model subgroup (DNME21–25 and DNME31–35) tests, especially on DNME31–35, where the accuracy is much higher than other criteria, and the overall accuracy on other model subgroups tests is similar to the other four criteria.
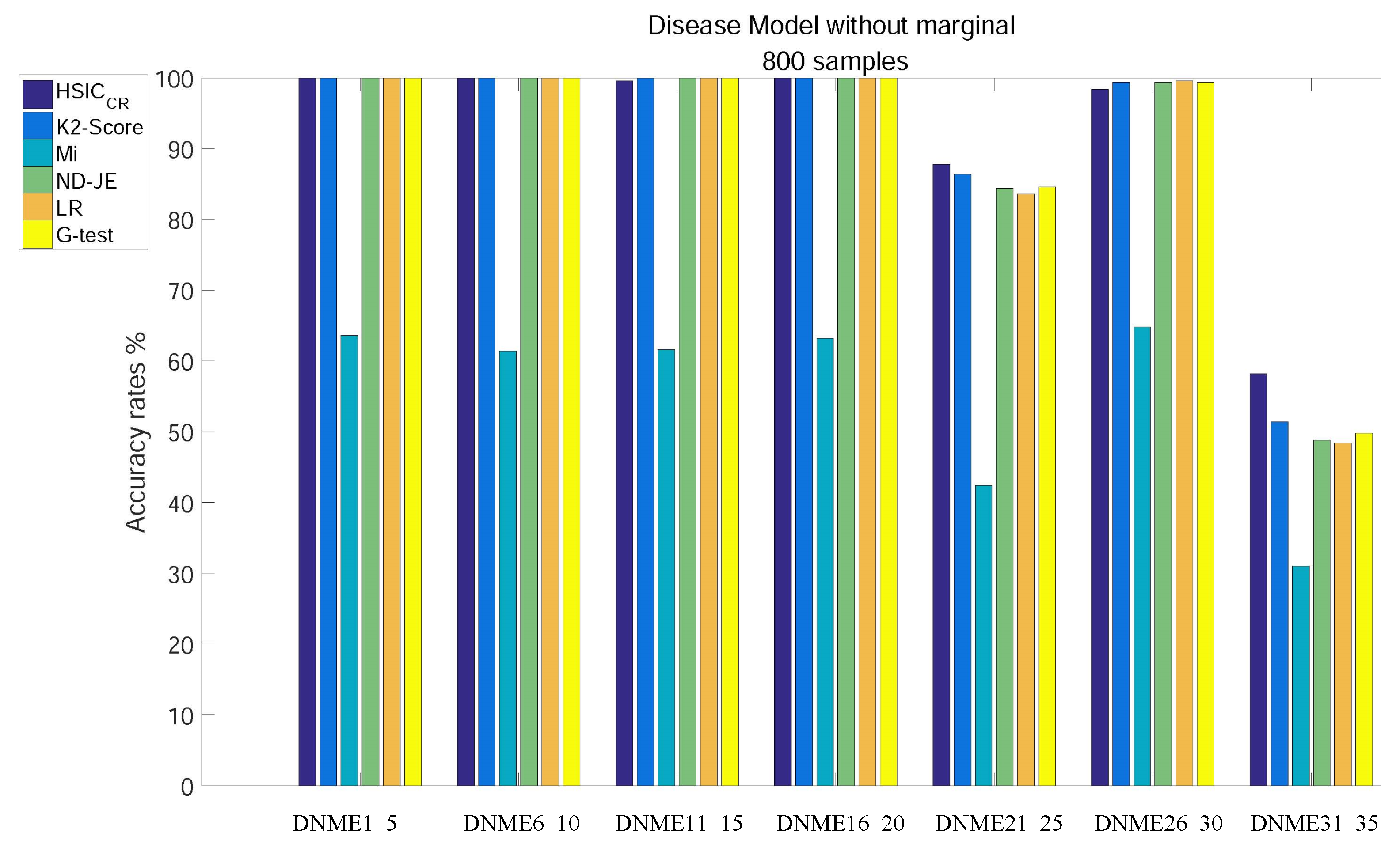
- Except for Mi, the accuracy of all criteria is close to 100% excluding tests on the two most difficult model subgroups (DNME21–25 and DNME31–35);
- Although the accuracy rate of Mi can be significantly improved with the increase in the size of samples, it is still relatively poor overall;
- With the number of samples increasing, there is still no change in the overall ranking, but the accuracy of the K2-Score on the DNME31–35 test rises to second;
- HSIC has the highest accuracy on the two most difficult model subgroup tests.
Disease Models with Marginal Effects
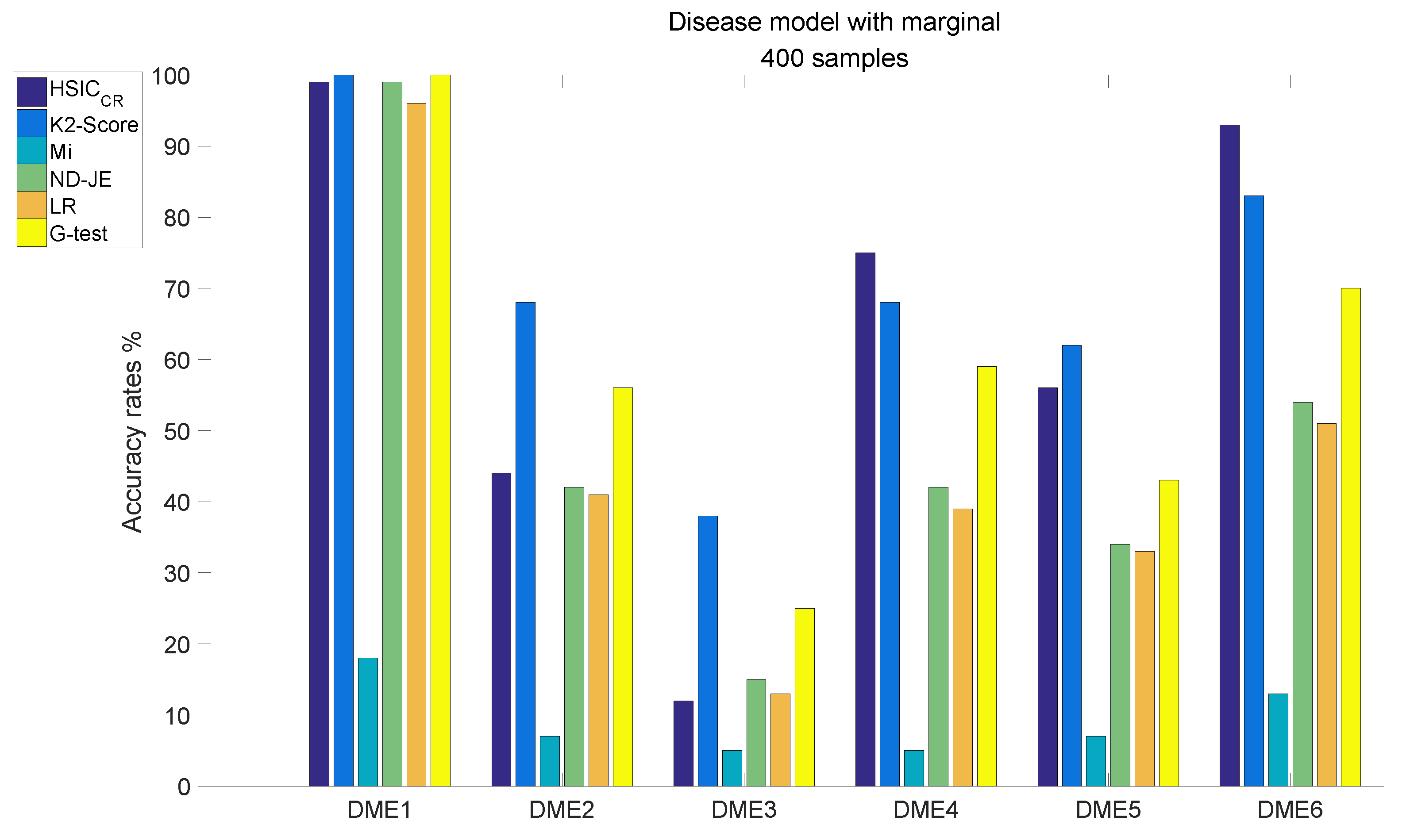
- The accuracy of all scoring criteria is close to 100% tested on DME1, except for Mi;
- Mi has extremely poor accuracy on all six models tests;
- Except for Mi, the accuracy rate of LR is worse than the other four criteria on DME2–6 tests, except that the accuracy on the DME3 test is nearly the same as that of HSIC;
- The accuracy rate of ND-JE ranks third on DME1 and DME3 tests, and fourth on the other four models tests;
- The accuracy rate of G-test ranks first on the DME1 test, second on DME2 and DME3 tests and third on the other four models tests;
- HSIC has the highest accuracy rate on DME4 and DME6 tests, its accuracy rate on the DME5 test is slightly worse than LR and the accuracy rate on DME1–2 tests ranks third, whereas the accuracy rate on the DME3 test is a little better than Mi;
- K2-Score has the highest accuracy rate on DME1–3 and DME5 tests, its accuracy rate on DME4 and DME6 tests ranks second and it significantly outperforms the others on the most difficult model (DME3) test (although its accuracy rate in DME3 is below 50%).
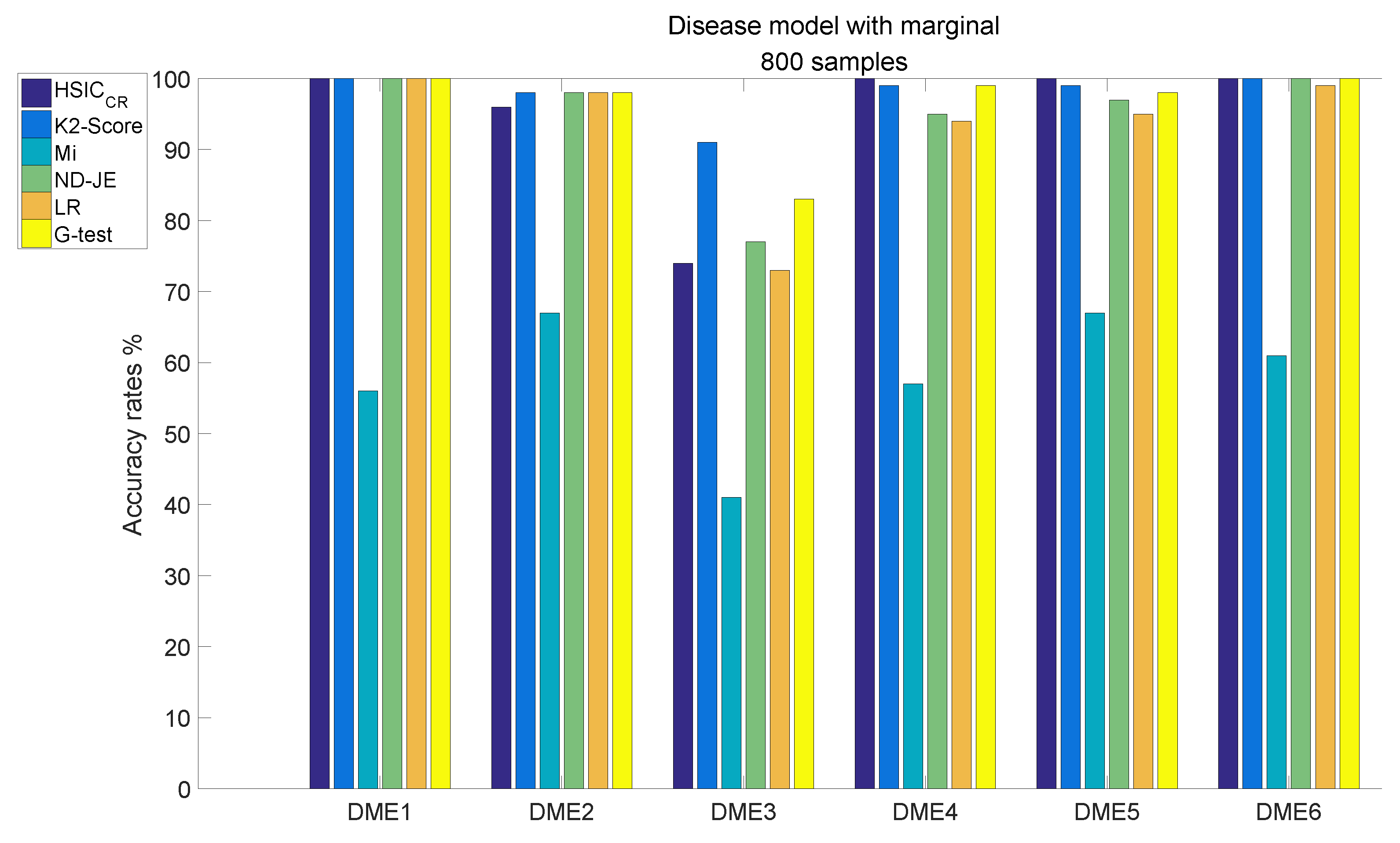
- Although the accuracy of Mi can be significantly improved with the increase in the size of samples, it is still relatively poor overall;
- The accuracy rates of the other five scoring criteria all exceed 95% tested by the models, except on DME3;
- K2-Score has the highest accuracy rate on the most difficult model test (over 90%), the accuracy rate of G-test ranks second (over 80 %) and the accuracy rates of ND-JE, HSIC and LR are not good enough, at just over .
4.2.2. Disease Models with k = 3
- The accuracy of all scoring criteria is close to 100% tested on DM2, DM3–4 and DM7–8;
- The accuracy of all scoring criteria is close to 80% on the DM1 test;
- The K2-Score has a poor accuracy rate on the most difficult model (DM5) test, whose accuracy is just close to ;
- The accuracy rates of the scoring criteria on the DM6 test are good enough, and the accuracy rates of the scoring criteria are close to 100%, except the K2-Score;
- HSIC has the highest accuracy rate on the DM5 test, and its accuracy rate is the only one that exceeds 60% among all scoring criteria.
4.2.3. The Running Time Analysis
4.3. Case Study: A Real Chronic Dialysis Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| SNP | single-nucleotide polymorphism |
| GWAS | genome-wide association analysis |
References
- Carlson, C.S.; Eberle, M.A.; Kruglyak, L.; Nickerson, D.A. Mapping complex disease loci in whole-genome association studies. Nature 2004, 429, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.H.; Hemani, G.; Haley, C.S. Detecting epistasis in human complex traits. Nat. Rev. Genet. 2014, 15, 722–733. [Google Scholar] [CrossRef]
- Guo, X.; Meng, Y.; Yu, N.; Pan, Y. Cloud computing for detecting high-order genome-wide epistatic interaction via dynamic clustering. BMC Bioinform. 2014, 15, 102. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, J.; Cai, Z.; Du, D.Z.; Pan, Y. Searching genome-wide multi-locus associations for multiple diseases based on bayesian inference. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 14, 600–610. [Google Scholar] [CrossRef]
- Gyenesei, A.; Moody, J.; Semple, C.A.; Haley, C.S.; Wei, W.H. High-throughput analysis of epistasis in genome-wide association studies with BiForce. Bioinformatics 2012, 28, 1957–1964. [Google Scholar] [CrossRef][Green Version]
- Liyan, S. The Research on Epistasis Detection Algorithm in Genome-wide Association Study. Ph.D. Thesis, Jilin University, Changchun, China, 2020. [Google Scholar]
- Ritchie, M.D.; Hahn, L.W.; Roodi, N.; Bailey, L.R.; Dupont, W.D.; Parl, F.F.; Moore, J.H. Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am. J. Hum. Genet. 2001, 69, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, X.; Feng, Y.; Guo, M.; Yu, G.; Wang, J. ELSSI: Parallel SNP–SNP interactions detection by ensemble multi-type detectors. Brief. Bioinform. 2022, 23, bbac213. [Google Scholar] [CrossRef] [PubMed]
- Tuo, S.; Liu, H.; Chen, H. Multipopulation harmony search algorithm for the detection of high-order SNP interactions. Bioinformatics 2020, 36, 4389–4398. [Google Scholar] [CrossRef]
- Sun, Y.; Shang, J.; Liu, J.X.; Li, S.; Zheng, C.H. epiACO—A method for identifying epistasis based on ant Colony optimization algorithm. BioData Min. 2017, 10, 23. [Google Scholar] [CrossRef]
- Tuo, S.; Zhang, J.; Yuan, X.; He, Z.; Liu, Y.; Liu, Z. Niche harmony search algorithm for detecting complex disease associated high-order SNP combinations. Sci. Rep. 2017, 7, 11529. [Google Scholar] [CrossRef]
- Aflakparast, M.; Salimi, H.; Gerami, A.; Dubé, M.; Visweswaran, S.; Masoudi-Nejad, A. Cuckoo search epistasis: A new method for exploring significant genetic interactions. Heredity 2014, 112, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.J.; Shen, H.B. MACOED: A multi-objective ant colony optimization algorithm for SNP epistasis detection in genome-wide association studies. Bioinformatics 2015, 31, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Jin, Y.; Olhofer, M.; Sendhoff, B. A reference vector guided evolutionary algorithm for many-objective optimization. IEEE Trans. Evol. Comput. 2016, 20, 773–791. [Google Scholar] [CrossRef]
- Shouheng, T.; Hong, H. DEaf-MOPS/D: An improved differential evolution algorithm for solving complex multi-objective portfolio selection problems based on decomposition. Econ. Comput. Econ. Cybernet. Stud. Res. 2019, 53, 151–167. [Google Scholar]
- Verzilli, C.J.; Stallard, N.; Whittaker, J.C. Bayesian graphical models for genomewide association studies. Am. J. Hum. Genet. 2006, 79, 100–112. [Google Scholar] [CrossRef]
- Cooper, G.F.; Herskovits, E. A Bayesian method for the induction of probabilistic networks from data. Mach. Learn. 1992, 9, 309–347. [Google Scholar] [CrossRef]
- Jiang, X.; Neapolitan, R.E.; Barmada, M.M.; Visweswaran, S. Learning genetic epistasis using Bayesian network scoring criteria. BMC Bioinform. 2011, 12, 89. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.S. Bayesian inference of epistatic interactions in case-control studies. Nat. Genet. 2007, 39, 1167–1173. [Google Scholar] [CrossRef]
- Paninski, L. Estimation of entropy and mutual information. Neural Comput. 2003, 15, 1191–1253. [Google Scholar] [CrossRef]
- Lin, J. Divergence measures based on the Shannon entropy. IEEE Trans. Inf. Theory 1991, 37, 145–151. [Google Scholar] [CrossRef]
- Bush, W.S.; Edwards, T.L.; Dudek, S.M.; McKinney, B.A.; Ritchie, M.D. Alternative contingency table measures improve the power and detection of multifactor dimensionality reduction. BMC Bioinform. 2008, 9, 238. [Google Scholar] [CrossRef] [PubMed]
- Neyman, J.; Pearson, E.S. On the use and interpretation of certain test criteria for purposes of statistical inference: Part I. Biometrika 1928, 20A, 175–240. [Google Scholar]
- Stamatis, D.H. Essential Statistical Concepts for the Quality Professional; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Pearl, J. Models, Reasoning and Inference; Cambridge University Press: Cambridge, UK, 2000; Volume 19. [Google Scholar]
- Schaid, D.J. Genomic similarity and kernel methods I: Advancements by building on mathematical and statistical foundations. Hum. Hered. 2010, 70, 109–131. [Google Scholar] [CrossRef]
- Gretton, A.; Bousquet, O.; Smola, A.; Schölkopf, B. Measuring statistical dependence with Hilbert-Schmidt norms. In Proceedings of the International Conference on Algorithmic Learning Theory, Singapore, 8–11 October 2005; Springer: Berlin/Heidelberg, Germany, 2005; pp. 63–77. [Google Scholar]
- Gretton, A.; Fukumizu, K.; Teo, C.; Song, L.; Schölkopf, B.; Smola, A. A kernel statistical test of independence. Adv. Neural Inf. Process. Syst. 2007, 20, 585–592. [Google Scholar]
- Kodama, K.; Saigo, H. KDSNP: A kernel-based approach to detecting high-order SNP interactions. J. Bioinform. Comput. Biol. 2016, 14, 1644003. [Google Scholar] [CrossRef]
- Urbanowicz, R.J.; Kiralis, J.; Sinnott-Armstrong, N.A.; Heberling, T.; Fisher, J.M.; Moore, J.H. GAMETES: A fast, direct algorithm for generating pure, strict, epistatic models with random architectures. BioData Min. 2012, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Chuang, L.Y.; Lin, Y.D. Multiobjective multifactor dimensionality reduction to detect SNP–SNP interactions. Bioinformatics 2018, 34, 2228–2236. [Google Scholar] [CrossRef]
- Chen, J.B.; Yang, Y.H.; Lee, W.C.; Liou, C.W.; Lin, T.K.; Chung, Y.H.; Chuang, L.Y.; Yang, C.H.; Chang, H.W. Sequence-based polymorphisms in the mitochondrial D-loop and potential SNP predictors for chronic dialysis. PLoS ONE 2012, 7, e41125. [Google Scholar] [CrossRef]
- Yang, C.H.; Kao, Y.K.; Chuang, L.Y.; Lin, Y.D. Catfish Taguchi-based binary differential evolution algorithm for analyzing single nucleotide polymorphism interactions in chronic dialysis. IEEE Trans. Nanobiosci. 2018, 17, 291–299. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
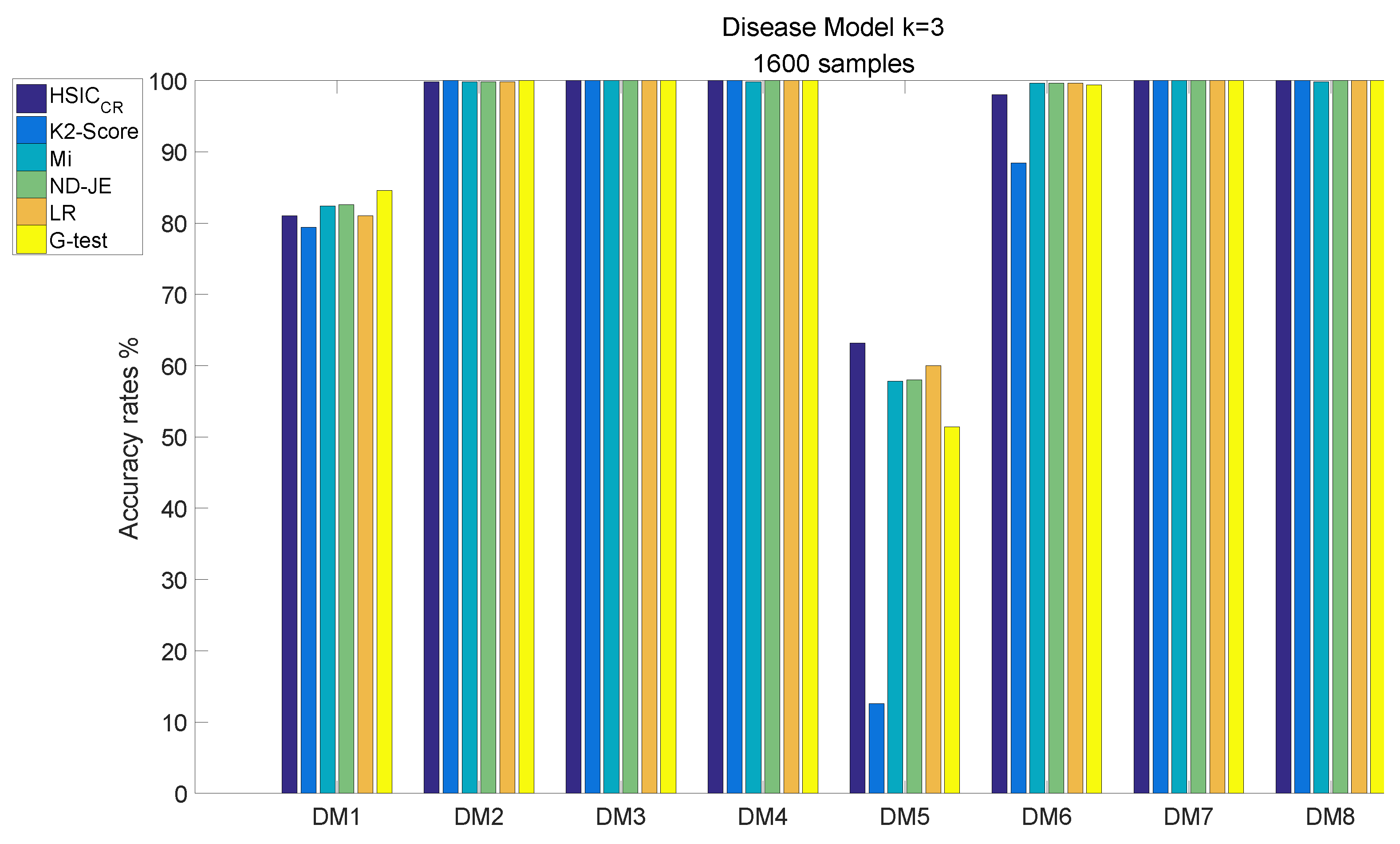{kind=link}
| Scoring Criterion | 400 Samples | 800 Samples | Total (%) | DNME31–35 (%) |
|---|---|---|---|---|
| HSIC | 2535 | 3220 | 5755 (82.2%) | 445 (44.5%) |
| K2-Score | 2479 | 3186 | 5665 (80.9%) | 336 (33.6%) |
| G-test | 2443 | 3169 | 5612 (80.2%) | 314 (31.4%) |
| ND-JE | 2440 | 3163 | 5603 (80.0%) | 301 (30.1%) |
| LR | 2437 | 3158 | 5595 (79.9%) | 297 (29.7%) |
| Mi | 494 | 1971 | 2465 (35.2%) | 192 (19.2%) |
| Scoring Criterion | 400 Samples | 800 Samples | Total (%) | DME3 (%) |
|---|---|---|---|---|
| K2-Score | 419 | 586 | 1005 (83.8%) | 159 (79.5%) |
| HSIC | 379 | 570 | 949 (79.1%) | 86 (43%) |
| G-test | 353 | 578 | 931 (77.6%) | 108 (54%) |
| ND-JE | 286 | 567 | 853 (71.1%) | 92 (46%) |
| LR | 273 | 559 | 832 (69.3%) | 86 (43%) |
| Mi | 55 | 349 | 404 (33.7%) | 48 (24%) |
| Scoring Criterion | Total (%) | DM5 (%) |
|---|---|---|
| HSIC | 3710 (92.8%) | 316 (63.2%) |
| LR | 3702 (92.6%) | 300 (60%) |
| ND-JE | 3700 (92.5%) | 290 (58%) |
| Mi | 3696 (92.4%) | 289 (57.8%) |
| G-test | 3677 (91.9%) | 257 (51.4%) |
| K2-Score | 3402 (85.1%) | 63 (12.6%) |
| Scoring Criterion | 2-Order (s) | 3-Order (s) |
|---|---|---|
| HSIC | 120.2426 | 80.3045 |
| LR | 77.3055 | 55.4438 |
| ND-JE | 134.922 | 76.5377 |
| Mi | 78.3 | 55.6138 |
| G-test | 77.6589 | 56.5221 |
| K2-Score | 125.0655 | 81.4237 |
| SNP | D-Loop Position | ||||
|---|---|---|---|---|---|
| 1∼5 | A16051G | T16086C | T16092M | T16093C | C16108T |
| 6∼10 | C16111T | T16126C | G16129A | T16136C | T16140C |
| 11∼15 | G16145A | C16148T | T16157C | A16162G | A16164G |
| 16∼20 | C16167T | T16172C | T16209C | T16217C | C16218T |
| 21∼25 | T16223C | A16227G | C16234T | A16235G | T16243M |
| 26∼30 | C16248T | T16249C | C16256T | C16257W | C16260T |
| 31∼35 | C16261T | C16266D | A16272G | G16274A | C16278T |
| 36∼40 | C16290T | C16291T | C16295T | C16297T | C16298T |
| 41∼45 | C16304T | A16309G | T16311C | A16316G | G16319A |
| 46∼50 | T16324C | C16327T | A16335G | C16355T | T16356C |
| 51∼55 | T16357C | T16362C | G16390A | A16399G | A16463G |
| 56∼60 | C16519T | A93G | G103A | T146M | C150T |
| 61∼65 | C151T | T152C | A153G | G185A | A189G |
| 66∼70 | C194T | T195C | T199C | A200G | T204C |
| 71∼75 | G207A | A210G | T217C | A234G | A235G |
| 76∼77 | T317C | C461T |
| Rank | Combination | HSIC |
|---|---|---|
| 1 | 41, 21 | 0.035922 |
| 2 | 52, 21 | 0.033105 |
| 3 | 41, 17 | 0.019069 |
| 4 | 56, 21 | 0.018961 |
| 5 | 68, 39 | 0.018545 |
| 6 | 21, 19 | 0.017254 |
| 7 | 60, 21 | 0.014506 |
| 8 | 17, 8 | 0.011645 |
| 9 | 17, 14 | 0.0097405 |
| 10 | 75, 36 | 0.0095467 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, J.; Zeng, J.; Wang, X.; Li, G.; Zhu, J.; Wang, F.; Tang, D. HSICCR: A Lightweight Scoring Criterion Based on Measuring the Degree of Causality for the Detection of SNP Interactions. Mathematics 2022, 10, 4134. https://doi.org/10.3390/math10214134
Zheng J, Zeng J, Wang X, Li G, Zhu J, Wang F, Tang D. HSICCR: A Lightweight Scoring Criterion Based on Measuring the Degree of Causality for the Detection of SNP Interactions. Mathematics. 2022; 10(21):4134. https://doi.org/10.3390/math10214134
Chicago/Turabian StyleZheng, Junxi, Juan Zeng, Xinyang Wang, Gang Li, Jiaxian Zhu, Fanghong Wang, and Deyu Tang. 2022. "HSICCR: A Lightweight Scoring Criterion Based on Measuring the Degree of Causality for the Detection of SNP Interactions" Mathematics 10, no. 21: 4134. https://doi.org/10.3390/math10214134
APA StyleZheng, J., Zeng, J., Wang, X., Li, G., Zhu, J., Wang, F., & Tang, D. (2022). HSICCR: A Lightweight Scoring Criterion Based on Measuring the Degree of Causality for the Detection of SNP Interactions. Mathematics, 10(21), 4134. https://doi.org/10.3390/math10214134