
A Model for the Proliferation–Quiescence Transition in Human Cells

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
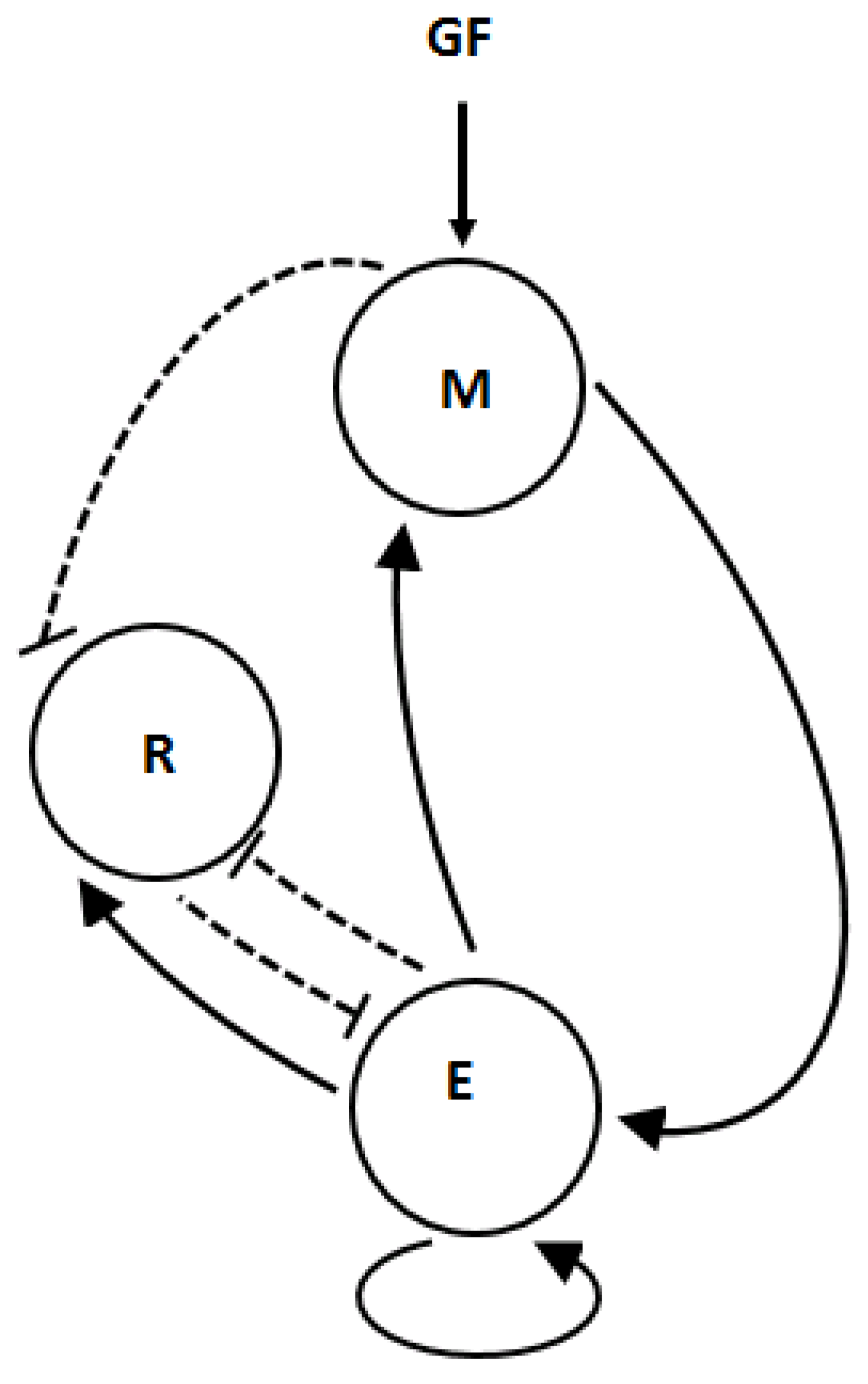
2.1. Proliferation–Quiescence Dynamics Model
- 1
- The production of is through mass action by extra-cellular growth signals and by , which is modelled using Michaelis–Menten kinetics, whereas its decay is modelled by mass action. Yao et al. [2] considered Michaelis–Menten kinetics for the production of through extra-cellular growth signals.
- 2
- The activation of is enhanced by and its inhibition is intensified by and , which are both modelled using mass action. It is pertinent to note that, in this study, we assume conservation of mass for the family of proteins, which was not considered in [2]. In addition, we assume self-activation and inhibition of using the Hill function with . On the contrary, Yao et. al. [2] modelled the production and depletion of using Michaelis–Menten kinetics and mass action, respectively, and self-activation and de-activation were not considered.
- 3
- is synthesised, cf. [40], with the use of Michaelis–Menten kinetics and its synthesis is enhanced by with the use of a Hill function with , while its decay is enhanced by with the use of mass action. On the contrary, the authors in [2] did not consider constitutive synthesis of which has been observed experimentally as indicated in [40].
2.2. Mathematical Analysis of the Model
2.3. Non-Dimensionalisation
2.4. Positivity, Boundedness and Existence and Uniqueness of Solutions
2.5. Steady States
2.6. Linear Stability Analysis
- (A).
- , and
- (B).
- and
- (C).
- and , then (M*, E*, R*) is asymptotically stable.
3. Results
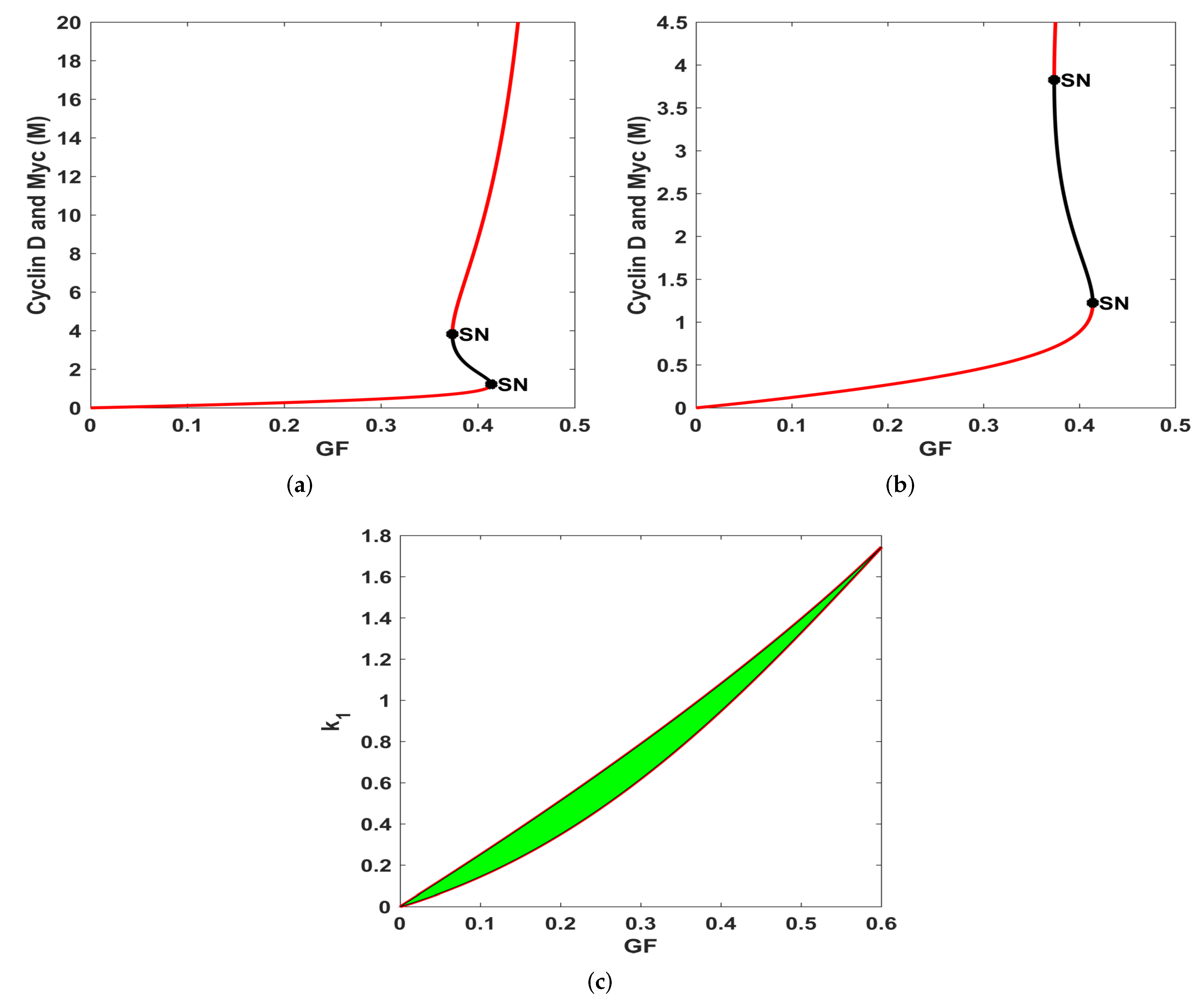
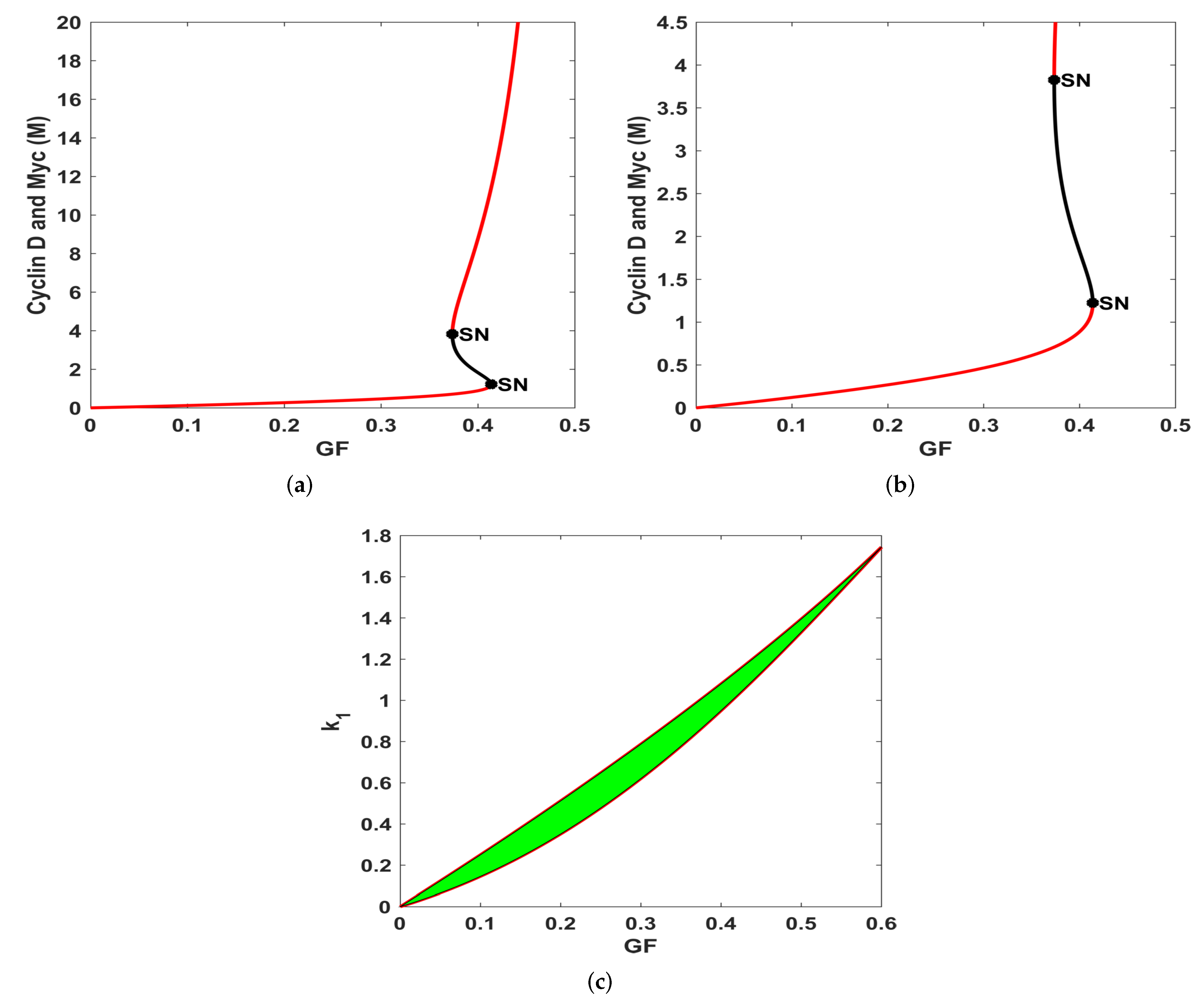
3.1. Bifurcation Analysis
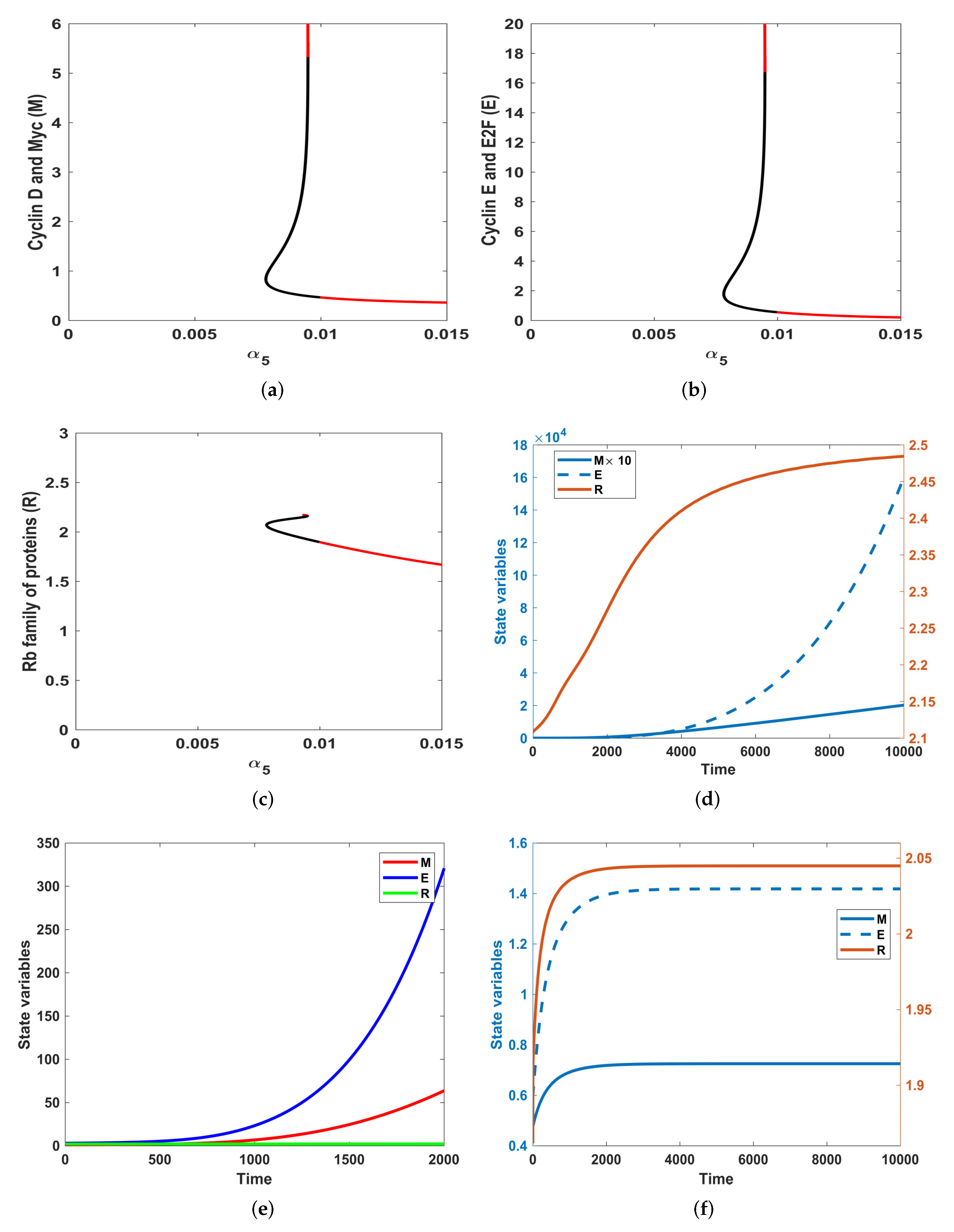
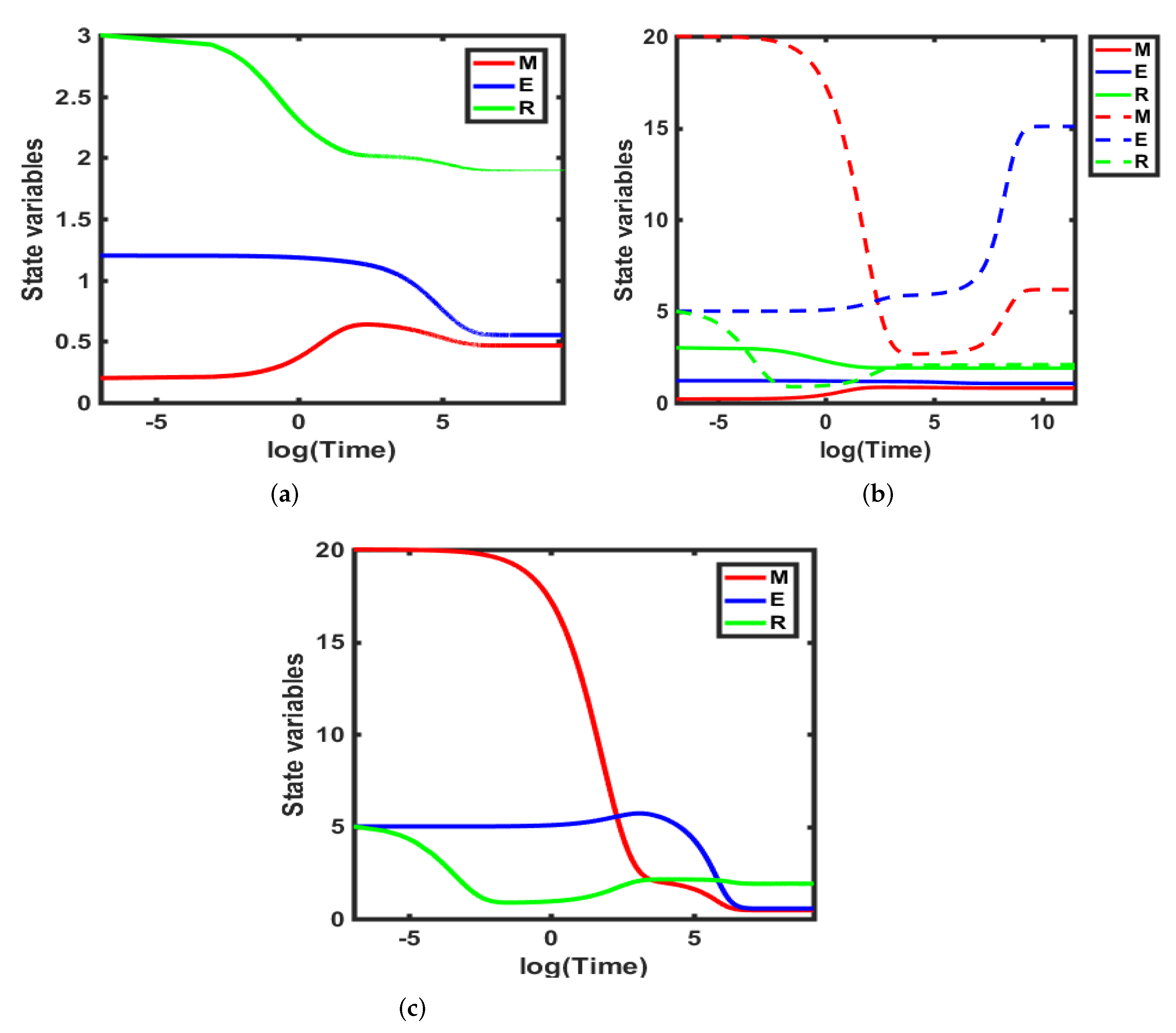
3.2. Numerical Simulations
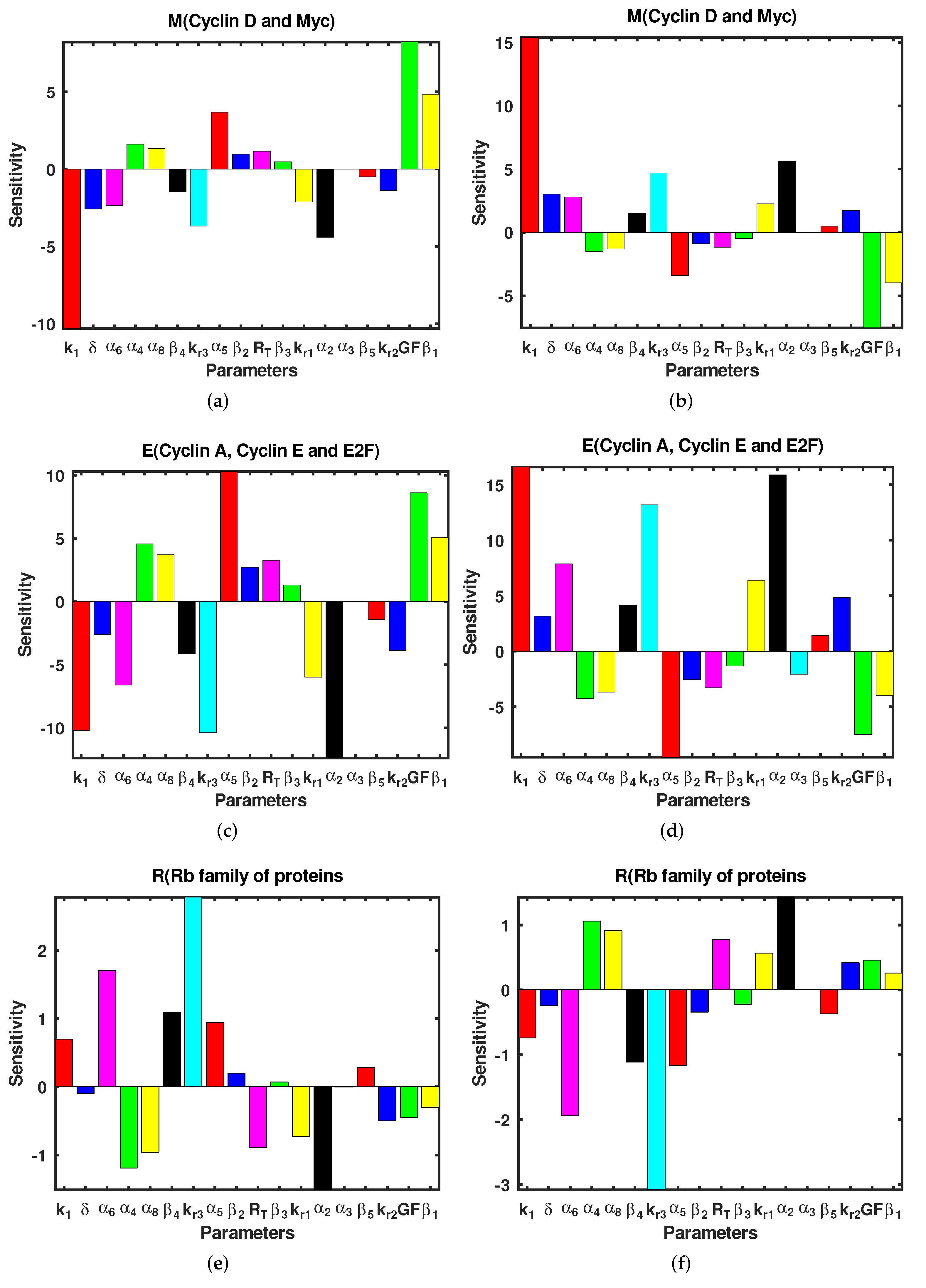
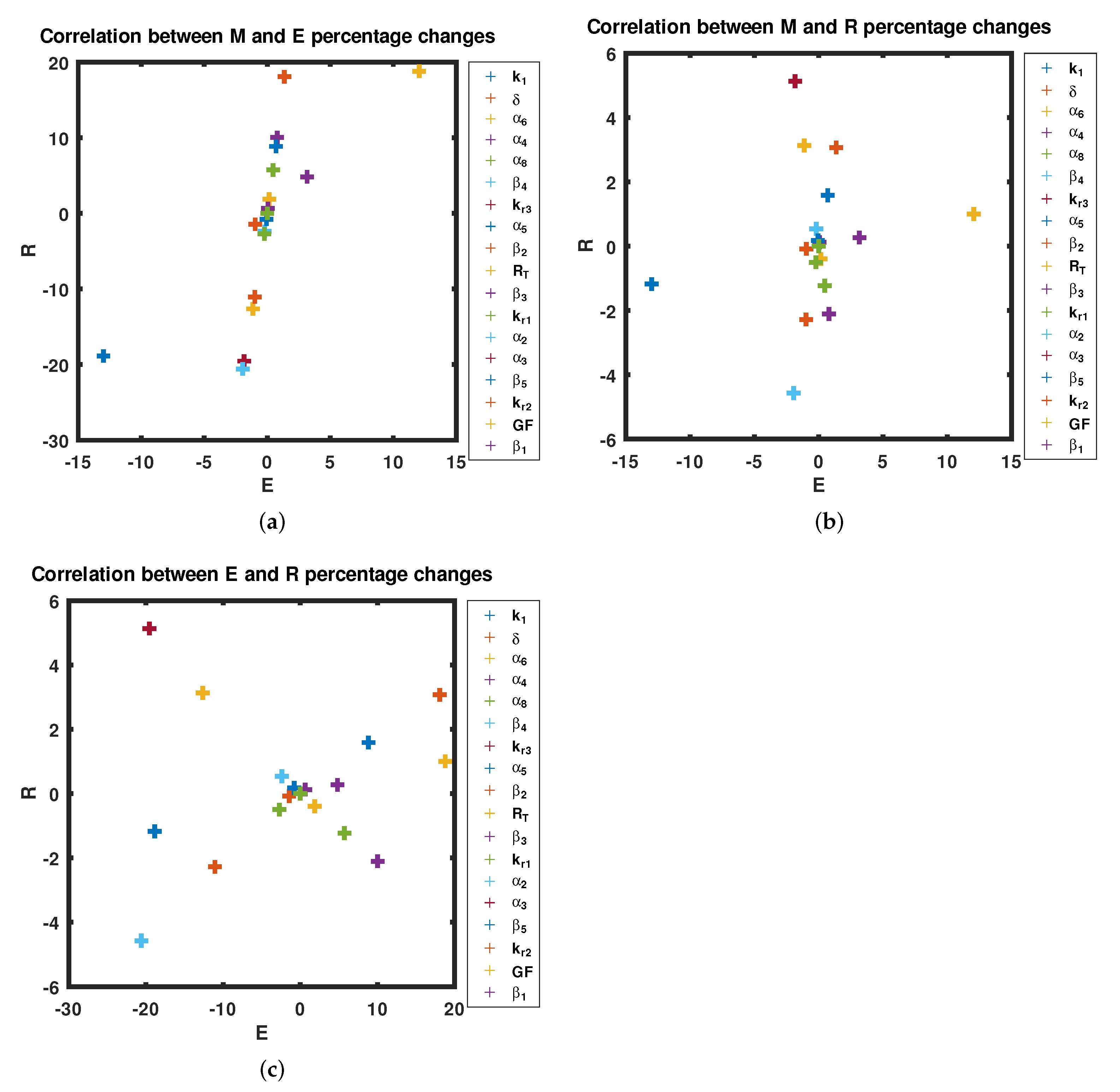
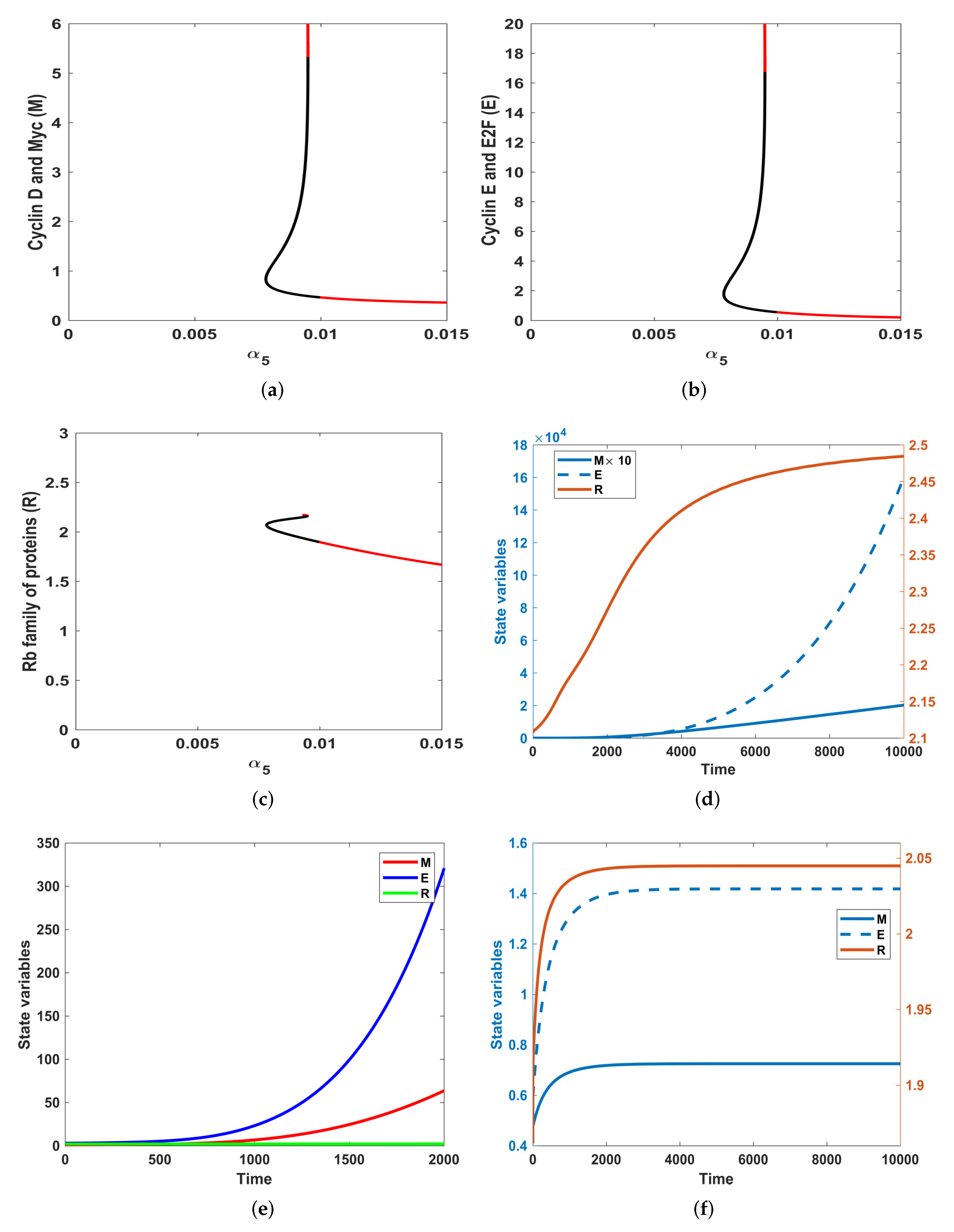
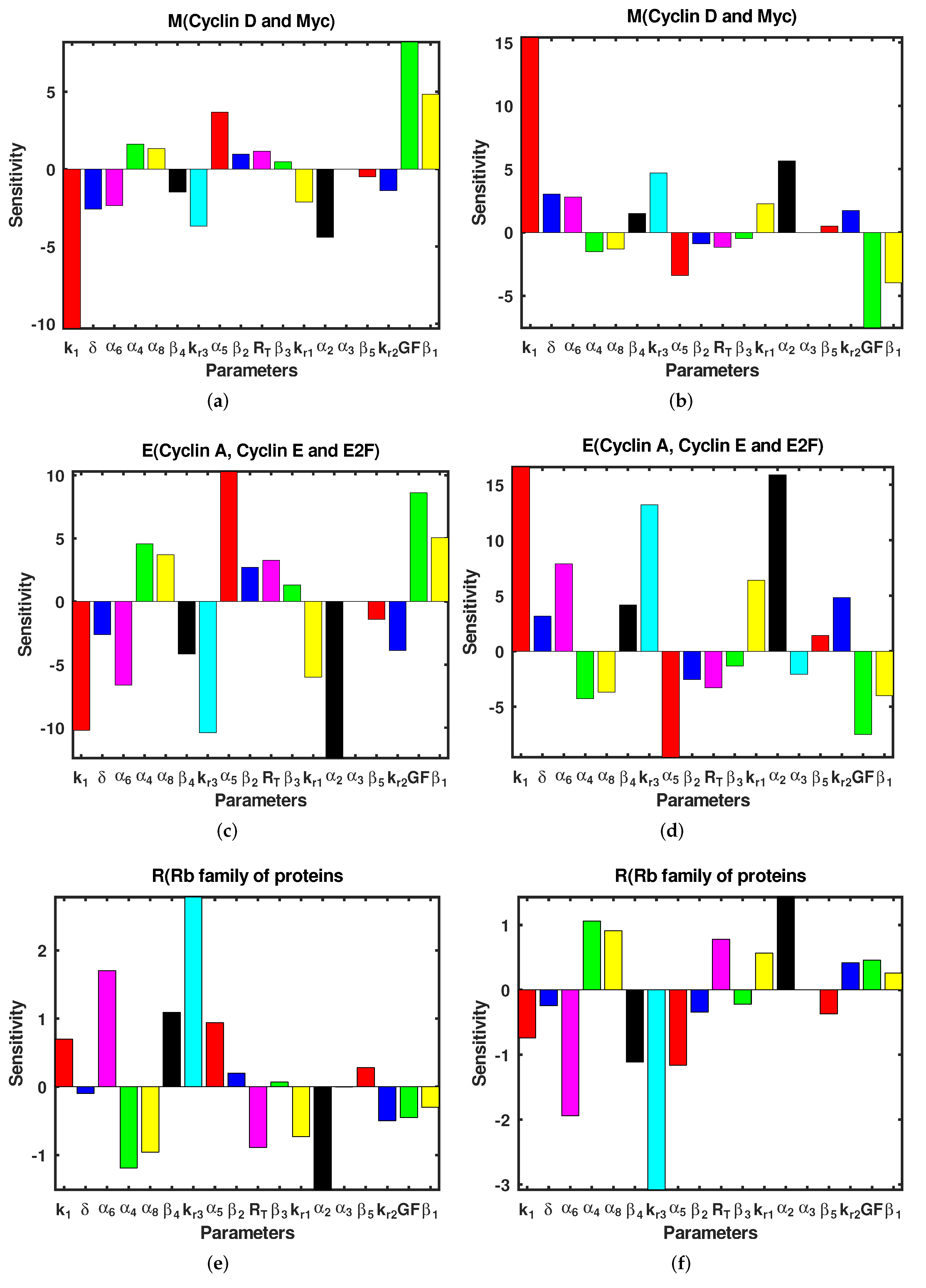
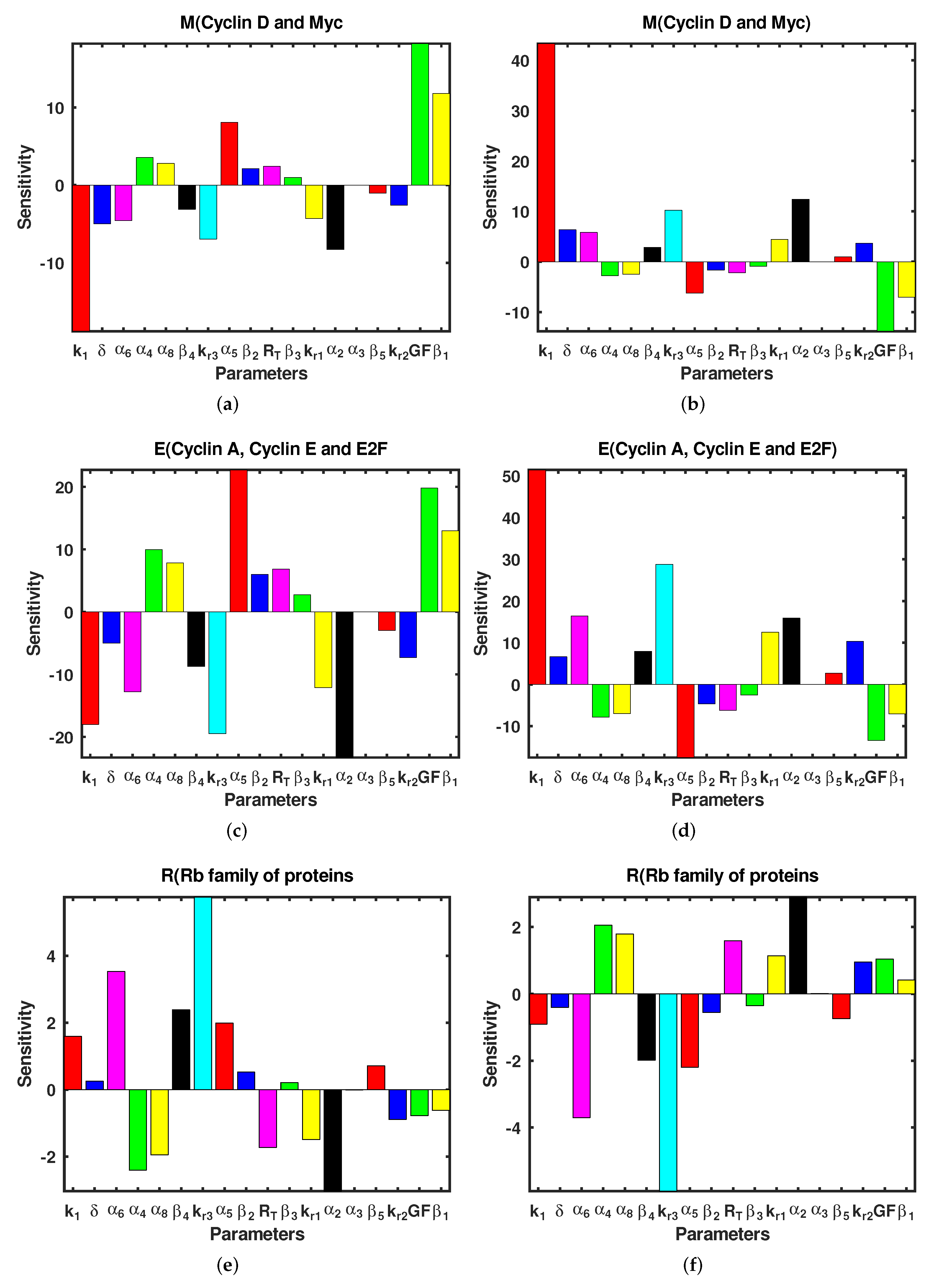
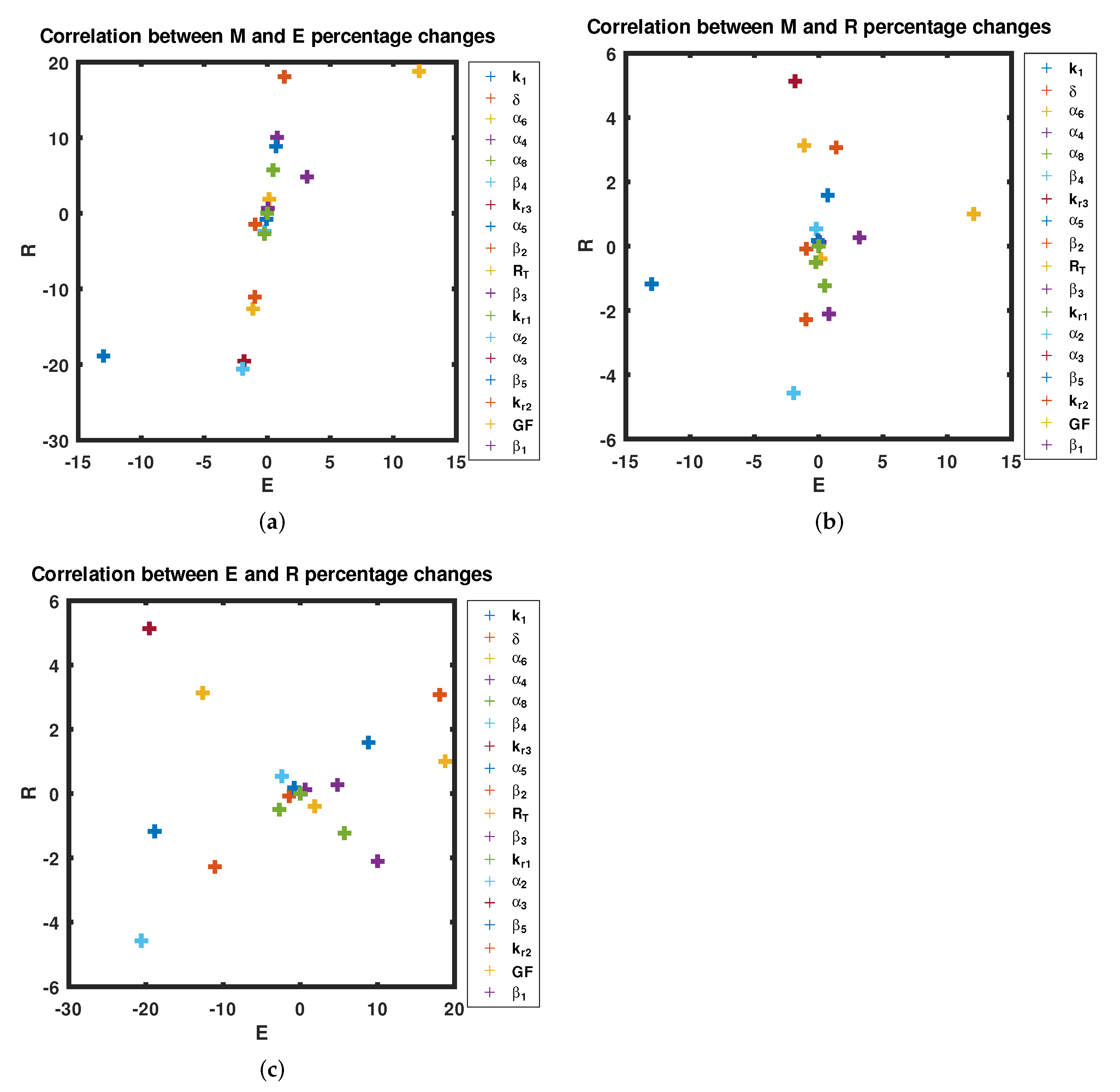
3.3. Sensitivity Analysis
4. Discussion
- Based on the concept of first principles, we investigated the dynamical potential of growth factors in the regulation of the cell-cycle entry.
- A mathematical model for the simplified network was constructed based on the model proposed in Yao et al. [2]. While previous studies modelled all links using Michaelis–Menten functions only [2], we used mass action, and Michaelis–Menten and Hill functions, resulting in a simpler model. In addition, we considered the R species to exist either in hyper-phosphorylated or hypo-phosphorylated form and that their total concentration is conserved.
- By varying the growth factor signal values through bifurcation analysis, numerical simulations illustrated that the magnitude of the value of the growth factor plays a critical role in regulating cell-cycle entry. Through bifurcation analysis, we deduced the existence of three consecutive dynamical behaviours, namely, stability, bi-stability and stability.
- Numerical simulations performed with different growth factor values validated the results derived from the bifurcation analysis. In particular, the biological interpretation of the uniform steady state can be established as follows:
- For , System (1) is asymptotically stable, indicating the regime in which cells are in a quiescent state. In this state, cells feature low levels of Cyclin D, Myc and high levels of R species.
- On the other hand, in the range , System (1) exhibits bi-stability, marking the position of the restriction point, as deduced in Yao et al. [42]. This point sets a high threshold separating quiescence from proliferation and acts as a barrier against unregulated and accidental cell growth. In addition, it provides a low-maintenance mechanism ensuring that the cell cycle proceeds, albeit later due to changes in the extracellular environment which is crucial for maintaining genome integrity.
- For values of , the system generates a stable dynamical behaviour where a cell is in the proliferation mode marking the higher steady state value. This state features high levels of Cyclin D, Myc and low levels of the family of proteins.
However, it remains to investigate the conditions under which the system exhibits excitable and oscillatory dynamics as observed in a different model proposed in [62], but that would be investigated in a subsequent work. While Yao et al. [42] identified a basic gene circuit underlying resettable bi-stable switch controlling cell-cycle entry, we obtained a range of values of the growth factor concentration for the three dynamical regimes.
5. Limitations
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Fujimaki, K.; Mitchell, G.C.; Kwon, J.S.; Croce, K.D.; Langsdorf, C.; Zhang, H.H.; Yao, G. Exit from quiescence displays a memory of cell growth and division. Nat. Commun. 2017, 8, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, G. Modelling mammalian cellular quiescence. Interface Focus 2014, 4, 20130074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harashima, H.; Dissmeyer, N.; Schnittger, A. Cell cycle control across the eukaryotic kingdom. Trends Cell Biol. 2013, 23, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.K.; Munger, K.; Adler, F.R. A mathematical model of cell cycle dysregulation due to human papilloma virus infection. Bull. Math. Biol. 2017, 79, 1564–1585. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef]
- Naetar, N.; Soundarapian, V.; Litovchick, L.; Goguen, K.L.; Sablina, A.A.; Bowman-Colin, C.; Sicinski, P.; Hahn, W.C.; DeCaprio, J.A.; Livingstone, D.M. PP2A-mediated regulation of Ras signaling in G2 is essential for stable quiescence and normal G1 length. Mol. Cell 2014, 54, 932–945. [Google Scholar] [CrossRef] [Green Version]
- Nasmyth, K. Viewpoint: Putting the cell cycle in order. Science 1996, 274, 1643–1645. [Google Scholar] [CrossRef]
- Pardee, A.B. A restriction point for control of normal cell proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Weiss, J.N.; Maclellan, R. Regulation of mammalian cell cycle: A model of the G1-to-S transition. J. Physiol. Cell Physiol. 2003, 284, C349–C364. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R. The Biology of Cancer. In Garland Science; WW Norton & Company: New York, NY, USA, 2013; ISBN 978-0815342205. [Google Scholar]
- Zetterberg, A.; Larsson, O. Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc. Natl. Acad. Sci. USA 1985, 82, 5365–5369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, N.; Vinod, P.K. Mathematical modeling of reversible transition between quiescence and proliferation. PLoS ONE 2018, 13, e0198420. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevins, J.R. The Rb/E2F and cancer. Hum. Mol. Genet 2001, 10, 699–703. [Google Scholar] [CrossRef]
- Nevins, J.R. E2F: A link between the Rb tumor suppressor protein and viral oncoproteins. Science 1992, 258, 424–429. [Google Scholar] [CrossRef]
- Yao, G.; Lee, T.J.; Mori, S.; Nevins, J.R.; You, L. A bistable Rb-E2F switch underlies the restriction point. Nat. Cell Biol. 2008, 10, 476–482. [Google Scholar] [CrossRef]
- Burns, F.J.; Tannock, I.F. On existence of a G0-phase in cell cycle. Cell Tissue Kinet. 1970, 3, 321. [Google Scholar]
- De Maertelaer, V.; Galand, P. Some properties of a “G0” -model of the cell cycle. II. Natural constraints on the theoretical model in exponential growth conditions. Cell Tissue Kinet. 1975, 8, 11–22. [Google Scholar] [CrossRef]
- Shields, R.; Smith, J.A. Cells regulate their proliferation through alterations in transition probability. J. Cell. Physiol. 1977, 91, 345–355. [Google Scholar] [CrossRef]
- Smith, J.A.; Martin, L. Do cells cycle? Proc. Natl Acad. Sci. USA 1973, 70, 1263–1267. [Google Scholar] [CrossRef] [Green Version]
- Castor, L.N. A G1 rate model accounts for cell cycle kinetics attributed to transition probability. Nature 1980, 287, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.L. Does the variability of cell cycle result from one or many chance events? Nature 1980, 286, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Drtja, T.P. A human DNA segment with properties of the gene that predisposes to retinobalstoma and osteosarcoma. Nature 2004, 323, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.J.; Chen, K.C.; Novak, B. Sniffers, buzzers, toggles and blinkers: Dynamics of regulatory and signaling pathways in the cell. Curr. Opin. Cell. Biol. 2003, 15, 221–231. [Google Scholar] [CrossRef]
- Aguda, B.D.; Tang, Y. The kinetic origins of the restriction point in the mammalian cell cycle. Cell Prolif. 1999, 32, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.S.; Dolnik, M.; Collins, J.J. A theory for controlling cell cycle dynamics using a reversibility binding inhibitor. Proc. Natl. Acad. Sci. USA 1998, 95, 14190–14195. [Google Scholar] [CrossRef] [Green Version]
- Goldbeter, A. A minimal cascade model for the mitotic oscillator involving cyclin and cdc kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 9107–9111. [Google Scholar] [CrossRef] [Green Version]
- Kohn, K.W. Functional capabilities of molecular network components controlling the mammalian G1/S cell cycle phase transition. Oncogene 1998, 16, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Norel, R.; Agur, Z. A model for the adjustment of the mitotic clock by cyclin and MPF levels. Science 1991, 251, 1076–1078. [Google Scholar] [CrossRef]
- Nurse, P. The incredible life and times of biological cells. Science 2000, 289, 1711–1716. [Google Scholar] [CrossRef] [Green Version]
- Obeyesekere, M.N.; Knudsen, E.S.; Wang, J.Y.J.; Zimmernan, S.O. A mathematical model of the regulation of the G1 phase of Rb+/+ and Rb−/− mouse embryonic fibroblasts and an osteosarcoma cell line. Cell Prolif. 1997, 30, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Thron, C.D. Bistable biochemical switching and the control of the events of cell cycle. Oncogene 1997, 15, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyson, J.J.; Novak, B. Regulation of the eukaryotic cell: Molecular antagonism hysteresis, and irreversible transitions. J. Theor. Biol. 2001, 210, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Tyson, J.J.; Novak, B. Checkpoints in the cell cycle from modeler’s perspective. Prog. Cell Cycle Res. 1995, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Novak, B.; Tyson, J.J. Modeling the control of DNA replication in fission yeast. Proc. Natl. Acad. Sci. USA 1997, 94, 9147–9152. [Google Scholar] [CrossRef] [Green Version]
- Thron, C.D. A model for a bistable biochemical trigger of mitosis. Biophys. Chem. 1996, 57, 239–251. [Google Scholar] [CrossRef]
- Aguda, B.D. A quantitative analysis of the kinetics of the G2 DNA damage checkpoint system. Proc. Natl. Acad. Sci. USA 1999, 96, 11352–11357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frolov, M.V.; Dyson, N.J. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 2004, 117, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Heldt, F.S.; Barr, A.R.; Cooper, S.; Bakal, C.; Novak, B. A comprehensive model for the proliferation-quiescence decision in response to endogenous DNA damage in human cells. Proc. Natl. Acad. Sci. USA 2017, 115, 2532–2537. [Google Scholar] [CrossRef] [Green Version]
- Attwooll, C.; Denchi, E.L.; Helin, K. The E2F family: Specific functions and overlapping interests. EMBO J. 2004, 23, 4709–4716. [Google Scholar] [CrossRef]
- Yao, G.; Tan, C.; West, N.; Nevins, J.T.; You, L. Origin of bistability underlying mammalian cell cycle entry. Mol. Syst. Biol. 2011, 7, 485. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V.; Pardee, A.B. The restriction point of the cell cycle. Cell Cycle 2002, 1, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Gierer, A.; Meinhardt, H.A. A theory of biological pattern formation. Kybernetik 1972, 12, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamps, D.; Koch, J.; Juma, V.O.; Campillo-Funollet, E.; Graessl, M.; Banerjee, S.; Mazel, T.; Chen, X.; Wu, Y.; Portet, S.; et al. Optogenetic Tuning Reveals Rho Amplification-Dependent Dynamics of a Cell Contraction Signal Network. Cell Rep. 2020, 33, 108467. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.C.; Nevins, J.R. Signaling network that link cell proliferation and cell fate. J. Biol. Chem. 2002, 277, 11617–11620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alon, U. An Introduction to Systems Biology: Design Principles of Biological Circuits; Chapman and Hall/CRC: New York, NY, USA, 2006; ISBN 9781439837177. [Google Scholar]
- Michaelis, L.; Menten, M.L. Die Kinetik der Invertinwirkung. Biochem. Z. 1913, 49, 333–369. [Google Scholar]
- Murray, J.D. Mathematical Biology I: An Introduction of Interdisciplinary Applied Mathematics; Springer: New York, NY, USA, 2002; pp. 257–271. [Google Scholar]
- Henley, A.S.; Dick, F.A. The retinoblsatoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.-B. Ordinary Differential Equations with Applications, 2nd ed.; National Hua University: Hsinchu, Taiwan, 2013; ISBN 978-981-4452-92-2. [Google Scholar]
- Fine, B.; Rosenberger, G. The Fundamental Theorem of Algebra; Springer: New York, NY, USA, 1997. [Google Scholar]
- Ermentrout, B. Simulating, Analyzing, and Animating Dynamical Systems: A Guide to XPPAUT for Researchers and Students; SIAM: Philadelphia, PA, USA, 2002; ISBN 978-0-89971-506-4. [Google Scholar]
- Shampine, L.F.; Reichdt, M.W. MATLAB ODE Suite. SIAM J. Sci. Comp. 1997, 18, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Dormand, J.R.; Prince, P.J. A family of embedded Runge-Kutta formulae. J. Comp. Appl. Math. 1980, 6, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Juma, V.O. Data-Driven Mathematical Modeling and Simulations of Rho-Myosin Dynamics. Ph.D. Thesis, University of Sussex, Sussex, UK, 2019. [Google Scholar]
- Juma, V.O.; Dehmelt, L.; Portet, S.; Madzvamuse, A. A mathematical analysis of an activator-inhibitor Rho GTPase model. J. Comput. Dyn. 2021, 9, 133. [Google Scholar] [CrossRef]
- Zagkos, L.; Mc Auley, M.; Roberts, J.; Kavallaris, N.I. Mathematical models of DNA methylation dynamics: Implications for health and ageing. J. Theor. Biol. 2019, 462, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-J.; Trame, M.N.; Lesko, L.J.; Schmidt, S. Sobol sensitivity analysis: A tool to guide the development and evaluation of systems phamacology models. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Saltelli, A.; Chan, K.; Scott, E.M. Sensitivity Analysis: Wiley Series in Probability and Statistics; Jon Wiley: Chichester, UK, 2000; ISBN 978-0-470-74382-9. [Google Scholar]
- Hampy, D.M. A review of techniques for parameter sensitivity analysis of environmental models. Environ. Monit. Assess. 1994, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Liu, H.; Hao, J.; Liu, Z. Dynamical behaviors of RB − E2F pathway including negative feedback loops involving miR449. PLoS ONE 2012, 7, e43908. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Value | Units | Reference |
|---|---|---|---|---|
| Growth factors activation rate | 1 | [17] | ||
| Growth factors concentration | varies | U | [42] | |
| Inhibition rate of M | 1.001 | [17] | ||
| Michaelis–Menten constant | 1 | U | [42] | |
| Activation rate of M | 1 | [42] | ||
| Activation of R protein family | 1 | U | [42] | |
| Inhibition rate of R by E | 1 | U | Estimate | |
| Inhibition rate of R by M | 1 | U | Estimate | |
| R baseline inhibition rate | 1 | U | Estimate | |
| Total concentration of R | 5 | U | [40] | |
| E self activation rate | 0.02 | U | [17] | |
| Activation rate E by M | 0.02 | [17] | ||
| R baseline inhibition rate | 1 | U | Estimate | |
| E constitutive activation rate | 0.001 | [40] | ||
| Michaelis–Menten constant | 0.92 | U | [42] | |
| Inhibition rate of E by R | 0.01 | U | [42] | |
| Michaelis–Menten constant | 0.05 | U | [42] | |
| Michelis–Menten constant | 1 | U | [42] | |
| Michaelis–Menten constant | 1 | U | [42] |
| 5% Increase in Parameter | % Change in | % Change in | % Change in |
|---|---|---|---|
| −10.330 | −10.186 | 0.7016 | |
| −2.5797 | −2.6158 | −0.0973 | |
| −2.3573 | −6.6279 | 1.7049 | |
| 1.6213 | 4.5563 | −1.1907 | |
| 1.3212 | 3.7135 | −0.96101 | |
| −1.4816 | −4.1652 | 1.0916 | |
| −3.6855 | −10.361 | 2.78 | |
| 3.6754 | 10.331 | 0.9486 | |
| 0.9689 | 2.7222 | 0.1993 | |
| 1.1586 | 3.2559 | −0.8912 | |
| 0.4682 | 1.3150 | 0.0671 | |
| −2.1313 | −5.9929 | −0.73 16 | |
| −4.4144 | −12.411 | −1.5102 | |
| 0.0008 | 0.00217 | −0.0044 | |
| −0.50358 | −1.4171 | 0.2834 | |
| −1.3792 | −3.8783 | −0.5012 | |
| 8.2181 | 8.6153 | −0.4477 | |
| 4.8355 | 5.0458 | −0.30391 |
| 5% Decrease in Parameter | % Change in | % Change in | % Change in |
|---|---|---|---|
| 15.379 | 16.574 | −0.74247 | |
| 3.0549 | 3.1573 | −0.23624 | |
| 2.7989 | 7.8667 | −1.9380 | |
| −1.5130 | −4.2539 | 1.0604 | |
| −1.3083 | −3.6785 | 0.91636 | |
| 1.4856 | 4.1750 | −1.1118 | |
| 4.6941 | 13.194 | −3.0811 | |
| −3.3974 | −9.5522 | −1.1635 | |
| −0.90100 | −2.5340 | −0.34861 | |
| −1,1643 | −3.2747 | 0.77670 | |
| −0.47058 | −1.3243 | −0.22096 | |
| 2.2750 | 6.3943 | 0.56813 | |
| 5.6528 | 15.889 | 1.4395 | |
| −0.00076 | −2.0707 | 0.00434 | |
| 0.50506 | 1.4196 | −0.37425 | |
| 1.7225 | 4.8407 | 0.41722 | |
| −7.5303 | −7.4886 | 0.46275 | |
| −3.9715 | −4.0111 | 0.25848 |
| 10% Increase in Parameter | % Change in | % Change in | % Change in |
|---|---|---|---|
| −18.792 | −18.005 | 1.5896 | |
| −4.9970 | −5.0238 | 0.25366 | |
| −4.5627 | −12.828 | 3.5291 | |
| 3.5450 | 9.9639 | −2.4070 | |
| 2.7918 | 7.8469 | −1.9470 | |
| −3.1078 | −8.7368 | 2.3944 | |
| −6.9382 | −19.505 | 5.7536 | |
| 8.0605 | 22.659 | 1.9911 | |
| 2.1256 | 5.9736 | 0.52836 | |
| 2.4269 | 6.8214 | −1.7297 | |
| 0.9800 | 2.7539 | 0.2105 | |
| −4.3112 | −12.122 | −1.4863 | |
| −8.2964 | −23.324 | −3.0332 | |
| 0.0017 | 0.0047 | −0.0093 | |
| −1.0545 | −2.9658 | 0.7076 | |
| −2.6078 | −7.3322 | −0.8934 | |
| 18.296 | 19.8 | −0.7814 | |
| 11.815 | 12.677 | −0.6242 |
| 10% Decrease in Parameter | % Change in | % Change in | % Change in |
|---|---|---|---|
| 43.284 | 51.402 | −0.90841 | |
| 6.340764 | 6.6298 | −0.40674 | |
| 5.8216 | 16.364 | −3.7086 | |
| −2.7912 | −7.8475 | 2.0483 | |
| −2.4764 | −6.9631 | 1.7939 | |
| 2.8264 | 7.9439 | −1.9852 | |
| 10.236 | 28.774 | −5.9032 | |
| −6.2295 | −17.514 | −2.2045 | |
| −1.6616 | −4.6717 | −0.56234 | |
| −2.2183 | −6.2377 | 1.5914 | |
| −0.89676 | −2.5224 | −0.34685 | |
| 4.44444 | 12.49 | 1.1354 | |
| 12.394 | 34.839 | 2.8958 | |
| −0.0014110 | −0.0038423 | 0.0081792 | |
| 0.95936 | 2.6969 | −0.73757 | |
| 3.6812 | 10.3 47 | 0.94703 | |
| −13.836 | −13.470 | 1.0361 | |
| −7.0325 | −7.0261 | 0.41178 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mapfumo, K.Z.; Pagan’a, J.C.; Juma, V.O.; Kavallaris, N.I.; Madzvamuse, A. A Model for the Proliferation–Quiescence Transition in Human Cells. Mathematics 2022, 10, 2426. https://doi.org/10.3390/math10142426
Mapfumo KZ, Pagan’a JC, Juma VO, Kavallaris NI, Madzvamuse A. A Model for the Proliferation–Quiescence Transition in Human Cells. Mathematics. 2022; 10(14):2426. https://doi.org/10.3390/math10142426
Chicago/Turabian StyleMapfumo, Kudzanayi Z., Jane C. Pagan’a, Victor Ogesa Juma, Nikos I. Kavallaris, and Anotida Madzvamuse. 2022. "A Model for the Proliferation–Quiescence Transition in Human Cells" Mathematics 10, no. 14: 2426. https://doi.org/10.3390/math10142426
APA StyleMapfumo, K. Z., Pagan’a, J. C., Juma, V. O., Kavallaris, N. I., & Madzvamuse, A. (2022). A Model for the Proliferation–Quiescence Transition in Human Cells. Mathematics, 10(14), 2426. https://doi.org/10.3390/math10142426