1. Introduction
A large number of molecules are contained in human body fluids, and these molecules are promising as biomarkers for disease diagnosis and therapeutic monitoring [
1,
2,
3]. Among these molecules, one of the essential types of biomarkers is the secreted proteins. Because of this, discovering secreted protein is an important step toward secreted protein biomarker identification. In recent years, although many secreted proteins have been identified through experiments, it remains a challenge to identify new secreted proteins in some human body fluids [
4,
5]. To facilitate the detection of secreted proteins, several computational approaches have been proposed to predict whether a protein is secreted into a specific fluid [
6,
7,
8,
9,
10]. These efforts can accelerate the detection of secreted proteins and avoid many unnecessary wet experiments. Nowadays, secreted protein discovery by computational methods has become a well-studied topic in bioinformatics.
Among these approaches, the most successful method uses a support vector machine (SVM) and protein features [
6,
7]. First, the features of each protein are computed based on their sequence by using some computational tools and websites [
11,
12]. Second, a feature selection method is used to choose some representative features from those features. Finally, the SVM classifier is used to differentiate the secreted proteins from not secreted proteins based on features selected previously. Although this approach is fast and effective, its weak representative ability limits the performance of secreted protein discovery. Another effective approach is using deep learning and protein sequences [
5]. Compared with the previous SVM-based approach, this approach can usually learn more complex features from protein sequences using a convolutional neural network (CNN), long short-term memory (LSTM), etc. [
13]. These complex features enhance the representative ability of this approach and promote higher performance. However, deep learning always requires a large amount of data. Due to the limited number of secreted proteins in some human body fluids, the performance in many fluids may suffer from overfitting. Furthermore, for some human body fluids, such as sweat, the number of secreted proteins is too small to learn representative features for prediction. Therefore, an effective approach urgently needs to be presented to obtain a more accurate prediction and enable computational detection in some human body fluids.
These available approaches ignore relationships between different human body fluids. Typically, a protein can be secreted into several human body fluids, which may be related. Therefore, predictions of computational methods in different human body fluids may also be related. When designing a computational approach to secreted protein discovery, relationships between different fluids need to be considered. Multi-task learning is a machine learning method that exploits relationships between tasks to improve the performances of all tasks [
14,
15]. Thus, we could use the multi-task learning method to take the relationships between human body fluids into account. Prediction of whether a protein is secreted into a specific fluid is regarded as a task. In addition, a shared network for different tasks is beneficial in preventing overfitting [
16,
17,
18]. However, several problems occurred in employing multi-task learning in multi-fluid secreted protein discovery. First of all, many of these human fluids have a really poor number of secreted proteins. As a result, positive samples may be less than negative samples. Even predicting a secreted protein to a specific fluid, the imbalanced dataset is still a problem and needs to be solved [
5]. All the datasets for different human body fluids need to be considered simultaneously. In addition, the performance of these human body fluids may conflict with each other [
19]. Performance of some fluids might get hurt if they were not coordinated well. To obtain decent performance in all human body fluids, all these problems must be solved.
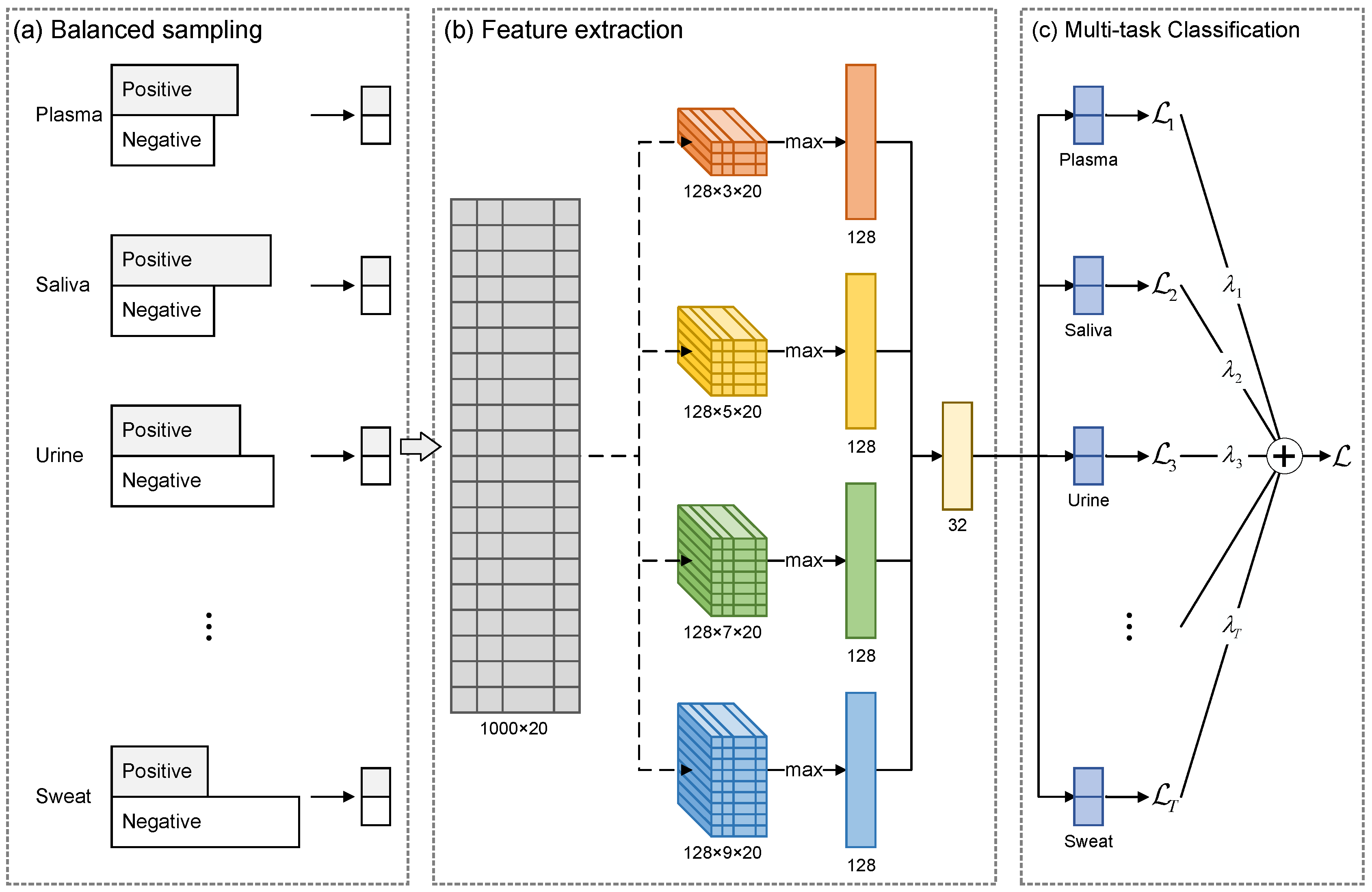
In this paper, we propose MultiSec, a novel approach that takes advantage of progress in multi-task learning and improves the state-of-the-art performance in secreted protein discovery. MultiSec was designed to simultaneously predict the probability that a protein is secreted into each of 17 human body fluids based on its sequence. This approach is composed of three modules: a balanced sampling module that generates balanced samples for each human body fluid during training, a lightweight convolutional neural network architecture to extract deep features for proteins, and a multi-task classification module that calculates the probabilities for protein to be secreted proteins in 17 human body fluids. Finally, we trained MultiSec on 17 human body fluids, including plasma, saliva, urine, cerebrospinal fluid (CSF), seminal fluid, amniotic fluid, tear fluid, bronchoalveolar lavage fluid (BALF), milk, synovial fluid, nipple aspirate fluid (NAF), cervical–vaginal discharge (CVF), pleural effusion (PE), sputum, exhaled breath condensate (EBC), pancreatic juice (PJ), and sweat. MultiSec achieved a more accurate prediction in all human body fluids with area under the ROC curves of 0.89–0.98. Comparison benchmarks on the independent testing datasets demonstrate that our approach outperforms other state-of-the-art approaches in all the compared human body fluids.
4. Disscussion
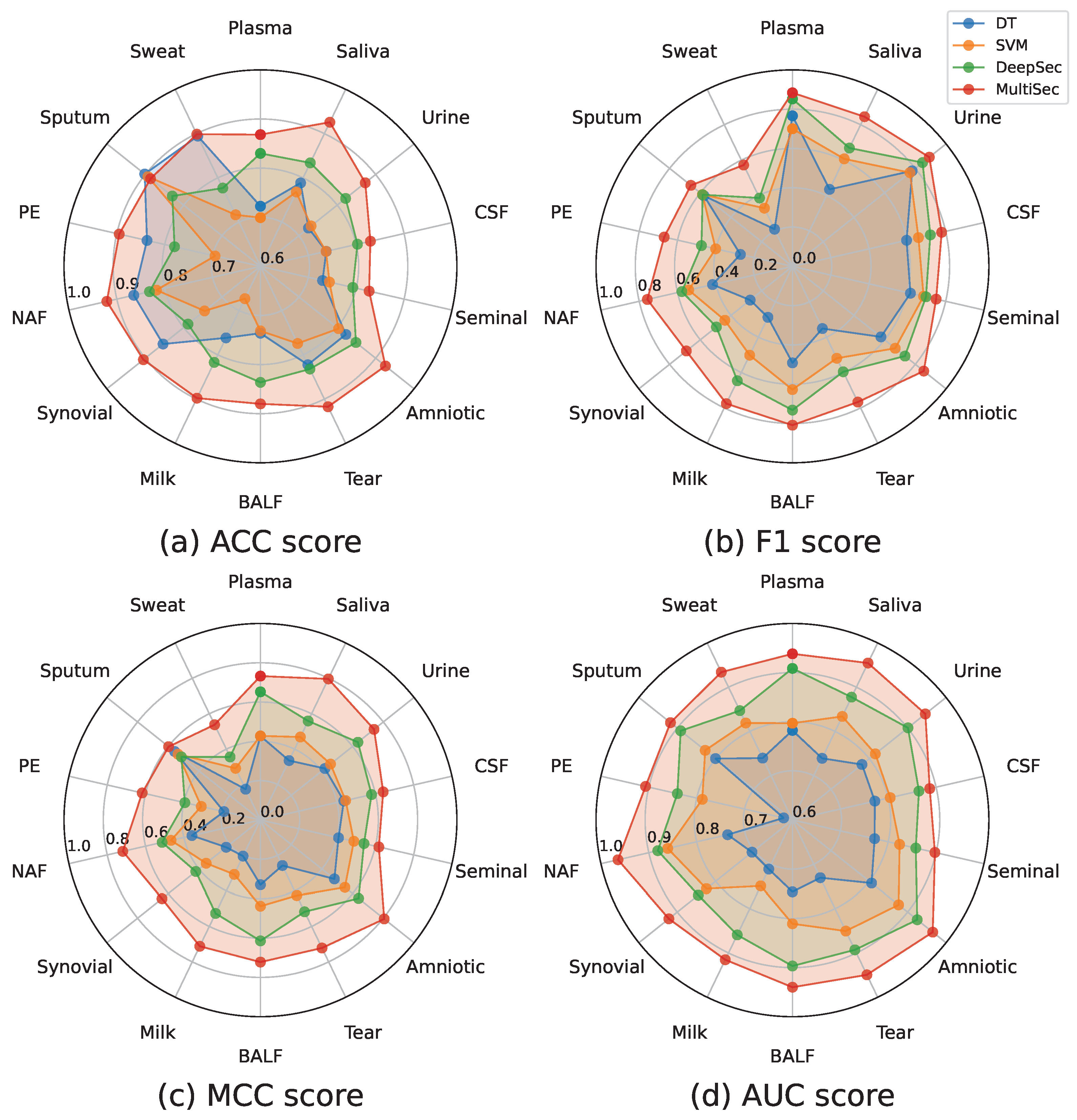
Comparison benchmarks presented in the previous section demonstrate that exploiting the relationships between different human body fluids can improve the discovery of secreted proteins. Furthermore,
Table A5 has shown that our method outperforms DeepSec by 6.08–23.75% on the MCC metric. All these comparisons indicate that MultiSec is the current best performing method and is superior to other state-of-the-art methods in secreted protein discovery.
To discover secreted protein in these 14 human body fluids, DeepSec needs to train 56 networks (1, 3, 1, 2, 2, 3, 5, 2, 3, 6, 6, 6, 6, and 10 for plasma, saliva, urine, cerebrospinal fluid, seminal fluid, amniotic fluid, tear fluid, bronchoalveolar lavage fluid, milk, synovial fluid, nipple aspirate fluid, pleural effusion, sputum, and sweat). Therefore, DeepSec always costs many computational resources. However, our novel approach can discover secreted proteins in all 17 human body fluids by using only a single network. Furthermore, our approach also performs better than DeepSec in all 14 human body fluids. From the comparison with DeepSec, we can conclude that MultiSec improves performance in all the human body fluids and significantly reduces the number of networks and training time.
5. Conclusions
In summary, we present a novel approach MultiSec to predict whether a protein is secreted into each of 17 human body fluids based on its sequence. The new approach was designed to exploit relationships between different human body fluids via multi-task learning. Compared with other state-of-the-art approaches, the benchmarks show that MultiSec outperforms other approaches in all compared human body fluids. Furthermore, compared with DeepSec, our approach decreases many networks into only one to discover secreted protein in 17 human body fluids. Our improvements also confirm the relationships between different human body fluids exist and help to discover secreted proteins.
Afterward, MultiSec was used to identify potentially secreted proteins. With this approach, 1244–6742 potentially secreted proteins have been discovered from 17 human body fluids. Furthermore, 103 proteins are predicted to be secreted into all these fluids simultaneously. Among these proteins, 20 are reported with a relatively high probability for protein to be secreted into 17 human body fluids simultaneously. We believe these identified proteins are worthwhile for further study with biological experiments.
In the future, we would consider fusing more features, such as protein features and secondary structures, into our approach. In addition, due to the limited number of secreted proteins, computational approaches in secreted protein discovery are very easy to overfit. Therefore, a more effective network architecture is also worthwhile to discover. Furthermore, other protein prediction tasks may also be related to secreted protein discovery, such as single peptide identification, protein localization, etc. [
24,
29,
30]. We will also find more tasks to improve secreted protein discovery.




{kind=link}
{kind=link}