
SCAFG: Classifying Single Cell Types Based on an Adaptive Threshold Fusion Graph Convolution Network

Abstract
:1. Introduction
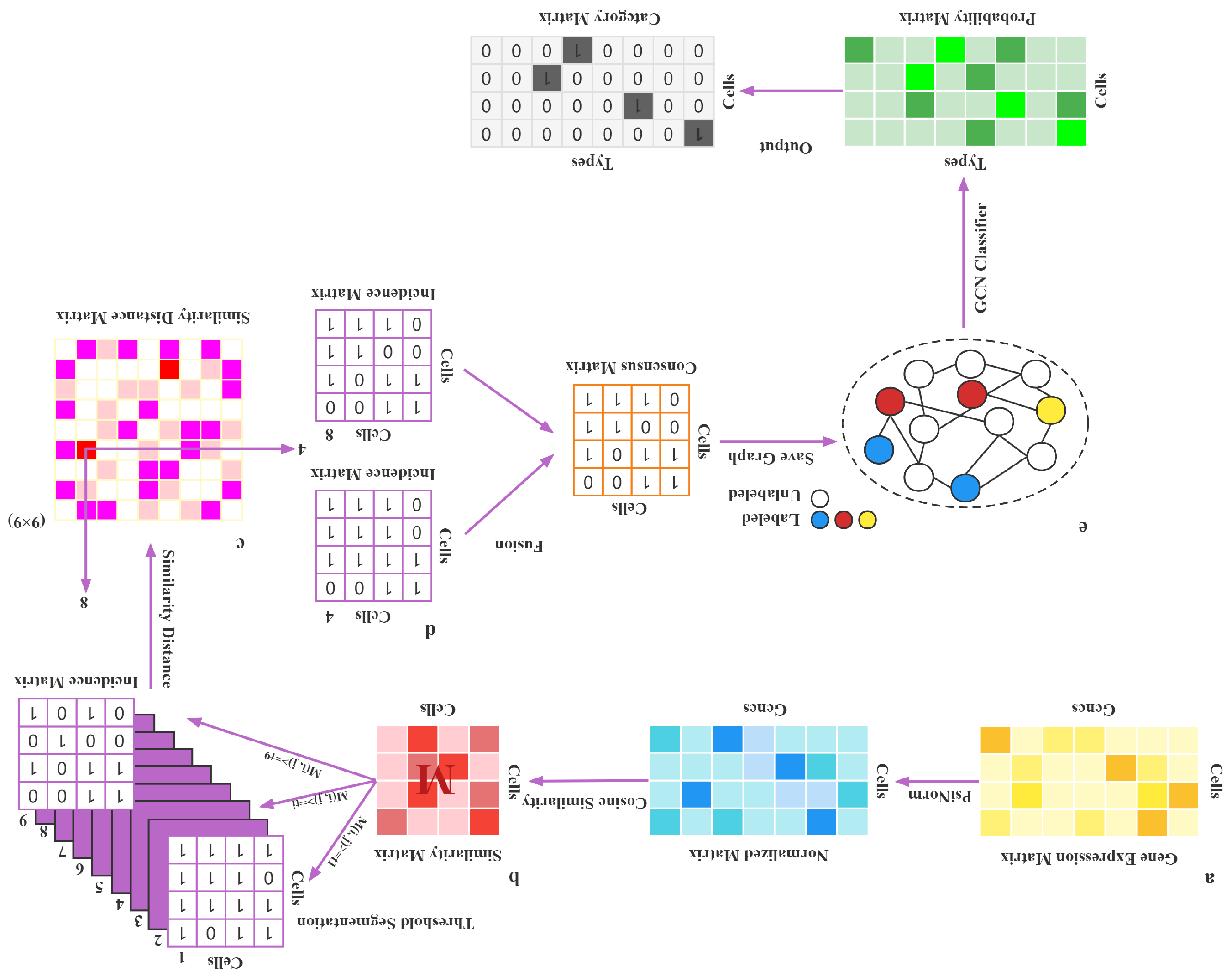
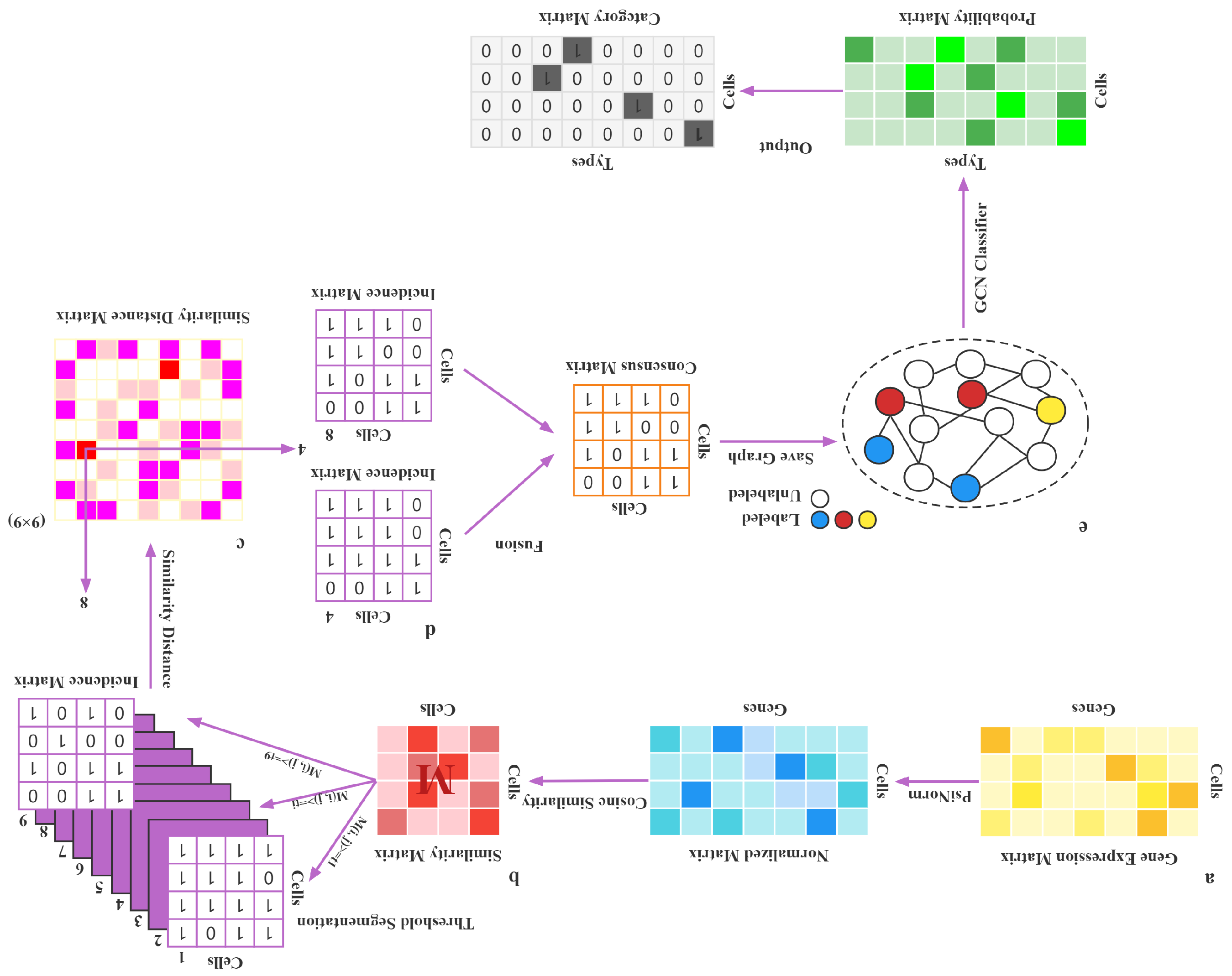
2. Materials and Methods
2.1. Real Datasets
2.2. Data Normalization
2.3. Cosine Similarity
2.4. Threshold Segmentation
2.5. Similarity Distance Between Matrices
| Algorithm 1 Construction of the Similarity Distance Matrix H |
| Require: Incidence matrix: ; |
| For each in S, they have the same dimension ; |
| Ensure: |
| 1: for do |
| 2: for do |
| 3: |
| 4: Calculate : |
| 5: for do |
| 6: for do |
| 7: if then |
| 8: |
| 9: end if |
| 10: end for |
| 11: end for |
| 12: return ; |
| 13: end for |
| 14: end for |
| 15. return H |
2.6. Fusion
| Algorithm 2 Incidence Matrix Fusion to Form the Consensus Matrix Q |
| Require: Similarity Distance Matrix ; |
| Ensure: |
| 1: for do |
| 2: for do |
| 3: if then |
| 4: |
| 5: |
| 6: |
| 7: end if |
| 8: end for |
| 9: end for |
| 10: return |
| 11: Select the incidence matrix |
| 12: for do |
| 13: for do |
| 14: if then |
| 15: |
| 16: else |
| 17: |
| 18: |
| 19: end if |
| 20: |
| 21: end for |
| 22: end for |
| 23: return Q |
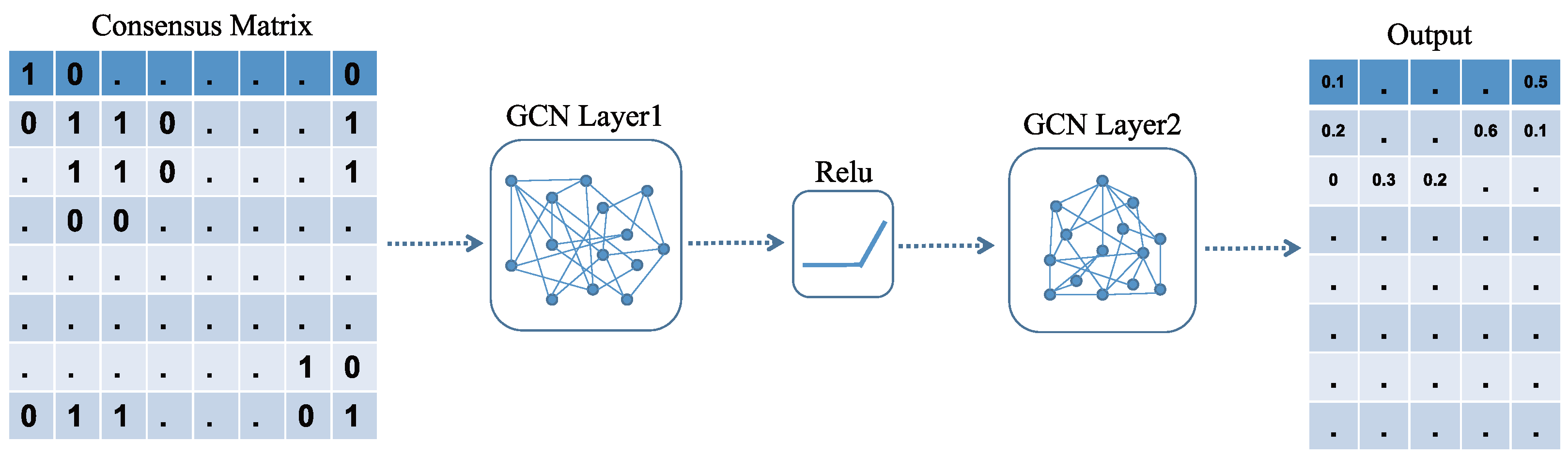
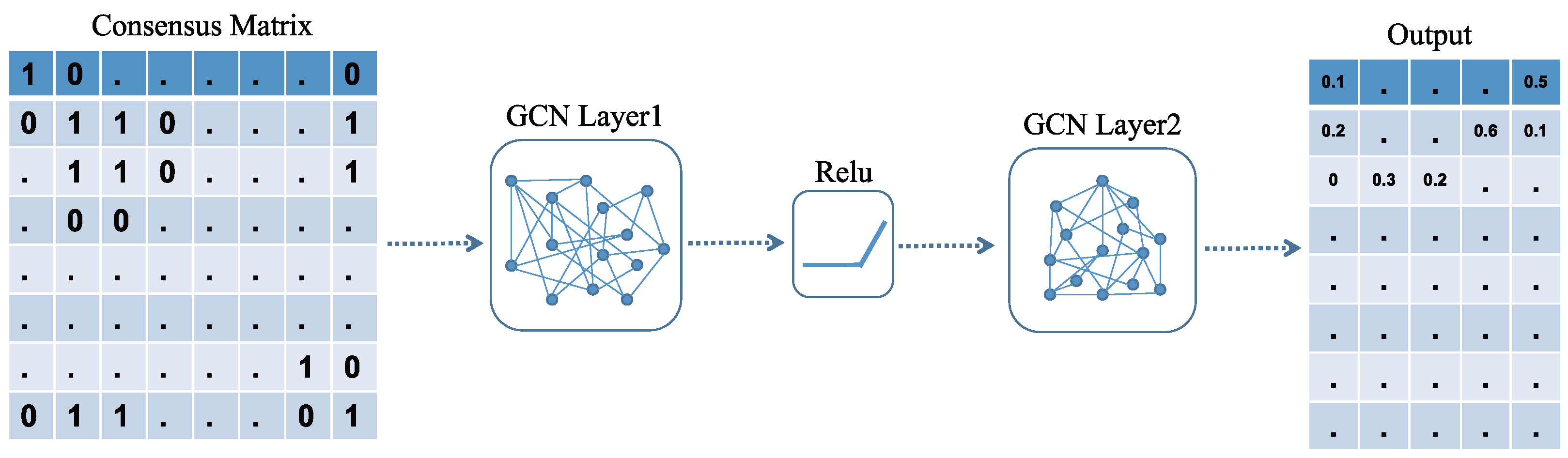
2.7. Construction of the GCN
3. Results and Discussion
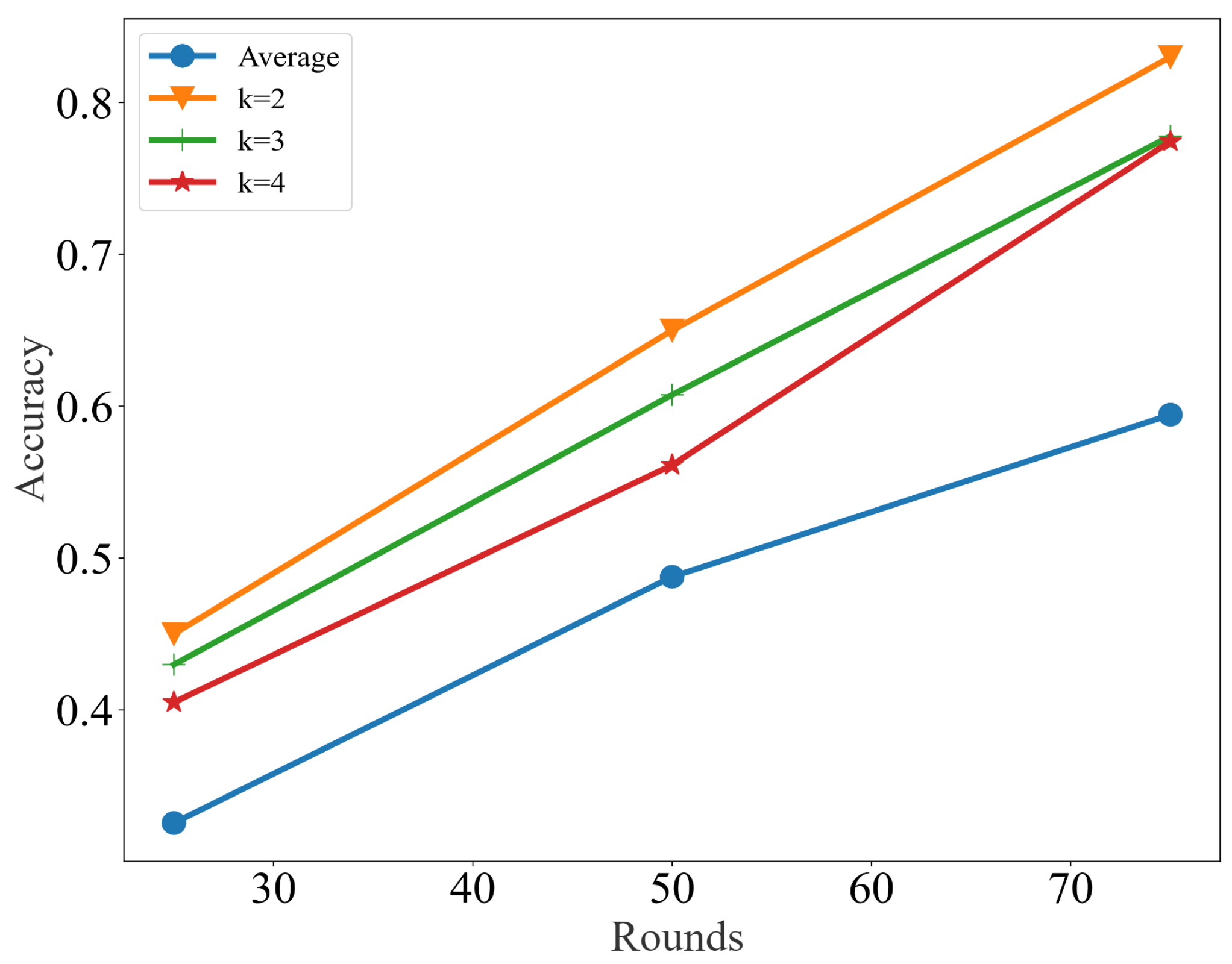
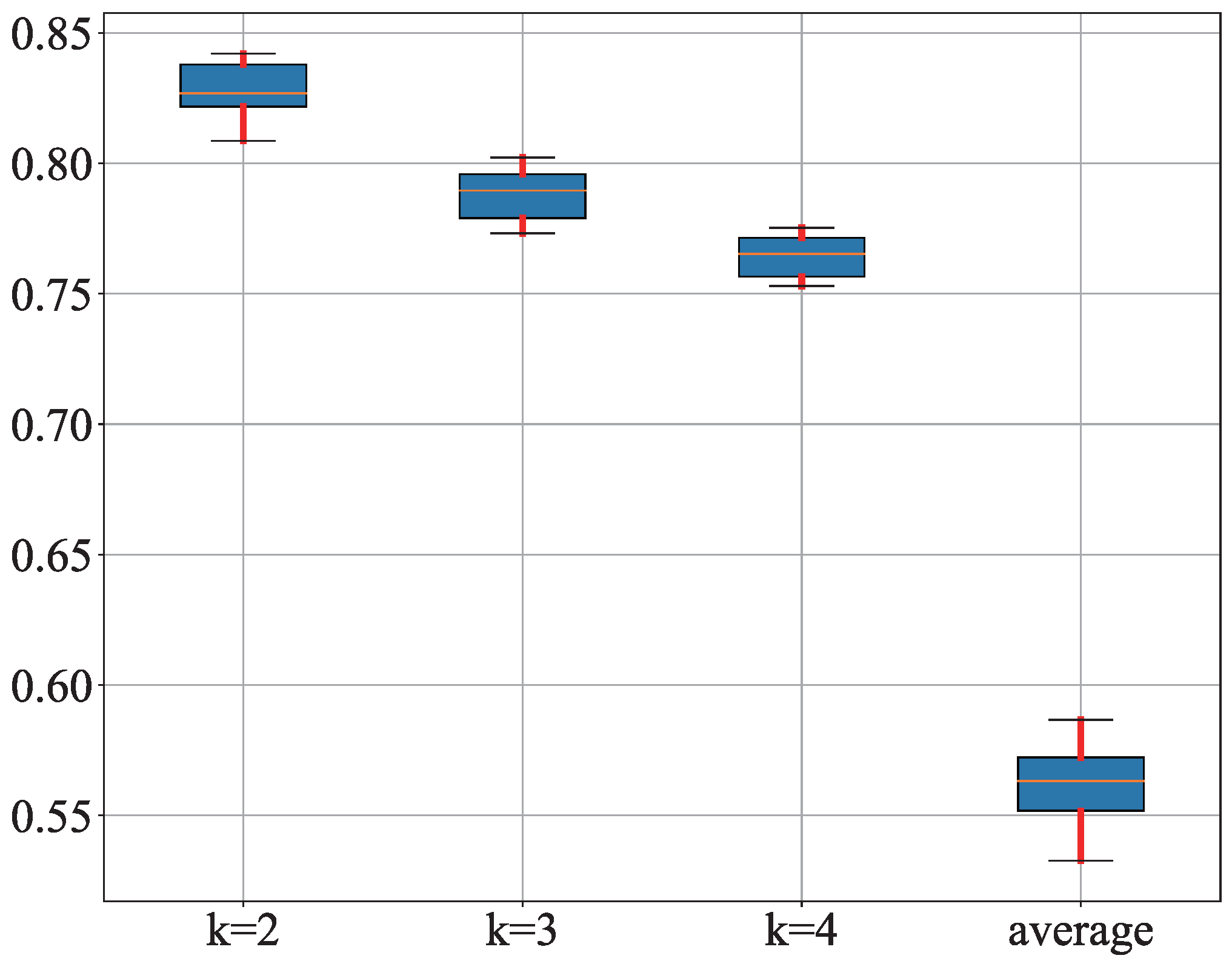
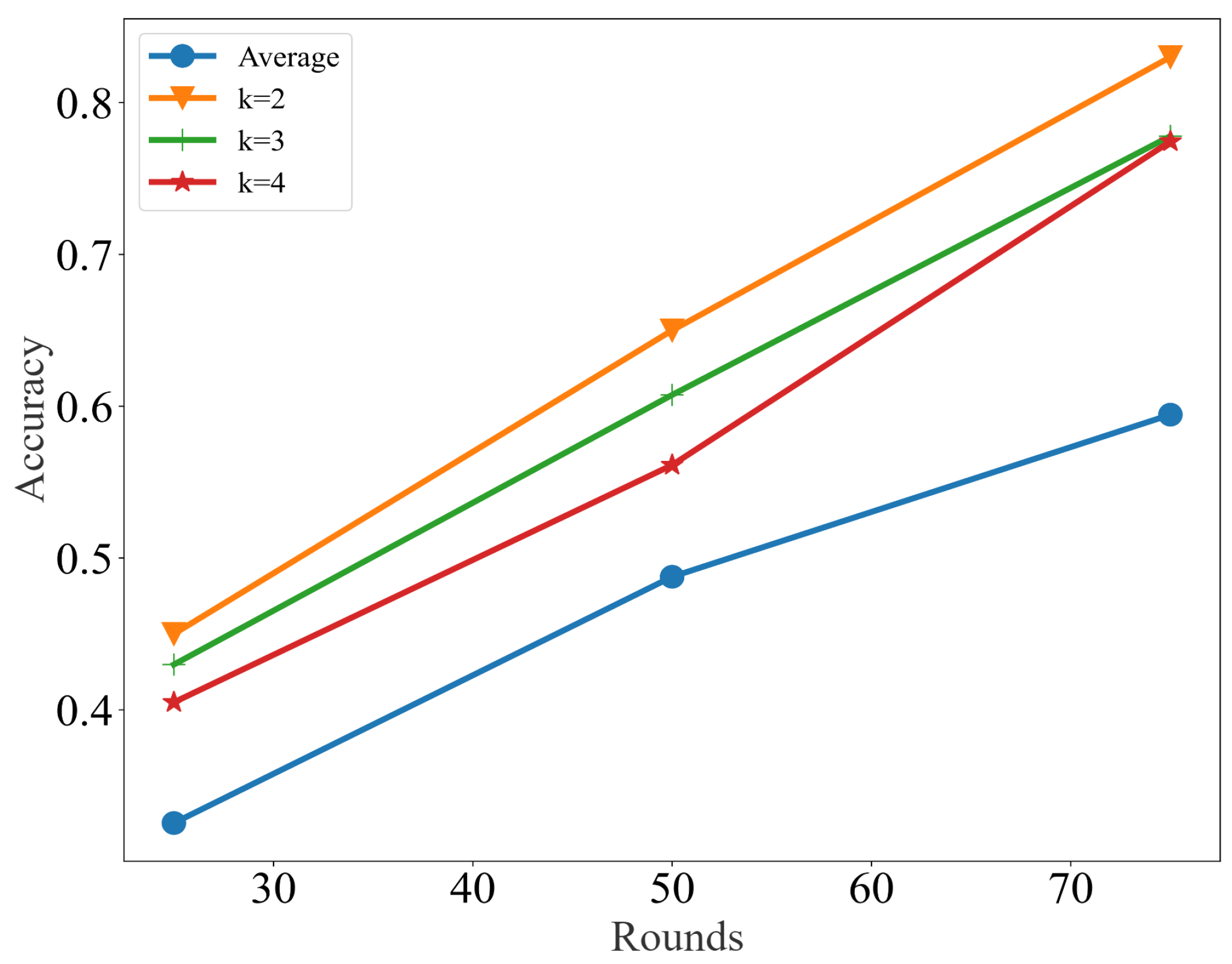
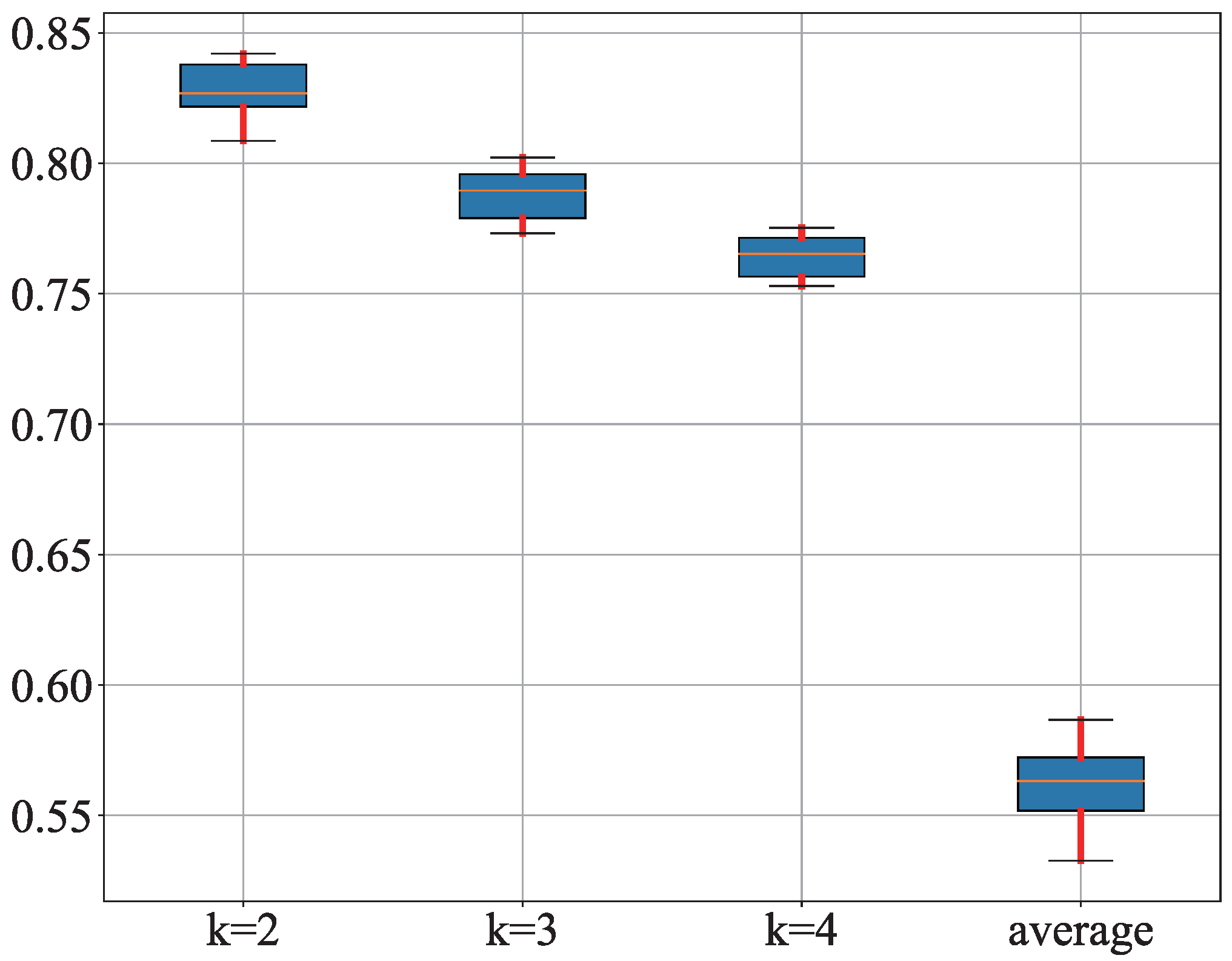
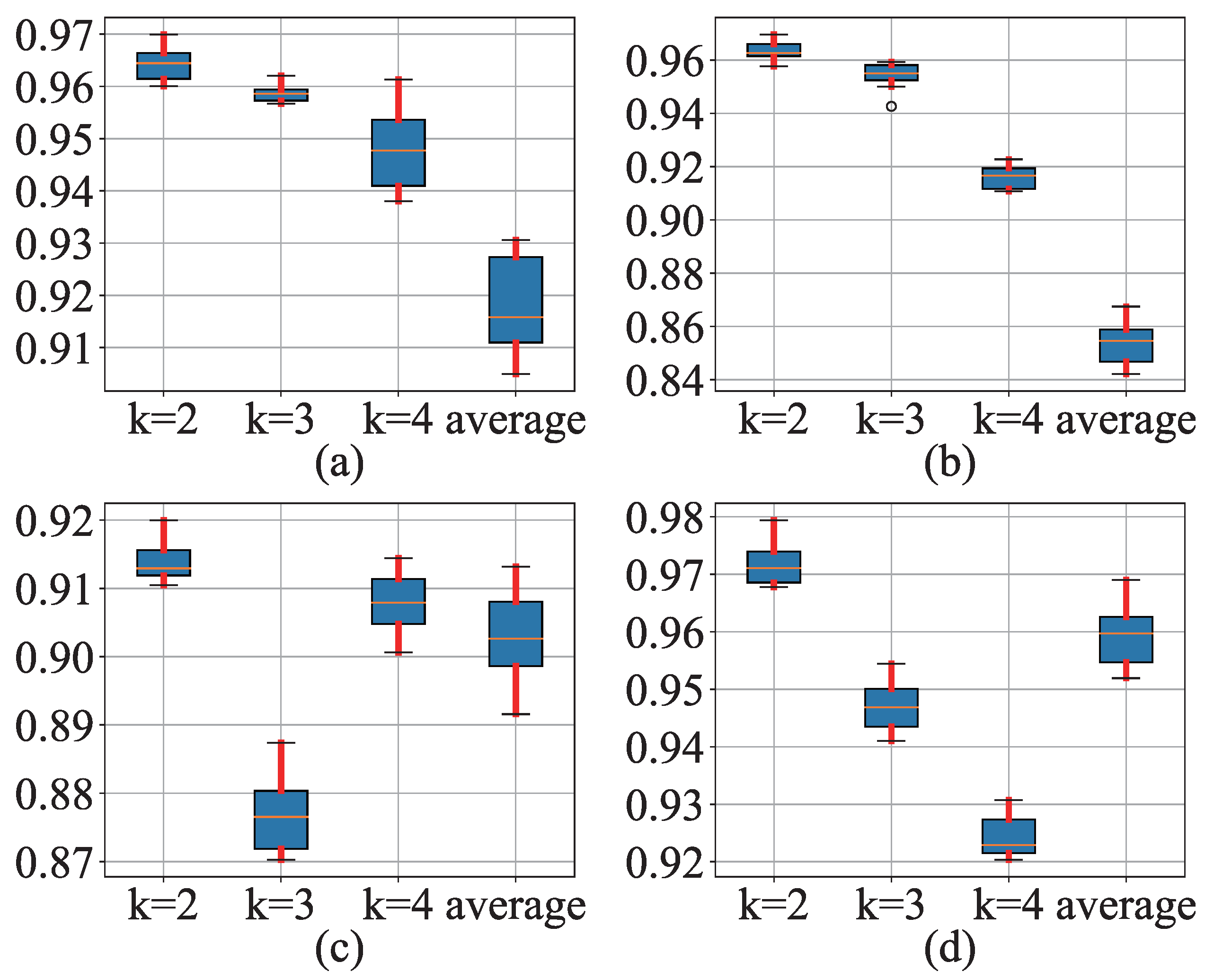
3.1. Results
3.2. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bissell, M.J.; Hall, H.G.; Parry, G. How does the extracellular matrix direct gene expression? J. Theor. Biol. 1982, 99, 31–68. [Google Scholar] [CrossRef]
- Barry, F.; Boynton, R.E.; Liu, B.; Murphy, J.M. Chondrogenic differentiation of mesenchymal stem cells from bone marrow: Differentiation-dependent gene expression of matrix components. Exp. Cell Res. 2001, 268, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Luo, P.; Lu, Y.; Wu, F.X. Identifying cell types from single-cell data based on similarities and dissimilarities between cells. BMC Bioinform. 2021, 22, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Grira, N.; Crucianu, M.; Boujemaa, N. Unsupervised and semi-supervised clustering: A brief survey. A Rev. Mach. Learn. Tech. Process. Multimed. Content 2004, 1, 9–16. [Google Scholar]
- Qi, R.; Wu, J.; Guo, F.; Xu, L.; Zou, Q. A spectral clustering with self-weighted multiple kernel learning method for single-cell RNA-seq data. Brief. Bioinform. 2021, 22, bbaa216. [Google Scholar] [CrossRef]
- Chi, M.; Bruzzone, L. Semisupervised classification of hyperspectral images by SVMs optimized in the primal. IEEE Trans. Geosci. Remote Sens. 2007, 45, 1870–1880. [Google Scholar] [CrossRef]
- Odena, A. Semi-supervised learning with generative adversarial networks. arXiv 2016, arXiv:1606.01583. [Google Scholar]
- Rosenberg, C.; Hebert, M.; Schneiderman, H. Semi-supervised self-training of object detection models. In Proceedings of the 2005 Seventh IEEE Workshops on Applications of Computer Vision (WACV/MOTION’05), Breckenridge, CO, USA, 5–7 January 2005. [Google Scholar]
- Blum, A.; Mitchell, T. Combining labeled and unlabeled data with co-training. In Proceedings of the Eleventh Annual Conference on Computational Learning Theory, Madison, WI, USA, 24–26 July 1998; pp. 92–100. [Google Scholar]
- Tian, F.; Gao, B.; Cui, Q.; Chen, E.; Liu, T.Y. Learning deep representations for graph clustering. In Proceedings of the AAAI Conference on Artificial Intelligence, Québec, QC, Canada, 27–31 July 2014; Volume 28. [Google Scholar]
- Schaeffer, S.E. Graph clustering. Comput. Sci. Rev. 2007, 1, 27–64. [Google Scholar] [CrossRef]
- Vapnik, V. Principles of risk minimization for learning theory. Adv. Neural Inf. Process. Syst. 1991, 4, 832–838. [Google Scholar]
- Fan, W.; Peng, H.; Luo, S.; Fang, C.; Li, Y. SCEC: A novel single-cell classification method based on cell-pair ensemble learning. In Proceedings of the International Conference on Intelligent Computing, Shenzhen, China, 12–15 August 2021; Springer: Berlin, Germany, 2021; pp. 433–444. [Google Scholar]
- Valentine, G. Images of danger: Women’s sources of information about the spatial distribution of male violence. Area 1992, 24, 22–29. [Google Scholar]
- Lee, C.; Landgrebe, D.A. Decision boundary feature extraction for neural networks. IEEE Trans. Neural Netw. 1997, 8, 75–83. [Google Scholar]
- Wu, H.; Prasad, S. Semi-supervised deep learning using pseudo labels for hyperspectral image classification. IEEE Trans. Image Process. 2017, 27, 1259–1270. [Google Scholar] [CrossRef]
- Wang, W.; Tan, H.; Sun, M.; Han, Y.; Chen, W.; Qiu, S.; Zheng, K.; Wei, G.; Ni, T. Independent component analysis based gene co-expression network inference (ICAnet) to decipher functional modules for better single-cell clustering and batch integration. Nucleic Acids Res. 2021, 49, e54. [Google Scholar] [CrossRef]
- Son, N.H. From optimal hyperplanes to optimal decision trees. Fundam. Inform. 1998, 34, 145–174. [Google Scholar] [CrossRef]
- Yao, X. Evolving artificial neural networks. Proc. IEEE 1999, 87, 1423–1447. [Google Scholar]
- Gregory, S. Finding overlapping communities in networks by label propagation. New J. Phys. 2010, 12, 103018. [Google Scholar] [CrossRef]
- Cui, W.; Zhou, H.; Qu, H.; Wong, P.C.; Li, X. Geometry-based edge clustering for graph visualization. IEEE Trans. Vis. Comput. Graph. 2008, 14, 1277–1284. [Google Scholar] [CrossRef]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Li, Q.; Han, Z.; Wu, X.M. Deeper insights into graph convolutional networks for semi-supervised learning. In Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018. [Google Scholar]
- Yan, S.; Xiong, Y.; Lin, D. Spatial temporal graph convolutional networks for skeleton-based action recognition. In Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018. [Google Scholar]
- Peng, H.; Fan, W.; Fang, C.; Gao, W.; Li, Y. SCMAG: A Semisupervised Single-Cell Clustering Method Based on Matrix Aggregation Graph Convolutional Neural Network. Comput. Math. Methods Med. 2021, 2021, 6842752. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Chu, L.F.; Leng, N.; Zhang, J.; Hou, Z.; Mamott, D.; Vereide, D.T.; Choi, J.; Kendziorski, C.; Stewart, R.; Thomson, J.A. Single-cell RNA-seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm. Genome Biol. 2016, 17, 1–20. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef]
- Leng, N.; Chu, L.F.; Barry, C.; Li, Y.; Choi, J.; Li, X.; Jiang, P.; Stewart, R.M.; Thomson, J.A.; Kendziorski, C. Oscope identifies oscillatory genes in unsynchronized single-cell RNA-seq experiments. Nat. Methods 2015, 12, 947–950. [Google Scholar] [CrossRef]
- Borella, M.; Martello, G.; Risso, D.; Romualdi, C. PsiNorm: A scalable normalization for single-cell RNA-seq data. Bioinformatics 2022, 38, 164–172. [Google Scholar] [CrossRef]
- Huang, A. Similarity measures for text document clustering. In Proceedings of the Sixth New Zealand Computer Science Research Student Conference (NZCSRSC2008), Christchurch, New Zealand, 14–18 April 2008; Volume 4, pp. 9–56. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Datasets | Number of Cells | Class | Number of Genes | Data Sources | References |
|---|---|---|---|---|---|
| Chung | 563 | 13 | 57915 | GSE75688 | Chung et al. [26]. |
| Chu | 1018 | 7 | 19097 | GSE75748 | Chu et al. [27] |
| Patel | 430 | 6 | 5948 | GSE57872 | Patel et al. [28]. |
| Xin | 1600 | 8 | 39851 | GSE81608 | Xin et al. [29]. |
| Ning | 460 | 4 | 19084 | GSE64016 | Ning et al. [30]. |
| Dataset | Iteration | Label Propagation | Label Spreading | Self-Training | GCN | SCAFG |
|---|---|---|---|---|---|---|
| 25 | 36.6 | 47.5 | 36.6 | 32.3 | 46.4 | |
| Chung | 50 | 44.6 | 56.1 | 49.8 | 44.6 | 64.2 |
| 75 | 47.7 | 59.2 | 54.2 | 58.8 | 82.2 | |
| 25 | 44.1 | 53.2 | 58.8 | 64.2 | 70.1 | |
| Chu | 50 | 58.3 | 55.6 | 69.5 | 86.7 | 92.3 |
| 75 | 61.5 | 56.9 | 73.2 | 89.2 | 96.4 | |
| 25 | 50.6 | 46.2 | 49.7 | 65.1 | 76.7 | |
| Patel | 50 | 67.1 | 58.3 | 59.4 | 73.8 | 89.9 |
| 75 | 70.6 | 64.8 | 65.2 | 77.6 | 96.3 | |
| 25 | 70.2 | 64.6 | 60.1 | 80.9 | 87.4 | |
| Xin | 50 | 76.2 | 74.5 | 68.5 | 82.2 | 88.1 |
| 75 | 80.1 | 74.8 | 74.2 | 83.6 | 91.2 | |
| 25 | 51.2 | 56.7 | 46.8 | 82.6 | 91.1 | |
| Ning | 50 | 58.4 | 69.2 | 63.1 | 85.4 | 93.3 |
| 75 | 62.4 | 74.6 | 69.2 | 89.8 | 97.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, H.; Li, Y.; Zhang, W. SCAFG: Classifying Single Cell Types Based on an Adaptive Threshold Fusion Graph Convolution Network. Mathematics 2022, 10, 3407. https://doi.org/10.3390/math10183407
Peng H, Li Y, Zhang W. SCAFG: Classifying Single Cell Types Based on an Adaptive Threshold Fusion Graph Convolution Network. Mathematics. 2022; 10(18):3407. https://doi.org/10.3390/math10183407
Chicago/Turabian StylePeng, Haonan, Yuanyuan Li, and Wei Zhang. 2022. "SCAFG: Classifying Single Cell Types Based on an Adaptive Threshold Fusion Graph Convolution Network" Mathematics 10, no. 18: 3407. https://doi.org/10.3390/math10183407
APA StylePeng, H., Li, Y., & Zhang, W. (2022). SCAFG: Classifying Single Cell Types Based on an Adaptive Threshold Fusion Graph Convolution Network. Mathematics, 10(18), 3407. https://doi.org/10.3390/math10183407